

Abstract
Although a diversity of proteins is known to form amyloid fibers, their common mechanisms are not clear. Here, we show that an intrinsically unfolded protein (U), represented by a disulfide-deficient variant of hen lysozyme with no tertiary structure, forms an amyloid-like fibril after prolonged incubation. Using variable pressure NMR along with sedimentation velocity, circular dichroism, and fluorescence measurements, we show that, before the fibril formation, the protein forms a pressure-dissociable, soluble assemblage (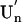 ) with a sedimentation coefficient of 17 S and a rich intermolecular β-sheet structure. The reversible assemblage is characterized with a Gibbs energy for association of –23.3 ± 0.8 kJ·mol–1 and a volume increase of 52.7 ± 11.3 ml·mol–1 per monomer unit, and involves preferential interaction of hydrophobic residues in the initial association step. These results indicate that amyloid fibril formation can proceed from an intrinsically denatured protein and suggest a scheme
) with a sedimentation coefficient of 17 S and a rich intermolecular β-sheet structure. The reversible assemblage is characterized with a Gibbs energy for association of –23.3 ± 0.8 kJ·mol–1 and a volume increase of 52.7 ± 11.3 ml·mol–1 per monomer unit, and involves preferential interaction of hydrophobic residues in the initial association step. These results indicate that amyloid fibril formation can proceed from an intrinsically denatured protein and suggest a scheme 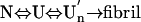 as a common mechanism of fibril formation in amyloidogenic proteins, where two-way arrows represent reversible processes, one-way arrow represents an irreversible process, and N, U, and
as a common mechanism of fibril formation in amyloidogenic proteins, where two-way arrows represent reversible processes, one-way arrow represents an irreversible process, and N, U, and 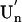 represent, respectively, the native conformer, the unfolded monomeric conformer, and the soluble assemblage of unfolded conformers.
represent, respectively, the native conformer, the unfolded monomeric conformer, and the soluble assemblage of unfolded conformers.
Formation of amyloid-like fibers, reported for a diversity of proteins, is frequently considered a generic property of proteins (ref. 1 and references therein, and refs. 2 and 3). Partially folded intermediates are often considered to be generally involved in fibrillogenesis (4–6). For example, naturally occurring amyloidogenic variants of human lysozyme are less stable and less cooperatively folded than WT lysozyme, and more prone to partial unfolding (7). Amyloid fiber formations under conditions that promote partial unfolding are reported for human and hen WT lysozymes (8, 9). However, use of denaturants, alcohol, transient heating, etc. to attain partially unfolded state often complicates the system and makes detailed thermodynamic analyses unfeasible.
Also for WT hen lysozyme, reduced forms as well as early folding intermediates have a strong tendency to aggregate (10), but little is known about these association reaction compared with the wealth of information about the folding–unfolding reactions for the same protein. In this study, we characterize the association reaction of an unfolded lysozyme variant (0SS), which is a genetically engineered disulfide-deficient variant of hen lysozyme. 0SS has all eight cysteinyl residues replaced by alanine or serine, diminished secondary structure (compared to WT) and no tertiary structure in aqueous buffer with no denaturant (11). Unlike disulfide-reduced or disulfide-reduced and alkylated proteins, 0SS is chemically stable, free of bulky alkylating groups. Soluble in aqueous buffer, 0SS is a good model for a denatured state of lysozyme that can be studied without the complications of added denaturants or extremes of temperature or pH.
Recently, hydrostatic pressure has gained attention as a means of dissociating protein assemblies and amyloid fibrils (12–18). Using a newly developing method (19–21) in high pressure NMR spectroscopy, we show here that pressure dissociates the soluble, amyloid-fibrillogenic precursor assembly of 0SS completely and reversibly. Preliminary residue-specific information about the intermolecular interactions in the 0SS assembly is also reported.
Materials and Methods
0SS Variant. Hen lysozyme 0SS variant was produced in Escherichia coli, and purified as described (11, 22). Cys-6 is replaced by Ser, and the other seven Cys residues at positions 30, 64, 76, 80, 94, 115, and 127 are replaced by Ala in 0SS. Reoxidation procedure is not necessary for this variant. N-terminal Met is attached unexcised. 15N-labeled 0SS was prepared as described (23).
