

Summary
The spindle assembly checkpoint (SAC) delays anaphase until all chromosomes are bi-oriented on the mitotic spindle. Under current models, unattached kinetochores transduce the SAC by catalyzing the intramitotic production of a diffusible APC/CCdc20 inhibitor. Here we show that nuclear pore complexes (NPCs) in interphase cells also function as scaffolds for anaphase-inhibitory signaling. This role is mediated by Mad1-Mad2 complexes tethered to the nuclear basket, which activate soluble Mad2 as a binding partner and inhibitor of Cdc20 in the cytoplasm. Displacing Mad1-Mad2 from nuclear pores accelerated anaphase onset, prevented effective correction of merotelic errors, and increased the threshold of kinetochore-dependent signaling needed to halt mitosis in response to spindle poisons. A heterologous Mad1-NPC tether restored Cdc20 inhibitor production and normal M phase control. We conclude that nuclear pores and kinetochores both emit “wait anaphase” signals that preserve genome integrity.
Introduction
A defining feature of all eukaryotes is the nuclear envelope (NE), which divides the cell into spatially and functionally distinct compartments. Macromolecular traffic across the NE is mediated by nuclear pore complexes (NPCs), large transmembrane cylinders formed from ∼30 nucleoporins (Nups), and soluble transport receptors that shuttle cargoes in response to the Ran GTPase (Hetzer and Wente, 2009; Stewart, 2007). Mounting evidence also implicates NPCs in modes of regulation that are distinct from nuclear transport. For example, some actively transcribed genes in yeast are tethered to NPCs via bridging complexes that also recruit transcription factors and mRNA processing enzymes, thereby enhancing gene expression at multiple levels (Dieppois and Stutz, 2010; Strambio-De-Castillia et al., 2010). By comparison, metazoan Nups stimulate transcription by interacting with target loci within the nuclear interior (Capelson and Hetzer, 2009). Further redistribution and repurposing occurs during mitosis, as NPC disassembly and nuclear envelope breakdown (NEBD) enable the Nup107-160 complex, Nup358/RanBP2, and the exportin Crm1 to relocalize at kinetochores, where these proteins regulate microtubule dynamics in conjunction with RanGTP (Arnaoutov et al., 2005; Joseph et al., 2004; Zuccolo et al., 2007).
Another example of NPC-to-kinetochore migration involves the Mad1-Mad2 complex. This heterodimer acts as the terminal transducer of the spindle assembly checkpoint (SAC) that delays anaphase until all kinetochores are bound by microtubules (Foley and Kapoor, 2013; Musacchio and Salmon, 2007). During interphase Mad1 and Mad2 are docked at the nucleoplasmic side of the NPC, principally through interactions with a conserved family of coiled-coil proteins (Tpr in vertebrates, Megator in flies, and Mlp1/2 in yeast) that make up the nuclear basket (Campbell et al., 2001; Lee et al., 2008; Lince-Faria et al., 2009; Scott et al., 2005). This arrangement persists until NEBD, when the Mad1-Mad2 complex is recruited to unattached kinetochores by upstream components of the SAC, including the Mps1, Aurora B, and Bub1 kinases, and the Rod-Zw10-Zwilch complex (Foley and Kapoor, 2013; Musacchio and Salmon, 2007). Compelling evidence indicates that Mad1 binding shifts Mad2 from its “open” (O or N1) to “closed” (C or N2) conformation, which not only stabilizes the heterodimer but also endows it with prion-like activity, whereby it can induce a similar structural change in soluble O-Mad2 (Musacchio and Salmon, 2007; Yu, 2006). As C-Mad2, this pool can bind Cdc20, a key activator of the anaphase-promoting complex or cyclosome (APC/C), a large ubiquitin ligase (Pines, 2011). In conjunction with a second Cdc20 inhibitor, BubR1, and its cofactor Bub3, C-Mad2 and Cdc20 form one or more mitotic checkpoint complexes (MCCs; (Fang, 2002; Sudakin et al., 2001; Tang et al., 2001)) that inhibit APC/C-mediated proteolysis of securin and cyclin B, thereby delaying sister chromatid separation and mitotic exit (Foley and Kapoor, 2013; Musacchio and Salmon, 2007).
In contrast, Mad1 and Mad2's role at interphase NPCs remains ill-defined. One hypothesis, namely that one or both SAC mediators modulates traffic across the NE, is supported by the finding that yeast Mad1 cycles between kinetochores and NPCs to inhibit Kap121-mediated nuclear import during this organism's closed mitosis (Cairo et al., 2012). However, higher organisms synchronize both NPC disassembly and kinetochore assembly with the start of M phase, eliminating opportunities for equivalent crosstalk (Cheeseman and Desai, 2008; Hetzer and Wente, 2009). While the Nups responsible for recruiting Mad1 and Mad2 to the NE have been suggested to support SAC activity in metazoans, how this occurs is still unclear and controversial (Lee et al., 2008; Lince-Faria et al., 2009).
We used genetic and computational methods to investigate the functions and regulation of human Mad1. Here we show that NPC tethering allows the Mad1-Mad2 dimer to initiate MCC assembly before cells reach mitosis (Sudakin et al., 2001). By proactively inhibiting APC/CCdc20, the NPC-derived “wait anaphase” signal buffers its intramitotic counterpart, which is regulated by kinetochore-microtubule attachment and established after NEBD. Together the two systems make the SAC more sensitive and robust, and facilitate the correction of mitotic errors that are invisible to the SAC. Collectively our results define a new role of the interphase NE in signal transduction and genome maintenance that outlasts its disassembly.
Results
Mitotic timing and checkpoint defects in MAD1L1-null cells
Because Mad1 RNAi often fails to elicit a SAC defect (Fava et al., 2011), we used adeno-associated virus (AAV)-mediated gene targeting to modify the MAD1L1 locus in human retinal pigment epithelial cells, such that exon 12 was either flanked by loxP sites or deleted outright (Fig. S1A-B). Next, MAD1L1flox/Δ cells or controls were infected with an adenovirus expressing Cre recombinase. Over the next 3 to 6 days Mad1 was depleted (Fig. 1A), which in turn abolished Mad2's targeting to kinetochores (Fig. 1B) and mitotic arrest in response to spindle poisons like nocodazole (Fig. S1C).
Figure 1. Gene deletion reveals Mad1's dual roles in mitotic timing and checkpoint enforcement.
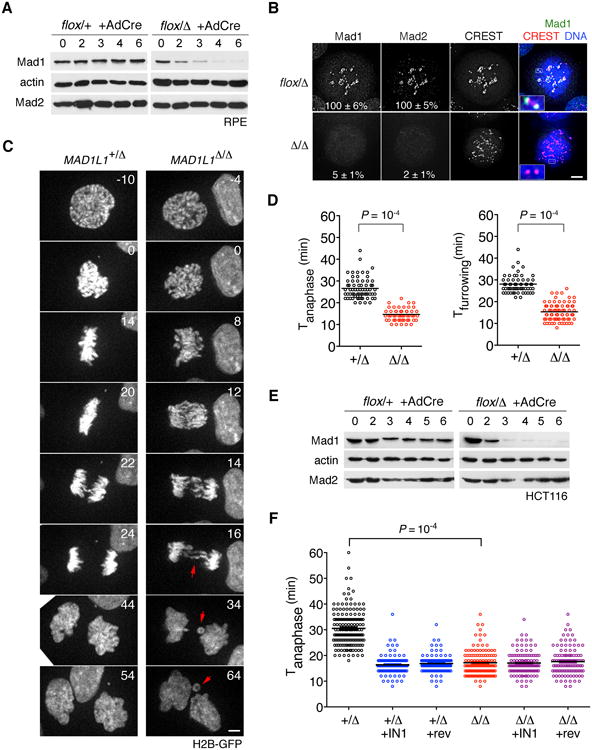
(A) RPE cells in which one or both MAD1L1 alleles had been targeted with AAV vectors (see Figure S1) were infected with AdCre and sampled for 6 days thereafter. (B) Cells in (A) were treated with nocodazole and MG132 for 90 min. Maximum-intensity projections and magnified views of kinetochores (insets) are shown. Mad1 and Mad2 signals were normalized against CREST. Scale bar, 5 μm. (C) Cells expressing H2B-GFP were traced during an unperturbed mitosis. Time 0 denotes NEBD. Arrowheads highlight lagging chromatids and micronuclei. (D) The interval from NEBD to anaphase (left plot) or furrow ingression (right plot) was determined from timelapse recordings. See Movies S1 and S2. (E-F) HCT116 cells were modified at the MAD1L1 locus, infected with AdCre, and analyzed as above. Where indicated, Mps1-IN-1 or reversine was added. Datasets in (D) and (F) were compared by Student's t test.
