

SUMMARY
Inducible HSP70 (HSP70i) chaperones peptides from stressed cells, protecting them from apoptosis. Upon extracellular release, HSP70i serves an adjuvant function, enhancing immune responses to bound peptides. We questioned whether HSP70i differentially protects control and vitiligo melanocytes from stress and subsequent immune responses. We compared expression of HSP70i in skin samples, evaluated the viability of primary vitiligo and control melanocytes exposed to bleaching phenols, and measured secreted HSP70i. We determined whether HSP70i traffics to melanosomes to contact immunogenic proteins by cell fractionation, western blotting, electron microscopy and confocal microscopy. Viability of vitiligo and control melanocytes was equally affected under stress. However, vitiligo melanocytes secreted increased amounts of HSP70i in response to MBEH, corroborating with aberrant HSP70i expression in patient skin. Intracellular HSP70i colocalized with melanosomes, and more so in response to MBEH in vitiligo melanocytes. Thus whereas either agent is cytotoxic to melanocytes, MBEH preferentially induces immune responses to melanocytes.
Keywords: autoimmune, stress, heat shock protein, melanocyte, vitiligo
INTRODUCTION
Vitiligo is a skin disorder characterized by progressive depigmentation. Over 0.5% of the world population is affected by vitiligo, with a 25% increased incidence among women; depigmentation will progress by 1% of body surface area a year on average (Cedercreutz et al., 2010). Expanding lesions are frequently infiltrated by T cells reactive with melanocytes thus supporting an etiologic involvement of cell mediated autoimmunity (van den Wijngaard et al., 2000). Genetic evidence likewise supports a process involving a break in tolerance (Jin et al., 2012), leading to activated T cells trafficking to the skin. However, there is limited concordance between monozygotic twins, suggesting a role for environmental factors in disease precipitation (Spritz, 2010). Indeed, patients appear to develop vitiligo under stressful circumstances (Picardi et al., 2003; Cedercreutz et al., 2010).
Stress associated with psychological as well as mechanical/chemical insults can initiate or accelerate vitiligo (Picardi et al., 2003). Several chemicals induce vitiligo, some of which are used in bleaching creams (Boissy and Manga, 2004). These agents may induce oxidative stress, lending credibility to the observation that antioxidant enzymes such as superoxide dismutase and catechol-O-methyltransferase are overexpressed in vitiligo skin (Le Poole et al., 1994; Sravani et al., 2009). Differential gene expression data further suggest that melanocytes from non-lesional skin of vitiligo patients may be inherently sensitive to stress (Stromberg et al., 2008).
To understand why stress would translate into an immune response specifically targeting melanocytes, it may be important to take note of the natural target antigens that have been uncovered, which predominantly reside in melanosomes. These organelles are uniquely immunogenic (Sakai et al., 1997), and carry lysosome-like properties (Le Poole, 1993a; Orlow, 1995; Raposo et al., 2002). Moreover, the process of melanogenesis within these organelles is associated with generating hydrogen peroxide and free radicals during melanogenesis, providing a direct link to stress-induced disease (Mastore et al., 2005). Thus stress can be a consequence of the very organelles that form a source of melanocyte antigens. Taken together with morphologic abnormalities reported for vitiligo melanocytes also affecting melanosomes (Li et al., 2009; Boissy et al., 1991) it has long been postulated that melanocytes from pigmented vitiligo patient skin selectively succumb to oxidative stress.
Meanwhile, heat shock proteins (HSPs) are prime candidates to mediate the process that translates environmental factors and oxidative stress into an autoimmune disease. Including the inducible isoform of HSP70 (HSP70i), heat shock proteins are upregulated in response to cellular stress, preventing apoptosis by chaperoning proteins to prevent misfolding (Beere and Green, 2001). Our research is focused on HSP70i because of its unique role in vitiligo (Mosenson et al, 2013b). Human HSP70i is also known as HSP70A1A or HSP70A1B, proteins with the same amino acid sequence yet encoded by separate genes with different regulatory regions (Brocchieri et al, 2008). Among stress-induced 70kDa proteins in the cell, HSP70i is generally considered a cytoplasmic protein, similar to HSC70, the constitutively expressed isoform of the protein. To date, 17 proteins of the HSP70 family have been described for human cells to protect cellular proteins and nucleotide sequences in different cellular compartment from degradation in response to stress (Brocchieri et al, 2008). When found in the extracellular milieu, stress proteins serve as an alarm signal to the immune system by activating local antigen presenting cells (APCs) (Kammerer et al., 2002). HSP70i is unique in this regard, as it is reportedly secreted by live cells (Asea, 2007).
Several lines of evidence have since solidified a role for HSP70i in precipitating vitiligo, including the observation that wild-type and vitiligo-prone mice develop accelerated disease in response to HSP70i (Mosenson et al., 2012; Mosenson et al., 2013). It was also determined that mice lacking the HSP70i gene do not depigment, attesting to a requirement for the HSP in disease development (Mosenson et al., 2012). The importance of HSP70i in initiating vitiligo may be attributed to its potent adjuvant and chaperone properties (Zhang et al., 2006). This is supported by prior studies which have shown that HSP70i can chaperone immunogenic peptides derived from melanocyte differentiation antigens (Noessner et al., 2002).
Here we address the hypothesis that disease precipitation in vitiligo follows a process whereby HSP70i is in contact with immunogenic melanosomal peptides and preferentially exported from vitiligo melanocytes under stress, thereby effectively introducing an immune response to melanocytes. We thus exposed primary melanocytes to phenolic agents modeling factors that can precipitate disease, comparing the sensitivity of cells from vitiligo and healthy origin in viability assays. Colocalization of HSP70i and melanosomal proteins was tested by Western blotting of fractionated cells, accompanied by electron microscopic analysis of concentrated fractions. Differential melanosome trafficking of HSP70i in vitiligo melanocytes after treatment with phenolic agents was followed by confocal microscopy using software plugins, while stress protein secretion was measured by ELISA. Taken together, the experiments can help to understand the interplay between melanocytes and the environment to precipitate disease.
RESULTS
HSP70i is aberrantly expressed in vitiligo skin
Vitiligo is initiated and accelerated after stress. Heat shock proteins play a likely role in mediating the stress reponse, particularly in progressively depigmenting skin. Affected skin will overexpress HSP70 as compared to unaffected or control skin samples (Abdou et al, 2013). In said study, the antibodies used cannot distinguish between constitutive and inducible HSP70. As the inducible isoform is crucial for vitiligo (Mosenson et al, 2013), we evaluated expression of this specific isoform in vitiligo skin. We thus probed nonlesional, perilesional and lesional skin sections from four vitiligo patients with the HSP70i specific antibody SPA-811 (Figure 1A). HSP70i expression was observed mainly in the epidermis, with highest expression in the lesional sections. Lesional skin contained intense immunoperoxidase staining of all cells and extracellular matrix, whereas nonlesional skin was mostly devoid of HSP70i expression (Figure 1A). Due to these staining patterns, HSP70i expression was evaluated by blinded subjective quantification of immunoperoxidase intensity. It was determined that HSP70i expression is significantly higher in lesional (P = 0.004) and perilesional (P = 0.048) versus nonlesional skin (Figure 1B). These data support previous findings that HSP70 is overexpressed in vitiligo skin, specifically the inducible isoform.
Figure 1.

HSP70i overexpression in vitiligo skin. (A) Representative immunoperoxidase staining of HSP70i in vitiligo skin displaying expression predominantly located in the epidermis, with minimal cellular expression observed in nonlesional (left), and moderate to strong expression in perilesional (middle) and lesional (right) skin. (B) Subjective, blinded quantification of HSP70i expression in tissue sections. Immunoperoxidase intensity: 1=low, 2=medium, 3=high. Data are presented as mean ± SEM as calculated by Student's t-test. *P< 0.05, **P<0.01, n=7.
