

Abstract
Virotherapy is currently undergoing a renaissance, based on our improved understanding of virus biology and genetics and our better knowledge of many different types of cancer. Viruses can be reprogrammed into oncolytic vectors by combining three types of modification: targeting, arming and shielding. Targeting introduces multiple layers of cancer specificity and improves safety and efficacy; arming occurs through the expression of prodrug convertases and cytokines; and coating with polymers and the sequential usage of different envelopes or capsids provides shielding from the host immune response. Virus-based therapeutics are beginning to find their place in cancer clinical practice, in combination with chemotherapy and radiation.
When viruses were first recognized more than 100 years ago, the idea of using them to fight cancer, and in particular leukaemia, was considered1. Clinical reports of cancer regressions that were coincidental with natural virus infections continued through the first half of the twentieth century2,3. Based on these anecdotal observations, early clinical trials were performed in which bodily fluids that contained human or animal viruses were used to transmit infections to patients with cancer4. Often, the host immune response prevailed, but occasionally, in immunosuppressed patients, the infection persisted and the cancer regressed, although the morbidity that occurred as a result of the infection of normal tissues was unacceptable. Some of the clinical studies performed at that time seem alarming in the context of current ethical standards; however, these were desperate times for those afflicted with cancer3.
The advent of tissue culture in the 1950s and 1960s allowed viruses to be propagated in a more defined environment and cancers to be modelled by implanting cancer cells into rodents, which allowed pre-clinical experimentation with many types of human and animal viruses5–10. The opportunity to influence the evolution of viruses by adapting them to grow well only in specific cancer cells and then using them as therapy for equivalent cancers was promptly recognized and seized11–13, but success was again limited, and research activity in the field of oncolytic virotherapy diminished because alternative approaches to improve efficacy were not available.
At this crossroads, oncolytic virotherapy was limited by the lack of knowledge of the determinants of viral tropism and of ways to manipulate those determinants to generate viruses that were more specific for cancer cells. It was recognized that cancer cells were better environments for the replication of naturally oncolytic viruses, whereas non-transformed cells could control virus infections. The need to improve the characteristics of natural oncolytic viruses became clear as more extensive testing identified limited efficacy or dose-limiting toxicities3,14. Consequently, research moved towards reprogramming viruses to become increasingly cancer specific, and thus safer. However, progress was initially slow, because an understanding of the natural virus tropism determinants and the molecular environment of the target cancer cells had to be developed.
During the past 20 years, tropism determinants have been identified and characterized for many different virus families. In addition, we can now easily visualize and quantify viral spread using reporter genes15,16, and document how virus replication relates to therapeutic efficacy17. Most importantly, reverse-genetics systems have been developed for almost every virus family, allowing the generation of viruses with improved oncolytic properties. Finally, our understanding of cancer has also improved with the availability of diagnostic markers and sophisticated animal models. These advances allow researchers to generate viruses with various levels of specificity for the molecular eccentricities of cancer cells.
FIGURE 1 and TABLE 1 introduce the most relevant families of human DNA and RNA viruses that are currently used in, or are approaching, clinical trials. Recombinant DNA viruses in clinical trials include adenovirus (Ad), herpes simplex virus 1 (HSV1) and vaccinia virus. Other DNA viruses that are approaching clinical trials include myxoma virus. The only engineered oncolytic RNA virus that is currently in clinical trials is measles virus (MV); non-engineered strains of Newcastle disease virus (NDV) and reovirus are currently in Phase I–II clinical trials, and reprogrammed poliovirus and vesicular stomatitis virus (VSV) are in pre-clinical trials. As the major clinical trials of oncolytic viruses for cancer therapy have recently been reviewed18, we will focus here mainly on the vector developments that are preparing viruses for the next generation of trials. The first half of this Review will discuss the principles of retargeting viruses to cancer cells, which are primarily illustrated using an RNA virus, MV, and a DNA virus, Ad. The focus is exclusively on the genetic reprogramming of replicating viral vectors. The second half of this Review focuses on arming viruses and shielding them from the host immune response to improve oncolytic efficacy. We also address the clinical use of reprogrammed viruses in combination with chemotherapy and radiation, and discuss a five-step plan for reprogramming viruses into cancer therapeutics.
Figure 1. Oncolytic viruses that are currently used in cancer clinical trials.

The major characteristics of seven families of oncolytic viruses are summarized. Recombinant strains of all the DNA viruses shown are currently in clinical trials, whereas among the RNA viruses shown, only recombinant MV is in clinical trials; recombinant poliovirus and VSV are in pre-clinical trials and non-engineered strains of reovirus and NDV are in Phase I–II clinical trials. HSV1, herpes simplex virus 1; MV, measles virus; NDV, Newcastle disease virus; VSV, vesicular stomatitis virus.
Table 1.
Oncolytic viruses in, or approaching, clinical trials
| Virus (strain and modifications) | Tumour target (clinical phase and application) |
|---|---|
| Adenovirus 5 (dl1520 derivative) | Squamous cell carcinoma of head and neck (approved drug in China; intratumoural) |
| Adenovirus 5 (PSE-E1A and E3 deleted) | Prostate (I; prostatic) |
| Herpes simplex virus 1 (ICP34.5 defective) | Glioblastoma multiforme (II; intratumoural) |
| Vaccinia virus (thymidine kinase knockout and expressing granulocyte–macrophage colony-stimulating factor) | Advanced liver tumours (I–II; intratumoural) |
| Reovirus (reolysin) | Superficial tumours (I; intralesional) |
| Newcastle disease virus (PV701) | Bladder, squamous cell carcinoma of head and neck and ovarian (I–II; intravenous) |
| Measles virus (V protein knockout and expressing the reporter carcinoembryonic antigen or the effector sodium iodide symporter) | Ovarian (I; intratumoural), glioma (I–II; intratumoural) and myeloma (I; intravenous) |
Retargeting viral tropism
When considering the development of an oncolytic agent for a tumour that is derived from a specific cell type, two general strategies can be used. First, viruses with a natural tropism for that cell type can be considered. This principle is illustrated by the selection of HSV to treat glioblastoma. The goal then becomes to deprogramme the virus from harming normal neurons, while maintaining oncolytic efficacy. Alternatively, one can reprogramme a virus that has only a marginal tropism for the target cell type. This strategy benefits from the inherently low toxicity of the virus for normal cells, but demands precise insights into the host mechanisms that limit viral spread and the availability of targeting elements to overcome these limitations. The success of both approaches is dependent on the selectivity of the strategies that are used. In this section, we discuss four complementary classes of targeting modifications, as illustrated in FIG. 2: particle activation, which is dependent on cancer-specific proteases; entry through cancer-specific cell-surface molecules; control of viral transcription and replication by tissue-specific promoters; and the preferential spread of viruses that exploit cancer-specific molecular defects.
Figure 2. Four layers of specificity for retargeting viral tropism.

a | Virus particle activation can be reprogrammed to depend on proteases that are secreted by cancer cells. Activation occurs through matrix metalloproteases (MMPs) that are located in the tumour matrix. b | Recombinant viruses can be engineered to enter cells through a designated receptor rather than through the natural attachment protein. c | Viral transcription and replication can be made dependent on tissue- or cancer-specific promoters. d | Viruses with modifications or deletions of their immune-evasion proteins replicate preferentially in certain transformed cells. Not every targeting strategy can be applied to every virus, but more and more viruses with combined layers of specificity are being engineered for specific clinical trials.
We have selected the enveloped RNA virus MV (FIG. 3) and the icosahedral DNA virus Ad (FIG. 4) to illustrate how these alterations can be applied to viruses with different biological characteristics.
Figure 3. MV particle structure, genome organization and targeting approaches.