Characterization of 0SS Self-Association. Sedimentation equilibrium and sedimentation velocity measurements for the monomer state of 0SS were carried out at rotor speeds of 23,000 and 50,000 rpm, respectively, and at 20°C in a Hitachi CP100α ultracentrifuge with an ABS-8 UV monitor at 280 nm or at longer wavelengths. Because 0SS is essentially unfolded (in the absence of denaturant), the partial specific volume of denatured state, 0.700 ml·g–1 (24), was used in the calculation of the apparent molecular weight. Sedimentation velocity measurements for the associated state of 0SS in 50 mM sodium maleate (pH 2.0) at indicated protein concentrations were carried out at a rotor speed of 40,000 rpm and at 20°C in a Beckman Model E analytical ultracentrifuge, using charcoal-filled Epon double-sector cells with Schlieren optics. CD spectra were taken for 0SS incubated 1 day in 5 mM sodium maleate (pH 2.7)/45 mM sodium chloride at 25°C, by using a J-720 spectropolarimeter (Japan Spectroscopic, Tokyo). A cuvette of 0.1-mm optical path length was used. Strong absorbance in a far-UV region forced the concentration of maleate buffer to be reduced to 5 mM, and that of protein to <4 mg·ml–1. Sodium chloride was added to maintain the level of salt concentration. For thioflavine T fluorescence measurements, 3 μl of 8 mg·ml–1 0SS in 50 mM sodium maleate (pH 2.0) incubated for a specified period at 25°C was mixed with 297 μl of 20 μM thioflavine T in 20 mM potassium phosphate (pH 7.4)/100 mM sodium chloride. Fluorescence spectra were taken with an RF-5300PC spectrofluorimeter (Shimadzu) and a 5 × 5-mm cuvette, with an excitation wavelength of 450 nm and a bandwidth of 5 nm for both excitation and emission. A similar time course of variation in thioflavine T fluorescence spectra was observed for 4 mg·ml–1 0SS in 5 mM sodium maleate (pH 2.7)/45 mM sodium chloride, the condition used for CD measurements. Electron micrographs of 0SS assembly, stained with 2% uranyl acetate, were taken with a JEM-1200EX electron microscope (JEOL) operated at 80 kV and at a magnification of ×20,000.
NMR Measurements and Data Analysis. 0SS variant was dissolved in 90% H2O/10% D2O (vol/vol) containing 50 mM deuterated sodium maleate buffer (pH 2.0) at a concentration of 0.53 mM (7.6 mg·ml–1). All NMR spectra were measured at 20°C on a Bruker (Billerica, MA) DMX-750 spectrometer operating at frequencies of 750.13 MHz and 76.01 MHz for 1H and15N, respectively. An on-line pressure cell technique described earlier (21) was used to measure pressure-dependent NMR spectra. 1H chemical shifts were referenced to dioxan signal (3.75 ppm), and 15N chemical shifts were indirectly referenced to it. Data were processed with uxnmr and nmrpipe program packages (Bruker) running on a Silicon Graphics (Mountain View, CA) workstation. Spectra were zero-filled to give final matrix of 2,048 × 512 real data points and apodized with quadratic sine-bell function in both dimensions. Chemical shifts and cross-peak intensities were evaluated with nmrpipe by keeping the same threshold level throughout the experiments. Assignment of individual 15N/1H cross peaks of 0SS variant was carried out in the following way. First, individual 15N/1H signals of 0SS were readily assigned in 8M urea based on the reported assignments for a protein closely related to 0SS, namely reduced and S-methylated hen lysozyme in 8 M urea (25). Then, on one hand, at 1 bar (1 bar = 100 kPa), chemical shifts of 0SS variant were followed for individual cross peaks for decreasing urea concentration from 8 to 2 M and then extrapolated to 0 M urea, giving a set of individually assigned 15N and 1H chemical shifts at 0 M urea at 1 bar (from urea concentration variation). On the other hand, at 0 M urea, the chemical shifts were followed also for individual peaks for decreasing pressure from 2,000 to 300 bars and then extrapolated to 1 bar, again giving a set of unassigned individual 15N and 1H chemical shifts at 0 M urea at 1 bar (from pressure variation). By matching the two sets of chemical shifts at 0 M urea at 1 bar to each other, we could assign 55 cross peaks in 15N/1H heteronuclear single quantum correlation (HSQC) spectra of 0SS between 1 and 2,000 bars, of which 47 peaks did not suffer from merging with changing pressure. Detailed procedure of the assignment has been described (26).