Vitiligo and control melanocytes are equally sensitive to stress induced by phenolic agents
Whether melanocytes from non-lesional vitiligo skin are selectively sensitive to bleaching agents is a question that remains unanswered. We propose that vitiligo is an autoimmune disorder mediated by CTL killing of melanocytes rather than by direct cytotoxicity of reactive oxygen species (ROS) that arise from bleaching agent exposure. The data presented in Figure 1 suggested that the overexpression of HSP70i in vitiligo skin may play a protective role in response to stress. Thus, we measured the viability of primary melanocytes harvested from nonlesional vitiligo and from control skin in response to the phenolic compounds 4-tertiary butyl phenol (4-TBP) and monobenzyl ether of hydroquinone (MBEH). The melanocytes were exposed to 125, 250, and 500 μM of 4-TBP or MBEH for 72 hours, and viability was determined by MTT assay (Figure 2). All samples were compared relative to vehicle treated control cells. Treatment with 4-TBP reduced viability by approximately 4, 24, and 69% in response to 125, 250, and 500 μM concentrations of 4-TBP respectively for both control and non-lesional vitiligo skin-derived melanocyte cultures (Figure 2). Similarly, treatment with MBEH reduced viability by approximately 19, 32, and 62% for cells of either origin in response to 125, 250, and 500 μM concentrations, respectively (Figure 2). Our results indicate that melanocyte viability decreases as the concentration of 4-TBP and MBEH increases; however, there were no differences between control and vitiligo melanocytes in response to any of the treatments. These data were performed in duplicate with similar results, and demonstrate that vitiliginous melanocytes are not more resistant or susceptible to direct killing by bleaching agents.
Figure 2.
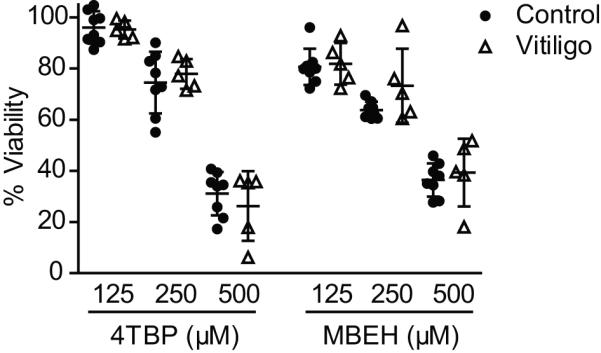
Cell viability of vitiliginous melanocytes. Primary melanocytes from control and vitiligo patients were plated at 10,000 cells per well in triplicate, and exposed to 125, 250, or 500 μM concentration of 4-tertiary butyl phenol (4-TBP) or monobenzyl ether of hydroquinone (MBEH) for 72 hours. Percentage viability was quantified in MTT assays, and compared relative to vehicle treated controls. Cell viability decreased as 4-TBP and MBEH dosage increased; however, no differences were observed between control and vitiligo melanocytes for any treatment. Data provided from two independent experiments with triplicate values for each experiment. Healthy control (n =8), vitiligo (n = 5). Data are presented as mean ± SEM as calculated by Student's t-test.
Intracellular HSP70i colocalizes in part with melanosomal proteins
The immunostaining patterns presented in Figure 1 suggested that HSP70i is uniquely expressed in affected vitiligo tissues. We hypothesize that prior to melanocyte death, HSP70i is found in in part within melanosomes, where it can bind melanocyte antigens and be secreted into the extracellular milieu by stressed melanocytes. To assess HSP70i and melanosomal antigen colocalization, melanocyte homogenates were fractionated by density gradient centrifugation (Figure 3A), and proteins within individual fractions were further separated by SDS-PAGE (Figure 3B). Blotted proteins were probed with the pAb SPA-820 which binds both the constitutive and inducible HSP70 isoforms. Intense immunoperoxidase detection (40.56% mean band intensity) of HSP70 by SPA-820 was observed in the melanosomal fractions (19–24) whereas weak or no HSP70 expression was detected (6.33% mean band intensity) in the non-melanosomal fractions (1–18) (Figure 3C). Fractions 7–9 and 20–22 were pooled, concentrated and processed for transmission electron microscopy (Figure 3B). Electron micrographs of fractions 20–22 support a high concentration of late melanosomes, coinciding with high content of HSP70. In order to delineate the presence of inducible HSP70 amongst these fractions, blotted proteins were probed with the HSP70i specific mAb SPA-810. Again, detection of HSP70i was limited to the melanosomal fractions (19–24; Figure 3D). In addition, the presence of the melanosomal antigen gp100 (Ab HMB45) was detected selectively amongst these fractions (Figure 3D). When comparing the proportion of HSP70i found in melanosomal fractions among vitiligo and control cells, we measured the intensity of HSP70i staining in melanosomal fractions of PIG1 and PIG3V cells as shown in Supplementary Figure 1. When calculated for proportionate numbers of cells loaded onto each gradient, a 1.44-fold higher HSP70i content was observed in melanosomal fractions of PIG3V cells, however, this increase represents a relatively lower fraction of intracellular expression overall when compared to PIG1 cells. These data indicate that HSP70i is in close proximity to melanocyte antigens in control and vitiligo melanocytes.
Figure 3.

HSP70i colocalizes with melanosomal fractions. (A) Adult melanocytes (HM162P7) were dounced and underloaded on an iodixanol gradient for ultracentrifugation. Dense bands containing melanin were observed in the melanosomal (20/21 and 23) fractions. (B, upper image) Collected fractions (non-melanosomal 7–9, and melanosomal 20–22) were fixed and analyzed by electron microscopy (EM). (B, lower image) Collected fractions were run on a Western Blot. SPA-820 antibody (which binds both constitutive and inducible HSP70 isoforms) reacted with the melanosomal (19–24) but not nonmelanosomal fractions (1–15). (C) Quantification of western blot band intensities shown in EM. Mean luminosities indicate stronger antibody reactivity in fractions 19–24 versus 1–18. (D) Western blot analysis of non-melanosomal (1, 2 and 18) and melanosomal (19–23) fractions probed with the antibody SPA-810 which only detects inducible HSP70 (70 kD), and HMB45 to the melanocyte antigen gp100 (HMB45 detects a 45 kD product of gp100).Only the melanosomal fractions (19–23) react with both HSP70i and gp100 antibodies. Purified HSP70i protein was probed as a control. Together these data indicate that HSP70i is localized within the melanosome containing fractions.
Vitiligo melanocytes specifically increase HSP70i colocalizing within the melanosome fractions in response to stress
To further examine the location of HSP70i within cultured melanocytes, fluorescently labeled antibodies to HSP70i (GFP) and TRP-1 (PE/Cy7) were detected by confocal microscopy (Figure 4). Images of 1.0 μm serial Z-slices indicated that both HSP70i and TRP-1 are ubiquitously distributed throughout the cytoplasm, but not in the nucleus (Figure 4A). HSP70i appears to have a punctate pattern directly outside the nucleus, whereas TRP-1 is mainly detected within the dendrites (Figure 4A). Next, primary vitiligo and control melanocytes were exposed to sub-lethal doses (125 μm) of 4-TBP or MBEH for 72 hours. We analyzed the presence of HSP70i (GFP) and the melanocyte antigen gp100 (PE/Cy7). The gp100 reactive antibody HMB45 was selected as it was previously shown to bind a 35 kD fragment of gp100 located predominantly in the melanosome (Harper et al., 2008). The confocal images displayed colocalization of HSP70i and gp100 as shown by Z-slices and merged images (Figure 4B). Interestingly, punctate staining of HSP70i was detected outside the melanocytes suggestive of its secretion (Figure 4B). Extracellular gp100 staining was also visible to a lesser extent, possibly due to degradation or loss of the HMB45 binding epitope (Figure 4B). No extracellular staining was observed in the control slides where no primary antibody was added (Supplementary Figure 2). Colocalization of HSP70i and gp100 was quantified using the ImageJ colocalization plug-in JACoP (Just Another Co-localization Plugin; Bolte and Cordelières, 2006), which calculates the distance between fluorescent pixels (green and red), and also assigns a pseudocolor representing gradation of colocalization (Figure 4B). The number of HSP70i/gp100 colocalized pixels was compared relative to the total number of HSP70i labeled pixels within the cell. When treated with bleaching phenols, but not with vehicle, the amount of HSP70i and gp100 colocalization differed between vitiligo and healthy melanocytes (Figure 4C). Vitiligo melanocytes treated with 4-TBP displayed a ~2-fold increased HSP70i/gp100 colocalization as compared to healthy melanocytes (Fig. 4C: P=0.0439). Importantly, vitiligo melanocytes treated with MBEH demonstrated the greatest colocalization (7.5%) as compared to either vhicle of 4-TBP treatment (Figure 4C; P = 0.0001). Together, these data demonstrate that HSP70i is located in part in close proximity to melanosomal proteins, in particular in vitiligo skin-derived melanocytes after MBEH exposure.