The MV genome has six genes that code for eight proteins. The first gene codes for the nucleocapsid protein (N) that encapsidates the genomic RNA. The last gene (L) codes for the polymerase protein that replicates and transcribes the genome together with the phosphoprotein (P), a polymerase cofactor. The matrix protein (M) organizes virus particle assembly. The two glycoproteins haemagglutinin (H) and the fusion (F) protein contact the receptor and execute membrane fusion, respectively. Two non-structural proteins that are coded by the P gene (C and V) control the innate immune response. a | Three-dimensional structure of MV H117. Residues that are necessary for signalling lymphocytic activation molecule (SLAM)-dependent or CD46-dependent fusion are in red. The site of addition of the single-chain fragment variable (scFv) is in black. b | A schematic of the P and V proteins that are encoded by the P gene. These proteins share their amino-terminal domain, but differ at the carboxyl terminus. The residues of the V and P common domain that are important for the interaction with STAT1 (signal transducer and activator of transcription 1) have been characterized. Three amino acids in a conserved hexapeptide are shown in purple. Data from the Horvath group indicate that STAT2 and MDA5 interact with different sequences in the unique cysteine-rich domain of V (A. Ramachandran, J-P. Parisien and C.M. Horvath, unpublished observations). c | Schematic of the MV F protein and amino acid sequences of its cleavage site. The standard F protein is cleaved into F1 and F2 fragments by furin, a ubiquitous protease. Furin cleavage occurs even after a hexameric peptide that codes for a matrix metalloproteinase 2 (MMP2) cleavage site is introduced (F-MMP), but the resulting F1 protein, which is extended by six residues, is inactive. Trimming of three amino-terminal residues by MMP2 cleavage confers function to F-MMP.
Figure 4. Adenovirus particle structure, genome organization and targeting approaches.

An icosahedral, non-enveloped adenovirus (Ad) particle is shown. The key genes in the viral genome that are relevant to the four targeting approaches discussed in the main text are indicated. a | Cancer-specific transcription and replication targeting are applied to the E1 and E4 genes. b | Cancer-specific proteolytic activation has not yet been attempted. c | Cancer-specific receptor attachment is mediated by genetic or chemical modification of the IX, penton, hexon or fibre proteins or genes. d | Preferential spread targeting can be based on over-expression of the ADP gene and the insertion of exogenous genes into the viral genome. ITR, inverted terminal repeat; MMP, matrix metalloproteinase; Pro, protease.
Activation through cancer-specific proteases
The replication and pathogenesis of almost every virus is dependent on interactions with host cell proteases. Importantly, enveloped viruses, such as paramyxoviruses, influenza and HIV-1, require protease cleavage of viral glyco-proteins for productive cell entry following receptor recognition19. Many cancer cells express proteases that could be exploited to enhance virus specificity. Desirable protease targets for oncolytic viruses that can be activated are those that are expressed preferentially and/or at high levels by cancer cells. Notably, matrix metalloproteinases (MMPs) are endopeptidases that are over-expressed in nearly every human cancer20.
Proof of principle for MMP protease activation was achieved using retroviruses by adding blocking ligands that contained MMP-cleavable linkers to the amino (N) terminus of retroviral glycoproteins21,22. In another approach that introduced only minimal structural modifications, the protease cleavage specificity of the fusion protein from MV and Sendai virus was changed from being dependent on the ubiquitous protease furin or the respiratory airway protease tryptase Clara, respectively, to being dependent on an MMP23,24 (FIG. 3a). Recombinant MV that expressed the modified fusion protein (MV-MMP) was unable to propagate or produce a cytopathic effect unless it was added to cells that expressed an MMP. In mice, MV-MMP retained full oncolytic activity when inoculated into MMP-positive subcutaneous cancers but, unlike wild-type MV, MV-MMP did not infect and kill susceptible mice after intracranial inoculation, which proved that the safety of the virus had been enhanced24.
These experiments illustrate the potential safety and efficacy benefits of retargeting oncolytic viruses to MMP-positive cancer cells at the level of virus particle activation. Another protease that can be targeted and that is secreted by invasive metastatic cancer cells is the urokinase-type plasminogen activator25. This retargeting strategy could be directly adapted to restrict cellular entry of other enveloped viruses that are currently being used in clinical trials, and which have fusion proteins that are dependent on protease activation, including HSV and other large DNA viruses and NDV among the paramyxoviruses. Additionally, it might be possible to modify the regulatory or structural proteins of viruses that have an icosahedral capsid to be dependent on pro-teases that are expressed preferentially and/or at high levels by cancer cells.
Entry through cancer-cell-specific receptors
Viruses bind to one or more host cell surface proteins during infection, and the tissue-specific expression of these proteins can determine viral tropism. A range of chemical and genetic engineering strategies have been tested to retarget the cell entry of both enveloped and non-enveloped viruses through designated cancer-cell-specific receptors26 (BOX 1), but certain approaches have been challenging. In particular, the structural constraints of the icosahedral symmetry that is typical of many viruses often makes displaying specificity domains incompatible with efficient particle assembly27. In addition, the biological characteristics of ligands and viruses can sometimes be incompatible, making it difficult to combine certain ligands with some viruses.
Box 1. Biochemical versus genetic modification of viruses.
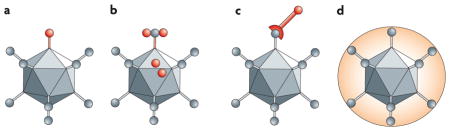
Reprogramming oncolytic viruses requires at least two steps: first, retargeting by adding new ligands to mediate binding to receptors that are expressed on cancer cells, and second, detargeting by blocking promiscuous binding to non-cancer cells. In genetic retargeting, an exogenous ligand is fused to, or replaces, the normal receptor-binding protein of the virus (see the figure, parts a and b). This genetic approach has the advantage that the injected virus and its progeny that are produced during oncolysis will all be targeted. However, genetically retargeting viruses is a complex, technically challenging process. As an alternative, viral particles can be modified chemically (see the figure, parts c and d): they can be conjugated to cell-targeting ligands by using antibody–virus interactions (for example, bi-specific antibodies), molecular bridges (for example, biotin–avidin) or frank covalent coupling with chemical cross-linkers (for example, bifunctional polyethylene glycol (PEG)). As these simple complexing approaches are combinatorial, they can target more than one receptor and avoid the functional complications of introducing foreign domains into viral proteins. However, these approaches only mediate targeting of the inoculated virus. Once progeny virions are produced in the tumour, they default to their genetically encoded tropism. This loss of targeting in progeny and the need to combine multiple Good Manufacturing Practice (GMP)-grade reagents for use in patients makes the biochemical approach more difficult for clinical translation than having one genetically targeted reagent.
Biochemical and genetic targeting are complementary and can be combined. For example, chemical modification could target tumour vasculature, allowing the genetically encoded virus tropism to target cancer cells. In addition, targeting can be performed using polymers such as PEG to ‘shield’ the virus from antibodies and other interactions (see the figure, part d). Therefore, both chemical and genetic engineering hold promise for targeting oncolytic viruses.
Usually, only one viral protein is involved in receptor binding, but other proteins support subsequent particle internalization or membrane fusion. This is true for non-enveloped viruses, such as Ad, which uses its fibre protein for cell binding and its penton base for cell entry (FIG. 4). The substantial efforts to retarget Ad have recently been reviewed27, and therefore we will focus instead on targeting strategies for MV as an archetypical enveloped oncolytic virus. Among enveloped viruses, receptor targeting is most advanced in the Paramyxoviridae, in which two different glycoproteins — haemagglutinin and the fusion protein — carry out receptor binding and membrane fusion, respectively28,29. FIGURE 3 illustrates the structure of an MV particle and the modifications to haemagglutinin that are necessary to achieve retargeting.
To retarget viral tropism at the level of receptor recognition, virus entry through a natural receptor (or receptors) must first be inactivated. The primary receptor for wild-type MV is the signalling lymphocytic activation molecule (SLAM; also known as CD150)30, but the MV vaccine strain also binds to the ubiquitous CD46 (also known as membrane cofactor protein (MCP))31,32. To identify the residues of MV haemagglutinin that selectively support SLAM- or CD46-dependent cell entry, an iterative mutagenesis strategy and functional receptor-dependent fusion assays were used. This identified several residues that are required for either SLAM- or CD46-dependent fusion33 (FIG. 3b). Recombinant viruses with mutations in these residues were obtained by reverse genetics and shown to be selectively receptor blind, recognizing either CD46 or SLAM33.
The next retargeting step requires that viral tropism be expanded to target receptor proteins that are expressed preferentially on cancer cells. Proof of principle for this step was achieved by displaying the small specificity domains epidermal growth factor (EGF) or insulin-like growth factor 1 (IGF1) on MV haemagglutinin. When these domains were added to the extra-cellular terminus of haemagglutinin34, recombinant viruses that expressed these extended proteins became replication competent in previously non-susceptible cells that expressed the EGF or IGF1 receptors.
The applicability of receptor targeting has been greatly expanded by using domains that confer antibody-like specificity. In practice, however, antibodies are difficult to engineer into viral proteins, as they are tetrameric, contain disulphide bonds and are usually substantially larger than the viral proteins to be modified. Researchers have therefore used single-chain fragment variable (scFv) antibodies, which comprise the antigen-binding variable regions of the heavy and light chains of antibodies and retain complete antigen specificity. ScFvs that are specific for every human protein can be relatively easily produced, making these molecules the gold standard for targeting.
