

Abstract
The three Akt isoforms are functionally distinct. Here we show that their phosphoproteomes also differ, suggesting that their functional differences are due to differences in target specificity. One of the top cellular functions differentially-regulated by Akt isoforms is RNA processing. IWS1, an RNA processing regulator, is phosphorylated by Akt3 and Akt1 at Ser720/Thr721. The latter is required for the recruitment of SETD2 to the RNA Pol II complex. SETD2 trimethylates histone H3 at K36 during transcription, creating a docking site for MRG15 and PTB. H3K36me3-bound MRG15 and PTB regulate FGFR-2 splicing, which controls tumor growth and invasiveness downstream of IWS1 phosphorylation. 21/24 non-small-cell-lung carcinomas we analyzed, express IWS1. More important, the stoichiometry of IWS1 phosphorylation in these tumors correlates with the FGFR-2 splicing pattern, and with Akt phosphorylation and Akt3 expression. These data identify a novel Akt isoform-dependent regulatory mechanism for RNA processing and demonstrate its role in lung cancer.
Keywords: Akt isoforms, PKB, IWS1, SETD2, MRG15, PTB, FGFR2, phosphoproteomics, RNA metabolism, alternative splicing, histone H3 K36 trimethylation, transcriptional elongation
INTRODUCTION
Akt is an AGC family serine-threonine protein kinase. It is activated by a variety of extracellular and intracellular signals and its activation depends on PtdIns-3,4,5-P3, and to a lesser extent on PtdIns-3,4-P2, both of which are products of the phosphoinositide 3-kinase (Franke et al., 1995; Manning and Cantley, 2007). Although the three Akt isoforms, Akt1, Akt2 and Akt3, have been treated by most of the earlier studies as a single entity, recent evidence supports their unique roles in the regulation of cell function (Chen et al., 2001; Cho et al., 2001; Ezell et al., 2012; Iliopoulos et al., 2009; Maroulakou et al., 2007; Polytarchou et al., 2011; Tschopp et al., 2005; Wan et al., 2011).
To determine whether the functional differences between Akt isoforms are due to signaling differences, we employed a system of immortalized lung fibroblasts, which were engineered to express one Akt isoform at a time but are otherwise identical (Ezell et al., 2012; Iliopoulos et al., 2009; Polytarchou et al., 2011). A phosphoproteomics screen of these cells, identified 606 proteins that are phosphorylated on Akt phosphorylation consensus sites in at least one of the Akt isoform-expressing cells. Analysis of the phosphoproteome data indeed identified signaling pathways and cellular functions preferentially regulated by one or another isoform. One of the differentially-regulated functions was the post-transcriptional processing of mRNA (RNA metabolism) (Moore and Proudfoot, 2009).
One of the differentially-phosphorylated proteins involved in the regulation of RNA metabolism is IWS1, which is phosphorylated at a conserved phosphorylation site, primarily by Akt3 and Akt1. IWS1 was originally identified in the yeast Saccharomyces Cerevisciae through its interaction with Spt6 (Krogan et al., 2002), a histone H3/H4 chaperone (Duina, 2011). Spt6 also binds the C terminal domain (CTD) of the large subunit of RNA Pol II, following the phosphorylation of the latter at Ser2 (Yoh et al., 2007). Recent studies in mammalian cells revealed that the Spt6/IWS1 complex contains two additional proteins, Aly/REF, an adaptor that contributes to nucleocytoplasmic RNA transport, and SetD2, a histone H3 trimethyl transferase (Yoh et al., 2007; Yoh et al., 2008). The same studies showed that this complex contributes both to alternative RNA splicing and to nucleocytoplasmic RNA transport.
The regulation of RNA splicing is a complex and poorly understood process. Although some of the splicing events are constitutive, occurring in all cell types independent of external signals, others, also known as alternative splicing events, are conditional. Recent estimates suggest that at least 90% of the metazoan genes are alternatively spliced (Wang et al., 2008). This gives rise to a proteome that is significantly more complex than the number of genes would suggest. Alternative splicing plays an important role in differentiation and development, as well as in the response of fully differentiated cells to various signals (Kalsotra and Cooper, 2011). Defects in alternative splicing have been linked to a host of human diseases, primarily neurodegenerative diseases and cancer (Cooper et al., 2009). Alternative splicing depends on the interplay of cis-acting RNA elements with trans-acting splicing factors (Chen and Manley, 2009), and on epigenetic cues, such as DNA methylation and histone modifications (Luco et al., 2011). However, very little is known about the control of this machinery by cellular signals (Zhou et al., 2012).
One of the genes that undergo alternative splicing during development and in some cancers is the gene encoding FGF Receptor-2 (FGFR-2) (Carstens et al., 2000; Warzecha et al., 2009). Alternative splicing gives rise to two isoforms of the receptor, the IIIc and the IIIb isoform, of which only the IIIc isoform is recognized by basic FGF (bFGF or FGF-2) (Miki et al., 1992). The IIIc isoform is expressed preferentially in mesenchymal cells, as opposed to epithelial cells (Luco et al., 2010; Orr-Urtreger et al., 1993) and promotes cell migration and invasiveness in response to FGF-2. Moreover, the FGFR-2 IIIb to IIIc switch has been linked to epithelial mesenchymal transition (EMT) and to a migratory and metastatic phenotype in cancer cells (Thiery and Sleeman, 2006). The FGFR-2 alternative splicing discussed above belongs to a subset of alternative splicing events which depend on histone H3 trimethylation at K36 in the body of the target gene and on the polypyrimidine tract binding protein (PTB) (Carstens et al., 2000; Luco et al., 2010).
The data presented in this report confirmed the differential phosphorylation of IWS1 at Ser720/Thr721 by Akt3 and Akt1 and showed that its phosphorylation at this site is required for the recruitment of SetD2 to the Spt6-IWS1-Aly/REF complex. In the absence of SetD2 recruitment to the complex in lung carcinoma cell lines, the trimethylation of histone H3 at K36 in the body of the FGFR-2 gene was impaired. This interfered with the recruitment of MRG15, an H3K36me3-binding protein and its interacting partner PTB, and shifted the splicing of FGFR-2 from the IIIc to the IIIb isoform. The shift in the alternative splicing of FGFR-2 interferes with cell migration and invasiveness in response to FGF-2 and suppresses the proliferation and invasiveness of tumor cells both in culture and in animals. Addressing the expression of FGFR-2 in a set of lung-derived normal and tumor samples revealed that whereas the overall expression was similar in both, there was a shift toward the IIIc isoform in the tumor samples. More important, the relative expression of the IIIc and IIIb isoforms in non-small-cell-lung-carcinomas (NSCLCs) correlated with the stoichiometry of IWS1 phosphorylation and the latter correlated with Akt phosphorylation and Akt3 expression. Finally, data from “Oncomine” show that the levels of expression of IWS1 in lung cancer correlate with the tumor stage. These findings combined, underpin the importance of this pathway in the pathogenesis of lung cancer. Overall, our data suggest that Akt isoform-dependent phosphorylation events are essential for RNA processing and provide novel insights into the role of Akt in carcinogenesis.