Figure 4.
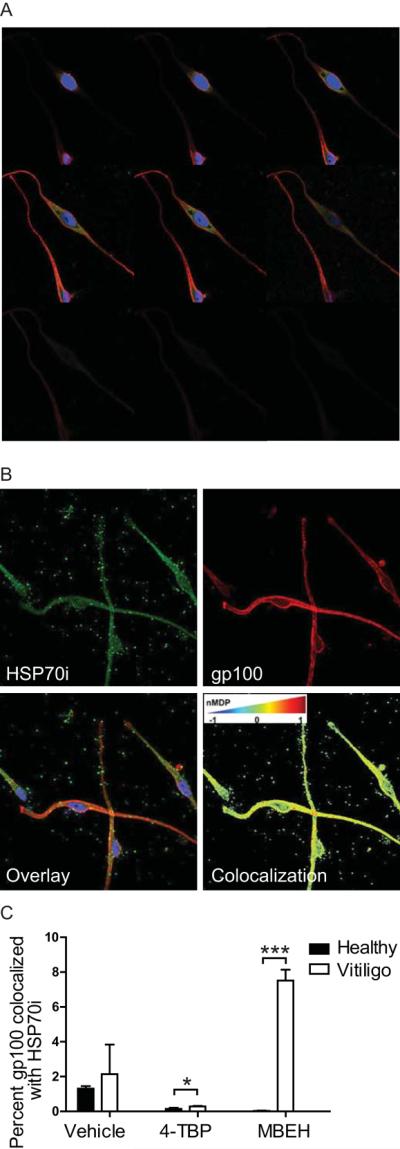
HSP70i is overexpressed in vitiliginous melanosomes after stress. (A) Representative serial 1.0 μM Z-sliced images of neonatal melanocytes (Mf0627P11) indicate cytoplasmic expression of HSP70i (SPA-811 Ab detected by FITC) and the melanocyte antigen TRP-1 (Ta99 Ab detected by PE/Cy7) throughout the cell, but not present in the nucleus (DAPI counterstained). (B) Representative 0.5 μM Z-slice images of neonatal melanocytes (Mf0627P11) probed with antibodies to HSP70i (SPA-811) and gp100 (HMB45). Individual channels for HSP70i (GFP), gp100 (PE/Cy7) and merged images are shown. Note extracellular detection of HSP70i/gp100. An image pseudocolored by the ImageJ plug-in JACoP indicates levels of HSP70i/gp100 colocalization as low (blue), middle (yellow) and high (red). Perinuclear red mapping indicates over-lapping between the two labels, suggestive of colocalization. (C) Vitiligo and neonatal melanocytes were treated with 250 μM bleaching agents (4-TBP and MBEH) for 24 hours followed by confocal microscopy. Five z-slices from representative treated melanocytes were analyzed for HSP70i/gp100 colocalization using JACoP, with an nMDP cutoff of 1 used in the calculations. Data is presented as HSP70i/gp100 colocalization relative to total HSP70i staining. Graphed is the normalized mean deviation production (nMDP) for 5, 1μm Z-slices for each sample. Healthy control (n =3), vitiligo (n = 3). Data are presented as mean ± SEM as calculated by Student's t-test.* P<0.05, ***P< 0.001
HSP70i is detected in melanosomes by electron microscopy
The confocal images presented in Figure 4 supported MBEH-enhanced melanosomal colocalization of HSP70i, however, differences in expression were minimal in melanocytes treated with 4-TBP (Figure 4C). This prompted us to assess HSP70i expression in response to 4-TBP by a more sensitive method. Healthy melanocytes were exposed to 125 μM 4-TBP for four hours, followed by immunostaining with gold particle-conjugated HSP70i antibodies. Detection was performed by transmission electron microscopy (Figure 5). The images display HSP70i (gold particles) throughout the cytoplasm, which are more abundant in the 4-TBP treated melanocytes (Figure 5). Labeling was quantified as described in Materials & Methods. The expression of HSP70i in control and 4-TBP treated melanocytes was 10.1 (± 4) and 25.2 (± 3) gold particles/50×10−6 nm2, respectively (P=0.0002), indicating increased expression of HSP70i overall. A small fraction colocalized with melanosomes as observed under higher magnification (Figure 5, insets). Together with the confocal images, these data demonstrate that part of HSP70i is found within melanosomes, and trafficking to the melanosome can be significantly enhanced in vitiligo melanocytes exposed to MBEH.
Figure 5.

Transmission electron microscope detection of HSP70i in 4-TBP treated melanocytes. Healthy melanocytes (Mf0632P2) were treated with 125 μM 4-TBP for 4 hours followed by immunostaining with gold particle-conjugated antibodies to HSP70i. The melanocytes were next fixed and scanned by transmission electron microscopy. Gold particles (red arrows) are observed in sections probed with anti-HSP70i which are more abundant (P < 0.0002) in 4-TBP treated melanocytes as shown in the bar graph below. HSP70i is seen throughout the cytoplasm in line with data shown in Figure 4, and occasionally juxtapositioned to melanosomes (insets). Bar equals 500 nm. (inset bars equal 40nm).
Primary vitiligo melanocytes secrete more HSP70i in response to MBEH treatment
The above data support that HSP70i is in close proximity to melanocyte antigens in cells under stress. Extracellular HSP70i is known to have adjuvant properties, thus we next compared HSP70i secretion of vitiligo and control melanocytes in response to chemical stress. It was previously published that immortalized vitiligo PIG3V cells secrete 5-fold more HSP70i in response to the bleaching agent 4-TBP compared to immortalized control PIG1 cells (Kroll et al., 2005). As data presented in Figures 4 and 5 demonstrated MBEH-induced trafficking of HSP70i to melanosomes, we likewise investigated the effects of this bleaching agent on HSP70i secretion For a comparison to earlier data, we exposed the same immortalized PIG1 and PIG3V cultures to MBEH. In addition, primary melanocyte cultures from healthy and vitiligo patient skin were exposed to MBEH. The PIG1 and PIG3V cells, as well as primary melanocytes from healthy and vitiligo patients were treated with 125μM of MBEH for 24 hours. Images indicate that exposure to a low dose of MBEH does not induce significant cell death in either the immortalized or primary cell lines (Figure 6A and B), similar to the results presented in Figure 2. The supernatants were then analyzed for HSP70i by high sensitivity ELISA, and measurements were compared relative to untreated samples. The results confirm that PIG1 and PIG3V cells secrete ~ 3-fold increased levels of HSP70i after MBEH treatment, comparable to reported findings using 4-TBP (Kroll et al., 2005). Importantly, PIG3V cells secrete significantly higher levels of HSP70i as compared to PIG1 cells with vehicle (P = 0.0145) and MBEH (P = 0.0053) treatment (Figure 6A). Importantly, primary vitiligo melanocytes secrete ~7-fold increased amounts of HSP70i after MBEH versus vehicle treatment, whereas no detectable levels of HSP70i were secreted by healthy melanocytes (P = 0.0031; Figure 6B). Meanwhile, HSP70i levels were similar in supernatants of primary vitiligo and healthy melanocytes after vehicle treatment (Figure 6B). Taken together, the data reveal that vitiligo melanocytes respond to stress by secreting greater amounts of HSP70i.