Having validated the penetrance of our system, we asked how Mad1 contributes to progression through an otherwise unperturbed mitosis. Previous studies have defined two modes through which anaphase can be delayed by the SAC network. The first and more familiar pathway uses kinetochores to activate Mad2 as a Cdc20 binding partner and inhibitor (Foley and Kapoor, 2013; Musacchio and Salmon, 2007). However, a large fraction of Mad2 and BubR1 are already bound to Cdc20 during interphase and appear to define the minimal length of M phase independently of kinetochores (Maciejowski et al., 2010; Malureanu et al., 2009; Meraldi et al., 2004; Sudakin et al., 2001). Notably, this interphase-specific inhibitor or “mitotic timer” still requires Mps1 (Maciejowski et al., 2010) but supposedly not Mad1 (Meraldi et al., 2004), raising the question of where and how Mad2 is activated during interphase. As a first step we analyzed mitotic timing in MAD1L1Δ/Δ cells via timelapse microscopy (Fig. 1C). In sharp contrast to the findings of Meraldi et al. (2004), anaphase and cytokinesis occurred 14 ± 0.3 and 15 ± 0.4 minutes after NEBD, versus 26 ± 0.6 and 28 ± 0.4 minutes in control cells (Fig. 1D and Movies S1-S2). Furthermore, ∼70% of MAD1L1–null cells displayed lagging chromatids or bridges (Fig. 1C, arrows), indicating that some chromosomes had disjoined without proper attachments to the spindle. To rule out cell type-specific effects we conducted similar studies in an unrelated cell line (HCT116; Fig. 1E and S1B) and again saw pronounced M phase acceleration and checkpoint override after MAD1L1 deletion (Fig. 1F and S1D). Interestingly, whereas the Mps1 inhibitors reversine and IN-1 (Kwiatkowski et al., 2010; Santaguida et al., 2010) shortened M phase in wildtype cells, they had no effect on MAD1L1-null cells (Fig. 1F), suggesting that Mad1 and Mps1 control mitotic timing through a common pathway.
Mad1 directs MCC assembly during interphase
Next we examined the dynamics of a key downstream target of APC/CCdc20, cyclin B. To avoid overexpression artefacts, a C-terminal Venus tag was knocked into the CCNB1 locus (Fig. 2A and S2A-C). Quantitative imaging revealed a two-pronged defect, as MAD1L1Δ/Δ cells accumulated less cyclin B1-Venus in late G2 phase, and then initiated its rapid phase of degradation at NEBD (Fig. 2A-B and Movie S3), rather than at metaphase (Clute and Pines, 1999). A similar profile has been observed in other timer-deficient mutants (Maciejowski et al., 2010; Malureanu et al., 2009) and suggests that Mad1 may be needed to inhibit Cdc20 before M phase onset. To test this hypothesis, control and MAD1L1-null cultures were depleted of mitotic cells via shakeoff (to greater than 99.5% purity) and used to immunopurify and quantitate interphase-specific MCCs as previously described (Maciejowski et al., 2010). Cdc20 binding to both Mad2 and BubR1 was severely compromised in the absence of Mad1 (Fig. 2C-D).
Figure 2. Mad1 directs MCC assembly in interphase cells and stabilizes cyclin B both before and after NEBD.
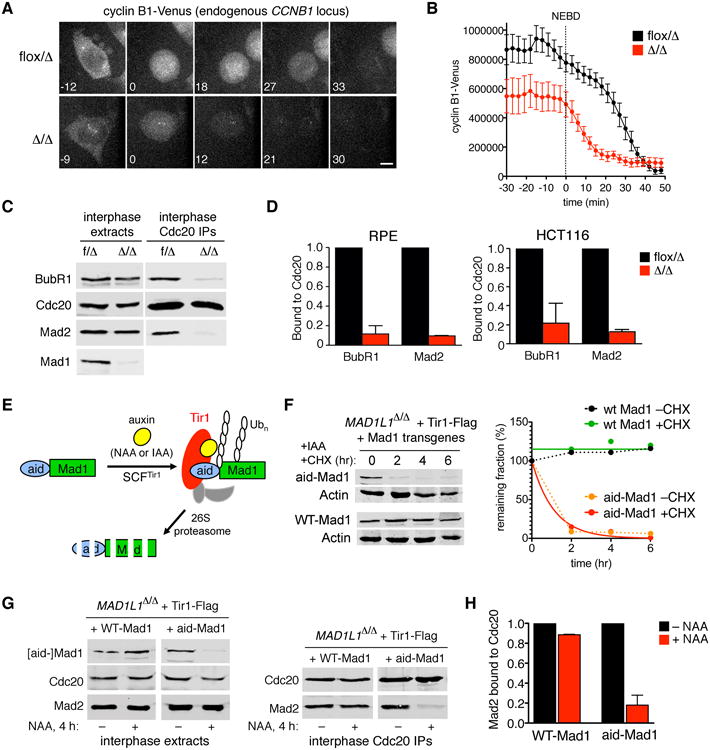
(A) Endogenous cyclin B1 was tagged with Venus (Figure S2A-C) and followed by spinning disk microscopy. Time 0 denotes NEBD. Scale bar, 5 μm. See Movie S3. (B) Cyclin B1-Venus profiles in control versus MAD1L1-null cells (n=10). (C-D) Mad1 controls interphase MCC assembly. Cdc20 and associated proteins were immunopurified from interphase extracts. Recovery of BubR1 and Mad2 was determined by quantitative blotting. Error bars denote SEM. (E) System for chemically induced proteolysis of Mad1. Upon auxin addition, aid-Mad1 is polyubiquitylated by SCFTir1, targeting it for destruction via the proteasome. (F) Cells were treated with 3-indoleacetic acid (IAA) in the presence or absence of cycloheximide. (G) Cells were treated with 1-naphthaleneacetic acid (NAA) for 4 hr, then processed as in (C). (H) Quantification of Mad2-Cdc20 complexes in (G). Error bars indicate SEM.
To determine if MCC loss reflected a direct and ongoing requirement for Mad1 in interphase, we used a chemical-genetic method to destroy Mad1 acutely. MAD1L1Δ/Δ cells were reconstituted with a human codon-optimized version of the auxin receptor and SCF ubiquitin-ligase adapter Tir1 (Nishimura et al., 2009) and wildtype or aid (auxin-inducible degron)-tagged Mad1 (Fig. 2E). Whereas aid-Mad1 was destabilized by auxins, wildtype Mad1 was immune (Fig. 2F-G). Crucially, a brief pulse of aid-Mad1 destruction was enough to liberate Cdc20 from Mad2 in interphase extracts (Fig. 2G-H), demonstrating that Mad1 is continuously required for MCC assembly in interphase cells.
