

Summary
Aims
DJ‐1 is a key redox‐reactive neuroprotective protein implicated in regulation of oxidative stress after stroke. However, the molecular mechanism, especially the role of mitochondrial function, by which DJ‐1 protects neural cells in stroke remains to be elucidated. The aim of this study was to reveal whether DJ‐1 translocates into the mitochondria in exerting neuroprotection against oxidative stress. In particular, we examined DJ‐1 secretion from primary rat neural cells (PRNCs) exposed to experimental stroke.
Methods
Primary rat neural cells were exposed to the oxygen–glucose deprivation (OGD), an established in vitro stroke model, and DJ‐1 translocation was measured by immunocytochemistry, and its secretion detected by ELISA.
Results
Under OGD, DJ‐1 translocated into the healthy mitochondria, and significant levels of DJ‐1 protein were detected. Treatment with anti‐DJ‐1 antibody reduced cell viability and mitochondrial activity, and increased glutathione level. Interestingly, OGD reversed the ratio of astrocyte/neuron cells (6/4 to 4/6).
Conclusions
Altogether, these results revealed that DJ‐1 participates in the acute endogenous neuroprotection after stroke via the mitochondrial pathway. That DJ‐1 was detected immediately after stroke and efficiently translocated into the mitochondria offer a new venue for developing neuroprotective and/or neurorestorative strategies against ischemic stroke.
Keywords: Mitochondria, Neuroprotection, Oxidative stress, Stem cells, Stroke
Introduction
Despite many scientific breakthroughs, stroke remains a major unmet clinical need. Largely due to deprivation of oxygen, glucose, and other nutrients following blood flow interruption into the brain, stroke presents with a necrotic infarcted core and an evolving ischemic penumbra, which is amenable to therapeutic intervention. The progression of the stroke penumbra toward becoming part of the infarcted core has been shown to be accompanied by multiple secondary cell death processes. In particular, oxidative stress has been demonstrated as a major exacerbating secondary cell death closely associated with stroke, as well as other neurological disorders including Alzheimer's disease and Parkinson's disease (PD). A relatively new key protein implicated in this oxidative stress cell death cascade is DJ‐1. Acting as a multifunctional redox‐sensitive protein, DJ‐1 can promote neuroprotection by dampening mitochondrial oxidative stress 1, molecular chaperoning of PD‐aggregating protein α‐synuclein 2, stimulating anti‐apoptotic and antioxidative gene expression 3, 4, and facilitating the prosurvival Akt while suppressing apoptosis signal‐regulating kinase (ASK1) pathways 5, 6, 7. Although primarily residing in the cytoplasm and nucleus, DJ‐1 translocates into mitochondria of many mammalian cells 1 and is secreted into the serum under pathologic conditions, including breast cancer, melanoma 8, 9, and, of high relevance to stroke, oxidative stress 1. This DJ‐1 translocation into the mitochondria has been shown to reduce aberrant formation of free radicals, that is, mitochondrial reactive oxygen species (ROS) 10, further highlighting the close association of DJ‐1 and stroke‐relevant oxidative stress. We hypothesized that in addition to DJ‐1 harboring an intracellular defense system against oxidative stress after stroke, the protein also functions as an extracellular signaling molecule thereby rendering neuroprotection to neighboring neural cells via paracrine and/or autocrine cues. We tested this hypothesis that DJ‐1 translocated into the mitochondria, with subsequent DJ‐1 protein secretion into the serum, using cultured primary rat neural cells (PRNCs; mixed astrocytes/neurons) exposed to an experimental stroke condition.
Materials and methods
Cell Culture and Oxygen–Glucose Deprivation (OGD)
Primary rat neural cells were obtained from BrainBit. According to the protocol, cells (4 × 104 cells/well) were suspended in 200 μL neural medium containing 2 mM L‐glutamine and 10 ng/mL leukemia inhibitory factor in the absence of antibiotics and grown in poly‐l‐lysine‐coated 96‐well (BD) at 37°C in humidified atmosphere containing 5% carbon dioxide in 40% of the neuron and 60% astrocyte cell population (determined immunocytochemically using vesicular glutamate transporter‐1). After 5 days in culture (approximately cell confluence of 70%), PRNCs were exposed to OGD as described previously with few modification 10. The cells were initially exposed to OGD medium (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 1 mM NaH2PO4, 26.2 mM NaHCO3, 0.01 mM glycine, 1.8 mM CaCl2 pH 7.4), then placed in an anaerobic chamber (Plas Labs) containing nitrogen (95%) and carbon dioxide (5%) for 15 min at 37°C, and finally, the chamber was sealed and incubated for 90 min at 37°C (hypoxic–ischemic condition). Control cells were incubated in same buffer containing 5 mM glucose at 37°C in a regular CO2 (5%) incubator (normoxic condition). OGD was terminated by adding 5 mM glucose to medium, and cell cultures reintroduced to the regular CO2 incubator (normoxic condition) at 37°C for 2 h, of which period represented a model of “reperfusion.” To confirm that extracellular DJ‐1 was secreted by PRNCs, anti‐DJ‐1 antibody (ratio 1:20; Abcam, Cambridge, MA, USA, ab76008) was added to the cell culture medium to capture extracellular DJ‐1 during the reperfusion period. Pilot studies, using proper controls, were performed to eliminate nonspecific IgG binding.