Figure 6.

Vitiligo melanocytes secrete more HSP70i in response to MBEH. (A) The immortalized melanocyte lines (vitiligo PIG3VP54 and control PIG1P96; n=3 measurements) and (B) primary healthy and vitiligo melanocytes were treated with 125 um MBEH for 24 hours. Supernatants from treated and untreated cultures were assessed for HSP70i content by high-sensitivity ELISA. The percent increase in HSP70i of MBEH and vehicle treated compared to untreated samples is indicated. Images of cell cultures (A and B) show no differences in cell density or death after MBEH treatment. A significant increase in HSP70i secretion was observed in vitiligo, but not control melanocytes after MBEH treatment. These data indicate that melanocytes obtained from vitiligo skin secrete more HSP70i in response to stress than healthy cells. Control (n =4 individual cultures), vitiligo (n = 3 individual cultures). Data are presented as mean ± SEM as calculated by Student's t-test. **P< 0.01, ***P< 0.001.
DISCUSSION
Controversies remain regarding the etiology of vitiligo. There is support for the contribution of overproduced free radicals in stressed vitiligo melanocytes, as well as for an autoimmune response wherein T cells specifically target melanocytic antigens. The data presented in this manuscript suggest that the above may not represent mutually exclusive mechanisms, but rather aspects of disease that complement each other, joining forces to initiate and perpetuate vitiligo development. This supports the convergence and melanocytorrhagy theories which state that autoimmunity, impaired redox status, and genetic predisposition all contribute to vitiligo development (Le Poole et al., 1993b; Kumar and Parsad, 2012).
It was reported that HSP70 is overexpressed in vitiligo perilesional and lesional skin compared to unaffected areas (Abdou et al, 2013). For that study, an antibody was used that cannot distinguish between constitutive and inducible HSP70. Here we were able to demonstrate that the inducible isoform of HSP70 (HSP70i) is more abundant in affected areas of vitiligo skin. As lesional tissue no longer contains melanocytes, it stands to reason that HSP70i overexpression cannot be limited to melanocytes. Indeed abundant expression is observed throughout the epidermis under stress, likely providing keratinocytes protection from undergoing apoptosis under these circumstances. The selective loss of melanocytes rather than keratinocytes from vitiligo skin may then be ascribed to a combination of relatively high turnover of keratinocytes and the fact that HSP70i can chaperone highly immunogenic melanosomal proteins from pigment cells in perilesional skin, alerting the immune system and recruiting melanocyte reactive T cells to the area. Hence our efforts to locate HSP70i within melanosomes. Indeed any stress to the skin may affect keratinocyte physiology as well, and will thus result in elevated expression of HSP70i. However, immunogenic melanosomal proteins are not epressed by keratinocytes and release of HSP70i has been reported primarily for cells of neuronal heritage (Mambula et al, 2007). Taken together, the data presented in the current manuscript provide several lines of support for the role of HSP70i in guiding melanosomal proteins outside the cell under stress, recruiting a melanocyte reactive immune response. This can ultimately help explain why bleaching phenol exposure can ultimately lead to progressive depigmentation.
Melanocytes obtained from vitiligo patients express intrinsic abnormalities including dilated endoplasmic reticulum profiles and abnormal melanosome compartmentalization (Boissy et al., 1991; Li et al., 2009). Moreover, others have reported that melanocytes from vitiligo patients have reduced calcium uptake, which is important for inhibiting redox activity (Schallreuter-Wood et al., 1996). Such observations suggest that vitiligo melanocytes are more sensitive to stress, and generate hydrogen peroxide and other free radicals, especially during melanogenesis. Increased production of H2O2 may increase the susceptibility of melanocytes to apoptosis/necrosis (Zhang et al., 2013). Both apoptosis and necrosis may be at play in vitiligo as opposing forms of cell death are induced by different phenolic compounds, among which only MBEH is FDA approved to pharmaceutically accelerate depigmentation in vitiligo patients (Hariharan et al, 2011). MBEH is also known to initiate a CTL mediated autoimmune response targeting melanocytes (van den Boorn et al, 2011). Although a preliminary study suggested that vitiligo melanocytes may be more sensitive to 4-TBP exposure than control cells, the most important data from that study is the restoration of cell viability in presence of catalase (Manga et al., 2006). Meanwhile, the sensitivity of cells to bleaching phenols is affected by donor pigmentation, donor age and culture conditions, and a comparison among multiple cultures is required to identify overall differences in viability upon exposure.
In our manuscript, we have chosen to include 2 model agents with known relevance to the vitiligo disease process in vivo, namely 4-TBP and MBEH. The current studies address the changes in HSP70i metabolism, required to provide adjuvant effects supporting melanocyte-reactive immune responses elicited by MBEH. Although any necrotic agent is expected to cause the same effects on HSP70i metabolism in cultured vitiligo melanocytes we have observed in response to MBEH, differences in subsequent depigmentation should be sought in (1) the ability of different necrotic agents to penetrate the skin in vivo to access melanocytes, as well as (2) differential consequences for the melanogenic pathway as in: conversion of phenolic bleaching agents within melanosomes to quinones that haptenize melanogenic enzymes to generate neoantigens, and overexpression of melanogenic proteins, both incentives for a melanocyte-reactive specific immune response.
In our current study, the data indicate that cell viability of vitiligo melanocytes is not different from that of healthy melanocytes in response to bleaching agents. It follows that melanocytes from vitiligo patients are not more likely to die directly from exposure to 4-TBP or MBEH than control melanocytes, however, they do secrete a greater amount of HSP70i under those conditions. Thus, bleaching agent-exposed vitiligo melanocytes are more likely to mount an immune response directed to the antigenic proteins chaperoned by HSP70i.
Patient surveys indicate that vitiligo is initiated and accelerated after stress, with ~50% of patients reporting a Koebner phenomenon (Cedercreutz et al., 2010). Heat shock proteins are produced in response to stress, and function to re-fold damaged proteins to prevent cell death (Beere and Greene, 2001). Given the proposed role of HSP70i as an instigator of immune responses in vitiligo (Mosenson et al., 2013), here we determined that inducible HSP70 is overexpressed in affected vitiligo tissue. This corresponds with our recently published data that HSP70i is a critical component in vitiligo development (Denman et al., 2008; Mosenson et al., 2012). Besides activating dendritic cells (DCs) to become lytic towards tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expressing melanocytes in human vitiligo, it was also shown that HSP70i overexpression can enhance vitiligo development in mice (Kroll et al., 2005; Mosenson et al., 2012). Moreover, a central role for HSP70i was determined by demonstrating that mice knockout for inducible Hsp70 are resistant to depigmentation after vaccination with melanocyte antigens (Mosenson et al., 2012).
We previously reported that vitiliginous melanocytes overproduce HSP70i in response to 4-TBP induced stress (Kroll et al., 2005). This appears counterintuitive since HSPs protect cells from apoptosis under stress. However, extracellular HSP70i serves as an “alarm signal” to activate immune responses (Kammerer et al., 2002). This illustrates the importance of HSP70i in immune activation as it is the primary HSP secreted by live cells of neuronal origin (Asea, 2007). Based on prior work we thus interpret our data to mean that immune targeting follows stress to melanocytes as a factor in vitiligo, in a process mediated by HSP70i secretion. We found that HSP70i would be differentially secreted by MBEH treated control and vitiliginous melanocytes. Using a high sensitivity ELISA we demonstrated that immortalized PIG3V vitiligo melanocytes show relatively increased HSP70i secretion compared to PIG1 immortalized normal melanocytes upon MBEH exposure, similar to data previously reported using 4-TBP (Kroll et al., 2005). Importantly, we also established that HSP70i is more abundantly secreted by primary vitiliginous melanocytes. These data suggest that vitiliginous melanocytes respond to stress by overproduction/secretion of HSP70i rather than by direct cell death. We propose that the increased HSP70i secretion by vitiligo melanocytes is subsequently followed by selective immune activation and recruitment of melanocyte-reactive cytotoxic T lymphocytes (CTLs), which ultimately contribute to vitiligo development.
