

Abstract
Purpose
To extend the results of a phase III trial in non-small cell lung cancer patients with adenocarcinomas harboring EML4-ALK fusion.
Experimental Design
we performed a co-clinical trial in a mouse model comparing the ALK inhibitor crizotinib to the standard-of-care cytotoxic agents docetaxel or pemetrexed.
Results
Concordant with the clinical outcome in humans, crizotinib produced a substantially higher response rate compared to chemotherapy, associated with significantly longer progression-free survival. Overall survival was also prolonged in crizotinib- compared to chemotherapy-treated mice. Pemetrexed produced superior overall survival compared to docetaxel, suggesting that this agent may be the preferred chemotherapy in the ALK population. Additionally, in the EML4-ALK-driven mouse lung adenocarcinoma model, HSP90 inhibition can overcome both primary and acquired crizotinib resistance. Furthermore, HSP90 inhibition, as well as the second-generation ALK inhibitor TAE684, demonstrated activity in newly developed lung adenocarcinoma models driven by crizotinib-insensitive EML4-ALK L1196M or F1174L.
Conclusions
Our findings suggest that crizotinib is superior to standard chemotherapy in ALK inhibitor-naïve disease and support further clinical investigation of HSP90 inhibitors and second-generation ALK inhibitors in tumors with primary or acquired crizotinib resistance.
Introduction
EML4-ALK fusion accounts for approximately 4% of non-small cell lung cancer (NSCLC) (1, 2). Crizotnib (3, 4), an FDA-approved inhibitor of anaplastic lymphoma kinase (ALK), demonstrated efficacy in a phase II clinical trial in lung cancer patients with tumors harboring EML4-ALK rearrangements (5). Despite striking activity in early studies, some reports have also noted impressive activity with chemotherapy in ALK-positive cancers (6, 7). Indeed, retrospective analyses have suggested that time to progression on crizotinib is statistically similar to that achieved by first-line platinum-based chemotherapy (2, 8, 9). Thus, it was unclear if crizotinib is superior to either first or second line chemotherapy in this subset of patients. These uncertainties were recently addressed by a phase III trial comparing crizotinib to chemotherapies in the second-line setting that definitively demonstrated the superiority of crizotinib (10).
Nonetheless, both primary and acquired resistance have been observed in patients treated with crizotinib (11–14). Up to 30% ALK-positive patients do not respond to crizotinib treatment, while those who respond initially will eventually develop acquired resistance after prolonged treatment. Secondary mutations in the ALK kinase domain have been identified in a subset of patients who become insensitive to crizotinib.
Co-clinical trials, in which highly faithful genetically engineered murine cancer models (GEMMs) are carefully randomized and used to mimic human clinical trials, have the potential to provide mechanistic insights that impact the analysis of the concurrent human study (15–20). In recent years, we and others have performed numerous treatment studies in GEMMs leading to identification of clinically relevant biomarkers and novel treatment methods, as well as successful prediction of clinical outcomes (21–26). In particular, we have previously described a co-clinical trial using a murine model recapitulating human NSCLC driven by an activating Kras mutation (21). The murine trial predicted the clinical superiority of combined selumetinib and docetaxel compared to docetaxel alone(27). Importantly, patient stratification and biomarker strategies identified from the murine trial have provided valuable insight for the design of subsequent clinical studies.
In the present analysis, we have performed a murine co-clinical study mimicking the phase III clinical trial in ALK-positive patients with advanced disease who had received prior platinum-doublet-based first-line treatment. In this study, patients were randomized to receive crizotinib or standard second-line therapy, including docetaxel or pemetrexed. Using novel murine models of EML4-ALK NSCLC, we determined the short- and long-term efficacy of crizotinib treatment compared to docetaxel or pemetrexed. The results demonstrate the predictive power of EML4-ALK-driven murine lung adenocarcinoma models and validate their use for studying additional treatments for the ALK population. Toward this end, we explored treatment with an HSP90 inhibitor and a second-generation ALK inhibitor to overcome either primary or acquired crizotinib resistance in order to anticipate their roles in the growing ALK armamentarium.