Measurement of Mitochondrial Activity, Cell Viability, and Oxidative Stress
Following reperfusion, reduction of 3‐(4, 5‐dimethyl‐2‐thiazolyl)‐2, 5‐diphenyltetrazolium bromide (MTT) by cellular dehydrogenases was used as a measure of mitochondrial activity as previously described 10. In addition, trypan blue (0.2%) exclusion method was conducted and mean viable cell counts were calculated in four randomly selected areas (1 mm2, n = 10) to reveal the cell viability after hypoxic–ischemic and normoxic condition. Briefly, within 5 min after adding trypan blue, we digitally captured under microscope (×200) 10 pictures (approximately 100 cells/picture) for each condition, then randomly selected five pictures, and counted the number of cells for each individual treatment condition (normoxic, hypoxic–ischemic, hypoxic–ischemic + DJ‐1 antibody). Normalized cell viability was calculated from the following equation: viable cells (%) = (1.00 – [Number of blue cells/Number of total cells]) × 100. As glutathione (GSH) has been validated as an antioxidant component of oxidative defense system in the eukaryotic cell 11 and that increased intracellular GSH level provides a measure of toxicological response precluding cell death 11, we performed GSH assay using manufacturer's protocol for GHS‐GloTM Glutathione Assay kit (Promega, Madison, WI, USA).
Measurement of Extracellular DJ‐1 Concentration
Following reperfusion, the DJ‐1 concentration of cell supernatant was measured by CircuLex DJ‐1/PARK‐7 ELISA Kit (MBL International Corporation, Nagano, Japan, CY‐9050) according to the manufacturer's instructions 12. Absorbance from each sample was measured using a Synergy HT plate reader (Bio‐Tex Instruments, INC., Winooski, VT, USA) at dual wavelengths of 450/540 nm.
Immunocytochemical Analysis
For visualization of mitochondrial membrane potential, PRNCs (8 × 104 cell/well) in 400 μL neural medium in poly‐l‐lysine 8‐chamber (BD) were incubated with 100 nM MitoTracker Red (Invitrogen, Eugene, OR, USA, M7512) for 30 min before cell fixation, washed with PBS 13, 14, and performed immunocytochemical analysis. PRNCs were fixed in 4% paraformaldehyde for 20 min at room temperature after OGD or non‐OGD treatment. The cells were washed five times for 10 min in PBS containing 0.1% Tween 20 (PBST) (Sigma, St. Louis, MO, USA). Then they were blocked by 5% normal goat serum (Invitrogen, Camarillo, CA, USA) in PBST for 60 min at room temperature. The cells were incubated with rabbit monoclonal anti‐DJ‐1 (1:100; Abcam, ab76008) and mouse monoclonal anti‐MAP2 (Neuronal marker, 1:500; Abcam, ab11267) with 5% normal goat serum for overnight at 4°C to examine whether DJ‐1 secreted from the neural cells under hypoxic–ischemic condition. In addition, the cells were incubated with rabbit monoclonal anti‐DJ‐1 (1:100; Abcam, ab76008) and mouse monoclonal anti‐ATP synthase (mitochondrial) β‐chain (1:200; Cell Signaling Technologies, Lake Placid, NY, USA, 05‐709) with 5% normal goat serum for overnight at 4°C to examine whether DJ‐1 translocated into the mitochondria. The cells were washed five times for 10 min in PBST and then soaked in 5% normal goat serum in PBST containing corresponding secondary antibodies, goat anti‐rabbit IgG‐Alexa 488 (green, 1:1000; Invitrogen, Eugene, OR, USA), and goat anti‐mouse IgG‐Alexa 594 (red, 1:1000; Invitrogen, Eugene, OR, USA) or goat anti‐mouse IgG‐Alexa 405 (blue, 1:200; Invitrogen, Eugene, OR, USA) for 90 min. Finally, cells were washed five times for 10 min in PBST and three times for 5 min in PBS, then processed for Hoechst 33258 (bisBenzimideH 33258 trihydrochloride, Sigma) for 30 min, washed in PBS, and cover‐slipped with Fluoromount (Sigma). Immunofluorescent images were visualized using Zeiss Axio Imager Z1. Control experiments were performed with the omission of the primary antibodies yielding negative results.