HSP70i binds proteins/peptides to serve many functions including protein folding, trafficking, and potentially MHCI/II loading (Srivastava, 2002). Within melanocytes, melanosomal proteins such as TRP-1 and gp100 are transported from endosomes to lysosomes by HSP70i to be transferred to LAMP-2a proteins for internalization (Terlecky, 1994; Agarraberes and Dice, 2001). Intriguingly, within vitiligo skin melanocytes express MHCII which is normally reserved to APCs (Le Poole et al., 1993c). Thus, melanocytes under stress may upregulate expression of HSP70i which cross-presents melanocyte-antigens to MHCII as has been observed in other cell types (Srivastava, 2002).
Besides these intracellular immune functions, HSP70i residing in melanosomes may also be externalized during melanosome transfer, potentially affecting antigen uptake, processing and presentation by DCs. Secretion of HSP70i by live cells was suggested to involve purging of lysosomal contents and proceed via formation of exosomes, leaving the cells as secretory-like granules (Evdonin et al., 2006). In melanoma, under minimal stress, low levels of HSP70i are transported to the membrane via endosomes, whereas under stress, elevated levels of expression lead to re-routing of HSP70i into lysosomes for subsequent secretion (Juhasz et al., 2013). Extracellular HSP70i can induce DCs to more actively phagocytize and cross-present antigen, and activate responder T cells (Srivastava, 2002), thus the peptides bound to HSP70i may invoke a targeted immune response. Because of these adjuvant properties, HSP70i has been used in melanoma vaccines (Zhang et al., 2006).
Stress proteins isolated from melanoma tumors will chaperone melanocyte-specific peptides critical for directing a targeted response towards these antigens (Noessner et al., 2002). This antigen-bound HSP70i can activate DCs leading to production and expansion of tumor reactive T cells. Not surprisingly, T cells isolated from both melanoma and vitiligo patients recognize many shared melanocyte antigens. Confocal microscopic analysis shows that a fraction of intracellular HSP70i is associated with melanosomes. The same is shown by electron microscopic and Western blot analysis of fractionated cells. This follows prior observations that constitutive HSP70 is responsible for shuttling lysosomal content across the organelle membrane (Agarraberes and Dice, 2001). Given the similarity in function of melanosomes within melanocytes and lysosomes in other cells (Orlow, 1995; Le Poole et al., 1993a; Raposo et al., 2002), a shuttle function for this other isoform may explain the melanosomal association and its observed increase under stress.
Intriguingly, melanocytes in perilesional vitiligo skin express MHCII (Le Poole 1993). As melanosomes are physiologically similar to lysosomes, which process antigens for presentation in the context of MHCII (Le Poole et al, 1993a; Orlow, 1995; Raposo et al, 2002). Furthermore, HSP70i can bind melanocyte antigens (Noessner et al., 2002), and can deliver antigens for presentation in the context of MHCII (Srivastava, 2002). Interestingly, we showed that HSP70i colocalizes with melanosomes to a greater extent after MBEH exposure. Thus stress likely induces trafficking of HSP70i to the melanosome where it may assist in protein folding, or antigen loading (Chi et al., 2006). This is coherent with the observation that a melanosome transport signal enhances immunogenicity of intracellular proteins (Wang et al., 1999). Greater colocalization occurred in response to MBEH, whereas 4-TBP gave little to no effect, supporting the very different responses of melanocytes to either form of stress (Hariharan et al., 2011). This is also in line with evidence that MBEH and 4-TBP induce melanocyte cell death by different mechanisms (Hariharan et al., 2011).
Overall, our data indicate that vitiliginous melanocytes are indirectly more sensitive to MBEH-induced stress through increased secretion of HSP70i and subsequent immune activation. MBEH and 4-TBP induce ROS (van den Boorn et al., 2011; Manga et al., 2006), which will in turn contribute to HSP70i production (Kim et al., 2005). Our data provide support for the concept that upregulated HSP70i then traffics in part to melanosomes where it can bind antigenic peptides (Wang et al., 1999; Noessner et al, 2002). HSP70i secretion likely involves an exosomal mechanism (Lancaster and Febbraio, 2005) that involves melanosome occlusions (Ando et al., 2012). Within the cells, HSP70i can contribute to MHC antigen loading and membrane transport (DeNagel and Pierce, 1992; Castelli et al., 2001). Upon secretion, the melanocyte-antigen bound HSP70i can activate DCs leading to targeted killing of melanocytes. Our data provide a mechanism by which stress differentially induces overexpression and melanosome localization of HSP70i within vitiligo melanocytes, leading to enhanced secretion of HSP70i-antigen complexes, subsequent DC activation and T cell recruitment. Thus stress may selectively initiate an autoimmune response in vitiligo patients which leads to melanocyte cytotoxic T lymphocyte killing.
MATERIALS AND METHODS
Preparation of bleaching agents
4-tertiary butyl phenol 4-TBP (Sigma, St. Louis, Missouri, USA) was prepared as a stock solution of 250 mmol/l in 70% ethanol and diluted to a working concentration of 250 μmol/l. Monobenzyl ether of hydroquinone MBEH (Sigma) was dissolved in 20% dimethyl sulfoxide (Sigma) and mixed with 70% ethanol for a stock concentration of 250 mmol/l. 4-TBP and MBEH were diluted with media to the listed working concentrations.
Cell Culture
For all experiments using primary melanocytes from control or from non-lesional vitiligo skin, cells were harvested from skin incubated overnight in (0.1 mg/ml trypsin (Mediatech, Manassas, VA, USA), 100 IU/ml penicillin,100 mg/ml streptomycin, 100 mg/ml amphotericin (Mediatech) prepared in Dulbecco's Phosphate Buffered Solution (DPBS; Mediatech). Melanocytes were maintained in melanocyte medium Ham's F-12 medium (Mediatech) with 2 mmol/l glutamine (Mediatech), 100 IU/ml penicillin,100 mg/ml streptomycin,100 mg/ml amphotericin (Mediatech), 0.1 mmol/l isobutylmethylxanthine (Sigma, St. Louis, Missouri, USA), 10 ng/ml 12-O-tetradecanoyl phorbol-13-acetate (Sigma), and 1% Ultroser G serum substitute (PALL Life Sciences, Port Washington, NY, USA). Informed consent was obtained from patients and all samples were obtained with approval from the Institutional Review Board at Loyola University, adopting the principles described in the Declaration of Helsinki. The source of cell cultures is further described in supplemental Tables 1 and 2.
Table 1.
Control melanocytes
| Name | Color | Age | Source | comments |
|---|---|---|---|---|
| HEMa-LP #5C061 | I | adult | Not provided | Commercial source |
| HEMa-LP #1183797 | I | adult | Not provided | Commercial source |
| Mf0862 | I | 0 | Foreskin | |
| Mf0880 | I | 0 | Foreskin | |
| Mf0885 | I | 0 | Foreskin | |
| Mf0887 | I | 0 | Foreskin | |
| Mf12387 | I | 0 | Foreskin | |
| Mf1005 | II | 0 | Foreskin | |
| Mf11234 | II | 0 | Foreskin | |
| PIG1 | II | 0 | Foreskin | Immortalized cell line |
| HM162 | III | 0 | Foreskin | |
| Mf0627 | III | 8 | Foreskin | |
| Mf0632 | III | 0 | Foreskin | |
| Mf0644 | III | 0 | Foreskin | |
| Mf0865 | III | 0 | Foreskin | |
| Mu09236 | III | 14 | Unknown | |
| Mf12385 | III | 0 | Foreskin |
Table 2.