Mad1-Mad2 dimers are required for interphase MCC assembly but not nuclear transport
In light of Mad1's association with nuclear pores, which have both transport-dependent and -independent roles in other processes (Capelson and Hetzer, 2009; Strambio-De-Castillia et al., 2010), we considered two different explanations for these results. First, Mad1 might control the transport of specific MCC subunits or regulators in humans, analogous to its role in inhibiting Kap121-mediated import in yeast (Cairo et al., 2012). Second, Mad1-Mad2 might activate soluble O-Mad2 for Cdc20 binding, using its intrinsic templating activity (Vink et al., 2006). To investigate these possibilities an N-terminal Venus tag was knocked into the MAD2L1 locus (Fig. S2D-F). Venus-Mad2 was predominantly enriched at the NE and nuclear interior, but also found in the cytoplasm (Fig. 3A, upper panels). Fluorescence loss in photobleaching (FLIP) was then used to probe the connectivity of these pools. Repeated bleaching of the nuclear interior led to extensive signal loss in the cytoplasm, demonstrating that Mad2 cycles between both compartments (Fig. 3B, upper left). In contrast NE-bound Mad2 was partially resistant to FLIP, in line with its low mobility under FRAP (Shah et al., 2004). As a result, cytosolic bleaching was less effective in inducing nuclear FLIP than the converse (Fig. 3B, upper right). Upon MAD1L1 deletion, Venus-Mad2 was no longer visible at the NE but continued to cycle between the nucleus and cytoplasm, resulting in symmetric sensitivity to FLIP (Fig. 3A-B, bottom panels). To confirm and extend these results, cells with unmodified MAD2L1 alleles were stained with a pan-reactive Mad2 polyclonal or a conformation-specific monoclonal that detects C-Mad2's catalytic interface (Fava et al., 2011). Both probes were strongly enriched at the NPCs of control cells but not MAD1L1Δ/Δ cells (Fig. 3C-D). Their absence was specific, as the localization and fractionation profiles of Cdc20 and other MCC subunits, Mps1, and nuclear basket Nups were unaffected by MAD1L1 deletion or Mps1 inhibition (Fig. 3C, 3E, and S3A-C). Consistently, MAD1L1 deletion did not affect the translocation of a model karyopherin-dependent cargo (Fig. S3D). Together these results indicate that Mad1 binds and presents C-Mad2 at NPCs, but does not control the transport of soluble Mad2 or other MCC-relevant cargoes.
Figure 3. Mad1-Mad2 heterodimers promote interphase MCC assembly independently of nuclear transport.

(A) Endogenous Mad2 was tagged with Venus (Figure S2D-F) and followed by fluorescence loss in photobleaching. Iteratively bleached regions are marked by white boxes. Signal intensities are displayed on a heatscale. (B) Signals in bleached cells (FLIP) or adjoining unbleached cells (CTRL) were determined from 10 cells per condition and plotted relative to the starting value. (C-D) Cells were stained with antibodies against Mad1, Tpr, total or closed Mad2, and mAb414. Single z-sections (interphase cells) or maximum-intensity projections (mitotic cells) are shown. (E) Fractionation of MCC subunits and regulators after aid-Mad1 degradation. Cells were treated with NAA, IAA, or DMSO for 3 hours, then fractionated prior to Western blotting. (F) Cdc20 and associated proteins were immunopurified from MAD1L1-null cells reconstituted with wildtype Mad1 (wt) or a mutant that cannot bind Mad2 (K541A L543A; Mad1AA). Error bars indicate SEM. (G) The NEBD-to-anaphase interval in Mad1wt and Mad1AA cells. (H) M phase duration in nocodazole-treated Mad1wtand Mad1AA cells. Scale bars, 5 μm. Datasets were compared by Student's t test. See also Figure S3.
To test the functional relevance of these observations, MAD1L1Δ/Δ cells were reconstituted with wildtype Mad1 or a catalytically dead mutant that cannot bind Mad2 (K541A L543A, hereafter Mad1AA (Sironi et al., 2002)). Although targeted to NPCs, Mad1AA did not recruit Mad2 (Fig. 3C), license its interaction with Cdc20 in the cytoplasm (Fig. 3F), or reinstate either basal mitotic timing or checkpoint controls (Figs. 3G-H). We conclude that both interphase and mitotic cells require Mad1-Mad2 dimers to produce their respective inhibitors.
Deletion of Mad1's N-terminus abrogates its NPC localization and uncouples the SAC's timer and checkpoint arms
We next sought Mad1 alleles that selectively abrogate its activity during interphase. A series of deletion mutants was expressed in MAD1L1-null cells and assayed for NPC localization, revealing the necessity and sufficiency of amino acids 1-274 (Fig. S4A-B). Hereafter we refer to this region as Mad1's NPD (nuclear pore-targeting domain; Fig. 4A). Further analysis mapped three separate activities to the NPD, including nuclear import and export signals between amino acids 1-89 and 180-274 (Fig. S4A) and an in vitro binding site for Tpr (Fig. 4B). A Mad1 mutant lacking amino acids 1-179 (Mad1ΔNP2; Fig. 4C-D) was analyzed as a separation-of-function allele. Despite forming a stable heterodimer with Mad2, Mad1ΔNP2 failed to support MCC assembly during interphase (Fig. 4E-F), but then regained activity at NEBD, as it was recruited to unattached kinetochores with the same kinetics and efficiency as wildtype Mad1 (Fig. 4G-H and Movie S4) and produced similar yields of MCCs in mitosis (Fig. 4I-J). To corroborate these findings we filmed cells during an unperturbed mitosis or in the presence of nocodazole to prevent kinetochore-microtubule attachment. Consistent with the molecular data presented above, Mad1ΔNP2 cells entered anaphase prematurely, yet arrested for many hours when challenged with nocodazole (Fig. 5A and Movies S5-S6), suggesting that Mad1's N-terminal domain is required for the timer arm (but not the checkpoint arm) of the SAC.
Figure 4. Mad1's N-terminus mediates its NPC recruitment and is required for MCC production during interphase.

(A) Overview of Mad1 architecture. The nuclear pore-targeting domain (NPD) was mapped in Figure S4. The Mad2-interaction domain (M2iD) and C-terminal domain (CTD) were described previously (Kim et al., 2012; Luo et al., 2002; Sironi et al., 2002). (B) In vitro pulldown assays. Tpr [1-775] was synthesized by in vitro translation, then tested for binding to recombinant Mad1 fragments (N [1-244], M [245-478], or C [484-718]) on Dynabeads. (C) FLAP-Madlwt and FLAP-Mad1ΔNP2 were immunoprecipitated with GFP-specific antibodies (detects FLAP tag) and blotted as shown. (D) Cells in (C) were immunostained after MAD1L1 deletion. Single z-sections and magnified views of NPCs are shown. (E) Cells in (C) were processed for interphase extracts before or after MAD1L1 deletion. Extracts were immunoprecipitated with GFP antibodies (middle panel) or Cdc20 antibodies (right panel). (F) Quantitation of Mad1-Mad2 and Mad2-Cdc20 binding. Error bars indicate SEM. (G) Spinning-disk imaging of MAD1L1-null cells reconstituted with FLAP-tagged Mad1wt or Mad1ΔNP2. Scale bars, 5 μm. See Movie S4. (H) Cells in (G) were treated with nocodazole. (I) Mitotic extracts and Cdc20 IPs were analyzed by Western blotting. (J) Quantitation of Mad2 and BubR1 binding to Cdc20. Error bars indicate SEM.
Figure 5. The premitotic “wait anaphase” signal enables error-free chromosome segregation.

(A) Spinning-disk imaging of MAD1L1-null HCT116 cells expressing Mad1wt or Mad1ΔNP2 and H2B-mCherry. Where indicated, cells were treated with nocodazole (noc) to engage the SAC. Arrowheads indicate lagging chromosomes. Scale bars, 5 μm. See Movies S5 and S6. (B) Frequency of lagging chromosomes. Where indicated, Mad1ΔNP2 cells were treated with the APC/C inhibitor proTAME to delay anaphase by 10 min. (C) Lagging chromatids correlate with early anaphase. Cells in (B) were classified according to the presence or absence of lagging chromatids. Cumulative anaphase entry was then plotted. Genotypes with minimal (≤5%) lagging are displayed as a single group. (D) Lagging chromatids arise from merotelic errors. Boxes highlight kinetochores attached to microtubules spanning both spindle poles. (E) Anaphases were scored for lagging chromosomes with (grey bars) or without (striped bars) confirmed merotelic attachments. (F-G) Mad1wt and Mad1ΔNP2 cells were hybridized with a chromosome 6-specific probe. Lymphoblastoid cell lines (LCLs) from healthy human donors were used as controls. (G) Mad1ΔNP2 cells exhibit a marked increase in non-diploid FISH signals (30%) as compared with Mad1wt cells (5%). P-values were computed by chi-squared test.