Measurement of OGD on the Ratio of Astrocytes and Neurons
Briefly, we digitally captured 20 pictures of immunocytochemically stained cells under the microscope (×200) of each treatment condition (normoxic and hypoxic–ischemic), randomly selected 10 pictures, and counted the number of MAP2‐positive cells with Hoechst, then the ratio of MAP2‐positive neuronal cells was calculated as follows: Neurons (%) = ([Number of MAP2‐positive cells/Number of Hoechst]) × 100. The number of astrocytic cells (i.e., the other cell population in the cultured PRNCs) was calculated by subtracting% MAP2‐positive neuronal cells from 100%. As we used PRNCs (mixed astrocytes/neurons), the specific neuronal MAP2 antibody labeled neuronal cells, while those non‐MAP2‐stained cells corresponded to astrocytic cells.
Statistical Analysis
The data were evaluated using one‐way analysis of variance (ANOVA) followed by post hoc compromised t‐tests. Statistical significance was preset at P < 0.05. Data are represented as means ± SD from quintuplicates of each treatment condition.
Results
DJ‐1 Protein Promotes Neuroprotection
We first confirmed whether PRNCs could secrete DJ‐1 in the hypoxic–ischemic condition. After being exposed to OGD, cell viability (F2,9 = 80.497, P < 0.0001) (Figure 1A) and mitochondrial activity (F2,11 = 273.593, P < 0.0001) (Figure 1B) were significantly decreased, on the other hand, GSH level was increased (F2,9 = 111.248, P < 0.0001) (Figure 1C) compared with normoxic condition. As shown in Figures 1D and 2, we detected DJ‐1 released from PRNCs (Figure 2, represented by arrow heads). To reveal whether the therapeutic role of DJ‐1 was accompanied by its extracellular secretion by PRNCs, we showed that treating the cell culture system with DJ‐1 antibody exacerbated the OGD‐induced reduction in cell viability and mitochondrial activity, despite further increasing GSH levels (Figure 1A–C), implicating that the extracellular DJ‐1, which was sequestered by the DJ‐1 antibody (F2,18 = 150.049, P < 0.0001) (Figure 1D), was required for the observed neuroprotection. The physiological relevance of these observations is that the loss of DJ‐1 perturbs the endogenous neuroprotection of rat neural cells against oxidative stress.
Figure 1.
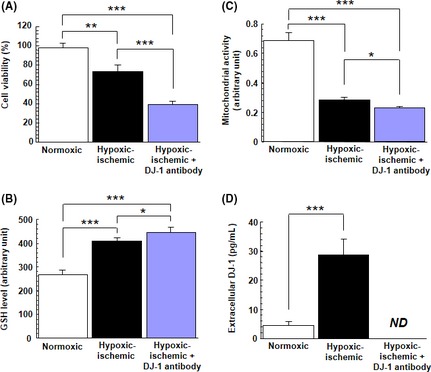
Effect of OGD on PRNCs. OGD altered PRNCs proliferation and DJ‐1 secretion. PRNCs were subjected to OGD for 90 min, followed by a 2‐h reperfusion period under normoxic condition. Under hypoxic–ischemic condition, cell viability tests using trypan blue exclusion method (A) and MTT assay (B) revealed that the number of viable PRNCs significantly decreased, concomitant with increased GSH production (C) and detection of secreted DJ‐1 (D). For panels C and D, both assays were normalized with cell number in each condition. *P < 0.05, **P < 0.01, ***P < 0.0001. ND: nondetectable by the CircuLex DJ‐1/PARK ELISA kit.
Figure 2.
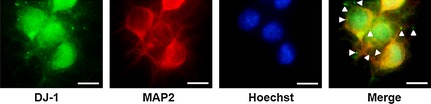
DJ‐1 secreted from PRNCs under hypoxic–ischemic condition. DJ‐1 (green), MAP2 (red), and Hoechst (blue). Arrow heads represent DJ‐1 that secreted from PRNCs following hypoxic–ischemic insult. Scale bars = 10 μm.