Enveloped viruses provide an optimal platform for applying scFvs, as both the ligands and viral envelope glycoproteins are naturally secreted through the same pathway. For example, a recombinant MV that displays an scFv which is specific for the carcinoembryonic antigen was generated and was shown to enter carcinoembryonic-antigen-expressing cells that were not susceptible to entry by wild-type MV35. Subsequently, MV that had been retargeted to bind CD20, a marker for non-Hodgkin’s lymphoma (NHL), and CD38, a marker for myeloma, were also generated using the functional display of scFvs, and were shown to be oncolytic in immunodeficient mice that contained CD20- or CD38-positive cancer xenografts36,37. Recently, the oncolytic efficacy of scFv-retargeted MV was demonstrated in an immunocompetent mouse model38.
Proof of principle for a completely retargeted MV was achieved by combining scFvs that were specific for EGF receptor (EGFR) or CD38 displayed on MV haemagglutinin with mutations that ablated productive interactions with the SLAM and CD46 receptors39. These retargeted viruses replicated in and killed cancer cells that expressed the targeted receptors, but were unable to infect cells that expressed SLAM or CD46. When administered intratumourally or intravenously to mice with human CD38- or EGFR-positive tumours, these viruses mediated targeted anti-tumour activity. These data provide an in vivo demonstration of antibody-directed tumour destruction by a retargeted oncolytic virus39.
ScFv-mediated cell entry through targeted receptors should be applicable to enveloped viruses with glyco-proteins that are folded in the same reducing compartment as scFvs, and indeed proof of principle for the applicability of scFvs in retargeting HSV has been achieved40. Some success has also been reported for other viruses with capsids that are produced within the cytoplasm, such as Ad, based on the display of scFvs41 and the CD40 ligand42. However, these approaches require complex re-engineering or selection of ligands for proper folding and disulphide replacement in this foreign non-reducing environment. In both cases, receptor retargeting holds promise for future generations of clinical oncolytic viruses.
Cancer-specific transcription and replication
Another layer of specificity for cancer cells can be applied at the level of virus replication, by two main approaches. In the first approach, key virus gene products are placed under the transcriptional control of tissue-specific or cancer-specific promoters, such that they are preferentially, if not exclusively, expressed in tumour cells. Reprogramming replication functions using such approaches tends to be simpler in DNA viruses, such as Ad, herpesviruses or poxviruses, as these viruses have large genomes and genome-packaging capacities, which facilitates the insertion of exogenous elements. The expression or interactions of the proteins that are encoded by the key Ad early genes E1A, E1B and E4, for example, has been modified. See REF. 43 for a detailed discussion of the biology of Ad early genes.
In a simplified view, the E1A protein transactivates other Ad promoters that are involved in the virus life cycle (FIG. 4a). E1A also drives cells into synthesis (S) phase to facilitate virus replication and interacts with more than ten proteins, including the retinoblastoma tumour suppressor (Rb) and p300. The E1B 55 kDa protein (E1B-55K) binds and destroys the tumour suppressor p53 to allow entry into S phase. The E1B 19 kDa protein prevents the cell from undergoing apoptosis and aborting virus replication. Manipulation of these proteins or their expression can restrict virus replication to targeted cancer cells.
A second approach to cancer-specific replication is to place the E1 or E4 genes under the control of tissue-specific or cancer-cell-specific promoters. For example, expression of E1A from the prostate-specific antigen promoter renders Ad replication and cell killing prostate specific (TABLE 1). For prostate cancer, conventional therapy often includes prostate ablation, which allows selection of a tissue-specific, rather than a cancer-specific, promoter44. Additionally, the telomerase promoter has been placed upstream of the E1 and/or E4 genes to drive oncolytic virus activity45,46. This promoter is an example of the burgeoning number of promoters that are being tested to reprogramme the replication of Ad and other viruses for oncolysis.
An interesting example of a virus with cancer specificity of disputed origin is ONYX-015, the archetypical attempt to generate cancer-specific oncolytic Ad. ONYX-015 is derived from the mutant Ad strain dl1520, in which E1B-55K is deleted47. It was originally thought that the deletion of E1B-55K would protect normal cells from infection by ONYX-015, as these cells express the tumour suppressor p53. By contrast, it was proposed that ONYX-015 would proliferate and kill cancer cells that lack p53 with no restraints. Although early work seemed to support this hypothesis48, later studies revealed that the situation is more complex, as ONYX-015 was cytopathic even in the presence of p53 (REF. 49). Tumour selectivity of ONYX-015 is currently mainly attributed to late mRNA export50,51. H101, an E1B-55K-deleted virus that is similar to ONYX-015, has recently been approved by the Chinese State Food and Drug Administration for use in combination with chemotherapy in the treatment of late-stage refractory nasopharyngeal cancer (TABLE 1). Approval of H101 or a related product by the United States Food and Drug Administration will be contingent on the availability of extensive survival and quality-of-life data. H101, like ONYX-015, is an important case study in applied virology and the only approved drug in the clinic18, but has only modest cancer selectivity. Both H101 and ONYX-015 also illustrate the need to better integrate observations in different experimental systems and ensure the cancer specificity of oncolytic viruses by applying overlapping layers of cancer specificity.
Exploiting cancer cell defects to balance attenuation
The most potent oncolytic viruses are arguably wild-type viruses. However, these viruses can kill normal cells and cause dose-limiting toxicities. Efforts have therefore been made to reduce the toxicity of most oncolytic viruses by attenuation52. For example, in a recent clinical trial that tested the use of herpesvirus in glioma, attenuation of the virus by inactivation of ICP34.5, a neurovirulence gene, and ICP6, the gene that encodes the large subunit of ribonucleotide reductase, made it safe (TABLE 1). As it is possible that this attenuation also reduces oncolytic efficacy53, selective expression of ICP34.5 through glioma-specific promoters is being developed54,55.
Attenuation is always relative to the target cell. Ideally, oncolytic viruses should be highly attenuated in normal cells, but maintain normal replication in cancer cells. Many viruses replicate well in those cancer cells that have accumulated defects in innate immunity functions, which should allow the viral proteins that are normally used to circumvent these protective measures to be modified without extensively sacrificing replication competence in tumour56. However, it is clear that completely ablating the expression of certain virus proteins attenuates viral replication, even in cancer cells, as these virus proteins often have multiple functions. Therefore, current attenuation strategies are moving towards conservative modification of virus proteins, through the mutation of single residues that are essential for specific interactions.
For example, many cancerous cells cannot produce interferon (IFN) or respond to IFN stimulation57–61. Such abnormalities make these cells highly susceptible to virus infection, and indeed it has been shown that VSV with mutations in the matrix protein, which is responsible for modulating the cellular IFN response, replicates preferentially in these cancer cells62. When this study was published, the mechanism by which the mutation operated was not completely understood, but current studies can profit from the identification of the cellular proteins that directly interact with certain virus proteins and, sometimes, from the knowledge of the structural basis of these interactions.
For example, the mechanisms that MV uses to control the IFN system are understood in some detail. IFN binds to neighbouring cells to activate cytoplasmic STAT (signal transducer and activator of transcription) proteins by phosphorylation. Activated STAT proteins function as transcription factors to upregulate the expression of antiviral proteins, thereby protecting the cell in advance of infection63–66. Viruses have evolved diverse mechanisms to block the IFN pathway at various points67,68. The innate immune defences of cells that are infected with wild-type MV are compromised by expression of the virus non-structural C and V proteins, which abrogate IFN signalling by inhibiting STAT phosphorylation69 and its translocation to the nucleus70,71.
In the first-generation oncolytic MVs that are currently being tested in clinical trials, the V protein carries a mutation that renders it non-functional, making the viruses highly susceptible to IFN72. Importantly, several human ovarian carcinoma cells have been shown to be capable of inducing IFN and controlling the replication of these viruses, thereby limiting their oncolytic efficacy. To increase oncolysis, IFN susceptibility was reversed by replacing the mutated gene with the wild-type sequence73. However, increasing oncolysis by including a wild-type gene in an oncolytic virus might enhance its ability to replicate in non-target cells, which raises a potential safety concern.