Vitiligo melanocyte cultures from non-lesional skin
| Name | Color | Age | Source | Gender | Disease | Activity | Duration | Comments |
|---|---|---|---|---|---|---|---|---|
| PIG3V | I | 34 | Unknown | female | 5% | regressing | 8 y | Immortalized cell line. UV-A and cortisone treatment |
| Mu08114 | I | 23 | Unknown | female | 2% | progressing | 8 y | No treatment |
| Ma13090 | I | 53 | Shoulder | male | 5% | stable | 18 y | No treatment |
| Mc1043 | II | 33 | Scalp | female | 3% | stable | 10 y | MNF |
| Mc1044 | II | 22 | Scalp | female | 3% | active | 3 y | Tacrolimus and excimer laser treatment |
| Mc1054 | II | 35 | Scalp | female | 15% | active | 3 y | Tacrolimus and excimer laser treatment |
| Mc10103 | II | 26 | Scalp | female | 2% | active | 19 | UVB-narrowband treatment |
| Mp12134 | III | 44 | Ankle | male | 0.1% | stable | 11 y | protopic |
Immunohistology
Four-millimeter punch biopsies were obtained from non-vitiligo control patients, or non-lesional and lesional areas of actively progressing vitiligo in consenting patients attending the Loyola Dermatology outpatient clinic. The skin samples were snap-frozen in OCT compound (Sakura Finetek, Torrence, CA, USA). Cryostat sections (8 mm) were fixed in cold acetone, and indirect immunoperoxidase staining procedures were performed essentially as described previously (Le Poole et al., 1993b). Briefly, tissue sections were treated with Super Block (ScyTek, West Logan, UT, USA) to prevent nonspecific antibody binding, and incubated with primary antibodies SPA-810 to human HSP70i (mouse monoclonal; Enzo Life Sciences formerly Stressgen, Farmingdale, NY, USA), followed by horseradish peroxidase–conjugated secondary antibodies (goat anti-mouse IgG1; Southern Biotech, Birmingham, AL, USA). Enzymatic detection was finalized with aminoethylcarbazole as a substrate (Sigma). Images were imported using Adobe Photoshop software (Adobe Systems Inc., San Jose, CA, USA). Immunohistochemical analysis was performed at least in triplicate for each staining and on each tissue sample, on at least three sections. Actual sample sizes are listed in the figure legends.
Melanocyte Fractions
HM162P7 melanocytes were dounced with a tight pestle (pestle `A'; Pierce, Rockford, IL, USA) on ice in hypotonic buffer presence of antipain, leupeptin, phosphoramidon, pefabloc and aprotinin before underlayering a 5–30% iodixanol gradient then spun for 18 hrs at 29,000 rpm. Resulting fractions were collected from the top. The same procedure was applied to immortalized PIG1P59 and PIG3VP24 melanocytes, loading homogenates from 9×106 and 7×106 cells, respectively, onto 12.5 ml gradients.
Western Blotting
Cell lysates were obtained from healthy melanocytes (HM162P7) and also immortalized cell lines PIG1P51 and PIG3VP24; P stands for passage) and protein content was measured using Bio-Rad Protein Assay reagent (Bio-Rad, Hercules, CA, USA), for equal loading at 20 μl for cellular proteins, or 5 μg of purified HSP70 per slot onto a 10% polyacrylamide minigel. After electrophoresis, separated proteins were transferred on to Immobilon-P membrane (Millipore, Billerica, MA, USA). The blots were incubated with SPA-820 to constitutive and inducible HSP70 (mouse monoclonal IgG1; Enzo Life Sciences). In a separate experiment blots were incubated with SPA-811 to inducible HSP70 (polyclonal rabbit; Enzo Life Sciences), and HMB45 to gp100 (monoclonal mouse IgG1; Dako,Carpinteria, CA, USA). After washing, blots were reacted with horseradish peroxidase labeled goat anti-mouse antiserum; Dako) or alkaline phosphatase labeled goat anti-rabbit antiserum Dako) conjugated secondary Abs. Blots were developed with aminoethylcarbazole (Sigma) as the substrate.
Electron Microscopy
After cell fractionation, HM162P7 fractions (7–9 and 20–22) were pooled then centrifuged at 10,000rpm for 10 minutes to pellet the cellular material. Gradient material was aspirated off and the cellular material re-suspended in 500ul of half-strength Karnovski's fixative for 30 minutes at room temperature. Material was re-centrifuged, fixative aspirated, 1 ml of 1% Agar in sodium cacodylate buffer added (Agar, low melting, SIGMA cat#A-5030) and refrigerated overnight. The Agar gelatin cast containing the pellet was processed in EPON 812 (Ted Pella, Inc., Redding, CA) by routine procedure (Boissy et al., 1991). For immunocytochemistry, Mf0632P2 melanocytes were cultured in Tissue-Tek chamber slides and treated with 4-TBP as previously described. Cells were then washed, fixed with half-strength Karnovski's fixative, and processed in EPON 812 (Ted Pella, Inc.) by routine procedure. Sections were obtained using a RMC-MT6000XL ultramicrotome and stained with uranyl acetate and lead citrate. For immunocytochemistry, sections were washed, treated with mouse anti-HSP70i at 1:5, wash, treated with goat anti-mouse antibody conjugated to 5 nm gold particles (Ted Pella, Inc.) at 1:10, and counterfixed and stained by routine procedures. All sections were viewed, and selected images were digitally photographed using a JEOL JEM-1230 transmission electron microscope. Gold labeling was quantified by counting particles per 50 × 10−6 nm2 in eight melanocytes per group and data analyzed by paired Student's t-test.
Confocal microscopy
Melanocytes isolated from healthy neonatal foreskin, or adult non-lesional vitiligo skin were plated at 10,000 cells per well on 8 well gelatin coated Lab-Tek chamber slides (Thermo Fisher Scientific, Rochester, NY, USA). Cells were maintained in melanocyte medium as described above. Cells were treated for 24 hours with 250 μM of either 4-TBP, MBEH, or untreated. The cells were fixed with with 3.7% formaldehyde (Polysciences) in 0.1 M PIPES, pH 6.8 [piperazine-N, N'-bis (2-ethanesulfonic acid)] (Sigma). Polyclonal rabbit anti HSP70i (SPA-811; Enzo Life Sciences) and monoclonal mouse anti-gp100 (HMB45; Dako) were incubated in 0.3% saponin (Sigma) in PBS. After washing, fluorescent staining was performed with FITC conjugated goat anti-rabbit, and PE/Cy7 goat anti-mouse secondary antibodies (Southern Biotech). Confocal images were captured using a Zeiss LSM 510 scanning electron microscope (Zeiss, Maple Grove, MN, USA). Image analysis was performed using Adobe Photoshop (Adobe Systems Inc.) and ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA) software. For visualizing colocalization, the following channels were used: FITC as green, and PE/Cy7 as red. Colocalization was quantified with the ImageJ plug-in JACoP (Just Another Co-localization Plugin; Bolte and Cordelières, 2006). Primary melanocytes: healthy neonatal (Mf0887P7; Mf0627P4; Mf11234P1) and adult vitiligo (Mu0885P11; Mc1044P11; Mu0885P4).
Cytoxicity assay
Cell viability was measured for both control and vitiligo skin-derived melanocyte cultures by 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assays (Bioassay system, Hayward, California, USA) according to the manufacturer's instructions. Briefly, 20,000 cells/well were plated in triplicate wells of a 96-well plate to attach overnight. Cells were either treated with vehicle alone or with 125, 250, or 500 μmol/l 4-TBP or MBEH for 72 h. MTT reagent (tetrazole) was added to the cells and incubated in a 37°C humidified chamber for 4 h. Tetrazole is converted to formazan in the mitochondria of living cells. The formazan crystals formed were solubilized in buffer (PBS) and the wavelength was read in a reader at 562 nm (BMG Labtech, Inc., Durham, North Carolina, USA). Cell viability was calculated as absorbance in % of the vehicle-treated control. Primary melanocytes included were healthy neonatal (Mf0887P2; Mf0627P6; Mf0644P1; Mf0865P4; Mf0862P2; Mf1005P5; Mf0880P2), healthy adult (Mu09236P3), and adult vitiligo (Mc10103P4; Mc1054P2; Mc1043P2; Mu08114P7; Mc1044 P4).