DJ‐1 Translocates into the Mitochondria under Hypoxic–Ischemic Condition
An equally important finding here is the localization of DJ‐1 in PRNCs as determined by immunofluorescent microscopy. First, the neuron cells were stained both with anti‐DJ‐1 antibody and anti‐ATP synthase β‐chain antibody under a normoxic condition (Figure 3A–C). Employing the anti‐ATP synthase β‐chain antibody, of which antigen is localized in the mitochondrial inner membrane and linkages with mitochondrial complex I, the results (Figure 3D–F) revealed that DJ‐1 translocated into the mitochondrial inner membrane following the hypoxic–ischemic insult (Figure 3F), but not in normoxic condition (Figure 3A–C).
Figure 3.
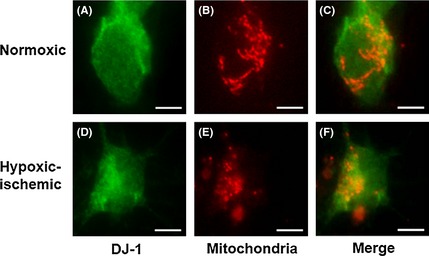
DJ‐1 translocated into the mitochondrial inner membrane following hypoxic–ischemic insult. DJ‐1 positive cells (A and D), mitochondria localization in PRNCs (B and E), and double‐labeled DJ‐1 and mitochondria (C and F). A–C were normoxic condition. D–F were hypoxic–ischemic condition. Scale bars = 5 μm.
DJ‐1 Selectively Translocates to the Healthy Mitochondria
Next, following confirmation that DJ‐1 translocated into the mitochondria of PRNCs after hypoxic–ischemic insult, we examined whether DJ‐1 selectively translocated into the healthy mitochondria, damaged mitochondria, or both. Employing a chemical reagent MitoTracker, which accumulates on healthy mitochondria, DJ‐1 was shown to translocate to the healthy mitochondria (white arrows; represented electrochemical active mitochondria) more than the damaged mitochondria (yellow arrows; which were not capable of permeating the dye MitoTracker) (Figure 4), indicating that DJ‐1 translocation is closely associated with preservation of functional mitochondria. DJ‐1 colocalized with active mitochondria but not with inactive mitochondria (yellow arrows).
Figure 4.
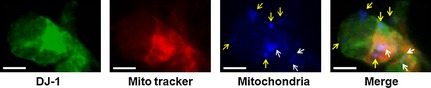
DJ‐1 selectively translocates to the electrochemical active mitochondria. For visualizing the mitochondrial membrane potential, cells were incubated with 100 nM MitoTracker (red) for 30 min before cell fixation (as described in Materials and methods), and then cells were immunostained for anti‐DJ‐1‐ (green) and anti‐ATP synthase (mitochondrial) β‐chain (blue). Arrows (yellow) indicated depolarized mitochondria (which were not capable of permeating the dye MitoTracker). Arrows (white) represented electrochemical active mitochondria (which possessed to permeate the dye MitoTracker). DJ‐1 colocalized with electrochemical active mitochondria but not with depolarized mitochondria (yellow arrows). Scale bars = 5 μm.
Effect of Hypoxic–Ischemic Condition on the Ratio of Astrocytes/Neurons
We next counted the number of MAP2‐positive neuronal cells and extrapolated the number of astrocytic cells in the cultured PRNCs under normoxic and hypoxic–ischemic conditions. Interestingly, quantitative analyses showed that the hypoxic ischemia significantly reversed the ratio of astrocytes/neurons (6/4) compared with normoxic condition (4/6) (P < 0.01) (Figure 5).
Figure 5.
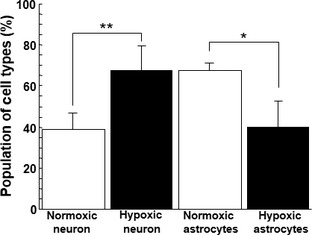
Effect of OGD on the ratio of astrocyte/neuron. Amount of astrocyte‐ and neuron‐cell was counted. *P < 0.01, **P < 0.001.