RESULTS
Differential regulation of the Akt-phosphoproteome by the three Akt isoforms
To investigate signaling differences between Akt isoforms, we performed a phosphoproteomics screen on an isogenic cellular platform, where Akt-null cells were engineered to express one Akt isoform at a time, or a combination of all three (Fig 1A and S1A). Cell lysates were digested with LysC and the resulting peptides were affinity purified with Akt phosphosubstrate antibodies. Enriched phosphopeptides were digested with trypsin, purified over StageTip and analyzed by mass spectrometry. Comparison of the z scores (Fig S1B) of individual phosphorylation sites revealed significant differences between the phosphoproteomes of Akt1, Akt2, Akt3 and Akt1/2/3 cells (Fig 1B). To validate these data, we examined the z scores of known phosphorylation events that were also detected in our screen (Fig 1C and Table S1) and we examined and confirmed a set of phosphorylation events first detected in the present study (Fig 1C, S1C–G). These analyses validated all phosphorylation events above MAD = −0.64. A Venn diagram of Akt1, Akt2, Akt3 and Akt1/2/3-mediated phosphorylation events, with z-scores above MAD = 2, identified sets of proteins that are differentially phosphorylated by one or another isoform (Fig S2A). The subcellular localization of these proteins revealed differences in distribution between isoform-specific targets (Fig S2B). Moreover, bioinformatics analyses suggested that Akt isoforms differentially regulate canonical pathways and cell functions (Fig 2A and 2B). One of the functions predicted to be differentially regulated by different Akt isoforms is RNA metabolism, which is represented by 25 proteins that are involved in different stages of the process (Fig 3C and S2C).
Figure 1. The phosphoproteomes of Akt1, Akt2, Akt3 and Akt1/2/3-expressing cells.

(A) Western blots of cell lysates of Akt-null (TKO) lung fibroblasts and their Akt isoform-expressing derivatives were probed with the indicated antibodies.
(B) Heat map of the relative abundance of all the phosphorylation events detected in the phosphoproteomics screen. The relative abundance of each phosphorylation event is presented as the robust z-score of the ratio Akt1/TKO, Akt2/TKO, Akt3/TKO or Akt1/2/3/TKO.
(C) Heat map of the abundance of phosphorylation of known Akt substrates (black) and Akt substrates that were identified in this screen (red). The phosphorylated residue is shown on the right.
Figure 2. The phosphoproteomes of Akt1, Akt2 and Akt3-expressing cells suggest functional differences between Akt isoforms. IWS1 phosphorylation by Akt1 and Akt3.

(A) Heat map showing the probability [-log(p value)] that a given canonical pathway may be regulated by Akt1, Akt2, Akt3 or Akt1/2/3. Analysis based on phosphorylation events with a robust z score ≥2 MAD.
(B) Probability that RNA trafficking and RNA posttranscriptional modifications may be regulated by Akt1, Akt2, or Akt3.
(C) Diagram of the sequential steps involved in RNA processing. Akt targets identified or confirmed by our screen are indicated.
(D) Sequence of the phosphorylated IWS1 peptide. The asterisks show the phosphorylated residues. The numbers show the median absolute deviation of the robust z scores.
(E) Conservation of the phosphorylated peptide and the adjacent sequence.
(F) Upper: Western blots of TKO lung fibroblasts and their Akt isoform-expressing derivatives were probed with anti-myc or anti-Hsp90 (loading control). Lower: The lung fibroblasts in the upper panel were transduced with the indicated constructs and their lysates were used for immunoprecipitation and immunoblotting with the indicated antibodies. Western blots of the same lysates were probed with anti-FLAG or anti-CREB, as indicated (G) HeLa cells transduced with lentiviral constructs of FLAG-IWS1 (wt or mutant) were treated with 5 μM MK2206 or DMSO for 2 hours. Anti-Flag-IWS1 immunoprecipitates were probed with the Akt phosphosubstrate antibody. Western blots of the same cell lysates were probed with the indicated antibodies.
(H) Left: In vitro kinase assays of myc-tagged Akt1, Akt2 or Akt3, using GST-IWS1wt (upper panel) and GSK3α/β (lower panel) recombinant proteins as substrates. Phosphorylation was detected with the Akt phosphosubstrate or the phospho-GSK3α/β (21/9) antibody. Right: In vitro kinase assay of myc-Akt1 using wt or mutant GST-IWS1 as substrates.
Figure 3. IWS1 phosphorylation at Ser720/Thr721 is required for SETD2 recruitment and H3K36me3 trimethylation in the FGFR-2 gene.

(A) NCI H522, NCI H1299 and T47D cell lysates, harvested before and after treatment with IGF-1, were probed with the indicated antibodies.
(B) Lysates of shControl, shIWS1, shIWS1/wt rescue and shIWS1/mutant rescue NCI H522, NCI H1299 and T47D cells, were probed with the indicated antibodies.
(C) Upper: Flag-IWS1 immunoprecipitates of shIWS1/wt rescue and shIWS1/mutant rescue NCI H1299 cells were probed with the Akt phosphosubstrate or the anti-FLAG (IWS1) antibody. Lower: The same lysates were probed with antibodies to phospho-IWS1, or total IWS1.
(D) SetD2, Spt6, REF/Aly and Flag-IWS1 immunoprecipitates from lysates of shIWS1/wt rescue and shIWS1/mutant rescue NCI H522 cells were probed with anti-Flag (IWS1), anti-RNA PolII and antiSetD2, as indicated. The same cell lysates were probed with the indicated antibodies.
(E) Schematic showing the alternatively-spliced exons 8 (IIIb) and 9 (IIIc) of the human FGFR-2 gene. The map position of the primer sets is indicated by square dots. TSS: Transcription start site.
(F) ChIP assays showing the binding of IWS1 to the FGFR-2 and GAPDH genes in the indicated shControl and shIWS1 cells. Bars show the mean fold enrichment in the binding of endogenous IWS1 in shControl, vs shIWS1 cells ± SD.
(G) ChIP of Flag-IWS1 in shIWS1/wt rescue and shIWS1/mutant rescue NCI H522 cells shows that both wt and mutant IWS1 bind exons IIIb and IIIc of FGFR-2. Bars show the mean fold enrichment in binding of Flag-IWS1 in these cells, relative to the parental cells ± SD. IWS1 binding is also presented as percentage of the input in figure S3D.
(H and I) ChIP assays showing the binding of HA-SetD2 to the FGFR-2 and GAPDH genes in the indicated shControl, shIWS1, shIWS1/wt rescue and shIWS1/mutant rescue cells, transduced with an HA-SetD2 lentiviral construct. Bars show the mean fold enrichment in binding of HA-SetD2 in shControl relative to shIWS1 cells or in shIWS1/wt rescue relative to shIWS1/mutant rescue cells ± SD. HA-SetD2 expression and binding is also presented as percentage of the input in figure S3E.
(J and K) ChIP assays showing the abundance of histone H3K36me3 in the FGFR-2 and GAPDH genes in the indicated cells. Bars show the mean fold enrichment in H3K36me3 in shControl relative to shIWS1 cells or in shIWS1/wt rescue relative to shIWS1/mutant rescue cells ± SD.