The premitotic “wait anaphase” signal enables merotelic error correction
Curiously, despite their checkpoint proficiency, Mad1ΔNP2 cells made frequent errors in chromosome segregation, as evidenced by lagging chromatids in nearly half of all anaphases (Fig. 5A, arrowheads, and Fig. 5B). Lagging chromatids most often originate from merotelic attachments, in which a single kinetochore is captured by microtubules emanating from both spindle poles (Gregan et al., 2011). Merotelic errors are uniquely dangerous because they interfere with anaphase chromosome dynamics yet do not trigger the SAC, which is satisfied by high kinetochore-microtubule occupancy and tension (Cimini, 2008; Salmon et al., 2005). Instead merotelic errors are corrected in a piecemeal manner throughout M phase (Bakhoum et al., 2009; Cimini et al., 2006). Consistently, we observed a strong correlation between early anaphase entry and induction of lagging chromatids in Mad1ΔNP2 cells and MAD1L1-null cells (Fig. 5C), as well as their co-suppression by low doses of the APC/C inhibitor proTAME (Zeng et al., 2010). Through studies on fixed cells, we confirmed that the vast majority of lagging chromatids were indeed connected to microtubule fibers spanning both spindle poles (Fig. 5D-E), as expected for unresolved merotelic attachment. About 30% of Mad1ΔNP2 cells ultimately acquired nondiploid chromosome counts (Fig. 5F-G), due to lagging chromatids that never reached the correct daughter cells and/or caused cytokinesis failure and tetraploidy (Sotillo et al., 2007; Weaver et al., 2007).
The premitotic “wait anaphase” signal enhances checkpoint establishment
A second role of the premitotic “wait anaphase” signal emerged when we combined its ablation with perturbations in Aurora B-dependent signaling at kinetochores. These experiments were motivated by an apparent paradox: on the one hand, cytological analyses place Aurora B at the apex of the SAC, as its kinase activity drives the kinetochore-specific recruitment and activation of Mps1 and all other SAC mediators (including Mad1-Mad2) at NEBD (Hewitt et al., 2010; Nijenhuis et al., 2013; Santaguida et al., 2011; Saurin et al., 2011; Vigneron et al., 2004). On the other hand, the functional integrity of the SAC is far more resistant to Aurora B inhibition than Mps1 inhibition, even when indexed to quantifiable and accepted indicators of SAC signaling at kinetochores. For example, concentrations of Mps1 inhibitors that decrease kinetochore-associated levels of Mps1 autophosphorylation, Mad1, or Mad2 by ∼80% prevent cells from arresting in mitosis when challenged with nocodazole (Hewitt et al., 2010; Kwiatkowski et al., 2010; Maciejowski et al., 2010; Santaguida et al., 2010). In sharp contrast, doses of Aurora B inhibitors that recapitulate these effects do not block checkpoint establishment (Santaguida et al., 2011; Saurin et al., 2011). However, we previously noted one important distinction between these two kinases, namely that Mps1 (but not Aurora B) is also required to produce MCCs during interphase (Maciejowski et al., 2010). We hypothesized that this extra pool of Cdc20 inhibitors could allow Aurora B-inhibited cells to respond to unattached kinetochores, despite inefficient signaling at the latter. To test this idea, cells reconstituted with Mad1wt or Mad1ΔNP2 were treated with ZM447439 (hereafter ZM), a well-characterized and specific inhibitor of Aurora B (Ditchfield et al., 2003). As expected ZM had no effect on basal mitotic timing or nocodazole-induced checkpoint arrest in Mad1wt cells. In sharp contrast, ZM exposure caused further mitotic acceleration in Mad1ΔNP2 cells, as well as wholesale collapse of the checkpoint (Fig. 6A-D and Movies S7-S8). Similar results were obtained when nocodazole levels were titrated to reduce on-kinetochore signaling without ZM (Fig. S5A).
Figure 6. The premitotic “wait anaphase” signal enhances SAC surveillance by decreasing the requirements for checkpoint signaling at kinetochores.
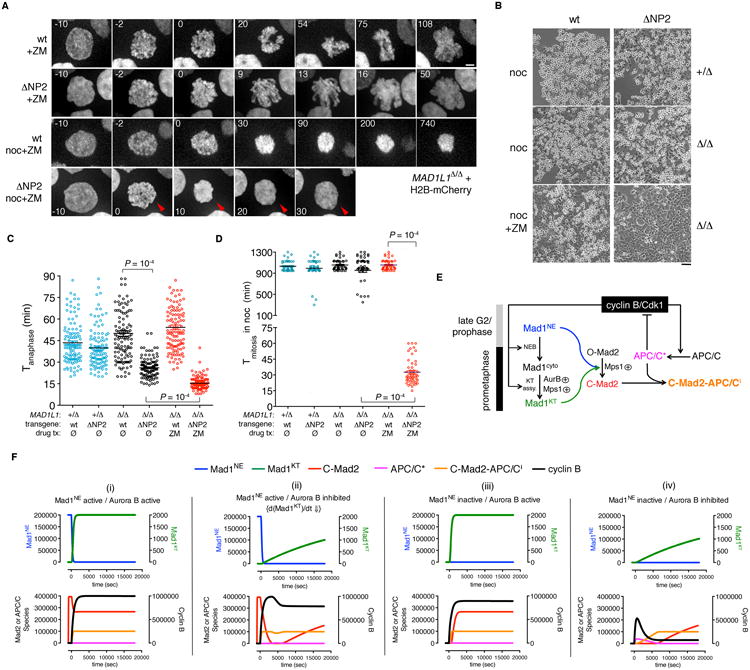
(A) MAD1L1-null HCT116 cells expressing Mad1wt or Mad1ΔNP2 and H2B-mCherry were treated with ZM ± nocodazole. Arrowheads indicate a cell that entered and exited mitosis within 30 minutes. Scale bar, 5 μm. See Movies S7 and S8. (B) Phase-contrast micrographs of MAD1L1-null cells reconstituted with Mad1wt or Mad1ΔNP2 and treated with nocodazole ± ZM for 15 hr. Scale bar, 20 μm. (C and D) Loss of the NPC-derived timer makes Aurora B limiting for anaphase, mitotic exit, and checkpoint enforcement. (C) Cells were filmed in the presence or absence of ZM as in Fig. 3G. Data were compared by one-way ANOVA. (D) Cells were filmed in the presence of nocodazole ± ZM as in Fig. 3H. Data were compared by one-way ANOVA. See Fig. S5A. (E) Proposed scheme for synergy between NE- and kinetochore-associated Mad1-Mad2 pools during checkpoint establishment. (F) ODE implementation of (E) results in simulated M phase arrest so long as Mad1 is tethered to nuclear pores before NEBD and/or rapidly targeted to kinetochores after NEBD, in agreement with wet experiments. See Fig. S5B and the Extended Experimental Procedures.
To understand these results in quantitative terms, we developed a computational model that describes cells entering M phase with persistently unattached kinetochores (Fig. 6E-F). Four events were parameterized: (1) during interphase, NE-bound Mad1-Mad2 heterodimers convert soluble 0-Mad2 into an active anaphase inhibitor (C-Mad2); (2) at M phase onset, cyclin B/Cdk1 triggers NPC disassembly and NEBD, thereby shedding Mad1-Mad2 into the cytosol; (3) Aurora B and Mps1 kinases target free Mad1-Mad2 to unattached kinetochores (Mad1KT), thereby initiating C-Mad2 production from these sites; (4) cyclin B/Cdk1 activates the APC/C ubiquitin ligase (APC/C*), which will degrade cyclin B and drive the system out of mitosis if unopposed by sufficient levels of C-Mad2. The in silico model's dynamics substantiated our findings in living cells, as the high stability of cyclin B in the unperturbed state (Fig. 6F, panel i) was preserved even when Mad1 activity at interphase NPCs (panel iii) or timely migration to prometaphase kinetochores (panel ii) were attenuated individually. In contrast, limiting both modes of Mad1 regulation (panel iv) caused cyclin B to become unstable, as premade C-Mad2 was no longer available to offset the delay in producing C-Mad2 de novo from kinetochores. Collectively these analyses indicate that the premade “wait anaphase” signal significantly lowers the threshold of on-kinetochore signaling needed to instigate a checkpoint arrest, allowing higher organisms to delay kinetochore assembly until NEBD (Cheeseman and Desai, 2008; Gascoigne and Cheeseman, 2013), when the negative feedback loop from APC/CCdc20 to Cdk1/cyclin B is also triggered (Pines, 2011).