Identification and characterization at the amino acid level of specific protein interactions, such as between the MV V protein and the IFN machinery, is therefore important. For MV, it is now known that tyrosine 110 and a few neighbouring amino acids in the N terminus of the phosphoprotein (P) (FIG. 3) are necessary for the interaction with STAT1 (REFS 72, 74). It is also known that the cysteine-rich domain of the V protein interacts directly with the helicase MDA5, thereby limiting the cellular recognition of viral RNA and IFN induction75 (FIG. 3). The residues of the V protein that are involved in this interaction are currently being mapped, together with other V residues that are important for the interaction with STAT2 (A. Ramachandran, J.P. Parisien and C.M. Horvath, personal communication).
Analogous to the first-generation oncolytic MVs, the oncolytic Ads ONYX-015 and H101 have some level of cancer-cell specificity owing to deletion of the E1B-55K gene, but both are attenuated in normal cells and cancer cells, which limits their effectiveness in the absence of combined therapy (for example, chemotherapy18). Retargeting the specificity of oncolytic Ad has therefore focused on introducing specific mutations or deletions in E1A to block its binding to Rb and/or p300; this would inhibit virus replication in normal cells, but would allow proliferation and killing in cancer cells without Rb function or with mutations in cell-cycle components76,77. These viruses maintain good potency against a range of tumours and have improved safety in animal models. Furthermore, combinations of E1A and E1B mutations have generated a novel Ad that seems to be more cancer-cell-specific than the single-mutant viruses44.
In summary, knowledge of the specific interactions between viral and cellular proteins lays the foundations to ablate individual replicative functions of oncolytic viruses (including immune evasion and inhibition of apoptosis) and attenuate replication in normal, but not in certain cancerous, cells. This balanced attenuation will generate oncolytic viruses that are specific for the unique IFN and/or cell-cycle deficiencies of a given cancer type and, eventually, for the tumours of individual patients with cancer.
Arming viruses
Arming oncolytic viruses that are inefficient in pre-clinical models or clinical trials with genes that encode prodrug convertases or therapeutic proteins can enhance their potency. This approach is particularly interesting when it is combined with other therapeutic modalities (discussed below). One intrinsic limitation of certain oncolytic approaches is that therapeutic gene delivery is limited to infected cells. If the effect of the encoded protein is cell autonomous (for example, a tumour suppressor protein such as p53), arming will only kill the primary infected cell. By contrast, if the protein is secreted, for example, a cytokine, it can have systemic efficacy, but also side effects. Other proteins, such as thymidine kinase (TK), can have bystander effects that kill neighbouring cells without acting systemically. Immunostimulatory arming proteins, including cancer antigens and cytokines, can also have systemic effects by activating immune responses against cancer cells. It is also possible that cell-autonomous cell-killing genes prime the immune system by generating apoptotic or necrotic cancer cell remnants that can be taken up by antigen-presenting cells for cross-presentation and activation of T cells against cancer cells.
Prodrug convertase transgenes
Prodrug convertases are enzymes that metabolize non-toxic substrates and convert them into lethal drugs that can act within the infected cell, locally or, in some cases, systemically. Early examples include the use of HSV TK to sensitize cells to the drug ganciclovir78,79. This approach adds tumour specificity, as only cells that are dividing (such as cancer cells) are killed by the TK-activated drug. Similar approaches have used other convertases, such as cytosine deaminase80, or have combined two convertases into one protein, such as the TK–cytosine deaminase hybrid protein81, to apply different layers of cancer specificity and tumour killing.
Pro-apoptotic transgenes
Arming oncolytic viruses with transgenes that are capable of inducing apoptosis is an attractive strategy for improving anti-cancer activity. In combination with replicative viruses, however, apoptosis is a double-edged sword. Premature apoptosis of infected cells can reduce virus progeny yields, thereby counteracting the oncolytic activity of the virus. By contrast, apoptosis that is induced at late stages of virus infection, when the progeny virions are matured, can improve virus release from infected cells82 and enhance progeny spread and anti-cancer efficacy. Oncolytic Ads that are engineered to express TRAIL (tumour-necrosis-factor-related apoptosis-inducing ligand) are more oncolytic than the parental viruses in cancer cell lines and in animal tumour models83–85. Arming oncolytic Ads with another pro-apoptotic transgene, p53, resulted in increased apoptosis in vitro, but not in increased anti-tumour activity in vivo86,87. Therefore, thorough analysis of the expression levels of pro-apoptotic transgenes and the sensitivity to apoptosis of target cancer cells is required to sustain synergistic interactions between the induction of apoptosis and virus-replication-mediated oncolysis.
Immuno-activating transgenes
A complementary approach is to disarm Ad in normal cells by expressing gene products that can further protect normal cells from the leaky toxicity of standard oncolytic Ad. Examples include the delivery of type I or II IFNs or cytokines such as granulocyte–macrophage colony-stimulating factor88. A more recent application arms an Ad in cancer cells and disarms it in normal cells. In this case, a KD3 Ad that was targeted by mutations in E1A was armed by ADP overexpression. When this KD3 virus was disarmed by expressing an IFN-α gene, its anti-tumour activity was higher than that of the parental virus14. IFN expression also suppressed virus replication in normal cells, but not cancer cells. When tested in vivo, the disarmed KD3 virus mediated better tumour killing and drastically reduced liver toxicity. Although IFN expression disarmed Ad that innately resisted IFN, it should be noted that this strategy cannot be applied to viruses that are highly susceptible to IFN expression. This selective arming approach provides proof of principle for similar modifications in Ad and other viruses, provided that they can propagate genomes with multiple gene additions.
Shielding oncolytic viruses
Therapy of metastatic disease in patients with intact immunity89 will remain a challenge, even for effectively reprogrammed oncolytic viruses. This fact can be under-appreciated when viruses are initially tested against human tumours that are grown in immunodeficient mice. Conversely, testing in an immunocompetent animal model has its own caveats, as animal cell tumours might not recapitulate the biology of their human counterparts. Increased therapeutic applicability would be possible with strategies that help viruses and host immunity co-exist or even interact synergistically.
Temporary immunosuppression
Given that many oncolytic viruses are derived from human viruses, the presence of pre-existing neutralizing antibodies in patients with cancer can rapidly inactivate incoming viruses after injection. Even when no antibodies are present, the first injection of an oncolytic virus will induce neutralizing antibodies that can quench the activity of subsequent injections (FIG. 5). Moreover, the innate and cellular immune responses will combat oncolytic viruses. Although this might contribute to the killing of infected cancer cells, it might also lead to the lysis of normal cells in which the virus is replicating. These cellular responses might also reduce the amount of virus that is produced by infected cells and limit virus spread to adjacent tumour cells.
Figure 5. Strategies to improve oncolytic virus efficacy.

a | Shielding the virus against antibodies. Pre-existing neutralizing antibodies in humans can interfere with efficacy. Changing virus serotypes and coating particles with shielding polymers can address the neutralizing-antibody problem. b | Transient immunosuppression of the host. Infected cells can be attacked by macrophages, T cells and natural killer cells. Transient immunosuppression interferes with the activation and ability of these cells to recognize and/or kill infected cells and restrict oncolytic efficacy.
One strategy to limit the effects of neutralizing antibodies is to develop virotherapy protocols for individuals with low levels of antibodies; for example, patients with multiple myeloma90. An alternative strategy uses the immunosuppressive side effects of chemotherapeutics such as cyclophosphamide (CPA) to increase oncolysis through combination therapy91. CPA is a prodrug that is activated in the liver to a potent DNA-alkylating agent. In addition to its use as a chemotherapeutic for some cancers, CPA is also immunosuppressive, and has therefore been used with several viruses to downregulate immune cells in the brain and facilitate oncolytic spread within tumours92. A more recent approach has used the immuno-suppressive agent rapamycin to augment oncolytic activity. During organ transplants, rapamycin can bind FK-binding protein 12 to inactivate the mTOR pathway, thereby blocking lymphocyte activation. When applied with the oncolytic poxvirus myxoma virus, rapamycin enabled the virus to infect and kill cancer cells that were normally refractory to virus activity and increased the oncolytic potency in vivo in immunocompetent animals93,94. It is important to note that, if complete and prolonged, immunosuppression can allow for some proliferation of the injected cancer cells95. Thus, the use of reprogrammed viruses with many different levels of targeting and replication controls reduces certain risks of complete and prolonged immunosuppression, but not all of them.