Akt1 and Akt3 phosphorylate IWS1 at Ser720/Thr721
One of the proteins we identified as a differentially phosphorylated Akt target was IWS1 (Fig 2D and 2E), which is known to contribute to the assembly of a transcriptional elongation complex on the Ser2-phosphorylated CTD of the large subunit of RNA polymerase II (Yoh et al., 2007; Yoh et al., 2008). Immunoprecipitation of IWS1 from Flag-IWS1-transduced lung fibroblasts expressing Akt1, Akt2 or Akt3 with the Akt phosphosubstrate antibody and probing the immunoprecipitates with an anti-Flag antibody, confirmed the Akt isoform-dependent phosphorylation of IWS1 at a site corresponding to the Akt phosphorylation consensus (Fig 2F). In agreement with this observation, the inhibition of all Akt activity in HELA cells with 5 μM of the Akt inhibitor MK2206, blocked IWS1 phosphorylation (Fig 2G). Finally, in vitro kinase assays using bacterially expressed wild type IWS1 and its phosphorylation site mutants as substrates, confirmed that IWS1 is phosphorylated directly by Akt and that Ser720/Thr721 is the only Akt phosphorylation site in this protein (see supplement for further discussion). Remarkably, IWS1 was phosphorylated by Akt3 and less efficiently by Akt1, but not Akt2, suggesting that isoform-dependent phosphorylation specificity may be the result of structural differences that directly affect substrate recognition (Fig 2H and S2D).
IWS1 phosphorylation at Ser720/Thr721 is required for the recruitment of SetD2 to the RNA polymerase II elongation complex
To determine the biological significance of IWS1 phosphorylation, we first examined its expression in a set of human tumor cell lines. This analysis identified the NSCLC cell lines NCI H522 and NCI H1299 as high expressors and T47D, a human mammary carcinoma cell line as a low expressor (Fig S3A). Western blotting confirmed that NCI H522 and NCI H1299 cells express higher levels of IWS1 than T47D cells. IGF1 stimulation of these cells revealed that IWS1 phosphorylation is regulated by receptor tyrosine kinase-generated signals and correlates with activation of Akt (Fig 3A).
In parallel experiments NCI H522, NCI H1299 and T47D cells were transduced with shIWS1-resistant constructs of FLAG-IWS1 or Flag-IWS1S720A/T721A. Subsequent infection of these cells with the shIWS1 lentiviral construct gave rise to cell lines in which the endogenous IWS1 was knocked down or replaced by exogenous wild type or mutant IWS1 (Fig 3B). The phosphorylation of the wild type protein was confirmed by probing IWS1 immunoprecipitates of wild type and mutant rescue cell lysates with the Akt phosphosubstrate antibody (Fig 3C, upper panel). Alternatively, lysates from the same cells were probed with the phospho-IWS1 and total IWS1 antibodies (Fig 3C, lower panel). The specificity of the phospho-IWS1 antibody was further validated by using it to probe lysates of NCI H1299 and T47D cells, before and after treatment with MK2206 (Fig S3B).
IWS1 interacts with Spt6, which binds the phosphorylated Ser2 in the CTD of RNA Pol II, Aly/REF and SetD2 (Yoh et al., 2007; Yoh et al., 2008). Co-immunoprecipitation experiments revealed that the Ser720/Thr721 phosphorylation mutant of IWS1 fails to bind SetD2 but continues to bind Aly/REF and SPT6. As a result, neither IWS1 nor RNA Pol II co-immunoprecipitate with SetD2 (Fig 3D). This finding was confirmed with the reciprocal immunoprecipitation experiment. An HA-SetD2 construct was introduced into shIWS1 NCI H1299 cells rescued with wild type or mutant IWS1. FLAG-IWS1 was immunoprecipitated from cells rescued with wild type IWS1, before and after treatment with MK2206, as well as from cells rescued with mutant IWS1 and western blots of the immunoprecipitates were probed with an anti-HA antibody. The results (Fig S3C) confirmed the data in figure 3D.
SetD2 catalyzes the trimethylation of histone H3 at K36, a histone mark linked to alternative splicing of a set of genes that include FGFR-2 (Kolasinska-Zwierz et al., 2009; Luco et al., 2010). Alternative splicing via an H3K36me3-regulated process (Luco et al., 2010), gives rise to the FGFR-2 isoforms IIIb and IIIc (Fig 3E), which encode receptors with different ligand specificities and different expression patterns (Luco et al., 2010; Miki et al., 1992). Replacement of endogenous IWS1 with its phosphorylation site mutant prevented the binding of SetD2 and decreased the abundance of histone H3K36me3 in the FGFR-2 gene. Chromatin immunoprecipitations (ChIP) revealed that IWS1 binds to exons IIIb and IIIc, but not to the transcription start site (TSS) of FGFR-2 (Fig 3F), and that IWS1 phosphorylation at Ser720/Thr721 is not required for the binding (Fig 3G and S3D). ChIP assays for SetD2 in cells transduced with an HA-SetD2 construct confirmed that IWS1 phosphorylation is required for SetD2 recruitment to the FGFR-2 gene (Fig 3H, 3I and S3E) and the trimethylation of histone H3 at K36 in exons IIIb and IIIc (Fig 3J and 3K).
IWS1 phosphorylation at Ser720/Thr721 regulates the alternative splicing of FGFR-2
High abundance of histone H3K36me3 in the IIIb exon is a signal for exon skipping, while low abundance of the same mark is a signal for exon inclusion (Kolasinska-Zwierz et al., 2009). As a result, knocking down SetD2 or IWS1, which is required for the recruitment of SetD2 to the elongation complex, increased the abundance of the IIIb relative to the IIIc transcripts, in NCI H522 and NCI H1299 cells, which express primarily FGFR-2 IIIc, but not in T47D cells, which express primarily FGFR-2 IIIb (Fig 4A, 4B, S4A and S4B). More important, the change in alternative splicing induced by the knockdown of IWS1 was rescued by the wild type IWS1 but not by IWS1S720A/T721A (Figure 4B). We conclude that IWS1 phosphorylation by Akt3 and Akt1 shifts the balance of FGFR-2 splicing toward FGFR-2-IIIc, by regulating the recruitment of SetD2 to the elongation complex. If the recruitment of SetD2 depends on the phosphorylation of IWS1, as our data indicate, one would expect that overexpression of SetD2 would not rescue the FGFR-2 alternative splicing phenotype in shIWS1 and shIWS1/mutant rescue cells. The experiment in figure S4C confirmed the prediction.
Figure 4. IWS1 phosphorylation at Ser720/Thr721 by Akt3 and Akt1 regulates the alternative splicing of the FGFR-2 gene.

(A) The SETD2 knockdown shifts the IIIb/IIIc ratio of the FGFR-2 mRNA toward the IIIb isoform. (Left) Real time RT-PCR for SETD2 in the indicated cell lines transduced with two different shSetD2 constructs or with an shControl construct. (Right). Real time RT-PCR showing the IIIb/IIIc FGFR-2 mRNA ratios in the cells on the left panel. Bars show the mean SETD2 levels or the mean IIIb/IIIc ratios in shSetD2, relative to shControl cells ± SD.