Materials and Methods
Mice and treatment
Generation of bi-transgenic mice with lung-specific doxycycline-inducible EML4-ALK expression was described previously(28). Mice were subjected to magnetic resonance imaging (MRI) 4–6 weeks after initiation of a doxycycline-containing diet to determine baseline tumor volume. Mice with appropriate tumor burden were randomized to three groups and treated with crizotinib, pemetrexed or docetaxel. Crizotinib was delivered via daily oral feeding at 100 mg/kg in water. A separate group of mice was treated with 50mg/kg crizotinib. Docetaxel was delivered by intra-peritoneal (i.p.) injection at 16 mg/kg every other day for the first two weeks of treatment, and 8 mg/kg every 3 days thereafter. These mice were also treated with the HSP90 inhibitor 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) administered at 20 mg/kg in PBS daily by i.p. injection. The EML4-ALK L1196M- and F1174L-driven mouse models were created using an engineered knock-in system described previously(28), but the Tet-O promoter was replaced by a loxp-flanked stop cassette (LSL). These mice were also treated with 17-DMAG, as well as the second-generation ALK inhibitor, TAE684, delivered by oral feeding at a daily dose of 25 mg/kg.
PET-CT
All FDG-PET/CT studies were performed using a preclinical small-animal PET/CT system (Siemens Inveon). All animals were subjected to identical dietary preparation, warming, injection, and anesthesia protocols, and all images were analyzed with standard data acquisition and image reconstruction parameters. Specifically, each mouse received approximately 14 MBq of 18F-fluorodeoxyglucose (FDG) via intraperitoneal administration. After a one-hour uptake period, mice were anesthetized by inhalation of a mixture of sevoflurane and oxygen. CT (5 minutes) and PET (10 minutes) data were collected, processed and analyzed.
MRI
Animals were anesthetized with 1.5%–2% isoflurane (IsoFlo; Abbott) in 100% oxygen. Both cardiac and respiratory gating were applied to minimize motion effects. Acquisition of the magnetic resonance signal was synchronized with the cardiac and respiratory cycles. MRI protocols optimized for assessing pulmonary parenchyma and vessels in normal mice were adapted for operation at 4.7 Tesla (BioSpec 47/40; Bruker BioSpin).
Quantification
Mouse lung cancer MRI scans were quantified by two operators using 3D Slicer software to determine tumor volume. To account for random variation between operators, Bland-Altman analysis was performed based on quantification results from the two operators on a total of sixteen MRI scan images, as previously described (21), and adapted in this study. Progressive disease (PD) was defined as a >30% tumor burden increase compared to baseline. Partial response (PR) was defined as a >30% decrease in tumor burden compared to baseline. Mice having tumor burden changes within +/− 30% of baseline were considered to have stable disease (SD). Overall survival (OS) was assessed with endpoints of death or moribund condition. Progression-free survival (PFS) was defined as the time to tumor re-growth exceeding 20% of the lowest tumor burden achieved after initial treatment, development of moribund condition or death.
Immunohistochemistry
Antibodies recognizing phospho-Akt (Ser473), and phospho-S6 (Thr389) were obtained from Cell Signaling Technology. Phospho-ERK1/2 (pT185/pY187) antibodies was purchased from Biosource International. Tissue processing and immunohistochemistry (IHC) were performed on tumor samples as previously described (21). To quantify IHC, staining intensity (0–4+) was assessed for 10 microscopic fields, and an average value was derived.
Results
Crizotinib improves initial response of EML4-ALK lung cancer compared to standard-of-care therapy
In an effort to determine sensitivity to commonly used chemotherapeutics, a large cohort of EML-ALK mice was generated and serially observed for tumor formation as previously described (28). One group of tumor-bearing mice was treated with 50 mg/kg/day crizotinib for 2 weeks, which led to stable disease, but did not result in tumor regression (Fig. 1A, Supplemental Figure 1). Additional mice were then assigned treatment with 100 mg/kg/day crizotinib, docetaxel or pemetrexed and tumor response assessed by serial MRI. Tumor regression was observed at the dose of 100 mg/kg/day in the majority of animals (Fig. 1B, Supplemental Fig. 1) with a high rate of partial response (68%; defined as >30% tumor regression) or stable disease (23%). These results indicate that the response of ALK-driven tumors to crizotinib may be dose-dependent and suggest that insufficient ALK inhibition may be one of the mechanisms for primary crizotinib resistance observed clinically. Notably, even at 100 mg/kg/day, very few mice (2/22) achieved near-complete remission (>90% tumor regression). Also of note, 2 of 22 mice exhibited primary resistance (Fig. 1B, Supplemental Fig. 1B). Consistent with the known mechanism of action (28), phospho-ERK, phospho-Akt, phospho-S6, and Stat3 protein expression were reduced in crizotinib-treated tumors (Fig. 1C). Taken together, these data show that crizotinib is highly active in a faithful murine model of ALK-driven NSCLC.