Biological and chemical shields
One approach to evade neutralizing antibodies is to change the capsid and therefore switch the serotype of the virus. For example, mice that are administered with Ad2 serotype vectors generate potent neutralizing antibodies against Ad2 that drastically reduce transgene expression after subsequent Ad2 administrations96. However, if an Ad2 vector is used for the first round of transduction and then an Ad5 vector is used for the second round, there is little reduction in transduction, because the Ad2-specific antibodies do not overtly neutralize the Ad5 serotype96. Similarly, in baboons, serotype switching between Ad2 and Ad5 vectors allowed repeat administration in the presence of neutralizing antibodies that were generated against the first vector97. More recent efforts have recruited Ads from uncommon human serotypes or from other species to evade pre-existing and vector-induced antibody and T-cell responses98,99. This ‘sheep in wolf’s clothing’ approach will evade pre-existing antibodies in humans, but each new vector will generate its own neutralizing antibodies, necessitating the use of additional serotypes for later injections.
Analogously, the serotype of enveloped viruses has been switched. In an effort to develop an effective AIDS vaccine based on live-attenuated VSV, boosting was accomplished using vectors that expressed glycoproteins from different VSV serotypes100. MV is a monotypic virus without serotypes, but it has been shown that the envelope glycoproteins of MV and the related morbillivirus canine distemper virus (CDV) can be exchanged to produce chimeric viruses that evade pre-existing immunity101 (J. Lampe, G. Ungerechts and R.C., unpublished observations).
An alternative approach is to chemically shield viruses with polymers such as polyethylene glycol (PEG) or poly-(N-(2-hydroxypropyl)methacrylamide) (pHPMA). These hydrophilic polymers can be chemically cross-linked to viruses to shield them from pre-existing antibody responses and reduce new antibody and T-cell responses102–107 (BOX 1). Although polymer coating can markedly reduce virus infection, at least for Ad, this is largely an in vitro effect that is not always replicated in vivo104. Indeed, shielding can, in some cases, enhance tumour infection by reducing virus uptake into normal tissues108. Chemical cross-linking of polymers to icosahedral capsids is a well-established technique, but how tractable different enveloped viruses will be to polymer coating remains to be determined.
An additional prospect for shielding viruses is the use of ex vivo infected cells as carriers for oncolytic viruses. The original delivery paradigm, which was based on the injection of virus directly into the bloodstream, has not yet efficiently confronted issues such as the natural tendency of viruses to traffic to the liver, spleen and lung. Success has been demonstrated with various cell types, including mesenchymal cells, T cells and monocytes109,110. Recent work has focused on myeloma cells as carriers for oncolytic VSV in the therapy of multiple myeloma, a disseminated malignancy that is marked by defined trafficking of malignant plasma cells. Myeloma cells were capable of delivering replication-competent VSV to sites of malignancy after supra-lethal doses of ionizing radiation in an orthotopic mouse model110. Cell carriers have the potential to increase the efficiency of oncolytic virotherapy by shielding the virus from the immune system and actively trafficking the virus to the sites of malignancy.
Current clinical trials: combination regimens
A fundamental paradigm of cancer therapy is that no single drug or treatment will cure cancer. Therefore, most therapeutic regimens for cancer are based on combinations of drugs, radiation and surgery to maximize patient survival. As oncolytic viruses have shown promise for cancer therapy, but have so far provided incomplete cancer cures, the field has moved towards combining these viruses with traditional therapies.
An excellent example is the improved efficacy of ONYX-015 for head and neck cancer when combined with 5-fluorouracil and cisplatin111. H101 has shown similar effects on head and neck cancer when combined with chemotherapy (reviewed in REF. 18). Additional promising results for H101 and chemotherapy have been reported in the press, but await peer review. Another approach that was championed by the Freytag group112 is the combination of oncolytic Ad with radiation and chemotherapy. In this case, oncolytic Ad that expressed two prodrug convertases was delivered into prostate and other types of cancers together with local radiation therapy with promising results112. Subsequent work has added more layers to combination therapy by additionally arming the virus with ADP113.
Combination regimens are also being developed for other viral systems. An interesting perspective is the reprogramming of oncolytic vectors to take advantage of various components of current cancer therapy regimens. For example, CD20 antibodies are commonly used in the treatment of CD20-positive NHL and, together with fludarabine phosphate and CPA, constitute the FCR regimen, a front-line treatment for selected NHL. As an alternative to the CD20 antibody, a CD20-targeted MV was generated. This vector was also armed with the prodrug convertase purine nucleotide phosphorylase, which converts fludarabine phosphate to a highly diffusible substance that is capable of efficiently killing bystander cells. The CD20-targeted and convertase-armed MV was shown to synergize with fludarabine and to be effective after systemic inoculation in a mantle cell lymphoma xenograft model114. The next logical step is to synchronize vector delivery with CPA administration to open a broader window of therapeutic opportunity. Thus, studies that are based on the combination of oncolytic viruses with proven therapeutics could facilitate the integration of these vectors into current cancer regimens.
The future: safety and efficacy
Five steps facilitate the reprogramming of viruses into effective cancer therapeutics. BOX 2 explains how these steps support the translation of promising therapeutics into clinical practice. To serve the needs of the patient with cancer, efficacy and safety go hand in hand. Engineering more effective, immunoevasive viruses must be performed with careful thought to protect not just the patient, but also his or her contacts.
Box 2. Five steps for reprogramming viruses into cancer therapeutics.
Know the virus
Characterize the tropism determinants of the virus of interest, if possible at the amino acid level. Detailed knowledge of as many relevant interactions between viral and cellular proteins as possible is the most important prerequisite for successful reprogramming.
Know the cancer
Virologists should be aware of the most promising cancers for possible treatment with oncolytic vectors. Preferred cancers include malignancies that are derived from cell types in which the parental virus spreads efficiently.
Move rapidly to pre-clinical models
Viruses kill cultivated cells efficiently, but the reduction of tumours in mice is a better test for efficacy. Keep focused on the final goal, work with reporter or tracker genes and define objective parameters of safety and efficacy.
Combine reprogramming principles
Work on different layers of targeting individually, and then combine them. Characterize how your targeted oncolytic virus can be shielded from the immune system. Arm it with a prodrug convertase that synergizes with a chemotherapeutic that is in clinical use and document the efficacy of different therapeutic regimens.
In the end, only efficacy and safety in humans count
To have experimental correlates of human efficacy would be wonderful, but without good animal models we are always guessing. Translate promising therapeutics from bench to bedside in a timely manner.
Careful thought must be given to which genes should be engineered into an infectious agent. The introduction of immunosuppressive or immune- skewing genes into replication-competent viruses needs to be weighed most heavily. The insertion of the interleukin-4 gene into the mousepox ectromelia virus provides a cautionary example of what can go wrong115: the virus suppressed the ability of the immune system to kill infected cells and blocked the production of memory immune responses, thereby enhancing virulence. The consequences of engineering a virus that harms the patient or spreads to other individuals are obvious and unacceptable.
Alternative approaches to safeguard the replication of oncolytic viruses include transient immuno-suppression, which can be withdrawn if problems arise, or drug therapies that can specifically and transiently block virus-specific immune responses, such as mono-clonal antibodies that target specific cells or immune proteins. Using replication-defective viruses that express potent transgenes to complement replication-competent, but less virulent, oncolytic viruses might also be safe and efficient116. Another important area of research is the development of oncolytic viruses with life cycles that can be terminated by a prodrug114.
In summary, oncolytic viruses hold great promise as potent, self-amplifying cancer therapeutics. Virotherapy is attractive because there is no cross-resistance with chemotherapy and radiation therapies. Ad H101 is the first reprogrammed virus to be approved as a cancer drug; it has been administered to hundreds of patients with head and neck carcinoma in China, in combination with chemotherapy, and survival statistics and clinical-benefit data might support the worldwide usage of this virus. In the next few years, oncolytic viruses with increasingly sophisticated targeting combinations should become available for clinical trials. The future is getting brighter for patients with cancer.
Acknowledgments
R.C. has been supported by the National Institutes of Health (grant CA90636) and the Alliance of Cancer Gene Therapy. M.A.B. has been supported by the Department of Defense (grant W81XWH-05-1-0269) and the Susan G. Komen Foundation (grant BCTR0504036).