Mad1-NPC tethering is crucial for generating the premitotic “wait anaphase” signal
Based on these insights, we re-examined how the nuclear basket protein Tpr contributes to mitotic regulation. Whereas Mad1 and Tpr were strongly enriched at NPCs, only Mad1 was found at kinetochores (Fig. S6A-B); likewise, Mad1-Tpr complexes were abundant in interphase but not mitotic extracts (Fig. S6C). Tpr knockdown displaced Mad1 from NPCs (Fig. S6A) and decreased MCC production during interphase (Fig. S6D), but did not affect the recruitment of Mad1, Mad2, and other SAC regulators to kinetochores after NEBD (Fig. S6E-I). In agreement with our studies on Mad1ΔNP2 cells, Tpr-depleted cells exhibited accelerated mitotic timing and frequent lagging chromatids, but still arrested in M phase when challenged with spindle poisons (Fig. S6J-L). However, this arrest was more easily undone by low doses of Mps1 inhibitors, consistent with the reduced buffer of premade MCCs.
Tpr associates with NPCs via a short segment that binds Nup153, an upstream Nup that links the nuclear ring and basket (Fahrenkrog et al., 2002; Hase and Cordes, 2003; Krull et al., 2004; Walther et al., 2001). We used this information to test whether Mad1's N-terminus controls the speed and fidelity of M phase specifically through its NPC-targeting activity, or through other unrelated function(s) of this domain. Briefly, we generated constructs in which Mad1's entire NPD was deleted (Mad1ΔNP3) or exchanged with Tpr's Nup153-binding segment (amino acids 305-513) to create a chimera (Mad1X) (Fig. 7A). Mad1X was targeted to the chromatin-facing side of the NPC, whereas Mad1ΔNP3 was diffusely nuclear (Fig. 7B). Crucially, Mad1X restored interphase MCC production (Fig. 7C), as well as normal mitotic timing, error-free chromosome segregation, and checkpoint robustness in MAD1L1-null cells (Fig. 7D-H). Likewise, Mad1X retained its NPC localization after Tpr RNAi (Fig. 7I) and rescued M phase timing and fidelity in this context as well (Fig. 7J-K). Together, these results show that Mad1-Mad2 must be targeted to NPCs in order to produce the premitotic Cdc20 inhibitor, which ensures that anaphase and mitotic exit are robustly coupled to the establishment and correction of kinetochore-microtubule attachments.
Figure 7. Artificial Mad1-NPC tethering supports interphase MCC assembly and restores normal mitotic timing, fidelity, and checkpoint robustness.

(A) Constructs for artificial Mad1-NPC tethering. (B) MAD1L1flux/Δ HCT116 cells expressing the constructs in (A) were infected with AdCre, then fixed and stained as shown. (C) Cells in (B) were processed for interphase extracts before and after MAD1L1 deletion. Mad2-Cdc20 binding was then quantified; error bars denote SEM. (D) Cells in (B) were followed by timelapse microscopy. For comparison, Mad1ΔNP2 cells were treated with proTAME to delay anaphase entry by ∼10 min (Fig. 5B-C). (E) Frequencies of lagging anaphase chromatids. Error bars indicate SEM. (F) Cumulative anaphase entry as a function of time after NEBD. Genotypes with minimal (≤5%) lagging are plotted as a single group. (G) Phase-contrast micrographs of MAD1L1-null cells reconstituted with Mad1wt, Mad1ΔNP2, or Mad1X and treated with nocodazole ± ZM for 15 hr. Scale bar, 20 μm. (H) Cells in (G) were transduced with an H2B-mCherry expression vector, then filmed in the presence of nocodazole ± ZM. (I) Mad1X remains NPC-bound after Tpr depletion. MAD1L1flux/Δ HCT116 cells expressing Mad1wt or Mad1X were transfected with Tpr-specific siRNA, then fixed and immunostained as in (B). (J) Cells expressing H2B-mCherry were transfected with siRNAs and followed by timelapse microscopy 72 hr later. P-values were computed by one-way ANOVA; n.s., not significant. (K) The frequency of lagging chromatids was determined from the recordings in (J). Error bars indicate SEM. Scale bars, 5 μm. See also Figure S6.
Discussion
Over the past decade it has become clear that NPCs are not only portals for macromolecular transport, but also scaffolds for organization, expression, and maintenance of the nuclear genome (Hetzer and Wente, 2009; Strambio-De-Castillia et al., 2010). We discovered that the SAC mediator Mad1 uses NPC-mediated scaffolding to control the speed and fidelity of mitosis, well before it or key mediators of kinetochore-microtubule attachment (e.g., the Ndc80 and Ska complexes) are recruited to centromeric chromatin (Gascoigne and Cheeseman, 2013). This mode of regulation involves tethering of Mad1-Mad2 heterodimers to the nuclear basket, via a specialized domain on the N-terminus of Mad1. Once positioned at NPCs, the Mad1-Mad2 complex templates the assembly of Cdc20-inhibitory complexes or MCCs in the interphase cytoplasm. By defining the minimum length of time a cell will spend in mitosis, these preformed MCCs enhance the correction of potential chromosome segregation errors and decrease the threshold of kinetochore-dependent MCCs needed to establish a productive SAC response to spindle poisons.
Our work elaborates on current models of SAC signaling, which emphasize the kinetochore's role as a catalytic platform or scaffold for generating an anaphase inhibitor (De Antoni et al., 2005; Fava et al., 2011; Maldonado and Kapoor, 2011; Meraldi et al., 2004; Musacchio and Salmon, 2007). While informative, these models did not explain how similar anaphase inhibitors are made in interphase mammalian cells and yeast strains without kinetochores (Fraschini et al., 2001; Maciejowski et al., 2010; Malureanu et al., 2009; Poddar et al., 2005; Sudakin et al., 2001), or why 100-fold more Mad1-Mad2 is positioned at NPCs throughout interphase (Campbell et al., 2001; Shah et al., 2004). Our results link these observations, as disrupting Madl's native NPC-tethering mechanism prevented MCC assembly during interphase but not mitosis, while an artificial Mad1-NPC tether reversed this defect. We propose that interphase-specific MCC production parallels other NPC-scaffolded processes (e.g., mRNA surveillance or SUMO homeostasis; (Strambio-De-Castillia et al., 2010)), in which tethering to the nuclear pore not only concentrates an enzyme, but also links its catalytic cycle to the translocation of an NPC-permeable substrate. Here we envision that soluble 0-Mad2 encounters a high concentration of Mad1-Mad2 and possibly Mps1 at the nuclear basket (Campbell et al., 2001; Liu et al., 2003), causing a fraction to be converted to C-Mad2 as it travels through the pore and encounters Cdc20 in the cytoplasm, thereby translating Mad2's nucleocytoplasmic shuttling into productive MCC assembly.
By decoupling the interphase- and mitosis-specific pathways for MCC production, we gained insight into their contributions to M phase regulation. In the context of an unperturbed mitosis, where robust spindle assembly pathways lead to rapid kinetochore-microtubule attachment and minimal MCC production at kinetochores (Collin et al., 2013; Dick and Gerlich, 2013), NPC-derived inhibitors are rate-limiting not only for anaphase onset, but also the correction of merotelic errors. In addition to promoting near-diploid aneuploidy or tetraploidy, unresolved merotelic errors can cause non-reciprocal translocations and possibly chromothripsis (complex genomic rearrangements localized in one or a few subchromosomal regions) (Crasta et al., 2012; Janssen et al., 2011; Jones and Jallepalli, 2012). According to one hypothesis, such rearrangements are driven by the post-mitotic entrapment of lagging chromosomes in micronuclei, which contain fewer NPCs and hence import essential replication factors inefficiently, leading to error-prone DNA synthesis (Crasta et al., 2012). However, our data reveal the existence of a separate premitotic mechanism by which NPCs mitigate merotely itself, thus preventing anaphase lagging and micronucleus formation in the first place.