Figure 1. Crizotinib is more efficacious against EML4-ALK lung cancer than chemotherapy.

A, Waterfall plot showing tumor burden changes in EML4-ALK tumor-bearing mice after three weeks of treatment with daily administration of 50 mg/kg crizotinib. MRI scans reflecting tumor volumes before and after treatment were quantified and compared. Each bar represents one mouse. B, Waterfall plots comparing the response of EML4-ALK-driven tumors to 3 weeks of treatment with 100mg/kg crizotinib or docetaxel or pemetrexed. C, Pharmacodynamic studies showing suppression of signaling pathways downstream of ALK, including pSTAT3, pAkt, pERK and pS6. Tumors were harvested within 24 hours after 2 doses of crizotinib at 100 mg/kg and assessed by immunohistochemistry. Quantification represents the average intensity over 10 microscopic fields, as described in the materials and methods. D, Summary of the treatment study in panel B.
The activity of cytotoxic agents was less impressive: no mice treated with docetaxel achieved partial response, but this outcome was achieved with 2 out of 8 mice treated with pemetrexed (Fig. 1B). However, both docetaxel and pemtrexed produced disease stabilization (Fig. 1D). Of note, using the commonly applied “disease control rate” (DCR) metric of complete response + partial response + stable disease (CR+PR+SD), the three study arms appeared comparable (Fig. 1D). These data recapitulate clinical results showing similar rate of disease control between crizotinib and plantinum-based chemotherapy. Nonetheless, the objective response rate to crizotinib was significantly higher than with docetaxel or pemetrexed (p<0.0005, t test), similar to what has been observed in patients with ALK-rearranged lung cancer and confirming the utility of this model to predict clinical outcome.
Prior work in GEMMs has clearly shown that tumor response does not necessarily translate into enhanced long-term survival(23). To establish the effects of crizotinib, docetaxel and pemetrexed on overall and progression-free survival, cohorts of tumor-bearing mice were followed on therapy until progression or to overall survival endpoints. The median survival on crizotinib was more than 17 weeks after treatment initiation, versus only 4.3 weeks in the docetaxel arm (Fig. 2A). Time to progression (TTP) also differed significantly between groups (p=0.0165, log-rank test), with 3.1 and 6 weeks for docetaxel and crizotinib, respectively(Fig. 2B). Murine EML4-ALK lung tumors exhibited a more heterogeneous response to pemetrexed: TTP in pemetrexed-treated mice (2 weeks) was not different from that of docetaxel (3.1 weeks), but pemetrexed treatment did confer a significant median survival advantage over docetaxel treatment (8.5 weeks with pemetrexed vs 4.3 weeks with docetaxel, p=0.05). Collectively, our data suggest that crizotinib is superior to docetaxel and pemetrexed in treating EML4-ALK driven lung cancer.
Figure 2. Crizotinib produces improved overall survival and progression-free survival in EML4-ALK lung cancer compared with chemotherapy.
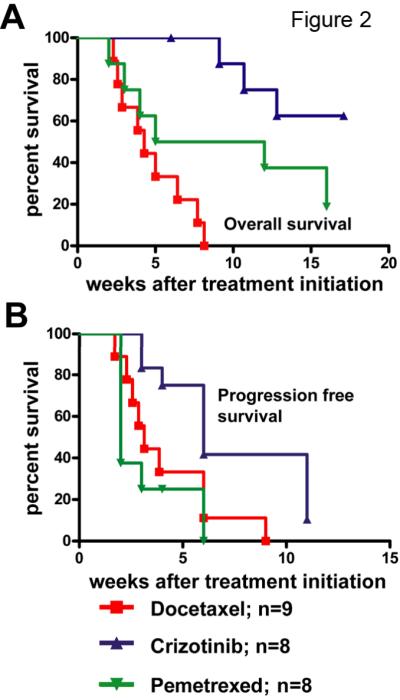
A, Kaplan-Meier curve showing overall survival of mice treated with crizotinib, docetaxel or pemetrexed. Total survival times are counted from treatment initiation. B, Kaplan-meier curve showing progression-free survival of mice treated with crizotinib, docetaxel or pemetrexed. Disease progression is defined as described in the methods.