Glossary
- Glioblastoma
The most common and aggressive type of primary brain tumour. Treatment can involve chemotherapy, radiotherapy and surgery, none of which provide a cure
- Matrix metalloproteinase
A proteolytic enzyme that degrades the extracellular matrix and has important roles in tissue remodelling and tumour metastasis
- Fibre protein
In adenoviruses, a trimeric antennae-like protein that projects from the virion and mediates initial cell-binding events by different serotypes with CAR, CD46 and, perhaps, other receptors
- Penton base
The pentameric protein base of the fibre trimer. For some adenoviruses, this protein mediates interactions with cellular integrins for binding and cell entry
- Non-Hodgkin’s lymphoma
A diverse group of cancers that arise from lymphocytes and have varying courses, treatments and prognoses
- Myeloma
A type of cancer of the plasma cells (immune-system cells in bone marrow that produce antibodies) that is often called multiple myeloma
Footnotes
DATABASES
Entrez Genome Project: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=genomeprj
CDV | HIV-1 | HSV1 | MV | NDV
FURTHER INFORMATION
Roberto Cattaneo’s homepage: http://mayoresearch.mayo.edu/mayo/research/staff/cattaneo_r.cfm
Michael A. Barry’s homepage: http://mayoresearch.mayo.edu/mayo/research/staff/barry_ma.cfm
Virology and Gene Therapy Ph.D. Program homepage: http://www.mayo.edu/mgs/vgt.html
References
- 1.Dock G. The influence of complicating diseases upon leukemia. Am J Med Sci. 1904;127:563–592. [Google Scholar]
- 2.Sinkovics J, Horvath J. New developments in the virus therapy of cancer: a historical review. Intervirology. 1993;36:193–214. doi: 10.1159/000150339. [DOI] [PubMed] [Google Scholar]
- 3.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–659. doi: 10.1038/sj.mt.6300108. A compelling narrative of the first 100 years of oncolytic viruses. [DOI] [PubMed] [Google Scholar]
- 4.Hoster H, Zanes R, Vonhaam E. The association of “viral” hepatitis and Hodgkin’s disease. Cancer Res. 1949;9:473–480. [PubMed] [Google Scholar]
- 5.Moore AE. Viruses with oncolytic properties and their adaptation to tumors. Ann NY Acad Sci. 1952;54:945–952. doi: 10.1111/j.1749-6632.1952.tb39969.x. [DOI] [PubMed] [Google Scholar]
- 6.Southam CM, Moore AE. Clinical studies of viruses as antineoplastic agents with particular reference to Egypt 101 virus. Cancer. 1952;5:1025–1034. doi: 10.1002/1097-0142(195209)5:5<1025::aid-cncr2820050518>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 7.Newman W, Southam CM. Virus treatment in advanced cancer: a pathological study of fifty-seven cases. Cancer. 1953;7:106–118. doi: 10.1002/1097-0142(195401)7:1<106::aid-cncr2820070112>3.0.co;2-l. A clinical study that seems alarming in the context of current ethical standards. [DOI] [PubMed] [Google Scholar]
- 8.Huebner RJ, Rowe WP, Schatten WE, Smith RR, Thomas LB. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer. 1956;9:1211–1218. doi: 10.1002/1097-0142(195611/12)9:6<1211::aid-cncr2820090624>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 9.Suskind RG, Huebner RJ, Rowe WP, Love R. Viral agents oncolytic for human tumors in heterologous host; oncolytic effect of Coxsackie B viruses. Proc Soc Exp Biol Med. 1957;94:309–318. doi: 10.3181/00379727-94-22931. [DOI] [PubMed] [Google Scholar]
- 10.Pond AR, Manuelidis EE. Oncolytic effect of poliomyelitis virus on human epidermoid carcinoma (hela tumor) heterologously transplanted to guinea pigs. Am J Pathol. 1964;45:233–249. [PMC free article] [PubMed] [Google Scholar]
- 11.Hammon WM, Yohn DS, Casto BC, Atchison RW. Oncolytic potentials of nonhuman viruses for human cancer. J Natl Cancer Inst. 1963;31:329–345. [PubMed] [Google Scholar]
- 12.Molomut N, Padnos M. Inhibition of transplantable and spontaneous murine tumours by the M-P virus. Nature. 1965;208:948–950. doi: 10.1038/208948a0. [DOI] [PubMed] [Google Scholar]
- 13.Yohn DS, Hammon WM, Atchison RW, Casto BC. Oncolytic potentials of nonhuman viruses for human cancer. II Effects of five viruses on heterotransplantable human tumors. J Natl Cancer Inst. 1968;41:523–529. [PubMed] [Google Scholar]
- 14.Shashkova EV, Spencer JF, Wold WS, Doronin K. Targeting interferon-α increases antitumor efficacy and reduces hepatotoxicity of E1A-mutated spread-enhanced oncolytic adenovirus. Mol Ther. 2007;15:598–607. doi: 10.1038/sj.mt.6300064. [DOI] [PubMed] [Google Scholar]
- 15.Doyle TC, Burns SM, Contag CH. In vivo bioluminescence imaging for integrated studies of infection. Cell Microbiol. 2004;6:303–317. doi: 10.1111/j.1462-5822.2004.00378.x. [DOI] [PubMed] [Google Scholar]
- 16.Piwnica-Worms D, Schuster DP, Garbow JR. Molecular imaging of host–pathogen interactions in intact small animals. Cell Microbiol. 2004;6:319–331. doi: 10.1111/j.1462-5822.2004.00379.x. [DOI] [PubMed] [Google Scholar]
- 17.Peng KW, et al. Pharmacokinetics of oncolytic measles virotherapy: eventual equilibrium between virus and tumor in an ovarian cancer xenograft model. Cancer Gene Ther. 2006;13:732–738. doi: 10.1038/sj.cgt.7700948. [DOI] [PubMed] [Google Scholar]
- 18.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nature Clin Pract Oncol. 2007;4:101–117. doi: 10.1038/ncponc0736. A current review of clinical trials of oncolytic viruses. [DOI] [PubMed] [Google Scholar]
- 19.Klenk HD, Garten W. Host cell proteases controlling virus pathogenicity. Trends Microbiol. 1994;2:39–43. doi: 10.1016/0966-842x(94)90123-6. [DOI] [PubMed] [Google Scholar]
- 20.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nature Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 21.Morling FJ, Peng KW, Cosset FL, Russell SJ. Masking of retroviral envelope functions by oligomerizing polypeptide adaptors. Virology. 1997;234:51–61. doi: 10.1006/viro.1997.8628. [DOI] [PubMed] [Google Scholar]
- 22.Peng KW, Vile R, Cosset FL, Russell S. Selective transduction of protease-rich tumors by matrix-metalloproteinase-targeted retroviral vectors. Gene Ther. 1999;6:1552–1557. doi: 10.1038/sj.gt.3300982. [DOI] [PubMed] [Google Scholar]
- 23.Kinoh H, et al. Generation of a recombinant Sendai virus that is selectively activated and lyses human tumor cells expressing matrix metalloproteinases. Gene Ther. 2004;11:1137–1145. doi: 10.1038/sj.gt.3302272. [DOI] [PubMed] [Google Scholar]
- 24.Springfeld C, et al. Oncolytic efficacy and enhanced safety of measles virus activated by tumor-secreted matrix metalloproteinases. Cancer Res. 2006;66:7694–7700. doi: 10.1158/0008-5472.CAN-06-0538. Demonstration of both the efficacy and safety of tumour protease-activated viruses. [DOI] [PubMed] [Google Scholar]
- 25.Andreasaen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57:25–40. doi: 10.1007/s000180050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nature Rev Genet. 2007;8:573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campos SK, Barry MA. Current advances and future challenges in adenoviral vector biology and targeting. Curr Gene Ther. 2007;7:189–204. doi: 10.2174/156652307780859062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navaratnarajah CK, Leonard VHL, Cattaneo R. Measles Virus. Springer; Heidelberg: in the press. [Google Scholar]
- 29.Lamb RA, Paterson RG, Jardetzky TS. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology. 2006;344:30–37. doi: 10.1016/j.virol.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tatsuo H, Ono N, Tanaka K, Yanagi Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 2000;406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 31.Dorig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- 32.Naniche D, et al. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol. 1993;67:6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vongpunsawad S, Oezgun N, Braun W, Cattaneo R. Selectively receptor-blind measles viruses: identification of residues necessary for SLAM- or CD46-induced fusion and their localization on a new hemagglutinin structural model. J Virol. 2004;78:302–313. doi: 10.1128/JVI.78.1.302-313.2004. Proof of principle for the concept of generating selectively receptor-blind viruses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider U, Bullough F, Vongpunsawad S, Russell SJ, Cattaneo R. Recombinant measles viruses efficiently entering cells through targeted receptors. J Virol. 2000;74:9928–9936. doi: 10.1128/jvi.74.21.9928-9936.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammond AL, et al. Single-chain antibody displayed on a recombinant measles virus confers entry through the tumor-associated carcinoembryonic antigen. J Virol. 2001;75:2087–2096. doi: 10.1128/JVI.75.5.2087-2096.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bucheit AD, et al. An oncolytic measles virus engineered to enter cells through the CD20 antigen. Mol Ther. 2003;7:62–72. doi: 10.1016/s1525-0016(02)00033-3. [DOI] [PubMed] [Google Scholar]