Discussion
The present study revealed a new molecular mechanism underlying DJ‐1‐mediated neuroprotection against hypoxic–ischemic insult in a cell culture paradigm. We demonstrated that DJ‐1 translocated into the mitochondria and that DJ‐1 is subsequently secreted by hypoxic–ischemic PRNCs. Interestingly, the hypoxic–ischemic condition reversed the ratio of astrocyte/neuron cells (6/4 to 4/6), suggesting DJ‐1's preferential rescue of neurons. These observations open new avenues of research and therapeutic development targeting DJ‐1 for treating stroke and other neurological disorders characterized by rampant mitochondrial deficits 15, 16, 17. This study also provided solid mechanistic evidence that DJ‐1 was secreted extracellularly and that when the DJ‐1 antibody is applied, DJ‐1 secretion was sequestered as evidenced by ELISA. This blockade of DJ‐1 resulted in reduction in mitochondrial activity and, eventually, cell viability under the OGD condition, altogether implicating the key role of mitochondrial translocation and extracellular secretion of DJ‐1 in neuroprotection against stroke.
DJ‐1 is Secreted under Hypoxic–Ischemic Injury
A major finding in the present study is the demonstration that neural cells secreted DJ‐1 under hypoxic–ischemic condition. This observation parallels similar reports that breast cancer and melanoma release DJ‐1 to the serum in vitro and in vivo 18, 19, 20. That significantly elevated levels of extracellular DJ‐1 were detected at the very early stage of OGD–reperfusion injury, and that antibody sequestration of this secreted protein resulted in exacerbation of oxidative stress and cell death, suggest an intimate involvement of DJ‐1 in the initial endogenous neuroprotective process in response to hypoxic–ischemic injury.
Mitochondrial Dysfunction Contributes to Increased Generation of ROS
Mitochondrial dysfunction is an important contributor to neurodegeneration 21, 22, including stroke 23. Hypoxic–ischemic cell death events may consist of mitochondria complex I spontaneously releasing ROS, a hallmark biochemical feature of oxidative stress 24. A cell death mechanism characterized by the collapse of mitochondrial membrane potential, which in turn triggers the aberrant disruption of the impermeability of the inner mitochondrial membrane has been shown to accompany oxidative stress 25, 26. Initial stroke‐induced ROS acts upon neighboring mitochondria, precipitating mitochondria permeability transition pore opening, and thereafter generating additional ROS 27. The translocation of DJ‐1 into the mitochondria due to stroke‐associated oxidative stress may correspond to a primary neuroprotective response in an effort to halt secondary cell death progression. Because mitochondrial complex I critically regulates oxidative stress and controls ATP production in eukaryotic cells, its dysfunction induces cell death 28, 29. DJ‐1 by translocating into the mitochondria achieves an efficient endogenous neuroprotection in mitigating mitochondrial defects. The observed DJ‐1 translocation may affect mitochondrial movement, promote cell–cell interaction, and enhance other procell survival processes. Moreover, we noted that the degrees of neuroprotective effects due to sequestering DJ‐1 on cell survival and MTT varied, thereby suggesting that in addition to ROS‐dependent pathways, DJ‐1 may arrest the OGD‐induced cell death via multipronged neuroprotective mechanisms.
Damaged Mitochondria can be Restored by Fusion with Intact Mitochondria
Constant fusion and fission events, mediated by conserved cellular machineries, can lead to the formation of a reticulated mitochondrial network 30, 31, 32. Rat primary neural cells, under hypoxic–ischemic condition, typically retract outgrowth processes with neighboring cells and lose contact with the extracellular matrix 33, 34. The functionality of damaged mitochondria can be restored by fusion with neighboring, intact mitochondria, assigning an important role for protein quality control to components regulating mitochondrial dynamics 35, 36, 37. The possibility that DJ‐1 activates a complex rearrangement of mitochondria allowing neural survival in response to cell injury warrants additional studies.
DJ‐1 Preferential Rescue of Neurons
In the present study, we assessed astrocyte and neuron viability at the acute time point post‐OGD when endogenous repair mechanisms 38 (presumably including DJ‐1 neuroprotection) are apparent, which likely contributed to the observed preferential rescue of neurons over astrocytes. However, without any therapeutic intervention, following the acute phase, the astrocytic cell death will dominate over the neuronal demise. The present hypoxia–ischemia protocol employed a 90‐min OGD followed by a 2 h reperfusion period and cell viability assays immediately thereafter, which corresponded to the acute phase. In contrast, Goldberg and Choi 39 observed that while neuronal cell death was detected within 60 min of OGD, the irreversible neuronal damage became only consistently evident when OGD exposure exceeds 4–6 h and with neuronal cell death assessment conducted a day later, which is more severe and prolonged (i.e., sub‐acute to chronic phase) compared with our protocol. In addition, Goldberg and Choi 39 used mixed neocortical cultures, containing both neurons and glia prepared from fetal mice at 14–17 days gestation. Altogether, our present results complement those reported by Goldberg and Choi 39, in that an evolving OGD‐induced astrocytic and neuronal cell death ensues over time characterized by varying ratios of astrocyte/neuron cell death with acute phase (accompanied by endogenous neuroprotection) showing neuronal rescue while the subacute/chronic phase (without therapeutic intervention) displaying neuronal degeneration. In the same vein, astrocyte viability may vary depending on the OGD phase and severity, and may mediate delayed neuronal cell death as we 38 and Goldberg and Choi 39 previously reported.