Crizotinib resistance can be overcome with HSP90 and second-generation ALK inhibitors
Although treatment with crizotinib attenuated signaling downstream of EML4-ALK (Fig. 1C), the rapid development of resistant disease (Fig. 2B) along with the two mice with tumors that did not respond or stabilize on crizotinib indicate that this model can be used to study both primary and acquired crizotinib resistance. In previous in vitro work, we demonstrated that the HSP90 inhibitor 17-DMAG more potently inhibits the growth of the ALK-dependent human H3122 NSCLC cell line than crizotinib (28), resulting in degradation of the ALK protein rather than direct inhibition of kinase activity. Additionally, in the ALK-driven GEMM, HSP90 inhibition and crizotinib achieved comparable overall survival benefit (27). Here, we used 18F-FDG uptake(21) to compare crizotinib treatment (100mg/kg) to treatment with 17-DMAG in this model. The response to crizotinib was heterogeneous (Fig. 3A). In contrast, treatment with 17-DMAG reduced FDG uptake in all tumors, and the magnitude of suppression was significantly greater in 17-DMAG-treated animals compared to crizotinib (Fig. 3B). Importantly, in animals treated with a brief exposure to crizotinib, immediate cross-over to treatment with 17-DMAG eliminated all residual 18F-FDG uptake, suggesting that HSP90 inhibition may eradicate cell populations with primary crizotinib resistance (Fig. 3C).
Figure 3. The HSP90 inhibitor 17-DMAG is a candidate to overcome primary crizotinib resistance.
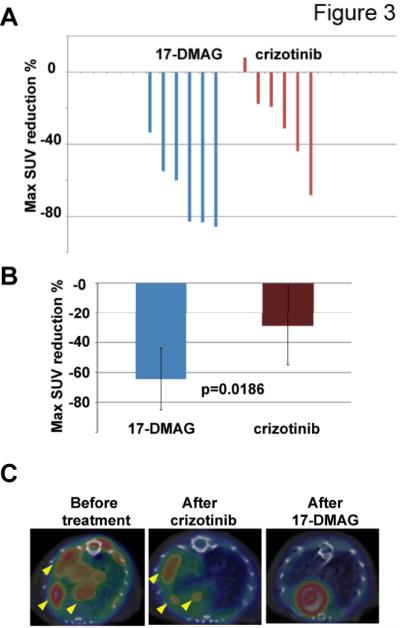
A, 17-DMAG suppresses tumor metabolism to a greater extent than crizotinib. PET-CT was used to assess changes in 18FDG uptake after 2 treatments of either 17-DMAG or crizotinib in EML4-ALK tumor bearing mice. SUVmax for each individual tumor nodule before and after treatment was compared to calculate relative changes. Each bar represents a discernible PET positive tumor nodule. B, Statistical summary of results presented in panel A. C, Representative images showing reduction but not complete elimination of 18FDG uptake after 2 doses crizotinib in an EML4-ALK tumor-bearing mouse. Further treatment with 17-DMAG completely ablates the PET signal.
To study whether HSP90 inhibition is efficacious in the setting of acquired resistance conferred by secondary kinase domain mutations identified in patients(11, 12), we developed another genetically-engineered mouse strain carrying an EML4-ALK fusion oncogene with a clinically-relevant L1196M gatekeeper mutation that confers resistance to crizotinib. Treatment for two weeks with 17-DMAG led to complete tumor regression (Fig. 4A). Similarly, four tumor-bearing mice that harbor a different mutation, EML4-ALK F1174L, also exhibited significant tumor reduction when treated with 17-DMAG, with TTP ranging from 6 to 11 weeks (Fig. 4B), a duration similar to the response seen to 17-DMAG in native EML4-ALK mice (unpublished results). We also evaluated TAE684, a highly potent second-generation ALK inhibitor that retains in vitro activity in the presence of secondary mutations that confer crizotinib resistance (24). As with 17-DMAG, treatment with TAE684 also led to complete tumor regression in the EML4-ALK L1196M model (Fig. 4A).