- 37.Peng KW, et al. Oncolytic measles virus displaying a single chain antibody against CD38, a myeloma cell marker. Blood. 2003;101:2557–2562. doi: 10.1182/blood-2002-07-2195. [DOI] [PubMed] [Google Scholar]
- 38.Ungerechts G, et al. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol Ther. 2007;15:1991–1997. doi: 10.1038/sj.mt.6300291. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura T, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nature Biotechnol. 2005;23:209–214. doi: 10.1038/nbt1060. The first demonstration of oncolytic efficacy after detargeting and retargeting at the receptor-recognition level. [DOI] [PubMed] [Google Scholar]
- 40.Zhou G, Roizman B. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13α2 receptor. Proc Natl Acad Sci USA. 2006;103:5508–5513. doi: 10.1073/pnas.0601258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hedley SJ, et al. An adenovirus vector with a chimeric fiber incorporating stabilized single chain antibody achieves targeted gene delivery. Gene Ther. 2006;13:88–94. doi: 10.1038/sj.gt.3302603. [DOI] [PubMed] [Google Scholar]
- 42.Belousova N, et al. Genetically targeted adenovirus vector directed to CD40-expressing cells. J Virol. 2003;77:11367–11377. doi: 10.1128/JVI.77.21.11367-11377.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24:7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- 44.Kim J, Kim JH, Choi KJ, Kim PH, Yun CO. E1A- and E1B-double mutant replicating adenovirus elicits enhanced oncolytic and antitumor effects. Hum Gene Ther. 2007;18:773–786. doi: 10.1089/hum.2006.167. [DOI] [PubMed] [Google Scholar]
- 45.Kim E, et al. Ad-mTERT-Δ19, a conditional replication-competent adenovirus driven by the human telomerase promoter, selectively replicates in and elicits cytopathic effect in a cancer cell-specific manner. Hum Gene Ther. 2003;14:1415–1428. doi: 10.1089/104303403769211637. [DOI] [PubMed] [Google Scholar]
- 46.Kuppuswamy M, et al. Oncolytic adenovirus that overproduces ADP and replicates selectively in tumors due to hTERT promoter-regulated E4 gene expression. Gene Ther. 2005;12:1607–1617. doi: 10.1038/sj.gt.3302581. [DOI] [PubMed] [Google Scholar]
- 47.Barker DD, Berk AJ. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology. 1987;156:107–121. doi: 10.1016/0042-6822(87)90441-7. [DOI] [PubMed] [Google Scholar]
- 48.Bischoff JR, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 49.Goodrum FD, Ornelles D. A p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J Virol. 1998;72:9479–9490. doi: 10.1128/jvi.72.12.9479-9490.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Shea CC, Soria C, Bagus B, McCormick F. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell. 2005;8:61–74. doi: 10.1016/j.ccr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 51.O’Shea CC, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 52.Chung RY, Saeki Y, Chiocca EA. B-myb promoter retargeting of herpes simplex virus γ34.5 gene-mediated virulence toward tumor and cycling cells. J Virol. 1999;73:7556–7564. doi: 10.1128/jvi.73.9.7556-7564.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markert JM, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 54.Kanai R, et al. Enhanced therapeutic efficacy of G207 for the treatment of glioma through Musashi1 promoter retargeting of γ34.5-mediated virulence. Gene Ther. 2006;13:106–116. doi: 10.1038/sj.gt.3302636. [DOI] [PubMed] [Google Scholar]
- 55.Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65:2832–2839. doi: 10.1158/0008-5472.CAN-04-3227. [DOI] [PubMed] [Google Scholar]
- 56.Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. A current and complete review of the diverse strategies that are adopted by viruses to control the IFN system. [DOI] [PubMed] [Google Scholar]
- 57.Linge C, Gewert D, Rossmann C, Bishop JA, Crowe JS. Interferon system defects in human malignant melanoma. Cancer Res. 1995;55:4099–4104. [PubMed] [Google Scholar]
- 58.Matin SF, et al. Impaired α-interferon signaling in transitional cell carcinoma: lack of p48 expression in 5637 cells. Cancer Res. 2001;61:2261–2266. [PubMed] [Google Scholar]
- 59.Ralph SJ, et al. Resistance of melanoma cell lines to interferons correlates with reduction of IFN-induced tyrosine phosphorylation. Induction of the anti-viral state by IFN is prevented by tyrosine kinase inhibitors. J Immunol. 1995;154:2248–2256. [PubMed] [Google Scholar]
- 60.Sun WH, et al. Interferon-α resistance in a cutaneous T-cell lymphoma cell line is associated with lack of STAT1 expression. Blood. 1998;91:570–576. [PubMed] [Google Scholar]
- 61.Wong LH, et al. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3γ. J Biol Chem. 1997;272:28779–28785. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- 62.Stojdl DF, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 63.Aaronson DS, Horvath CM. A road map for those who don’t know JAK–STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 64.Darnell JEJ. STATS and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 65.Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 67.Conzelmann KK. Transcriptional activation of α/β interferon genes: interference by nonsegmented negative-strand RNA viruses. J Virol. 2005;79:5241–5248. doi: 10.1128/JVI.79.9.5241-5248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia-Sastre A. Mechanisms of inhibition of the host interferon α-β-mediated antiviral responses by viruses. Microbes Infect. 2002;4:647–655. doi: 10.1016/s1286-4579(02)01583-6. [DOI] [PubMed] [Google Scholar]
- 69.Takeuchi K, Kadota SI, Takeda M, Miyajima N, Nagata K. Measles virus V protein blocks interferon (IFN)-α/β but not IFN-γ signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett. 2003;545:177–182. doi: 10.1016/s0014-5793(03)00528-3. [DOI] [PubMed] [Google Scholar]
- 70.Horvath CM. Weapons of STAT destruction. Interferon evasion by paramyxovirus V protein. Eur J Biochem. 2004;271:4621–4628. doi: 10.1111/j.1432-1033.2004.04425.x. [DOI] [PubMed] [Google Scholar]
- 71.Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol. 2003;77:7635–7644. doi: 10.1128/JVI.77.13.7635-7644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Devaux P, von Messling V, Songsungthong W, Springfeld C, Cattaneo R. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology. 2007;360:72–83. doi: 10.1016/j.virol.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 73.Haralambieva I, et al. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol Ther. 2007;15:588–597. doi: 10.1038/sj.mt.6300076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ohno S, Ono N, Takeda M, Takeuchi K, Yanagi Y. Dissection of measles virus V protein in relation to its ability to block alpha/beta interferon signal transduction. J Gen Virol. 2004;85:2991–2999. doi: 10.1099/vir.0.80308-0. [DOI] [PubMed] [Google Scholar]
- 75.Childs K, et al. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007;359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 76.Doronin K, et al. Tumor-specific, replication-competent adenovirus vectors overexpressing the adenovirus death protein. J Virol. 2000;74:6147–6155. doi: 10.1128/jvi.74.13.6147-6155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fueyo J, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 78.Boviatsis EJ, et al. Long-term survival of rats harboring brain neoplasms treated with ganciclovir and a herpes simplex virus vector that retains an intact thymidine kinase gene. Cancer Res. 1994;54:5745–5751. [PubMed] [Google Scholar]
- 79.Chase M, Chung RY, Chiocca EA. An oncolytic viral mutant that delivers the CYP2B1 transgene and augments cyclophosphamide chemotherapy. Nature Biotechnol. 1998;16:444–448. doi: 10.1038/nbt0598-444. [DOI] [PubMed] [Google Scholar]
- 80.Searle PF, et al. Nitroreductase: a prodrug-activating enzyme for cancer gene therapy. Clin Exp Pharmacol Physiol. 2004;31:811–816. doi: 10.1111/j.1440-1681.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- 81.Freytag SO, Rogulski KR, Paielli DL, Gilbert JD, Kim JH. A novel three-pronged approach to kill cancer cells selectively: concomitant viral, double suicide gene, and radiotherapy. Hum Gene Ther. 1998;9:1323–1333. doi: 10.1089/hum.1998.9.9-1323. [DOI] [PubMed] [Google Scholar]