Thermodynamic Analysis of Pressure Dissociation of an Assembly. This subject was treated previously by Royer (27), but we will give the essence concisely below. When n monomers reversibly associate to form a polymer,
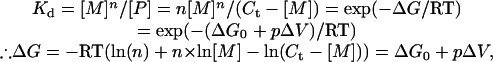 |
where Kd is a dissociation constant, [M] and [P] are number concentrations of monomer and polymer, respectively, and Ct is the total protein concentration (in monomer unit). We assumed that at 2,000 bars [M] equals to Ct (i.e., 100% monomeric). The slope of the plot of ΔG versus p gives ΔV, the volume change (per mol of polymer) on dissociation, and the intercept of the ordinate ΔG0, the stability of the assembly. (In the text, ΔV and ΔG0 values per mol of monomer are given.)
Results and Discussion
Self-Association of 0SS. Firstly, the dissociated state of 0SS was characterized by sedimentation equilibrium as well as sedimentation velocity measurements. In 20 mM sodium acetate (pH 4.0) the apparent Mr were 13,800 and 13,400 (±1,200) at 0SS concentrations of 0.11 and 0.21 mg·ml–1, respectively, in agreement with the expected Mr (14,200), indicating that at concentrations below ≈ 0.2 mg·ml–1, 0SS exists as a monomer in this solvent (for at least 1 week). Under these conditions, 0SS showed a sedimentation coefficient (s20,w) of 1.43 ± 0.03 S, which is significantly lower than that reported for WT lysozyme (1.6–2.1 S; Mr 14,300) (28). The decrease in the sedimentation coefficient indicates that the molecular shape of 0SS deviates from a spherical globule of WT lysozyme, consistent with the lack of disulfide cross links and higher-order structures.
In the present study of the assemblage of 0SS that includes high-pressure NMR measurements (described below), we primarily used sodium maleate buffer at acidic pH region because it has a low volume change on dissociation of proton, and therefore a minimal pH change on pressurization (29). At high protein concentrations [7.6 mg·ml–1 (Fig. 1A) and 3.8 mg·ml–1], the 0SS protein in 50 mM sodium maleate (pH 2.0) incubated for 1 day showed self-association: two well separated sedimenting boundaries with sedimentation coefficients of 1.22 ± 0.02 S and 17.3 ± 1.3 S were observed. Under the same conditions, WT hen lysozyme shows only a single sedimenting boundary of a monomeric state (1.7 ± 0.05 S). In light of the decrease in the sedimentation coefficient observed at pH 4.0 described above, the former 1.2 S peak was identified as monomeric 0SS. (Because maleate has strong absorbance in a UV region, a direct determination of the Mr of 0SS in 50 mM sodium maleate by means of a sedimentation equilibrium measurement with absorption detection was impractical.) As for the 17 S peak, the estimation of Mr highly depends on the shape/conformation of the assemblage. If we assume spherical globules for the 17 S assembly as well as WT lysozyme and the related dependence s ∝ Mr2/3, the assembly corresponds to ≈30 molecules of 0SS (30). On the other hand, if we assume, as another extreme of conformation, random coils for the 17 S assembly as well as the disordered 0SS and the related dependence s ∝ Mr1/2, it corresponds to ≈220 molecules of 0SS. Actually, considerable amount of β-structure is formed in the 17 S assembly as described below, and the latter assumption is not fully correct. However, even in the former assumption, deviation in shape of the 17 S assembly from a spherical globule, which is likely to exist, leads to an increased translational coefficient and therefore a reduced sedimentation coefficient, resulting in a higher Mr estimated for the 17 S assembly, compared with the case of a spherical globule.
Fig. 1.

Association of the 0SS variant. (A) A Schlieren pattern, taken 42 min after the start of sedimentation. Protein concentration was 7.6 mg·ml–1 in 50 mM sodium maleate (pH 2.0). (B) Far-UV CD spectra of the 0SS solution incubated for 1 day at 25°C in 5 mM sodium maleate (pH 2.7)/45 mM sodium chloride at protein concentrations of 1, 2, 3, and 4 mg·ml–1 (labeled on each spectrum).