DJ‐1 is a Key Protein in the Neural Survival Following Hypoxic–Ischemic Injury
The present data strongly support the hypothesis that DJ‐1 exerts significant control on both the dynamic cell structure and mitochondria function, requiring a balance between fission and fusion 40, 41, 42. Our results also complement recent reports of DJ‐1 targeting oxidative stress and inflammatory pathways 43, 44, 45. However, although we found increased levels of the antioxidant GSH at acute hypoxic–ischemic period, corresponding to DJ‐1 mitochondrial translocation, the sequestration of extracellular DJ‐1 with the antibody further led to GSH upregulation, but worsened cell viability and mitochondrial activity. We used PRNCs (mixed astrocytes/neurons), thus GSH concentrations reflected both cell types. However, our recent study 15 using the same DJ‐1 antibody could selectively reduce GSH in human neural progenitor cells (hNPCs), indicating direct DJ‐1 regulation of neuronal GSH. In the present study, DJ‐1 antibody could have selectively reduced neuronal GSH, but such a change might have been masked by its lack of inhibitory effects in astrocytes because of the present mixed cell culture setup. These observations highlight the importance of DJ‐1, in that while increased GSH levels have been widely shown as therapeutic against stroke, such neuroprotection is not recognized under conditions of DJ‐1 depletion. Increased DJ‐1 immunoreactivity in human stroke patients appears to highly correlate with robust expression of the glial fibrillary acidic protein‐labeled reactive astrocytes 45, further supporting the notion that DJ‐1 expression in stroke is accompanied by an active inflammatory component. Finally, whereas the present DJ‐1 nuclear translocation is observed in cultured PRNCs, it is tempting to speculate that in vivo DJ‐1 may similarly modulate the cell proliferation in neurogenic sites of lateral ventricle and hippocampal dentate gyrus. Along this line, we posit in agreement with recent reports 5, 46, 47, 48 that DJ‐1 is a major protein for neural cell survival in disease states, such as stroke and PD.
Conclusions
We demonstrated that under hypoxic–ischemic condition, DJ‐1 translocated into the healthy mitochondria. Moreover, significant levels of DJ‐1 protein were secreted by hypoxic–ischemic neural cells. DJ‐1 also preserves MTT‐ and GSH‐activity during oxidative stress. The present results provide evidence that DJ‐1 is closely involved in the early phase of neuroprotection against ischemic stroke via the mitochondrial pathway, suggesting that therapies designed to enhance efficient DJ‐1 translocation into the mitochondria may prove effective against ischemic stroke.
Conflict of Interest
The authors declare no conflict of interests.
Acknowledgments
This work was supported by NIH NINDS RO1 1R01NS071956‐01 (CVB), James and Esther King Biomedical Research Program 09KB‐01‐23123 (CVB) and 1KG01‐33966 (CVB), USF OR&I HSC‐18330 & COM HSC‐18300 (YK), and USF Department of Neurosurgery and Brain Repair Funds (CVB).
The first two authors contributed equally to this work.