In addition to fostering error correction, our studies reveal close cooperation between the “wait anaphase” signal emitted by interphase NPCs and pathways that target SAC mediators to nascent kinetochores once M phase begins. Insight into this role came from experiments in which we compared the effects of evicting Mad1-Mad2 from NPCs and inhibiting the Aurora B kinase, which promotes Mad1-Mad2's rapid migration to kinetochores at NEBD (Santaguida et al., 2011; Saurin et al., 2011). Interestingly, whereas neither perturbation prevented SAC establishment on its own, their combined imposition led to a fully penetrant defect. Through computational modeling, we were able to explain this result as arising from the extensive overlap between NPC- and kinetochore-regulated regimes of anaphase inhibition, which allows cells to initiate a checkpoint arrest earlier (i.e., with fewer kinetochore-generated MCCs) than would otherwise be required if kinetochores alone were responsible for APC/CCdc20 inhibition. While metazoan mitosis involves a graded transition between the two regimes at NEBD, we do not exclude the possibility that both could remain active during closed mitosis, thus explaining why yeast mutants without kinetochores still form MCCs in a G2/M-specific manner (Fraschini et al., 2001; Poddar et al., 2005).
Our discovery that Mad1-Mad2 uses both NPCs and kinetochores as sites for M phase regulation raises interesting questions about the evolutionary history of this arrangement. Comparative genomics suggests that the last eukaryotic common ancestor (LECA) already contained Mad1-Mad2 and other components of the SAC, as well as nucleoporins, soluble transport factors, and kinetochore proteins (Bapteste et al., 2005; DeGrasse et al., 2009; Meraldi et al., 2006; Vleugel et al., 2012). Moreover, extant eukaryotes from Opisthokonta, Archaeplastida, and Amoebozoa are known to recruit Mad1 and/or Mad2 to the NE (Ding et al., 2012; Iouk et al., 2002; Lince-Faria et al., 2009; Samereier, 2011). Given the early divergence of these taxa, we speculate that the NPC-derived “timer” emerged in a primitive eukaryote that segregated its chromosomes via DNA-membrane tethering as in prokaryotes (Toro and Shapiro, 2010), but was then co-opted by the new attachment site (the kinetochore) to buffer its evolving interactions with microtubules, creating the SAC. Similar buffering of “selfish” pericentromeric satellite repeats is thought to account for the recurrent positive selection of nucleoporins and karyopherins in Drosophila (Kusano et al., 2001; Larracuente and Presgraves, 2012; Phadnis et al., 2012; Tracy et al., 2010), highlighting the crucial and ongoing role of the nuclear transport machinery in maintaining chromosome stability on evolutionary timescales. Conversely, nucleoporins are highly overrepresented among oncogenic translocations (Capelson and Hetzer, 2009; Chow et al., 2012). For example, the N-terminus of Tpr undergoes frequent rearrangement with Met, Trk, and Raf in gastric and thyroid cancers, resulting in hyperactive tyrosine kinase fusions that are mislocalized to the cytoplasm (Kohler and Hurt, 2010). This segment of Tpr also induces lagging chromosomes when expressed on its own (Nakano et al., 2010), suggesting that these translocations fuel carcinogenesis not only through increased tyrosine kinase signaling, but also subversion of NPC-based defenses against chromosome instability.
Experimental Procedures
Gene targeting, retroviral transduction, and siRNA transfection
To target MAD1L1, CCNB1, and MAD2L1, gene-specific pAAV constructs, pRC, and pHelper were cotransfected into HEK293 cells using Lipofectamine Plus (Invitrogen). AAV particles were released by freeze-thaw and applied to RPE or HCT116 cells. After G418 selection or FACS sorting for Venus expression, correct recombinants were identified via genomic PCR and Southern blotting (Berdougo et al., 2009; Collin et al., 2013). To delete floxed sequences, CsCI-purified AdCre (Vector Development Lab, Baylor College of Medicine) was used at an MOI of 50. For retroviral transduction, constructs were cotransfected with pVSV-G into Phoenix cells. Supernatants were filtered, mixed 1:1 with fresh medium containing 20 μg/ml polybrene, and applied to target cells. Lipofectamine 2000 (Invitrogen) was used for mock or Tpr-specific siRNA transfections. Details of plasmid cloning, siRNA sequences, and expanded protocols can be found in the Extended Experimental Procedures.
Live-cell imaging, immunofluorescence microscopy, and interphase FISH
Cells in glass-bottomed dishes (MatTek) were imaged on a Nikon TE2000 widefield microscope or a Zeiss Axiovert 200 microscope with an UltraView spinning-disk confocal head. Photobleaching was performed on an LSM710 Live microscope, with 5 iterations at 100% power over twenty 2-min cycles. For immunofluorescence microscopy, cells on coverslips or chamber slides (Nunc) were fixed and permeabilized with 4% paraformaldehyde and 0.2% Triton X-100. 3% BSA was used as the blocking and antibody dilution buffer. After mounting in Prolong Gold (Invitrogen), specimens were imaged on a DeltaVision microscope (Applied Precision) and deconvolved in Softworx. For interphase FISH, a chromosome 6-specific probe was labeled with Spectrum Green-dUTP (Abbott Molecular) via nick translation. Cells fixed with methanokacetic acid (3:1) were dropped onto slides, hybridized with the probe, and imaged by epifluorescence microscopy (Jallepalli et al., 2001; Lengauer et al., 1997).
Cell synchronization, extract preparation, and quantitative blotting
Interphase extracts were prepared by depleting asynchronous cultures of mitotic cells via shakeoff. S- and G2-phase extracts were prepared by 20 hr treatment with thymidine or RO-3306. M phase extracts were prepared by 12-16 hr treatment with nocodazole or S-trityl-L-cysteine, followed by shakeoff. Whole-cell extracts were prepared by nitrogen cavitation (1250 psi, 45 min; Parr Instruments) in buffer B (140 mM NaCI, 30 mM Hepes, pH 7.8, 5% glycerol, 10 mM sodium pyrophosphate, 5 mM sodium azide, 10 mM NaF, 10 mM PMSF, 0.3 mM sodium orthovanadate, 20 mM β-glycerophosphate, 1 mM DTT, 0.2 mM microcystin, and 1× protease inhibitor cocktail). Alternatively, nuclear and cytoplasmic fractions were obtained via hypotonic lysis and centrifugation over sucrose cushions as described (Dieckmann et al., 2005; Scherl et al., 2002). Quantitative Western blotting was performed on an Odyssey scanner (Li-Cor) using IRDye-coupled antibodies.
Protein expression and in vitro pulldown assays
6His-tagged MacM fragments were expressed in the BL21(DE3) strain of E. coli, purified on Ni2+-charged Dynabeads, and dialyzed into S buffer (50 mM Na phosphate [pH 7.0], 50 mM NaCI, 1 mM EDTA, 1 mM ß-mercaptoethanol). A N-terminal segment of Tpr (1-775) in pET28 (Novagen) was used for in vitro translation (TnT Quick, Promega). Bead-bound 6His-Mad1 fragments were mixed with [35S]-Tpr in NP40 buffer (50 mM Tris [pH 8], 120 mM NaCI, 1% NP40) for 1 hr, washed, and eluted in 1× sample buffer. Reactions were analyzed by SDS-PAGE and autoradiography.
Mathematical simulations
The reaction scheme in Fig. 6E was implemented in MATLAB using an ordinary differential equation solver (ode15s). Reaction rates and protein numbers were taken from the literature and this study (Fig. S5B). Simulations were initiated without cyclin B activation. At time = 0, cyclin B activation was begun.
Supplementary Material
Highlights.