Figure 4. The HSP90 inhibitor 17-DMAG and the second-generation ALK inhibitor TAE684 overcome acquired crizotinib resistance.

A, EML4-ALK L1196M mutant responds to both 17-DMAG and TAE684. Representative MRI images showing tumor regression after 2 weeks treatment with either drug are shown. B, Both 17-DMAG and TAE684 can overcome crizotinib acquired resistance conferred by the secondary kinase domain mutation F1174L. Four mice with lung cancer driven by EML4-ALK F1174L (each indicated by a different color) were treated with 17-DMAG. Tumor burden was monitored by MRI at the indicated time points from time of treatment initiation. Treatment was switched to TAE684 at the time point marked by the red arrow, after development of acquired resistance to 17-DMAG. Tumor shrinkage followed by re-growth was observed at 11, 3 and 5 weeks after TAE684 administration, respectively, in 3 out of the 4 mice. C, 18FDG PET-CT study shows TAE684 can overcome acquired resistance to 17-DMAG in EML4-ALK lung cancer. Representative images are shown from a mouse treated with 17-DMAG until development of acquired resistance as reflected by recurrent 18FDG uptake (bottom left panel). Application of TAE684 successfully suppressed the recurrent 18FDG signal (bottom right panel). D, Upper panels, pharmacodynamic studies showing restoration of known signaling pathways downstream of ALK in F1174L mice that have become resistant to both 17-DMAG and TAE684 as single agents, including pSTAT3, pAkt, pEMK, pERK and pS6. Consistent areas within the same tumor nodule are shown. Lower panels. Tumors from untreated tumor-bearing mice sacrificed without any treatment applied were subjected to IHC. IHC quantification represents the average intensity over 10 microscopic fields.
We next investigated whether TAE684 can overcome crizotinib and 17-DMAG acquired resistance in both native EML4-ALK mice and EML4-ALK F1174L mutants. First, we treated native EML4-ALK mice with 17-DMAG until tumor re-growth occurred, indicated by rebound of 18F-FDG uptake on PET-CT. Subsequent treatment with TAE684 in these mice resulted in potent re-suppression of 18F-FDG uptake, indicating TAE684 can be used to circumvent resistance to HSP90 inhibitors in native ALK tumors (Fig. 4C). Similarly, when TAE684 was administered to the four EML4-ALK F1174L mice that had developed resistance to long-term 17-DMAG treatment, significant tumor regression was observed, as measured by changes in tumor burden on MRI (Fig. 4B). Surprisingly, however, 3 of the 4 mice experienced relapse soon after the initial response to TAE684, with TTP ranging from 3 to 11 weeks (Fig. 4B and Supplemental Fig. 2); the fourth mouse died one day after switching to TAE684 from unknown causes. The short TTP in these mice contrasts with our previous results in treatment-naïve native EML4-ALK mice that remained free of tumor recurrence for almost 2 years after an initial response to daily administration of TAE684(28). Pharmacodynamic studies of the EML4-ALK F1174L tumors with acquired TAE resistance suggest restoration of known ALK downstream signaling pathways (Fig. 4D). We also confirmed that these mice remain resistant to 17-DMAG. Interestingly, however, a 10-day treatment with the combination of 17-DMAG and TAE684 successfully eliminated most of these tumors that were resistant to either TAE684 or 17-DMAG as single agents (Supplemental Fig. 3).
Collectively, these results suggest that HSP90 inhibition may be a viable option for treating tumors with primary or acquired crizotinib resistance. Second-generation ALK inhibitors represent another strategy, but may exhibit reduced efficacy against tumors carrying secondary mutations conferring crizotinib resistance, compared to the long-term benefit seen when they are used in the setting of de novo treatment of native EML4-ALK tumors.