HSP70i ELISA
Melanocytes from vitiligo and non-vitiligo donors were plated in triplicate at 20,000 cells per well, and the immortalized cell lines PIG1 and PIG3V were plated in triplicate at 10,000 cells per well in a 96 well flat-bottom plate (Falcon). Cells were treated for 24 hours with either 125 μmol MBEH (prepared as above), vehicle (20% dimethyl sulfoxide diluted in 70% EtOH, and diluted to 1:1000 in melanocyte medium), or medium alone. All cells were maintained in a total volume of 150 μl. After 24 hours, cells were imaged, and 100 μl of supernatant was added to an HSP70i high sensitivity ELISA kit (Enzo Life Sciences) and HSP70i was detected according to manufactures instructions. Primary melanocytes: healthy adult melanocytes from Invitrogen (HEMa-LP C024-5C; lot# 5C061P6; lot# 1183797P3; Life Technologies, Carlsbad, CA, USA), healthy neonatal (Mf12385P5; Mf12387P5) and adult vitiligo (Mc1044P6; Ma13090P1; Mp12134P4). Immortalized cell lines: healthy PIG1P96 and vitiligo PIG3VP54.
Statistical Analyses
All data were presented as mean ± SEM unless otherwise indicated. Numeric data were analyzed for statistical significance using Student's unpaired one-tailed t-test with PRISM software (Graphpad, La Jolla, CA, USA). P values of <0.05 were considered statistically significant.
Supplementary Material
SIGNIFICANCE.
In response to bleaching phenols and other forms of stress, a subpopulation of individuals will develop vitiligo. It has been postulated that vitiligo melanocytes are intrinsically sensitive to stress. Here we turn our attention to stress proteins to help explain the differential responses of patient melanocytes under duress. We postulate that vitiligo melanocytes contribute to autoimmunity under stress by secretion of immune activating HSP70i chaperoning melanocyte antigens. Taken together, our data show that vitiligo melanocyte viability is not differentially affected by stress, yet preferential melanosome colocalization and HSP70i oversecretion can explain the autoimmune response that follows in vitiligo patients.
Acknowledgments
We thank the vitiligo and non-vitiligo patients who contributed tissue samples; Linda Fox (Loyola, IL) for her skillful assistance with confocal microscopy. These studies were supported by NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases grant RO1AR054749 to I.C.L.P.
REFERENCES
- Abdou AG, Maraee AH, Reyad W. Immunohistochemical expression of heat shock protein 70 in vitiligo. Ann Diagn Pathol. 2013;17:245–249. doi: 10.1016/j.anndiagpath.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001;114:2491–2499. doi: 10.1242/jcs.114.13.2491. [DOI] [PubMed] [Google Scholar]
- Ando H, Niki Y, Ito M, Akiyama K, Matsui MS, Yarosh DB, Ichihashi M. Melanosomes are transferred from melanocytes to keratinocytes through the processes of packaging, release, uptake, and dispersion. J Invest Dermatol. 2012;132:1222–1229. doi: 10.1038/jid.2011.413. [DOI] [PubMed] [Google Scholar]
- Asea A. Mechanisms of HSP72 release. J Biosci. 2007;32:579–584. doi: 10.1007/s12038-007-0057-5. [DOI] [PubMed] [Google Scholar]
- Beere HM, Green DR. Stress management - heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol. 2001;11:6–10. doi: 10.1016/s0962-8924(00)01874-2. [DOI] [PubMed] [Google Scholar]
- Boissy RE, Liu YY, Medrano EE, Nordlund JJ. Structural aberration of the rough endoplasmic reticulum and melanosome compartmentalization in long-term cultures of melanocytes from vitiligo patients. J Invest Dermatol. 1991;97:395–404. doi: 10.1111/1523-1747.ep12480976. [DOI] [PubMed] [Google Scholar]
- Boissy RE, Manga P. On the etiology of contact/occupational vitiligo. Pigment Cell Res. 2004;17:208–214. doi: 10.1111/j.1600-0749.2004.00130.x. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Brocchieri L, Conway de Macario E, Macario AJ. Hsp70 genes in the human genome: Conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC Evol Biol. 2008;8:19. doi: 10.1186/1471-2148-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli C, Ciupitu AM, Rini F, Rivoltini L, Mazzocchi A, Kiessling R, Parmiani G. Human heat shock protein 70 peptide complexes specifically activate antimelanoma T cells. Cancer Res. 2001;61:222–227. [PubMed] [Google Scholar]
- Cedercreutz K, Denman CJ, Klarquist J, Vaitla R, Boissy RE, Westerhof W, Hernandez C, Le Poole IC. Vitiligo etiology and treatment: Parameters derived from a patient survey. J Dermatol Nurses Assoc. 2010;6:265–272. [Google Scholar]
- Chi A, et al. Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. J Proteome Res. 2006;5:3135–3144. doi: 10.1021/pr060363j. [DOI] [PubMed] [Google Scholar]
- DeNagel DC, Pierce SK. A case for chaperones in antigen processing. Immunol Today. 1992;13:86–89. doi: 10.1016/0167-5699(92)90147-Y. [DOI] [PubMed] [Google Scholar]
- Denman CJ, McCracken J, Hariharan V, Klarquist J, Oyarbide-Valencia K, Guevara-Patino JA, Le Poole IC. HSP70i accelerates depigmentation in a mouse model of autoimmune vitiligo. J Invest Dermatol. 2008;128:2041–2048. doi: 10.1038/jid.2008.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evdonin AL, Martynova MG, Bystrova OA, Guzhova IV, Margulis BA, Medvedeva ND. The release of Hsp70 from A431 carcinoma cells is mediated by secretory-like granules. Eur J Cell Biol. 2006;85:443–455. doi: 10.1016/j.ejcb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Hariharan V, Toole T, Klarquist J, Mosenson J, Longley BJ, Le Poole IC. Topical application of bleaching phenols; in-vivo studies and mechanism of action relevant to melanoma treatment. Melanoma Res. 2011;21:115–126. doi: 10.1097/CMR.0b013e328343f542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper DC, Theos AC, Herman KE, Tenza D, Raposo G, Marks MS. Premelanosome amyloid-like fibrils are composed of only golgi-processed forms of Pmel17 that have been proteolytically processed in endosomes. J Biol Chem. 2008;283:2307–2322. doi: 10.1074/jbc.M708007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44:676–680. doi: 10.1038/ng.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz K, Thuenauer R, Spachinger A, Duda E, Horvath I, Vigh L, Sonnleitner A, Balogi Z. Lysosomal rerouting of Hsp70 trafficking as a potential immune activating tool for targeting melanoma. Curr Pharm Des. 2013;19:430–440. doi: 10.2174/138161213804143644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer R, Stober D, Riedl P, Oehninger C, Schirmbeck R, Reimann J. Noncovalent association with stress protein facilitates cross-priming of CD8+ T cells to tumor cell antigens by dendritic cells. J Immunol. 2002;168:108–117. doi: 10.4049/jimmunol.168.1.108. [DOI] [PubMed] [Google Scholar]