(B) IWS1 phosphorylation regulates FGFR-2 alternative splicing. Real time RT-PCR showing the IIIb/IIIc FGFR-2 ratios in the indicated shControl, shIWS1, shIWS1/wt rescue and shIWS1/mutant rescue cells. Bars show the mean IIIb/IIIc ratio in shIWS1, shIWS1/wt rescue and shIWS1/mutant rescue cells, relative to shControl cells ± SD.
(C) Akt inhibition interferes with histone H3K36 trimethylation in the FGFR-2 gene and promotes inclusion of the IIIb exon in the transcript. (Top) Left: Lysates of the indicated cells, harvested before and after a 2 hour treatment with MK2206 (5 μM) or DMSO, were probed with antibodies, as shown. Right: The FGFR-2 IIIb to IIIc ratio was measured by quantitative RT-PCR. Bars show the ratio in cells treated with MK2206 relative to cells treated with DMSO (mean ± SD). P-values were calculated with the student’s t test. (Bottom panels) ChIP assays measuring the abundance of H3K36me3 trimethylation in FGFR-2 in the indicated cell lines before and after a 2 hour treatment with 5 μM MK2206 or with DMSO. Bars show the fold enrichment of the abundance of H3K36me3 trimethylation in DMSO-treated vs Akt inhibitor-treated cells (mean ± SD).
(D) The knockdown of Akt1 and/or Akt3 promotes inclusion of the IIIb exon in the FGFR-2 transcript. Lysates of NCI H1299 cells harvested 48 hours after transfection with siAKT1, siAkt2, siAkt3 or siControl, were probed with the indicated antibodies (left). IIIb/IIIc ratios were measured by quantitative RT-PCR (right). Bars show the IIIb/IIIc ratio in siAkt vs si-control cells (mean ± SD).
(E) The Akt-independent mutant IWS1-DE reversed the MK2206-induced shift in FGFR-2 alternative splicing. RNA isolated from MK2206 or DMSO-treated NCI H522 and NCI H1299 cells transduced with the IWS1 phosphomimetic mutant IWS1-DE or the empty vector, was analyzed for the IIIb/IIIc ratio by quantitative RT-PCR. Bars show the IIIb/IIIc ratio in MK2206-treated, vs DMSO-treated cells (mean ± SD).
(F) FGFR-2 splicing shifts toward the IIIc transcript in response to IGF1. RNA from the cells in figure 3A was analyzed for the IIIb/IIIc ratio by quantitative RT-PCR. Bars show the IIIb/IIIc ratio in IGF1-treated vs untreated cells (mean ± SD).
(G and H) IWS1 phosphorylation is required for the binding of MRG15 to FGFR-2. ChIP assays showing the binding of MRG15 to the indicated sites in the FGFR-2 and GAPDH genes in shControl, shIWS1, shIWS1/wt rescue and shIWS1/mutant rescue NCI H522 (G) and NCI H1299 (H) cells. Bars show the mean fold enrichment in MRG15 binding in shControl vs shIWS1 or in shIWS1/wt rescue vs shIWS1/mutant rescue cells ± SD.
(I and J) IWS1 phosphorylation is required for the binding of PTB to FGFR-2. ChIP assays showing the binding of myc-PTB to the indicated sites in the FGFR-2 and GAPDH genes in shControl, shIWS1, shIWS1/wt rescue and shIWS1/mutant rescue NCI H522 (I) and NCI H1299 (J) cells, transfected with a myc-PTB construct. The expression of myc-PTB is shown in figure S4E. Bars show the mean fold enrichment in myc-PTB binding in shControl vs shIWS1 or in shIWS1/wt rescue vs shIWS1/mutant rescue cells ± SD. Myc-PTB binding is also presented as percentage of the input in figure S4E.
To address the dependence of FGFR-2 alternative splicing on Akt, we treated the cells with 5 μM MK2206, a dose that fully inhibits all Akt isoforms. Akt inhibition reduced H3K36 trimethylation in exons IIIb and IIIc and promoted exon IIIb inclusion in the FGFR-2 transcript (Fig 4C), suggesting that the IWS1 phosphorylation phenotype depends on the activity of Akt. Knocking down different Akt isoforms sequentially or in combination in the same cells not only confirmed this conclusion, but also showed that the skipping of exon IIIb is promoted primarily by Akt3 and Akt1 (Fig 4D).
The preceding data raised the question whether Akt regulates FGFR-2 splicing primarily by phosphorylating IWS1, or whether it also utilizes an alternative pathway. To address this question, we examined the splicing of FGFR-2 in MK2206 or DMSO-treated NCI H522 and NCI H1299 cells transduced with the phosphomimetic mutant IWS1-S720D/T721E (IWS1-DE) or with the empty vector (EV). The results showed that IWS1-DE rescues the MK2206-induced FGFR-2 alternative splicing phenotype (Fig 4E). The rescue of the MK2206-induced shift in alternative splicing by the Akt independent IWS1-DE mutant supports the hypothesis that Akt controls FGFR-2 alternative splicing by phosphorylating IWS1 and not through an alternative pathway. Support for this conclusion was provided by the experiment in figure S4D, which showed that none of the Akt isoforms can reverse the shIWS1 or shSetD2-induced shifts in FGFR-2 splicing, when overexpressed in NCI H522 cells. Although this conclusion is in agreement with all the data in this report however, it should be viewed with caution because Akt overexpression had no effect on FGFR-2 alternative splicing even in shControl cells (Fig S4D), perhaps because NCI H522 cells express sufficient levels of Akt to keep the pathway fully active.
If IGF1 promotes the phosphorylation of IWS1, perhaps through Akt (Fig 3A), and the phosphorylation of IWS1 shifts the alternative splicing of FGFR-2 toward the IIIc isoform, one would expect that treatment of NCI H522 and NCI H1299 cells with IGF1 would decrease the FGFR-2 IIIb/IIIc ratio. The experiment in figure 4F confirmed this prediction and provided evidence that Akt regulates FGFR-2 alternative splicing by receptor tyrosine kinase-induced signals.
IWS1 phosphorylation at Ser720/Thr721 is required for the binding of MRG15 and PTB to the FGFR-2 gene
Histone H3K36me3 provides a docking site for MRG15 (Zhang et al., 2006), a chromodomain protein that interacts with the polypyrimidine track binding protein (PTB) to translate the H3K36me3 mark into a signal that regulates alternative splicing (Luco et al., 2010). Consistent with the data on SetD2 binding and histone H3K36 trimethylation in the FGFR-2 gene (Fig 3), chromatin IPs in NCI H522 and NCI H1299 cells confirmed that knocking down IWS1 reduces the binding of both MRG15 and PTB and that their binding is restored by wild type, but not the mutant IWS1 (Fig 4G, 4H, 4I, 4J and S4E).