- 82.Mi J, Li Z, Ni S, Steinwaerder D, Lieber A. Induced apoptosis supports spread of adenovirus vectors in tumors. Hum Gene Ther. 2001;12:1343–1352. doi: 10.1089/104303401750270995. [DOI] [PubMed] [Google Scholar]
- 83.Sova P, et al. A tumor-targeted and conditionally replicating oncolytic adenovirus vector expressing TRAIL for treatment of liver metastases. Mol Ther. 2004;9:496–509. doi: 10.1016/j.ymthe.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 84.Dong F, et al. Eliminating established tumor in nu/nu nude mice by a TRAIL-armed oncolytic adenovirus. Clin Cancer Res. 2006;12:5224–5230. doi: 10.1158/1078-0432.CCR-06-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ren XW, et al. A tumor-specific conditionally replicative adenovirus vector expressing TRAIL for gene therapy of hepatocellular carcinoma. Cancer Gene Ther. 2006;13:159–168. doi: 10.1038/sj.cgt.7700868. [DOI] [PubMed] [Google Scholar]
- 86.Sauthoff H, et al. Modification of the p53 transgene of a replication-competent adenovirus prevents mdm2- and E1b-55kD-mediated degradation of p53. Cancer Gene Ther. 2006;13:686–695. doi: 10.1038/sj.cgt.7700936. [DOI] [PubMed] [Google Scholar]
- 87.Idema S, et al. AdΔ24 and the p53-expressing variant AdΔ24-p53 achieve potent anti-tumor activity in glioma when combined with radiotherapy. J Gene Med. 2007;9:1046–1056. doi: 10.1002/jgm.1113. [DOI] [PubMed] [Google Scholar]
- 88.Zhang JF, et al. Treatment of a human breast cancer xenograft with an adenovirus vector containing an interferon gene results in rapid regression due to viral oncolysis and gene therapy. Proc Natl Acad Sci USA. 1996;93:4513–4518. doi: 10.1073/pnas.93.9.4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vile R, Ando D, Kirn D. The oncolytic virotherapy treatment platform for cancer: unique biological and biosafety points to consider. Cancer Gene Ther. 2002;9:1062–1067. doi: 10.1038/sj.cgt.7700548. [DOI] [PubMed] [Google Scholar]
- 90.Peng KW, et al. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98:2002–2007. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]
- 91.Ikeda K, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nature Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 92.Lamfers ML, et al. Cyclophosphamide increases transgene expression mediated by an oncolytic adenovirus in glioma-bearing mice monitored by bioluminescence imaging. Mol Ther. 2006;14:779–788. doi: 10.1016/j.ymthe.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stanford MM, Barrett JW, Nazarian SH, Werden S, McFadden G. Oncolytic virotherapy synergism with signaling inhibitors: rapamycin increases myxoma virus tropism for human tumor cells. J Virol. 2007;81:1251–1260. doi: 10.1128/JVI.01408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lun XQ, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67:8818–8827. doi: 10.1158/0008-5472.CAN-07-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yamauchi K, et al. Induction of cancer metastasis by cyclophosphamide pretreatment of host mice: an opposite effect of chemotherapy. Cancer Res. 2008;68:516–520. doi: 10.1158/0008-5472.CAN-07-3063. [DOI] [PubMed] [Google Scholar]
- 96.Parks R, Evelegh C, Graham F. Use of helper-dependent adenoviral vectors of alternative serotypes permits repeat vector administration. Gene Ther. 1999;6:1565–1573. doi: 10.1038/sj.gt.3300995. [DOI] [PubMed] [Google Scholar]
- 97.Morral N, et al. Administration of helper-dependent adenoviral vectors and sequential delivery of different vector serotype for long-term liver-directed gene transfer in baboons. Proc Natl Acad Sci USA. 1999;96:12816–12821. doi: 10.1073/pnas.96.22.12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hemminki A, et al. A canine conditionally replicating adenovirus for evaluating oncolytic virotherapy in a syngeneic animal model. Mol Ther. 2003;7:163–173. doi: 10.1016/s1525-0016(02)00049-7. [DOI] [PubMed] [Google Scholar]
- 99.Xiang ZQ, et al. Oral vaccination of mice with adenoviral vectors is not impaired by preexisting immunity to the vaccine carrier. J Virol. 2003;77:10780–10789. doi: 10.1128/JVI.77.20.10780-10789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rose NF, et al. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell. 2001;106:539–549. doi: 10.1016/s0092-8674(01)00482-2. [DOI] [PubMed] [Google Scholar]
- 101.von Messling V, Zimmer G, Herrler G, Haas L, Cattaneo R. The hemagglutinin of canine distemper virus determines tropism and cytopathogenicity. J Virol. 2001;75:6418–6427. doi: 10.1128/JVI.75.14.6418-6427.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Croyle MA, Chirmule N, Zhang YA, Wilson JM. “Stealth” adenoviruses blunt cell-mediated and humoral immune responses against the virus and allow for significant gene expression upon readministration in the lung. J Virol. 2001;75:4792–4801. doi: 10.1128/JVI.75.10.4792-4801.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fisher KD, et al. Polymer-coated adenovirus permits efficient retargeting and evades neutralizing antibodies. Gene Ther. 2001;8:341–348. doi: 10.1038/sj.gt.3301389. [DOI] [PubMed] [Google Scholar]
- 104.Hofherr SE, Mok H, Gushiken FC, Lopez JA, Barry MA. Polyethylene glycol modification of adenovirus reduces platelet activation, endothelial cell activation, and thrombocytopenia. Hum Gene Ther. 2007;18:837–848. doi: 10.1089/hum.2007.0051. [DOI] [PubMed] [Google Scholar]
- 105.Lanciotti J, et al. Targeting adenoviral vectors using heterofunctional polyethylene glycol FGF2 conjugates. Mol Ther. 2003;8:99–107. doi: 10.1016/s1525-0016(03)00139-4. [DOI] [PubMed] [Google Scholar]
- 106.Mok H, Palmer DJ, Ng P, Barry MA. Evaluation of polyethylene glycol modification of first-generation and helper-dependent adenoviral vectors to reduce innate immune responses. Mol Ther. 2005;11:66–79. doi: 10.1016/j.ymthe.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 107.O’Riordan CR, et al. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum Gene Ther. 1999;10:1349–1358. doi: 10.1089/10430349950018021. [DOI] [PubMed] [Google Scholar]
- 108.Fisher KD, et al. Passive tumor targeting of polymer-coated adenovirus for cancer gene therapy. J Drug Target. 2007;15:546–551. doi: 10.1080/10611860701501014. [DOI] [PubMed] [Google Scholar]
- 109.Harrington K, Vile R. Virus smuggling, tax evasion and tumor assassination. Nature Med. 2006;12:507–509. doi: 10.1038/nm0506-507. [DOI] [PubMed] [Google Scholar]
- 110.Munguia A, Ota T, Miest T, Russell SJ. Cell carriers to deliver oncolytic viruses to sites of myeloma tumor growth. Gene Ther. 2008;15:797–806. doi: 10.1038/gt.2008.45. [DOI] [PubMed] [Google Scholar]
- 111.Khuri FR, et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nature Med. 2000;6:879–885. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- 112.Freytag SO, et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal readiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003;63:7497–7506. [PubMed] [Google Scholar]
- 113.Barton KN, et al. Second-generation replication-competent oncolytic adenovirus armed with improved suicide genes and ADP gene demonstrates greater efficacy without increased toxicity. Mol Ther. 2006;13:347–356. doi: 10.1016/j.ymthe.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 114.Ungerechts G, et al. Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res. 2007;67:10939–10947. doi: 10.1158/0008-5472.CAN-07-1252. An example of how oncolytic vectors can be reprogrammed to take advantage of various components of current cancer therapy regimens. [DOI] [PubMed] [Google Scholar]
- 115.Jackson RJ, et al. Expression of mouse interleukin-4 by a recombinant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J Virol. 2001;75:1205–1210. doi: 10.1128/JVI.75.3.1205-1210.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shashkova EV, Kuppuswarmy MN, Wold WS, Doronin K. Anticancer activity of oncolytic adenovirus vector armed with IFN-α and ADP is enhanced by pharmacologically controlled expression of TRAIL. Cancer Gene Ther. 2008;15:61–72. doi: 10.1038/sj.cgt.7701107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hashiguchi T, et al. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc Natl Acad Sci USA. 2007;104:19535–19540. doi: 10.1073/pnas.0707830104. [DOI] [PMC free article] [PubMed] [Google Scholar]