References
- 1. Canet‐Aviles RM, Wilson MA, Miller DW, et al. The parkinson's disease protein DJ‐1 is neuroprotective due to cysteine‐sulfinic acid‐driven mitochondrial localization. Proc Natl Acad Sci U S A 2004;101:9103–9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in parkinson's disease. Science 2003;302:819–822. [DOI] [PubMed] [Google Scholar]
- 3. Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ‐1, a cancer‐ and parkinson's disease‐associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci U S A 2006;103:15091–15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fan J, Ren H, Jia N, et al. DJ‐1 decreases Bax expression through repressing p53 transcriptional activity. J Biol Chem 2008;283:4022–4030. [DOI] [PubMed] [Google Scholar]
- 5. Gorner K, Holtorf E, Waak J, et al. Structural determinants of the c‐terminal helix‐kink‐helix motif essential for protein stability and survival promoting activity of DJ‐1. J Biol Chem 2007;282:13680–13691. [DOI] [PubMed] [Google Scholar]
- 6. Junn E, Taniguchi H, Jeong BS, Zhao X, Ichijo H, Mouradian MM. Interaction of DJ‐1 with Daxx inhibits apoptosis signal‐regulating kinase 1 activity and cell death. Proc Natl Acad Sci U S A 2005;102:9691–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang Y, Gehrke S, Haque ME, et al. Inactivation of drosophila DJ‐1 leads to impairments of oxidative stress response and phosphatidylinositol 3‐kinase/Akt signaling. Proc Natl Acad Sci U S A 2005;102:13670–13675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsuboi Y, Munemoto H, Ishikawa S, Matsumoto K, Iguchi‐Ariga SM, Ariga H. DJ‐1, a causative gene product of a familial form of parkinson's disease, is secreted through microdomains. FEBS Lett 2008;582:2643–2649. [DOI] [PubMed] [Google Scholar]
- 9. Waak J, Weber SS, Waldenmaier A, et al. Regulation of astrocyte inflammatory responses by the parkinson's disease‐associated gene DJ‐1. FASEB J 2009;23:2478–2489. [DOI] [PubMed] [Google Scholar]
- 10. Borlongan CV, Kaneko Y, Maki M, et al. Menstrual blood cells display stem cell‐like phenotypic markers and exert neuroprotection following transplantation in experimental stroke. Stem Cells Dev 2010;19:439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med 2009;30:42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bande MF, Santiago M, Blanco MJ, et al. Serum DJ‐1/PARK 7 is a potential biomarker of choroidal nevi transformation. Invest Ophthalmol Vis Sci 2012;53:62–67. [DOI] [PubMed] [Google Scholar]
- 13. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanaka M, Tanji K, Niida M, Kamitani T. Dynamic movements of Ro52 cytoplasmic bodies along microtubules. Histochem Cell Biol 2010;133:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaneko Y, Shojo H, Burns J, Staples M, Tajiri N, Borlongan CV. DJ‐1 ameliorates ischemic cell death in vitro possibly via mitochondrial pathway. Neurobiol Dis 2013;62C:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li B, Yu D, Xu Z. Edaravone prevents neurotoxicity of mutant l166p DJ‐1 in parkinson's disease. J Mol Neurosci 2013;51:539–549. [DOI] [PubMed] [Google Scholar]
- 17. Thomas KJ, McCoy MK, Blackinton J, et al. DJ‐1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet 2011;20:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Le Naour F, Misek DE, Krause MC, et al. Proteomics‐based identification of RS/DJ‐1 as a novel circulating tumor antigen in breast cancer. Clin Cancer Res 2001;7:3328–3335. [PubMed] [Google Scholar]
- 19. Miura K, Bowman ED, Simon R, et al. Laser capture microdissection and microarray expression analysis of lung adenocarcinoma reveals tobacco smoking‐ and prognosis‐related molecular profiles. Cancer Res 2002;62:3244–3250. [PubMed] [Google Scholar]
- 20. Kim YC, Kitaura H, Iguchi‐Ariga SM, Ariga H. DJ‐1, an oncogene and causative gene for familial parkinson's disease, is essential for SV40 transformation in mouse fibroblasts through up‐regulation of c‐Myc. FEBS Lett 2010;584:3891–3895. [DOI] [PubMed] [Google Scholar]
- 21. Saraiva LM, Seixas da Silva GS, Galina A, et al. Amyloid‐beta triggers the release of neuronal hexokinase 1 from mitochondria. PLoS One 2010;5:e15230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pivovarova NB, Andrews SB. Calcium‐dependent mitochondrial function and dysfunction in neurons. FEBS J 2010;277:3622–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen B, Friedman B, Cheng Q, et al. Severe blood‐brain barrier disruption and surrounding tissue injury. Stroke 2009;40:e666–e674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ten VS, Yao J, Ratner V, et al. Complement component c1q mediates mitochondria‐driven oxidative stress in neonatal hypoxic‐ischemic brain injury. J Neurosci 2010;30:2077–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flynn JM, Lannigan DA, Clark DE, Garner MH, Cammarata PR. RNA suppression of ERK2 leads to collapse of mitochondrial membrane potential with acute oxidative stress in human lens epithelial cells. Am J Physiol Endocrinol Metab 2008;294:E589–E599. [DOI] [PubMed] [Google Scholar]