The data in figure 4C confirmed that the regulation of FGFR-2 alternative splicing by SetD2 is IWS1 phosphorylation-dependent. Since the binding of MRG15 and PTB to target genes depends on histone H3K36 trimethylation, which is mediated by SetD2, these data suggest that MRG15 and PTB would also regulate FGFR-2 alternative splicing in an IWS1 phosphorylation-dependent manner. This prediction was confirmed with the experiment in figure S4F, which showed that the MK2206-induced increase in the IIIb/IIIc ratio is not rescued by MRG15, PTB, or SetD2. We conclude that the regulation of FGFR-2 alternative splicing by SetD2, MRG15 and PTB is strictly dependent on IWS1 phosphorylation by Akt.
Model of the regulation of PTB-dependent alternative splicing by IWS1 phosphorylation
Based on the data presented in figures 3 and 4, we propose that IWS1 phosphorylation regulates PTB-dependent alternative splicing via the mechanism described in the model in figure 5. According to this model, IWS1 phosphorylation is required for the recruitment of SetD2 to RNA Pol II. Inhibition of IWS1 phosphorylation prevents the trimethylation of histone H3 at K36 in the body of FGFR-2 and perhaps other target genes. This abrogates the recruitment of the histone H3K36me3-interacting protein MRG15 and its binding partner PTB and results in shifts in alternative splicing of target genes, such as FGFR-2.
Figure 5. Model of the regulation of FGFR-2 alternative splicing by IWS1 phosphorylation at Ser720/Thr721 by Akt1 or Akt3.
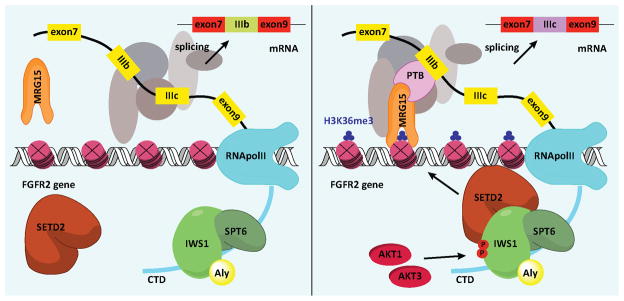
The phosphorylation of IWS1 attracts SetD2 to the Spt6/IWS1/Aly complex in the CTD of RNA Pol II. SetD2 in the complex trimethylates histone H3 at K36 during transcriptional elongation. MRG15 and its partner PTB bind histone H3K36me3. Binding of PTB to the IIIb exon promotes exclusion of this exon from the spliced transcript.
Alternative splicing of FGFR-2 induced by IWS1 phosphorylation at Ser720/Thr721 promotes cell migration, invasiveness and proliferation in response to FGF-2
The IIIb isoform of FGFR-2 is not recognized by FGF-2 (bFGF) whereas the IIIc isoform is (Miki et al., 1992). This suggested that by shifting FGFR-2 splicing from the IIIb to the IIIc isoform, the Akt1/Akt3-dependent IWS1 phosphorylation would promote cell migration and proliferation in response to FGF-2. Wound healing and transwell filter assays in NCI H1299 and NCI H522 cells confirmed that IWS1 phosphorylation indeed promotes both non-directional and directional cell migration in the presence of FGF-2 (Fig 6A, 6B, S5A and S5C). Similarly, matrigel invasion and cell proliferation assays confirmed that IWS1 phosphorylation promotes cell invasion and proliferation downstream of FGF-2 (Fig 6C, S5B, S5C and 6D). To determine whether the expression of the IIIc, as opposed to the IIIb isoform of FGFR-2, is responsible for the enhanced migration and proliferation of the shControl and the shIWS1/wt rescue cells, we infected shIWS1-transduced NCI H1299 cells with constructs expressing the IIIc or IIIb isoforms. shControl and shIWS1 cells not transduced with the IIIc or IIIb construct were used as controls. Using these cells we showed that whereas the expression of the IIIc isoform rescues fully the impaired migration and invasiveness of cells growing in FGF-2-supplemented media (Fig 6E, 6F and 6G), the expression of the IIIb isoform does not (Fig 6I, 6J and 6K). The IIIc isoform also rescued the impaired proliferation phenotype of the same cells, although only partially (Fig 6H). A repeat of the cell proliferation experiment in cells growing in DMEM supplemented with 10% serum, revealed that cell proliferation under these conditions is also regulated by IWS1 phosphorylation at Ser720/Thr721 (Fig 6L, 6M and 6N). Despite that, FGFR-2 IIIc overexpression did not rescue the impaired proliferation induced by IWS1 knockdown (Fig S5D), suggesting that although IWS1 phosphorylation regulates the proliferation of cells growing both in FGF-2 and in serum-supplemented media, the mechanisms by which it functions in the two culture conditions are not the same. Thus, whereas IWS1 phosphorylation regulates cell proliferation of cells growing in FGF-2-supplemented media in part by shifting the alternative splicing pattern of FGFR-2, in cells growing in serum-supplemented media, it may function by regulating the transcriptional elongation or splicing of other target genes.
Figure 6. IWS1 phosphorylation at Ser720/Thr721 in lung carcinoma cell lines promotes cell migration, invasiveness and proliferation.

(A) Wound healing in serum-free media supplemented with FGF-2. Assays were carried out on confluent monolayers of the indicated NCI H1299 cells. The bars show the fraction of the width of the wound that was covered by the migrating cells 24 hours later (mean ± SD).
(B) Directional migration of the cells in A toward FGF-2 was measured with the transwell assay. Bars show the numbers of migrating cells (mean ± SD) at 16h.
(C) Cell invasiveness of the cells in A in FGF-2-free and FGF-2-containing semisolid media. Bars show the numbers of invading cells (mean ± SD) at 16h.
(D) Growth curves of the cells in A, in media supplemented with 1% FBS and 20 ng/ml FGF-2. Live cell numbers (mean ± SD) were measured with the MTT assay in five independent cultures of each cell type.
(E, F and G) The IIIc isoform of FGFR-2 rescued the cell migration (E and F) and invasiveness (G) defects in shIWS1 NCI H1299 cells. Data presented as in A, B and C.
(H) Growth curves of the cells in E, in media supplemented with 1% FBS and 20 ng/ml FGF-2. Live cell numbers (mean ± SD) were measured with the MTT assay in five independent cultures of each cell type.
(I, J and K) The IIIb isoform of FGFR-2 failed to rescue the cell migration (I and J) and invasiveness (K) defects in shIWS1 NCI H1299 cells. Data presented as in A, B and C.
(L, M and N) Growth curves of the indicated NCI H522, NCI H1299 and T47D cells in media supplemented with 10% FBS. Live cell numbers (mean ± SD) were measured with the MTT assay in five independent cultures for each cell type.
Experiments in figures S4C and S4F showed that overexpression of SetD2 in NCI H522 and NCI H1299 cells cannot rescue the FGFR-2 splicing defect induced by blocking the IWS1 phosphorylation or by inhibiting the activity of Akt. These data predict that SetD2 should not rescue the impaired migration and invasiveness phenotype in cells in which the expression or phosphorylation of IWS1 was blocked. The experiments in figure S5E confirmed the prediction. Given our observation that the splicing phenotype of the IWS1 phosphorylation block is not affected by SetD2 overexpression, it is unlikely that it will be affected by MRG15 or PTB. However, the overexpression of these molecules may affect the biological phenotype because they may have additional activities unrelated to RNA splicing.