Discussion
Recently, development of numerous target-specific small molecule inhibitors and antibodies has resulted in improved anti-tumor responses with fewer adverse events than standard-of-care cytotoxic chemotherapy. Clinical application of these therapeutic agents is generally limited by the need for lengthy clinical trials to assure their safety and efficacy. Additionally, this process is complicated by newer patient stratification criteria and combination therapy strategies. However, the implementation of co-clinical trials promises to speed drug development by improving evaluation of treatments and better matching them to disease subtypes. Using a well-defined murine EML4-ALK lung cancer model, we demonstrate that crizotinib is superior to the conventional chemotherapy agents docetaxel and pemetrexed in terms of response and survival. Docetaxel treatment did not result in any tumor regression, whereas pemetrexed offered a modest survival benefit over docetaxel, producing a few partial responses. Thus, while a subset of ALK-driven tumors may respond to pemetrexed treatment, crizotinib appears a better choice.
We also noted primary resistance to crizotinib in EML4-ALK tumors, as found in phase II clinical trials (5). Of note, amplification of the ALK locus has been reported in NSCLC patients. However, it is not clear whether there is overexpression of the wild type or the translocated ALK locus, or whether overexpression of ALK is associated with primary crizotinib resistance. Nonetheless, crizotinib has a relatively narrow structural window for ALK binding and inhibition. Both ALK amplification and additional mutations perturbing crizotinib binding will render ALK-driven tumors insensitive to crizotinib (12, 14). We hypothesize that EML4-ALK mice with primary resistance to crizotinib may have increased ALK expression that helps to surpass the crizotinib effective threshold. Increasing the crizotinib dose to 150 mg/kg/day for two weeks can further reduce 18F-FDG uptake in tumors and produce partial response in primary resistant mice (data not shown), providing additional evidence for this hypothesis. We are currently conducting a more detailed study to further explore this possibility using genetically engineered mouse ALK NSCLC models that express different levels of ALK.
Results of our previous work and the results shown here demonstrate that both 17-DMAG and TAE684 can overcome primary resistance, offering potential clinical approaches to ALK-driven tumors. A recently completed phase II clinical trial demonstrated that the HSP90 inhibitor ganetespib has activity in crizotinib-naïve ALK-positive NSCLC patients, providing additional support for use of HSP90 inhibition in this population (29). Notably, although still under clinical investigation, it has been reported that EGFR activation or compound mutation of oncogenes such as KRAS may also contribute to primary resistance to crizotinib in the clinic (11, 12). Therefore, careful examination of ALK amplification status and other pathways in patients with tumors with primary crizotinib resistance will aid treatment selection.
Secondary mutations conferring crizotinib resistance in patients have been recapitulated in ALK-dependent cell lines cultured in continuously increasing concentrations of drug (14). These mutations occur in different locations in the ALK kinase domain; most are responsible for direct or indirect contact with crizotinib, and some can stabilize the active conformation of ALK. ALK fusion proteins carrying secondary mutations retain their dependence on HSP90 for stability (30, 31) Here, we have demonstrated that mouse tumors driven by such proteins are sensitive to 17-DMAG, providing further evidence for HSP90 inhibition as a strategy in the setting of acquired crizotinib resistance. Recently, the activity of ganetespib was described in a patient with ALK-rearranged NSCLC harboring a secondary crizotinib resistance G1269A mutation (29).
We have also evaluated the second-generation ALK inhibitor TAE684 in our models. Many of the altered residues that cause crizotinib resistance do not exhibit a significant role in binding of second-generation ALK inhibitors such as TAE684 and AP26113, which differ in chemical structure from crizotinib. Consequently, many crizotinib-resistant mutants remain sensitive to second-generation inhibitors in cell line assays, albeit at reduced potency compared to native ALK (12, 14). Among these secondary mutations, the ALK F1174L mutant confers both enhanced oncogenic potency and decreased crizotinib affinity (14). We have shown that the enhanced in vitro profile of TAE684 translates into response in mice genetically engineered to harbor the EML4-ALK F1174L mutation, even in the setting of resistance to 17-DMAG. However, responses in resistant tumors were short-lived. Although EML4-ALK F1174L is inhibited by TAE684, its sensitivity in vitro is 10-fold less than that of native ALK(14). We propose that the combination of reduced drug potency against ALK proteins carrying the secondary F1174L mutation, as well as the increased oncogenic activity conferred by this mutation, affects the response to TAE684. These results suggest that inhibitors specific for individual secondary mutations may improve efficacy in the crizotinib-resistant setting. In addition, considering the superior durability of response to TAE684 in crizotinib-naïve vs. resistant tumors, use of the more potent ALK inhibitor as first-line treatment may better delay the development of additional resistance mutations than the current sequence in which the weak inhibitor, crizotinib, precedes use of the more potent agent. Although TAE684 is not being developed clinically, the related LDK378 has demonstrated preliminary activity in both crizotinib-resistant and -naïve patients and has entered Phase 2 studies to assess the rate and duration of response in both populations (32).