The apparent concentration of the 17 S assembly at the total 0SS concentration of 7.6 mg·ml–1, calculated from the area of the peak and that of the 1.2 S peak (which fully appears at the centrifugation time of 230 min), after correction for radial dilution, is ≈4.0 mg·ml–1. At the total 0SS concentration of 3.8 mg·ml–1 it is reduced to 1.1 mg·ml–1. These concentrations for the polymeric state are probably underestimates, because corrections to the Johnston–Ogston effect (31), which leads to an apparently lower concentration for the fast-sedimenting component, is not presently taken into account. [Because the hydrostatic pressure in the ultracentrifuge cell (32) under the present centrifugal acceleration (<54 bars) was by far below the pressure range (200–2,000 bars), which is effective for dissociation of the 17 S state revealed by the present high-pressure NMR study (described below), the dissociation of the polymeric state with the pressure deriving from centrifugation is not expected to occur significantly.] The formation of the 17 S assembly therefore seems to become significant in the total 0SS concentration range higher than ≈3 mg·ml–1.
CD studies show that the formation of β-structure is involved in the self-association of 0SS that takes place in the concentration range suggested above. The far-UV CD spectrum at 1 mg·ml–1 0SS concentration in 5 mM sodium maleate (pH 2.7)/45 mM sodium chloride (Fig. 1B, spectrum 1) is identical to the spectrum taken for the monomer state of 0SS [0.07 mg·ml–1 in 20 mM sodium acetate (pH 4.0) (11)], showing the characteristics of an intrinsically denatured protein with a small amount of fluctuating secondary structure. The sharp NMR resonance lines of a dilute 0SS solution (data not shown) at positions for a typical denatured polypeptide chain indicates that the molecule is well hydrated and mobile. When 0SS concentration is increased up to 4 mg·ml–1, through the concentration range described previously, a negative CD band centered at 217 nm appears (Fig. 1B, spectra 2–4). The difference spectrum between the spectra 4 and 1, or 3 and 1, closely matched the CD spectrum for β-structure. These results indicate that intermolecular β-sheet structure is developed in the 17 S assembly. This feature is also observed in sodium acetate buffer (pH 4.0) in the same 0SS concentration range.
Fibril Formation. Solutions of the 17 S material show some thioflavine T fluorescence, with an emission maximum around 482 nm (Fig. 2A, the spectra for 0–2 days), diagnostic of amyloid fiber formation (33). However, fibrillar aggregate is not observed by electron microscopy for 0SS solutions incubated for 0–2 days only. On prolonged incubation over weeks to months, 0SS solutions develop fibrous aggregates that are morphologically similar to amyloid protofibrils (34, 35) (Fig. 2B). The fibril formation is observed not only in sodium maleate buffer (pH 2.0–2.7) but also in sodium acetate buffer (pH 4.0), as well as in Tris·HCl (pH 7.5) and in the 0SS concentration range 2.5–10 mg·ml–1. Accompanied with fibril formation, the 482-nm thioflavine T fluorescence band becomes prominent (Fig. 2 A). The characteristic red shift in Congo red absorbance and an intensified CD negative band at 217 nm were also observed. Thus, the amyloid-like fibrils of 0SS are formed apparently starting from an unfolded state and passing through a soluble precursor species of limited size with a spontaneous, intermolecular build-up of β-structure. No specific, partially unfolded conformer is involved as a prerequisite for this process.
Fig. 2.

(A) Thioflavine T fluorescence spectra of the 0SS solution incubated for (from bottom to top) 0, 1, 2, and 9 days, and 9 months at 25°C, in 50 mM sodium maleate (pH 2.0) at 8.0 mg·ml–1 protein concentration. (B) Electron microgram of the 0SS fibrous aggregate formed after 8 months of incubation in 20 mM sodium acetate (pH 4.0) at a protein concentration of 2.5 mg·ml–1. (Scale bar, 100 nm.) The fraction of thin and curved fibrils was higher in sodium maleate buffer than in sodium acetate buffer.
Dissociation of the 0SS Soluble Assemblage by Pressure. In 50 mM deuterated sodium maleate buffer (pH 2.0) and at the 15N-labeled 0SS concentration of 7.6 mg·ml–1, the 15N/1H HSQC spectra shows very low intensities for individual peaks (Fig. 3, spectrum at 30 bars). However, when pressure is applied to the NMR sample as described (19–21), the cross peaks appear at very low pressures at positions expected for a typical unfolded and hydrated protein. Cross-peak intensities progressively increase with rising pressure, reaching full values at 2,000 bars. A notable feature of the data is that the spectral changes are fully reversible, indicating that pressure reversibly dissociates the 17 S assembly into monomers.