The role of IWS1 phosphorylation at Ser720/Thr721 in lung cancer
To determine the role of IWS1 phosphorylation in oncogenesis, shControl, shIWS1, shIWS1/wt rescue and shIWS1/mutant rescue NCI-H1299 cells were injected subcutaneously in nude mice. The inoculated animals were monitored for tumor growth and they were sacrificed 4 weeks later. The results confirmed that IWS1 phosphorylation promotes both the growth and invasiveness of the tumor cells (Fig 7A and S6A). Immunohistochemical analysis of the xenografts with the validated anti-phospho-IWS1 antibody (Fig 3C and Fig S3B) confirmed the IWS1 phosphorylation in vivo (Fig 7B).
Figure 7. IWS1 phosphorylation at Ser720/Thr721 in NSCLC.

(A) IWS1 phosphorylation at Ser720/Thr721 promotes tumor growth and invasiveness in a xenograft NSCLC model in nude mice. Nude mice were injected subcutaneously with shControl, shIWS1, shIWS1/wt rescue and shIWS1/mutant rescue NCI H1299 cells. (N=10 mice/group). (Top) Tumor growth is expressed as mean tumor volume ± SE. (Bottom) Frequency of macroscopic invasion of the adjacent tissue by the tumor cells. Combined data from two experiments.
(B) Phospho-IWS1 (Ser720) staining of tumor xenografts. Formalin-fixed, paraffin-embedded tumor samples from the experiment in A were stained with a non-commercially available anti-phospho-IWS1 (Ser720) antibody from Cell Signaling (see Fig 3C lower panel and figure S3B). Secondary antibody was HRP-labelled. Tissues were counterstained with hematoxylin.
(C) FGFR-2 IIIc/IIIb exon ratio (left) and FGFR-2 expression (right) in normal human lung and lung cancer specimens, were determined by quantitative RT-PCR and presented as box plots.
(D and E) The shift of FGFR-2 splicing toward IIIc in NSCLCs correlates with IWS1 phosphorylation at Ser720/Thr721 and the latter correlates with Akt phosphorylation and Akt3 expression. (D) Lysates of 2 normal lung and 24 NSCLC samples were probed with the indicated antibodies. (E) Upper: The relative stoichiometry of IWS1 phosphorylation in the 21 NSCLCs expressing IWS1, was estimated from the phospho-IWS1/total IWS1 band density ratios and it was used to place the tumors in an ascending order of IWS1 phosphorylation abundance. Middle: IIIb/IIIc exon ratios in the tumors in the upper panel. The IIIb/IIIc ratio in normal lung was given the value of 1. Bars show the ratios in tumors vs normal lung, with some tumors exhibiting values greater and some lower than 1. Lower: The relative stoichiometry of IWS1 phosphorylation in the same tumors correlates with phospho-Akt (Ser473) (Left) and Akt3 expression (Right).
Western blotting of cell lysates derived from a set of human lung specimens (2 normal and 24 NSCLCs), revealed that IWS1 is not expressed in normal lung, but is expressed in 21/24 NSCLCs (Fig 7D). Immunohistochemical staining of a normal lung and NSCLC tissue array (17 and 23 samples respectively) with the anti-phospho-IWS1 antibody also revealed increased phospho-IWS1 nuclear staining in a fraction of the tumors (Fig S6B). Based on these findings, which suggest a potential role for IWS1 in NSCLC, we proceeded to address the expression of FGFR-2 IIIb and IIIc in normal lung and NSCLC samples. The results revealed that the overall FGFR-2 expression is similar in both, but it shifts toward IIIc in the tumor samples (Fig 7C). More important, the shift correlates with the phosphorylation of IWS1 and the latter correlates with the phosphorylation of Akt and the expression of Akt3. Plotting the relative stoichiometry of IWS1 phosphorylation (p-IWS1-(S720)/total IWS1) against the FGFR-2 IIIb/IIIc ratio in the 21 NSCLC samples that express IWS1, revealed that whereas tumors with high relative levels of IWS1 phosphorylation have almost universally low IIIb/IIIc ratios, tumors with low relative levels of IWS1 phosphorylation exhibit IIIb/IIIc ratios that are randomly distributed (Fig 7D and 7E) (p=0.0002, by Levene’s test for equality of variances). Plotting the relative stoichiometry of IWS1 phosphorylation against phospho-Akt (Ser 473), or Akt3 (Fig 7E), also revealed statistically significant correlations (p=0.00636 and p=0.00745 respectively, by Spearman’s rank order correlation). Collectively, these data provide strong support to the hypothesis that the phosphorylation of IWS1 plays a major role in the regulation of FGFR-2 alternative splicing in primary human NSCLCs and are consistent with our other data showing that the knockdown of Akt1 and Akt3 shifts the splicing of FGFR-2 from the IIIc to the IIIb isoform. Since a low IIIb/IIIc ratio has been shown to promote EMT and tumor cell invasiveness (Thiery and Sleeman, 2006), these data support the hypothesis that Akt promotes cancer progression by phosphorylating IWS1. This conclusion is further supported by data from the oncomine database (Bild et al., 2006), which reveal a positive correlation between IWS1 expression with tumor stage (Fig S6C).
DISCUSSION
Linking functional differences between Akt isoforms to differences in target specificity
In this paper we focused on a phosphoproteomics screen, which was carried out in a set of lung fibroblast cell lines that express different Akt isoforms but are otherwise identical. The results revealed significant differences in target specificity between Akt isoforms and identified a host of signaling pathways and cellular functions that may be differentially regulated by them (Fig 2A). Some of the predictions, such as the regulation of insulin and IGF signaling primarily by Akt2 are in agreement with prior literature (Cho et al., 2001). Based on these data it is expected that Akt inhibitors may have widely variable therapeutic effects and toxicity, depending on their relative specificity against the three Akt isoforms.
Regulation of RNA processing by Akt isoforms
One of the top functions differentially regulated by Akt isoforms is RNA metabolism, with 25 proteins involved in various steps of the RNA maturation process being phosphorylated by at least one of the three isoforms (Fig 2B and 2C). IWS1, one of these proteins, is phosphorylated primarily by Akt3 and Akt1 and is a component of a transcriptional elongation complex, which includes Spt6, Aly/REF and SetD2 and assembles on the Ser2 phosphorylated CTD of RNA Pol II (Yoh et al., 2007; Yoh et al., 2008). Data in this report showed that IWS1 phosphorylation is required for the recruitment of SetD2 to the complex and the trimethylation of histone H3 at K36 in the body of the target genes. Subsequent recruitment of the histone H3K36me3-intracting protein MRG15 and its binding partner PTB results in shifts in the alternative splicing of target genes, one of which is FGFR-2.