- 26. Reddy PV, Rao KV, Norenberg MD. The mitochondrial permeability transition, and oxidative and nitrosative stress in the mechanism of copper toxicity in cultured neurons and astrocytes. Lab Invest 2008;88:816–830. [DOI] [PubMed] [Google Scholar]
- 27. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ros‐induced ros release: An update and review. Biochim Biophys Acta 2006;1757:509–517. [DOI] [PubMed] [Google Scholar]
- 28. Taira T, Saito Y, Niki T, Iguchi‐Ariga SM, Takahashi K, Ariga H. DJ‐1 has a role in antioxidative stress to prevent cell death. EMBO Rep 2004;5:213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Foti R, Zucchelli S, Biagioli M, et al. Parkinson disease‐associated DJ‐1 is required for the expression of the glial cell line‐derived neurotrophic factor receptor RET in human neuroblastoma cells. J Biol Chem 2010;285:18565–18574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rabinowitz JD, White E. Autophagy and metabolism. Science 2010;330:1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barsoum MJ, Yuan H, Gerencser AA, et al. Nitric oxide‐induced mitochondrial fission is regulated by dynamin‐related gtpases in neurons. EMBO J 2006;25:3900–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parone PA, Martinou JC. Mitochondrial fission and apoptosis: An ongoing trial. Biochim Biophys Acta 2006;1763:522–530. [DOI] [PubMed] [Google Scholar]
- 33. Pallast S, Arai K, Pekcec A, et al. Increased nuclear apoptosis‐inducing factor after transient focal ischemia: A 12/15‐lipoxygenase‐dependent organelle damage pathway. J Cereb Blood Flow Metab 2010;30:1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci 2008;31:151–173. [DOI] [PubMed] [Google Scholar]
- 35. Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010;9:447–464. [DOI] [PubMed] [Google Scholar]
- 36. Reyes RC, Parpura V. Mitochondria modulate Ca2+‐dependent glutamate release from rat cortical astrocytes. J Neurosci 2008;28:9682–9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krebiehl G, Ruckerbauer S, Burbulla LF, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of parkinson's disease‐associated protein DJ‐1. PLoS One 2010;5:e9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Borlongan CV, Yamamoto M, Takei N, et al. Glial cell survival is enhanced during melatonin‐induced neuroprotection against cerebral ischemia. FASEB J 2000;14:1307–1317. [DOI] [PubMed] [Google Scholar]
- 39. Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: Calcium‐dependent and calcium‐independent mechanisms of neuronal injury. J Neurosci 1993;13:3510–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiong H, Wang D, Chen L, et al. Parkin, PINK1, and DJ‐1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest 2009;119:650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leadsham JE, Miller K, Ayscough KR, et al. Whi2p links nutritional sensing to actin‐dependent Ras‐cAMP‐PKA regulation and apoptosis in yeast. J Cell Sci 2009;122:706–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, Hinshaw JE. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol 2011;18:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aleyasin H, Rousseaux MW, Phillips M, et al. The parkinson's disease gene DJ‐1 is also a key regulator of stroke‐induced damage. Proc Natl Acad Sci U S A 2007;104:18748–18753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yanagisawa D, Kitamura Y, Inden M, et al. DJ‐1 protects against neurodegeneration caused by focal cerebral ischemia and reperfusion in rats. J Cereb Blood Flow Metab 2008;28:563–578. [DOI] [PubMed] [Google Scholar]
- 45. Mullett SJ, Hamilton RL, Hinkle DA. DJ‐1 immunoreactivity in human brain astrocytes is dependent on infarct presence and infarct age. Neuropathology 2009;29:125–131. [DOI] [PubMed] [Google Scholar]
- 46. Aron L, Klein P, Pham TT, Kramer ER, Wurst W, Klein R. Pro‐survival role for parkinson's associated gene DJ‐1 revealed in trophically impaired dopaminergic neurons. PLoS Biol 2010;8:e1000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vasseur S, Afzal S, Tardivel‐Lacombe J, Park DS, Iovanna JL, Mak TW. DJ‐1/PARK7 is an important mediator of hypoxia‐induced cellular responses. Proc Natl Acad Sci U S A 2009;106:1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yan H, Pu XP. Expression of the parkinson's disease‐related protein DJ‐1 during neural stem cell proliferation. Biol Pharm Bull 2010;33:18–21. [DOI] [PubMed] [Google Scholar]