Mad1-Mad2 directs mitotic checkpoint complex (MCC) assembly during interphase
Interphase MCC assembly requires Mad1's nuclear pore-targeting domain and Tpr
Loss of interphase MCCs reduces mitotic timing, fidelity, and checkpoint robustness
Artificially tethering Mad1-Mad2 to nuclear pores restores normal M phase controls
Acknowledgments
We thank N. Gray, J. Hanover, A. Musacchio, and E. Nigg for generous gifts of reagents, M. Leversha (MSKCC Cytogenetics Core) for performing FISH, and A. North (Bio-Imaging Resource Center, Rockefeller University) and K. Manova (MSKCC Molecular Cytology Core) for help with confocal imaging. We are grateful to F. Cubizolles for help in preparing the Venus-Mad2 targeting construct and E. Foley for assistance with live-cell imaging. P.C. was funded via a BBSRC project grant. This work was supported by NIH grant R01GM094972 to P.V.J.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnaoutov A, Azuma Y, Ribbeck K, Joseph J, Boyarchuk Y, Karpova T, McNally J, Dasso M. Crm1 is a mitotic effector of Ran-GTP in somatic cells. Nat Cell Biol. 2005;7:626–632. doi: 10.1038/ncb1263. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Thompson SL, Manning AL, Compton DA. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol. 2009;11:27–35. doi: 10.1038/ncb1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E, Charlebois RL, MacLeod D, Brochier C. The two tempos of nuclear pore complex evolution: highly adapting proteins in an ancient frozen structure. Genome Biol. 2005;6:R85. doi: 10.1186/gb-2005-6-10-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdougo E, Terret M, Jallepalli P. Functional dissection of mitotic regulators through gene targeting in human somatic cells. Methods Mol Biol. 2009;545:21–37. doi: 10.1007/978-1-60327-993-2_2. [DOI] [PubMed] [Google Scholar]
- Cairo LV, Ptak C, Wozniak RW. Mitosis-Specific Regulation of Nuclear Transport by the Spindle Assembly Checkpoint Protein Mad1p. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.10.017. [DOI] [PubMed] [Google Scholar]
- Campbell MS, Chan GK, Yen TJ. Mitotic checkpoint proteins HsMAD1 and HsMAD2 are associated with nuclear pore complexes in interphase. J Cell Sci. 2001;114:953–963. doi: 10.1242/jcs.114.5.953. [DOI] [PubMed] [Google Scholar]
- Capelson M, Hetzer MW. The role of nuclear pores in gene regulation, development and disease. EMBO Rep. 2009;10:697–705. doi: 10.1038/embor.2009.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman I, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol. 2008;9:33–46. doi: 10.1038/nrm2310. [DOI] [PubMed] [Google Scholar]
- Chow KH, Factor RE, Ullman KS. The nuclear envelope environment and its cancer connections. Nat Rev Cancer. 2012;12:196–209. doi: 10.1038/nrc3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim Biophys Acta. 2008;1786:32–40. doi: 10.1016/j.bbcan.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Cimini D, Wan X, Hirel CB, Salmon ED. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr Biol. 2006;16:1711–1718. doi: 10.1016/j.cub.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–87. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- Collin P, Nashchekina O, Walker R, Pines J. The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol. 2013;15:1378–1385. doi: 10.1038/ncb2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Antoni A, Pearson C, Cimini D, Canman J, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon E, et al. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol. 2005;15:214–225. doi: 10.1016/j.cub.2005.01.038. [DOI] [PubMed] [Google Scholar]
- DeGrasse JA, DuBois KN, Devos D, Siegel TN, Sali A, Field MC, Rout MP, Chait BT. Evidence for a shared nuclear pore complex architecture that is conserved from the last common eukaryotic ancestor. Mol Cell Proteomics. 2009;8:2119–2130. doi: 10.1074/mcp.M900038-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick AE, Gerlich DW. Kinetic framework of spindle assembly checkpoint signalling. Nat Cell Biol. 2013;15:1370–1377. doi: 10.1038/ncb2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieckmann R, CoutÈ Y, Hochstrasser D, Diaz JJ, Sanchez JC. Purification of Nucleoli From Lymphoma Cells and Solubilization of Nucleolar Proteins for 2-DE Separation. The Proteomics Protocols Handbook. 2005:79–85. [Google Scholar]
- Dieppois G, Stutz F. Connecting the transcription site to the nuclear pore: a multi-tether process that regulates gene expression. J Cell Sci. 2010;123:1989–1999. doi: 10.1242/jcs.053694. [DOI] [PubMed] [Google Scholar]
- Ding D, Muthuswamy S, Meier I. Functional interaction between the Arabidopsis orthologs of spindle assembly checkpoint proteins MAD1 and MAD2 and the nucleoporin NUA. Plant molecular biology. 2012;79:203–216. doi: 10.1007/s11103-012-9903-4. [DOI] [PubMed] [Google Scholar]
- Ditchfield C, Johnson V, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor S. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenkrog B, Maco B, Fager AM, Koser J, Sauder U, Ullman KS, Aebi U. Domain-specific antibodies reveal multiple-site topology of Nup153 within the nuclear pore complex. J Struct Biol. 2002;140:254–267. doi: 10.1016/s1047-8477(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Fang G. Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol Biol Cell. 2002;13:755–766. doi: 10.1091/mbc.01-09-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava LL, Kaulich M, Nigg EA, Santamaria A. Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. EMBO J. 2011;30:3322–3336. doi: 10.1038/emboj.2011.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol. 2013;14:25–37. doi: 10.1038/nrm3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraschini R, Beretta A, Sironi L, Musacchio A, Lucchini G, Piatti S. Bub3 interaction with Mad2, Mad3 and Cdc20 is mediated by WD40 repeats and does not require intact kinetochores. Embo J. 2001;20:6648–6659. doi: 10.1093/emboj/20.23.6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Cheeseman IM. CDK-dependent phosphorylation and nuclear exclusion coordinately control kinetochore assembly state. J Cell Biol. 2013;201:23–32. doi: 10.1083/jcb.201301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregan J, Polakova S, Zhang L, Tolic-Norrelykke IM, Cimini D. Merotelic kinetochore attachment: causes and effects. Trends Cell Biol. 2011;21:374–381. doi: 10.1016/j.tcb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase ME, Cordes VC. Direct interaction with nup153 mediates binding of Tprto the periphery of the nuclear pore complex. Mol Biol Cell. 2003;14:1923–1940. doi: 10.1091/mbc.E02-09-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzer MW, Wente SR. Border control at the nucleus: biogenesis and organization of the nuclear membrane and pore complexes. Dev Cell. 2009;17:606–616. doi: 10.1016/j.devcel.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt L, Tighe A, Santaguida S, White AM, Jones CD, Musacchio A, Green S, Taylor SS. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J Cell Biol. 2010;190:25–34. doi: 10.1083/jcb.201002133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iouk T, Kerscher O, Scott RJ, Basrai MA, Wozniak RW. The yeast nuclear pore complex functionally interacts with components of the spindle assembly checkpoint. J Cell Biol. 2002;159:807–819. doi: 10.1083/jcb.200205068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallepalli P, Waizenegger I, Bunz F, Langer S, Speicher M, Peters J, Kinzler K, Vogelstein B, Lengauer C. Securin is required for chromosomal stability in human cells. Cell. 2001;105:445–457. doi: 10.1016/s0092-8674(01)00340-3. [DOI] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- Jones MJ, Jallepalli PV. Chromothripsis: chromosomes in crisis. Dev Cell. 2012;23:908–917. doi: 10.1016/j.devcel.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J, Liu ST, Jablonski SA, Yen TJ, Dasso M. The RanGAP1-RanBP2 complex is essential for microtubule-kinetochore interactions in vivo. Curr Biol. 2004;14:611–617. doi: 10.1016/j.cub.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Kim S, Sun H, Tomchick DR, Yu H, Luo X. Structure of human Mad1 C-terminal domain reveals its involvement in kinetochore targeting. Proc Natl Acad Sci U S A. 2012;109:6549–6554. doi: 10.1073/pnas.1118210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler A, Hurt E. Gene regulation by nucleoporins and links to cancer. Mol Cell. 2010;38:6–15. doi: 10.1016/j.molcel.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Krull S, Thyberg J, Bjorkroth B, Rackwitz HR, Cordes VC. Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol Biol Cell. 2004;15:4261–4277. doi: 10.1091/mbc.E04-03-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano A, Staber C, Ganetzky B. Nuclear mislocalization of enzymatically active RanGAP causes segregation distortion in Drosophila. Dev Cell. 2001;1:351–361. doi: 10.1016/s1534-5807(01)00042-9. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski N, Jelluma N, Filippakopoulos P, Soundararajan M, Manak MS, Kwon M, Choi HG, Sim T, Deveraux QL, Rottmann S, et al. Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat Chem Biol. 2010;6:359–368. doi: 10.1038/nchembio.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM, Presgraves DC. The selfish Segregation Distorter gene complex of Drosophila melanogaster. Genetics. 2012;192:33–53. doi: 10.1534/genetics.112.141390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Sterling H, Burlingame A, McCormick F. Tpr directly binds to Mad1 and Mad2 and is important for the Mad1-Mad2-mediated mitotic spindle checkpoint. Genes Dev. 2008;22:2926–2931. doi: 10.1101/gad.1677208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer C, Kinzler K, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- Lince-Faria M, Maffini S, Orr B, Ding Y, Claudia F, Sunkel CE, Tavares A, Johansen J, Johansen KM, Maiato H. Spatiotemporal control of mitosis by the conserved spindle matrix protein Megator. J Cell Biol. 2009;184:647–657. doi: 10.1083/jcb.200811012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chan G, Hittle J, Fujii G, Lees E, Yen T. Human MPS1 kinase is required for mitotic arrest induced by the loss of CENP-E from kinetochores. Mol Biol Cell. 2003;14:1638–1651. doi: 10.1091/mbc.02-05-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Tang Z, Rizo J, Yu H. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell. 2002;9:59–71. doi: 10.1016/s1097-2765(01)00435-x. [DOI] [PubMed] [Google Scholar]
- Maciejowski J, George KA, Terret ME, Zhang C, Shokat KM, Jallepalli PV. Mps1 directs the assembly of Cdc20 inhibitory complexes during interphase and mitosis to control M phase timing and spindle checkpoint signaling. J Cell Biol. 2010;190:89–100. doi: 10.1083/jcb.201001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado M, Kapoor TM. Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat Cell Biol. 2011;13:475–482. doi: 10.1038/ncb2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malureanu L, Jeganathan K, Hamada M, Wasilewski L, Davenport J, van Deursen J. BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/C(Cdc20) in interphase. Dev Cell. 2009;16:118–131. doi: 10.1016/j.devcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Draviam V, Sorger P. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 2004;7:45–60. doi: 10.1016/j.devcel.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Meraldi P, McAinsh AD, Rheinbay E, Sorger PK. Phylogenetic and structural analysis of centromeric DNA and kinetochore proteins. Genome Biol. 2006;7:R23. doi: 10.1186/gb-2006-7-3-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Salmon E. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Nakano H, Funasaka T, Hashizume C, Wong RW. Nucleoporin translocated promoter region (Tpr) associates with dynein complex, preventing chromosome lagging formation during mitosis. J Biol Chem. 2010;285:10841–10849. doi: 10.1074/jbc.M110.105890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijenhuis W, von Castelmur E, Littler D, De Marco V, Tromer E, Vleugel M, van Osch MH, Snel B, Perrakis A, Kops GJ. A TPR domain-containing N-terminal module of MPS1 is required for its kinetochore localization by Aurora B. J Cell Biol. 2013;201:217–231. doi: 10.1083/jcb.201210033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Phadnis N, Hsieh E, Malik HS. Birth, death, and replacement of karyopherins in Drosophila. Molecular biology and evolution. 2012;29:1429–1440. doi: 10.1093/molbev/msr306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. Cubism and the cell cycle: the many faces of the APC/C. Nat Rev Mol Cell Biol. 2011;12:427–438. doi: 10.1038/nrm3132. [DOI] [PubMed] [Google Scholar]
- Poddar A, Stukenberg PT, Burke DJ. Two complexes of spindle checkpoint proteins containing Cdc20 and Mad2 assemble during mitosis independently of the kinetochore in Saccharomyces cerevisiae. Eukaryotic cell. 2005;4:867–878. doi: 10.1128/EC.4.5.867-878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon ED, Cimini D, Cameron LA, DeLuca JG. Merotelic kinetochores in mammalian tissue cells. Philos Trans R Soc Lond B Biol Sci. 2005;360:553–568. doi: 10.1098/rstb.2004.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samereier M. Cell Biology. Potsdam, DE: University of Potsdam; 2011. Functional analyses of microtubule and centrosome-associated proteins in Dictyostelium discoideum. [Google Scholar]
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol. 2010;190:73–87. doi: 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Vernieri C, Villa F, Ciliberto A, Musacchio A. Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. EMBO J. 2011;30:1508–1519. doi: 10.1038/emboj.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurin AT, van der Waal MS, Medema RH, Lens SM, Kops GJ. Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nature communications. 2011;2:316. doi: 10.1038/ncomms1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherl A, Coute Y, Deon C, Calle A, Kindbeiter K, Sanchez JC, Greco A, Hochstrasser D, Diaz JJ. Functional proteomic analysis of human nucleolus. Mol Biol Cell. 2002;13:4100–4109. doi: 10.1091/mbc.E02-05-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RJ, Lusk CP, Dilworth DJ, Aitchison JD, Wozniak RW. Interactions between Mad1p and the nuclear transport machinery in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:4362–4374. doi: 10.1091/mbc.E05-01-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JV, Botvinick E, Bonday Z, Furnari F, Berns M, Cleveland DW. Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr Biol. 2004;14:942–952. doi: 10.1016/j.cub.2004.05.046. [DOI] [PubMed] [Google Scholar]
- Sironi L, Mapelli M, Knapp S, De Antoni A, Jeang KT, Musacchio A. Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of a ‘safety belt’ binding mechanism for the spindle checkpoint. EMBO J. 2002;21:2496–2506. doi: 10.1093/emboj/21.10.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe S, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol. 2007;8:195–208. doi: 10.1038/nrm2114. [DOI] [PubMed] [Google Scholar]
- Strambio-De-Castillia C, Niepel M, Rout MP. The nuclear pore complex: bridging nuclear transport and gene regulation. Nat Rev Mol Cell Biol. 2010;11:490–501. doi: 10.1038/nrm2928. [DOI] [PubMed] [Google Scholar]
- Sudakin V, Chan G, Yen T. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 2001;154:925–936. doi: 10.1083/jcb.200102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Bharadwaj R, Li B, Yu H. Mad2-lndependent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell. 2001;1:227–237. doi: 10.1016/s1534-5807(01)00019-3. [DOI] [PubMed] [Google Scholar]
- Toro E, Shapiro L. Bacterial chromosome organization and segregation. Cold Spring Harbor perspectives in biology. 2010;2:a000349. doi: 10.1101/cshperspect.a000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy C, Rio J, Motiwale M, Christensen SM, Betran E. Convergently recruited nuclear transport retrogenes are male biased in expression and evolving under positive selection in Drosophila. Genetics. 2010;184:1067–1076. doi: 10.1534/genetics.109.113522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron S, Prieto S, Bernis C, Labbe J, Castro A, Lorca T. Kinetochore localization of spindle checkpoint proteins: who controls whom? Mol Biol Cell. 2004;15:4584–4596. doi: 10.1091/mbc.E04-01-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink M, Simonetta M, Transidico P, Ferrari K, Mapelli M, De Antoni A, Massimiliano L, Ciliberto A, Faretta M, Salmon E, et al. In vitro FRAP identifies the minimal requirements for Mad2 kinetochore dynamics. Curr Biol. 2006;16:755–766. doi: 10.1016/j.cub.2006.03.057. [DOI] [PubMed] [Google Scholar]
- Vleugel M, Hoogendoorn E, Snel B, Kops GJ. Evolution and function of the mitotic checkpoint. Dev Cell. 2012;23:239–250. doi: 10.1016/j.devcel.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Walther TC, Fornerod M, Pickersgill H, Goldberg M, Allen TD, Mattaj IW. The nucleoporin Nup153 is required for nuclear pore basket formation, nuclear pore complex anchoring and import of a subset of nuclear proteins. EMBO J. 2001;20:5703–5714. doi: 10.1093/emboj/20.20.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver B, Silk A, Montagna C, Verdier-Pinard P, Cleveland D. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Yu H. Structural activation of Mad2 in the mitotic spindle checkpoint: the two-state Mad2 model versus the Mad2 template model. J Cell Biol. 2006;773:153–157. doi: 10.1083/jcb.200601172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh DC, Hathaway N, Dimova N, Cuny GD, King RW. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell. 2010;18:382–395. doi: 10.1016/j.ccr.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccolo M, Alves A, Galy V, Bolhy S, Formstecher E, Racine V, Sibarita JB, Fukagawa T, Shiekhattar R, Yen T, et al. The human Nup107-160 nuclear pore subcomplex contributes to proper kinetochore functions. EMBO J. 2007;26:1853–1864. doi: 10.1038/sj.emboj.7601642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.