- Kim YH, Park EJ, Han ST, Park JW, Kwon TK. Arsenic trioxide induces Hsp70 expression via reactive oxygen species and JNK pathway in MDA231 cells. Life Sci. 2005;77:2783–2793. doi: 10.1016/j.lfs.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Kroll TM, Bommiasamy H, Boissy RE, Hernandez C, Nickoloff BJ, Mestril R, Le Poole IC. 4-tertiary butyl phenol exposure sensitizes human melanocytes to dendritic cell-mediated killing: Relevance to vitiligo. J Invest Dermatol. 2005;124:798–806. doi: 10.1111/j.0022-202X.2005.23653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Parsad D. Melanocytorrhagy and apoptosis in vitiligo: Connecting jigsaw pieces. Indian J Dermatol Venereol Leprol. 2012;78:19–23. doi: 10.4103/0378-6323.90942. [DOI] [PubMed] [Google Scholar]
- Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: A novel secretory pathway for cellular stress proteins. J Biol Chem. 2005;280:23349–23355. doi: 10.1074/jbc.M502017200. [DOI] [PubMed] [Google Scholar]
- Le Poole IC, van den Wijngaard RM, Westerhof W, Verkruisen RP, Dutrieux RP, Dingemans KP, Das PK. Phagocytosis by normal human melanocytes in vitro. Exp Cell Res. 1993a;205:388–395. doi: 10.1006/excr.1993.1102. [DOI] [PubMed] [Google Scholar]
- Le Poole IC, van den Wijngaard RM, Westerhof W, Dutrieux RP, Das PK. Presence or absence of melanocytes in vitiligo lesions: An immunohistochemical investigation. J Invest Dermatol. 1993b;100:816–822. doi: 10.1111/1523-1747.ep12476645. [DOI] [PubMed] [Google Scholar]
- Le Poole IC, Mutis T, van den Wijngaard RM, Westerhof W, Ottenhoff T, de Vries RR, Das PK. A novel, antigen-presenting function of melanocytes and its possible relationship to hypopigmentary disorders. J Immunol. 1993c;151:7284–7292. [PubMed] [Google Scholar]
- Le Poole IC, van den Wijngaard RM, Smit NP, Oosting J, Westerhof W, Pavel S. Catechol-O-methyltransferase in vitiligo. Arch Dermatol Res. 1994;286:81–86. doi: 10.1007/BF00370732. [DOI] [PubMed] [Google Scholar]
- Li Y, Chen F, Lin F, Guan C, Wei X, Wan Y, Xu A. VIT1/FBXO11 knockdown induces morphological alterations and apoptosis in B10BR mouse melanocytes. Int J Mol Med. 2009;23:673–678. doi: 10.3892/ijmm_00000179. [DOI] [PubMed] [Google Scholar]
- Manga P, Sheyn D, Yang F, Sarangarajan R, Boissy RE. A role for tyrosinase-related protein 1 in 4-tert-butylphenol-induced toxicity in melanocytes: Implications for vitiligo. Am J Pathol. 2006;169:1652–1662. doi: 10.2353/ajpath.2006.050769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastore M, Kohler L, Nappi AJ. Production and utilization of hydrogen peroxide associated with melanogenesis and tyrosinase-mediated oxidations of DOPA and dopamine. Febs j. 2005;272:2407–2415. doi: 10.1111/j.1742-4658.2005.04661.x. [DOI] [PubMed] [Google Scholar]
- Mosenson JA, et al. Mutant HSP70 reverses autoimmune depigmentation in vitiligo. Sci Transl Med. 2013;5:174ra28. doi: 10.1126/scitranslmed.3005127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosenson JA, Zloza A, Klarquist J, Barfuss AJ, Guevara-Patino JA, Poole IC. HSP70i is a critical component of the immune response leading to vitiligo. Pigment Cell Melanoma Res. 2012;25:88–98. doi: 10.1111/j.1755-148X.2011.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosenson JA, Eby JM, Hernandez C, Le Poole IC. A central role for inducible heat shock protein 70 in autoimmune vitiligo. Exp Dermatol. 2013 Sep;22(9):566–9. doi: 10.1111/exd.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noessner E, et al. Tumor-derived heat shock protein 70 peptide complexes are cross-presented by human dendritic cells. J Immunol. 2002;169:5424–5432. doi: 10.4049/jimmunol.169.10.5424. [DOI] [PubMed] [Google Scholar]
- Orlow SJ. Melanosomes are specialized members of the lysosomal lineage of organelles. J Invest Dermatol. 1995;105:3–7. doi: 10.1111/1523-1747.ep12312291. [DOI] [PubMed] [Google Scholar]
- Picardi A, Pasquini P, Cattaruzza MS, Gaetano P, Melchi CF, Baliva G, Camaioni D, Tiago A, Abeni D, Biondi M. Stressful life events, social support, attachment security and alexithymia in vitiligo. A case-control study. Psychother Psychosom. 2003;72:150–158. doi: 10.1159/000069731. [DOI] [PubMed] [Google Scholar]
- Raposo G, Fevrier B, Stoorvogel W, Marks MS. Lysosome-related organelles: A view from immunity and pigmentation. Cell Struct Funct. 2002;27:443–456. doi: 10.1247/csf.27.443. [DOI] [PubMed] [Google Scholar]
- Sakai C, Kawakami Y, Law LW, Furumura M, Hearing VJ., Jr Melanosomal proteins as melanoma-specific immune targets. Melanoma Res. 1997;7:83–95. doi: 10.1097/00008390-199704000-00001. [DOI] [PubMed] [Google Scholar]
- Schallreuter-Wood KU, Pittelkow MR, Swanson NN. Defective calcium transport in vitiliginous melanocytes. Arch Dermatol Res. 1996;288:11–13. doi: 10.1007/BF02505036. [DOI] [PubMed] [Google Scholar]
- Spritz RA. The genetics of generalized vitiligo: Autoimmune pathways and an inverse relationship with malignant melanoma. Genome Med. 2010;2:78. doi: 10.1186/gm199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sravani PV, Babu NK, Gopal KV, Rao GR, Rao AR, Moorthy B, Rao TR. Determination of oxidative stress in vitiligo by measuring superoxide dismutase and catalase levels in vitiliginous and non-vitiliginous skin. Indian J Dermatol Venereol Leprol. 2009;75:268–271. doi: 10.4103/0378-6323.48427. [DOI] [PubMed] [Google Scholar]
- Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: Chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- Stromberg S, Bjorklund MG, Asplund A, Rimini R, Lundeberg J, Nilsson P, Ponten F, Olsson MJ. Transcriptional profiling of melanocytes from patients with vitiligo vulgaris. Pigment Cell Melanoma Res. 2008;21:162–171. doi: 10.1111/j.1755-148X.2007.00429.x. [DOI] [PubMed] [Google Scholar]
- Terlecky SR. Hsp70s and lysosomal proteolysis. Experientia. 1994;50:1021–1025. doi: 10.1007/BF01923456. [DOI] [PubMed] [Google Scholar]
- van den Boorn JG, et al. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J Invest Dermatol. 2011;131:1240–1251. doi: 10.1038/jid.2011.16. [DOI] [PubMed] [Google Scholar]
- van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, Tigges B, Westerhof W, Das P. Local immune response in skin of generalized vitiligo patients. destruction of melanocytes is associated with the prominent presence of CLA+ T cells at the perilesional site. Lab Invest. 2000;80:1299–1309. doi: 10.1038/labinvest.3780138. [DOI] [PubMed] [Google Scholar]
- Wang S, Bartido S, Yang G, Qin J, Moroi Y, Panageas KS, Lewis JJ, Houghton AN. A role for a melanosome transport signal in accessing the MHC class II presentation pathway and in eliciting CD4+ T cell responses. J Immunol. 1999;163:5820–5826. [PubMed] [Google Scholar]
- Zhang Y, Liu L, Jin L, Yi X, Dang E, Yang Y, Li C, Gao T. Oxidative stress-induced calreticulin expression and translocation: New insights into the destruction of melanocytes. J Invest Dermatol. 2013 doi: 10.1038/jid.2013.268. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wang W, Li Q, Huang W. Fusion protein of ATPase domain of Hsc70 with TRP2 acting as a tumor vaccine against B16 melanoma. Immunol Lett. 2006;105:167–173. doi: 10.1016/j.imlet.2006.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.