Regulation of alternative RNA splicing by Akt isoforms
The alternative splicing of FGFR-2 gives rise to two isoforms, IIIb and IIIc (Carstens et al., 2000; Warzecha et al., 2009). IIIb is expressed primarily in epithelial cells, while IIIc, which is the isoform induced by IWS1 phosphorylation, is expressed primarily in mesenchymal cells and in invasive and metastatic tumors (Luco et al., 2010; Orr-Urtreger et al., 1993). Of these isoforms, only IIIc is recognized by FGF-2 (Miki et al., 1992). In agreement with these data, lung carcinoma cell lines in which the endogenous IWS1 was replaced with the phosphorylation site mutant, exhibit defects in FGF-2-induced cell proliferation, migration and invasion. More important, the defects in cell migration and invasiveness were fully rescued by FGFR-2 IIIc but not IIIb, while the defect in cell proliferation was rescued partially and only when the cells were grown in FGF-2-supplemented media. IWS1 phosphorylation and SetD2 recruitment are expected to target more genes, in addition to FGFR-2 (Luco et al., 2010). Moreover, in some of these genes they may promote transcriptional elongation by preventing cryptic intragenic transcription, with or without an effect on RNA splicing (Carvalho et al., 2013). Based on these considerations, we propose that IWS1 phosphorylation may promote tumor cell proliferation in culture and in animals by regulating RNA splicing, transcriptional elongation, or both on a host of cellular genes.
It has been firmly established that splicing occurs co-transcriptionally and that it is influenced by the imprinting of the pre-mRNA, during the RNA synthesis and processing steps that precede splicing (Moore and Proudfoot, 2009). To explain how the process of transcription may affect splicing, two models have been proposed: the kinetic model, which suggests that slowing the rate of transcription promotes the inclusion of weak exons into the mature mRNA product, and the chromatin modification model, which proposes that histone and DNA modifications regulate the assembly of protein complexes that control splicing (Luco et al., 2011), although they may also control nucleosome density and the rate of transcriptional elongation.
Our data support the chromatin modification model (Fig 5) and they show how the chromatin-regulated RNA splicing machinery is under the control of Akt signals. However, the link between chromatin modifications and alternative splicing may be more complex than the model indicates. This is suggested by the following observations. In mesenchymal cells, which express the IIIc isoform of FGFR-2, histone H3 trimethylation at K36 is accompanied by the downregulation of histone H3K4 and H3K27 trimethylation, and by histone H3K4 mono and dimethylation (Luco et al., 2010). More important, overexpression of the histone H3K4 methyltransferase ASH2 in these cells, increases the usage of the normally repressed exon IIIb, suggesting that H3K4 trimethylation may also contribute to the regulation of PTB-dependent alternative splicing (Luco et al., 2010). Other studies provided evidence linking MRG15 to the acetylation of histone H4 at K16 (Conrad et al., 2012) and perhaps K12 (Pena et al., 2011) during transcriptional elongation. MRG15 is a component of several chromatin-modifying complexes, which could give rise to these chromatin modifications. Thus, it has been shown that MRG15 bound to the H3K36me3 mark in the body of transcribed genes (Zhang et al., 2006), also interacts with the histone H3K4me3/me2 demethylase KDM5B (Xie et al., 2011), the histone H4K16 acetyltransferase complex NuA4 (mammalian Tip60) (Cai et al., 2003), the histone H4K16 deacetylase complex Rpd3S (mammalian HDCA2) (Kumar et al., 2011), and VEZF1 (Gowher et al., 2012), a known regulator of alternative splicing. Some of these interactions may take place in the context of larger complexes, such as the LAF and RLAF complexes (Moshkin et al., 2009). It has not been determined yet whether these histone modifications are also observed in the context of the Akt3/Akt1-dependent shift in the alternative splicing of FGFR-2. If they do, the system we established will allow us to determine whether they play an obligatory role in this process, and to address the epistatic relationships between them.
Relevance of IWS1 phosphorylation in lung cancer
It is now universally accepted that Akt plays a major role in oncogenesis. Experiments, both in culture and in animals, presented in this report, revealed that IWS1 phosphorylation by Akt3 and Akt1 regulates RNA splicing and perhaps transcriptional elongation and represents an important event in lung cancer. Analysis of sets of normal lung and lung cancer samples for the expression of FGFR-2 revealed that the overall expression is similar in both. However, the expression was shifted toward the IIIc isoform in the tumor samples. More important, the shift toward the IIIc isoform was found to correlate with the stoichiometry of IWS1 phosphorylation, which in turn correlates with Akt phosphorylation and Akt3 expression. Overall, this study identifies perturbations in RNA splicing and perhaps transcriptional elongation as a novel mechanism by which Akt promotes oncogenesis, it shows that Akt isoforms contribute to oncogenesis as upstream regulators of FGF-2 signaling, and provides direct evidence linking IWS1 to lung cancer.
In summary, data presented in this report link Akt isoform-specific signaling with chromatin modifications occurring in the body of the gene encoding FGFR-2, and perhaps other actively transcribed target genes. These chromatin modifications in turn, regulate alternative splicing and transcriptional elongation. By regulating these processes, IWS1 phosphorylation profoundly alters the biology of tumor cells. Interruption of IWS1 phosphorylation suppresses the proliferation, migration and invasiveness of tumor cells both in culture and in animals. Overall, this study provides new insights into the regulation of alternative splicing, a process that plays a critical role in differentiation and development and in a multitude of human diseases, including cancer.
EXPERIMENTAL PROCEDURES
For details, see supplement
Cells, culture, growth factors and Akt inhibitors
Mouse lung fibroblasts were described previously (Iliopoulos et al., 2009). Culture conditions, cell transfections and infections are described in the supplement.
Phosphoproteomics screen and data analysis
The phosphoproteomics screen and data analysis are presented in the supplement. Data are available in: https://tuftsmedicalcenter.resultsbuilderstage.com/Research-Clinical-Trials/Institutes-Centers-Labs/Molecular-Oncology-Research-Institute/Labs-Facilities/Tsichlis-Lab/Akt-phopshoproteomics.aspx
siRNAs, shRNAs and expression constructs. Cloning and site-directed mutagenesis
The origin of the siRNAs, shRNAs and expression constructs is described in Supplementary Table S3. Cloning and site-directed mutagenesis were carried out using standard procedures.
Immunoprecipitation, Immunoblotting and in vitro kinase assays
Immunoprecipitation, immunoblotting and in vitro kinase assays are described in the supplement. The phosphorylation substrates (IWS1 wt and IWS1S720A/T721A) were expressed as GST fusions in E Coli, BL21. GSK3α/β was purchased from Cell Signaling Technology (Danvers, MA).
Real-time RT-PCR
cDNA was synthesized from 1.0 μg of Trizol-extracted total cell RNA, using oligo-dT priming and the Retroscript reverse transcription kit from Ambion. Gene expression was quantified by real time RT-PCR.
Chromatin Immunoprecipitation (ChIP)
ChIP assays, using the Millipore kit (cat no. 17-295), were done as described in the supplement.