Our data also suggest promise for the strategy of combining ALK and HSP90 inhibitors. We found that application of crizotinib and 17-DMAG in rapid succession ablated FDG-PET activity in tumors driven by native EML4-ALK. This result is similar to the recent demonstration that combined crizotinib and HSP90 inhibition was superior to either agent alone against NCI-H3122 xenografts (29). Additionally, the combination of TAE684 and 17-DMAG successfully caused regression of tumors resistant to the agents individually. Therefore, the addition of an HSP90 inhibitor to crizotinib in ALK inhibitor-naïve patients and to a second-generation ALK inhibitor in crizotinib-resistant patients may substantially improve efficacy.
In summary, we have demonstrated that a GEMM model of EML4-ALK-driven lung cancer is predictive of clinical outcome, providing additional evidence for the utility of mouse co-clinical trials. The results support the selection of crizotinib prior to pemetrexed or docetaxel in the treatment of ALK-rearranged NSCLC. This model, as well as those harboring secondary crizotinib resistance mutations, will allow the evaluation of additional second generation ALK inhibitors, HSP90 inhibitors and other compounds targeting ALK-driven signaling that will ultimately help prioritize treatments beyond crizotinib and guide the development of future clinical trials.
Supplementary Material
Statement of translational relevance:
Our study extends the results of a phase III trial in non-small cell lung cancer patients with adenocarcinomas harboring EML4-ALK fusion. We performed a co-clinical trial in a mouse model comparing the ALK inhibitor crizotinib to the standard-of-care agents docetaxel or pemetrexed. Concordant with the clinical outcome in humans, crizotinib produced a higher response rate compared to chemotherapy, associated with significantly longer progression-free and overall survival. We also demonstrated that pemetrexed produced superior overall survival compared to docetaxel. Additionally, in the EML4-ALK-driven mouse lung adenocarcinoma model, HSP90 inhibition can overcome both primary and acquired crizotinib resistance. Furthermore, HSP90 inhibition as well as a second-generation ALK inhibitor, TAE684, demonstrated activity in lung adenocarcinoma models driven by crizotinib-insensitive EML4-ALK L1196M or F1174L. Our findings suggest that crizotinib is superior to standard chemotherapy in ALK inhibitor-naïve disease and support clinical investigation of HSP90 inhibitors and second-generation ALK inhibitors in tumors with primary or acquired crizotinib resistance.
Acknowledgments
Dr Wong owns equity in, receives compensation from G1Therapeutics, serves as a consultant to Molecular MD, and sponsored research agreement with AstraZeneca, Infinity Pharmaceuticals and Millennium Pharmaceuticals.
Footnotes
Author contributions ZC, OM, TT, KC, YW, XT, AA, and SW performed experiments and data analyses. PAJ, NES, JAE, GIS, ALK and KKW conceived and supervised all aspects of the project. ZC, NES, JAE, GIS, ALK and KKW wrote the manuscript. EA provided critical reagents.