Cell proliferation, migration and invasion assays
Cell proliferation was monitored with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Non-directional migration was measured with the wound healing assay and directional migration with the transwell filter assay (Kottakis et al., 2011) In vitro invasion was measured using BD BioCoat Matrigel Invasion Chambers (BD Biosciences)
Tumor xenografts
Nude mice injected subcutaneously with 2×106 shControl, shIWS1, shIWS1wt rescue or shIWS1mut rescue NCI H1299 cells, were monitored for a total of 4 weeks..
Immunohistochemistry
A human lung tumor tissue array and xenograft tumors were stained with the specific anti-phospho-IWS1 (Ser720) antibody as described in the supplement.
Supplementary Material
HIGHLIGHTS.
Phosphoproteomics screen shows differences in target specificity between Akt isoforms
Akt3 and Akt1 phosphorylate IWS1 at the conserved Ser720/Thr721 site
Phospho-IWS1 promotes the recruitment of SetD2 to the RNA Pol II elongation complex
Phospho-IWS1 controls FGFR2 alternative splicing and contributes to carcinogenesis
Acknowledgments
We thank numerous colleagues, listed in the extended experimental procedures section, for providing constructs. Animal experiments were carried out at the Dana-Farber Cancer Institute, the former institutional affiliation of C.P., M.H. and D.I. We thank the Tufts tumor bank for normal and tumor lung specimens. We also thank James Baleja for interesting discussions, Alexandra Touroutoglou for assisting in the statistical analysis of the tumor data and Philip Hinds, Claire Moore and Kevin Struhl for reviewing the manuscript prior to submission. This work was supported by NIH grant RO1CA57436 to PNT. PNT wishes to dedicate this paper to the memory of his mentor and friend Dr Jane F. Desforges who passed away on September 7, 2013.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Cai Y, Jin J, Tomomori-Sato C, Sato S, Sorokina I, Parmely TJ, Conaway RC, Conaway JW. Identification of new subunits of the multiprotein mammalian TRRAP/TIP60-containing histone acetyltransferase complex. J Biol Chem. 2003;278:42733–42736. doi: 10.1074/jbc.C300389200. [DOI] [PubMed] [Google Scholar]
- Carstens RP, Wagner EJ, Garcia-Blanco MA. An intronic splicing silencer causes skipping of the IIIb exon of fibroblast growth factor receptor 2 through involvement of polypyrimidine tract binding protein. Mol Cell Biol. 2000;20:7388–7400. doi: 10.1128/mcb.20.19.7388-7400.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, Carmo-Fonseca M, de Almeida SF. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 2013 doi: 10.1093/nar/gks1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Conrad T, Cavalli FM, Holz H, Hallacli E, Kind J, Ilik I, Vaquerizas JM, Luscombe NM, Akhtar A. The MOF chromobarrel domain controls genome-wide H4K16 acetylation and spreading of the MSL complex. Dev Cell. 2012;22:610–624. doi: 10.1016/j.devcel.2011.12.016. [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duina AA. Histone Chaperones Spt6 and FACT: Similarities and Differences in Modes of Action at Transcribed Genes. Genet Res Int. 2011;2011:625210. doi: 10.4061/2011/625210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezell SA, Polytarchou C, Hatziapostolou M, Guo A, Sanidas I, Bihani T, Comb MJ, Sourvinos G, Tsichlis PN. The protein kinase Akt1 regulates the interferon response through phosphorylation of the transcriptional repressor EMSY. Proc Natl Acad Sci U S A. 2012;109:E613–621. doi: 10.1073/pnas.1115029109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Gowher H, Brick K, Camerini-Otero RD, Felsenfeld G. Vezf1 protein binding sites genome-wide are associated with pausing of elongating RNA polymerase II. Proc Natl Acad Sci U S A. 2012;109:2370–2375. doi: 10.1073/pnas.1121538109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, Tsichlis PN. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal. 2009;2:ra62. doi: 10.1126/scisignal.2000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12:715–729. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell. 2011;43:285–298. doi: 10.1016/j.molcel.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Ahn SH, Zhong G, Kobor MS, Cagney G, Emili A, Shilatifard A, Buratowski S, Greenblatt JF. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol. 2002;22:6979–6992. doi: 10.1128/MCB.22.20.6979-6992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar GS, Xie T, Zhang Y, Radhakrishnan I. Solution structure of the mSin3A PAH2-Pf1 SID1 complex: a Mad1/Mxd1-like interaction disrupted by MRG15 in the Rpd3S/Sin3S complex. J Mol Biol. 2011;408:987–1000. doi: 10.1016/j.jmb.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007;67:167–177. doi: 10.1158/0008-5472.CAN-06-3782. [DOI] [PubMed] [Google Scholar]
- Miki T, Bottaro DP, Fleming TP, Smith CL, Burgess WH, Chan AM, Aaronson SA. Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc Natl Acad Sci U S A. 1992;89:246–250. doi: 10.1073/pnas.89.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Moshkin YM, Kan TW, Goodfellow H, Bezstarosti K, Maeda RK, Pilyugin M, Karch F, Bray SJ, Demmers JA, Verrijzer CP. Histone chaperones ASF1 and NAP1 differentially modulate removal of active histone marks by LID-RPD3 complexes during NOTCH silencing. Mol Cell. 2009;35:782–793. doi: 10.1016/j.molcel.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Bedford MT, Burakova T, Arman E, Zimmer Y, Yayon A, Givol D, Lonai P. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2) Dev Biol. 1993;158:475–486. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- Pena AN, Tominaga K, Pereira-Smith OM. MRG15 activates the cdc2 promoter via histone acetylation in human cells. Exp Cell Res. 2011;317:1534–1540. doi: 10.1016/j.yexcr.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polytarchou C, Iliopoulos D, Hatziapostolou M, Kottakis F, Maroulakou I, Struhl K, Tsichlis PN. Akt2 regulates all Akt isoforms and promotes resistance to hypoxia through induction of miR-21 upon oxygen deprivation. Cancer Res. 2011;71:4720–4731. doi: 10.1158/0008-5472.CAN-11-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, Michaelis T, Frahm J, Hemmings BA. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development. 2005;132:2943–2954. doi: 10.1242/dev.01864. [DOI] [PubMed] [Google Scholar]
- Wan M, Leavens KF, Saleh D, Easton RM, Guertin DA, Peterson TR, Kaestner KH, Sabatini DM, Birnbaum MJ. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab. 2011;14:516–527. doi: 10.1016/j.cmet.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Pelz C, Wang W, Bashar A, Varlamova O, Shadle S, Impey S. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 2011;30:1473–1484. doi: 10.1038/emboj.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Cho H, Pickle L, Evans RM, Jones KA. The Spt6 SH2 domain binds Ser2-P RNAPII to direct Iws1-dependent mRNA splicing and export. Genes Dev. 2007;21:160–174. doi: 10.1101/gad.1503107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Lucas JS, Jones KA. The Iws1:Spt6:CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 2008;22:3422–3434. doi: 10.1101/gad.1720008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Du J, Sun B, Dong X, Xu G, Zhou J, Huang Q, Liu Q, Hao Q, Ding J. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res. 2006;34:6621–6628. doi: 10.1093/nar/gkl989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell. 2012;47:422–433. doi: 10.1016/j.molcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.