References
- 1.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–3322. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 4.McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–3395. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 5.Shaw AT, Engelman JA. ALK in Lung Cancer: Past, Present, and Future. J Clin Oncol. 2013;31:1105–1111. doi: 10.1200/JCO.2012.44.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camidge DR, Kono SA, Lu X, Okuyama S, Baron AE, Oton AB, et al. Anaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J Thorac Oncol. 2011;6:774–780. doi: 10.1097/JTO.0b013e31820cf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw AT, Varghese AM, Solomon BJ, Costa DB, Novello S, Mino-Kenudson M, et al. Pemetrexed-based chemotherapy in patients with advanced, ALK-positive non-small cell lung cancer. Ann Oncol. 2013;24:59–66. doi: 10.1093/annonc/mds242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jangchul Park CK. Junichi Shimizu, Yoshitsugu Horio, Kimihide Yoshida, Tetsuya Mitsudomi, et alChemosensitivity and clinical features of EML4-ALK-positive patients with advanced non-small cell lung cancer. J Clin Oncol. 2012;(suppl):e18145. abstr. [Google Scholar]
- 9.Takeda M, Okamoto I, Sakai K, Kawakami H, Nishio K, Nakagawa K. Clinical outcome for EML4-ALK-positive patients with advanced non-small-cell lung cancer treated with first-line platinum-based chemotherapy. Ann Oncol. 2012;23:2931–2936. doi: 10.1093/annonc/mds124. [DOI] [PubMed] [Google Scholar]
- 10.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 11.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4:120ra117. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011;78:999–1005. doi: 10.1111/j.1747-0285.2011.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7:645–658. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- 16.Gutmann DH, Hunter-Schaedle K, Shannon KM. Harnessing preclinical mouse models to inform human clinical cancer trials. J Clin Invest. 2006;116:847–852. doi: 10.1172/JCI28271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richter SH, Garner JP, Wurbel H. Environmental standardization: cure or cause of poor reproducibility in animal experiments? Nat Methods. 2009;6:257–261. doi: 10.1038/nmeth.1312. [DOI] [PubMed] [Google Scholar]
- 18.Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 19.Singh M, Johnson L. Using genetically engineered mouse models of cancer to aid drug development: an industry perspective. Clin Cancer Res. 2006;12:5312–5328. doi: 10.1158/1078-0432.CCR-06-0437. [DOI] [PubMed] [Google Scholar]
- 20.Singh M, Murriel CL, Johnson L. Genetically engineered mouse models: closing the gap between preclinical data and trial outcomes. Cancer Res. 2012;72:2695–2700. doi: 10.1158/0008-5472.CAN-11-2786. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483:613–617. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts PJ, Usary JE, Darr DB, Dillon PM, Pfefferle AD, Whittle MC, et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res. 2012;18:5290–5303. doi: 10.1158/1078-0432.CCR-12-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010;28:585–593. doi: 10.1038/nbt.1640. [DOI] [PubMed] [Google Scholar]
- 25.Weaver Z, Difilippantonio S, Carretero J, Martin PL, El Meskini R, Iacovelli AJ, et al. Temporal molecular and biological assessment of an erlotinib-resistant lung adenocarcinoma model reveals markers of tumor progression and treatment response. Cancer Res. 2012;72:5921–5933. doi: 10.1158/0008-5472.CAN-12-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh M, Couto SS, Forrest WF, Lima A, Cheng JH, Molina R, et al. Anti-VEGF antibody therapy does not promote metastasis in genetically engineered mouse tumour models. J Pathol. 2012;227:417–430. doi: 10.1002/path.4053. [DOI] [PubMed] [Google Scholar]
- 27.Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 28.Chen Z, Sasaki T, Tan X, Carretero J, Shimamura T, Li D, et al. Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res. 2010;70:9827–9836. doi: 10.1158/0008-5472.CAN-10-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, et al. A Multicenter Phase II Study of Ganetespib Monotherapy in Patients with Genotypically Defined Advanced Non-Small Cell Lung Cancer. Clin Cancer Res. 2013;19:3068–3077. doi: 10.1158/1078-0432.CCR-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108:7535–7540. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, et al. Targeted Inhibition of the Molecular Chaperone Hsp90 Overcomes ALK Inhibitor Resistance in Non-Small Cell Lung Cancer. Cancer Discov. 2013;3:430–43. doi: 10.1158/2159-8290.CD-12-0440. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw AT, R.M., Kim D, Felip E, Chow L, Camidge R, et al. Clinical activity of the ALK inhibitor LDK378 in advanced, ALK-positive NSCLC. J Clin Oncol. 2013;(suppl):8010. abstr. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.