Fig. 3.

15N/1H-HSQC spectra of uniformly 15N-labeled 0SS variant at various pressures indicated. Spectra at 30, 400, (800, 1,200, and 1,600 not shown), and 2,000 bars were taken at an increasing pressure cycle, and those at 600, 300, and 200 bars were taken at a decreasing pressure cycle. A total of 55 cross peaks were assigned. The cross peaks labeled in the spectrum at 600 or 400 bars diminish conspicuously with decreasing pressure as detailed in Fig. 5.
Despite variation in cross-peak intensity with increasing pressure, the width of individual peaks is essentially conserved, indicating that exchange between monomeric and assembled states of 0SS is slow on the NMR time scale (<10–2 s–1). Thus, the normalized volume of individual peaks represents the fraction of monomer at a given pressure. Volumes of 47 cross peaks among the 55 peaks assigned in 15N/1H HSQC spectra are plotted against pressure in Fig. 4A. Thermodynamic analysis of averaged normalized intensity vs. pressure gives 23.3 ± 0.8 kJ·mol–1 for the stability of the assembly (ΔG0, per monomer unit) and –52.7 ± 11.3 ml·mol–1 for the change in partial molar volume (ΔV, per monomer unit) on dissociation of the assembly (Fig. 4B). Substantial decrease in partial molar volume suggests that the assembly has loose intermolecular packing with voids and/or ionic bonds, which are disrupted on dissociation, resulting in electrostriction. To the authors' knowledge, this is the first report of the volume change on self-association of a denatured protein. The two values of ΔG0 and ΔV do not change >3% when n, the estimated number of monomeric units in the assembly, is varied between 30 and 200. The volume change is comparable to the change reported for dissociation of dimeric or tetrameric proteins (15, 27, 36), as well as for multisubunit assemblies (37, 38) in which subunit proteins are folded. Essentially the same values of ΔG0 and ΔV (24.0 ± 0.8 kJ·mol–1 and –53.7 ± 9.3 ml·mol–1, respectively) are obtained when the change with pressure of peak intensity in one-dimensional 1H-NMR spectra (methyl proton signals at 0.9 ppm) are analyzed.
Fig. 4.

Variation in NMR peak volumes with pressure and thermodynamic analysis of the dissociation reaction. (A) Relative peak volumes of 47 individual residues as a function of pressure, calculated from 15N/1H-HSQC spectra. Open circles represent mean values at each pressure. (B) Free-energy change for dissociation versus pressure, calculated for the association reaction of the order n = 33. The line was obtained by regression of the data within 10–90% of the transition width.
Preferential Involvement of Hydrophobic Residues in Association. On closer inspection, however, small but reproducible spectral changes are detected as a function of pressure. The sharp methyl signal in the 1H NMR spectrum broadens slightly at low pressure (data not shown). The measurement of diffusion rate by means of transverse relaxation under magnetic field gradient shows that at 30 bars the diffusion constant for the monomer is decreased to 0.54 of that at 2,000 bars, suggesting that limited intermolecular interactions may occur at low pressure even among monomeric species. Moreover, in HSQC spectra with decreasing pressure, several specific cross peaks diminish conspicuously (Fig. 3): V29, W63, A64, I124 peaks almost disappear at 400 bars, and I55, A90, W108, V109, A110, W111, A127 peaks almost disappear at 300 bars. The peak volume for each of these residues at 300 bars relative to that at 2,000 bars is reduced to <0.014, significantly lower than the averaged value over all of the assigned peaks (Fig. 5).
Fig. 5.

Relative HSQC peak volumes of 47 residues at 300 bars plotted against residue positions. The dotted horizontal line indicates the averaged value (0.134) over all of the assigned peaks. For the 11 residues labeled, the relative volume is <0.014 (filled short bars). The horizontal bars at bottom labeled with roman numerals indicate the regions for the six hydrophobic clusters reported in figure 2B of ref. 39 (each bar corresponds to the residue region spanned by the half-height-width of each Gaussian distribution modeled for R2 relaxation rates).
These residues are hydrophobic and belong, collectively, to five of the six hydrophobic clusters identified in the unfolded state of lysozyme based on the deviations in NMR relaxation behavior from a random coil model (25, 39, 40) (Fig. 5). The unfolded construct, reduced and S-methylated lysozyme, used in the NMR relaxation study is closely similar to 0SS in the present study. Intramolecular networks of the hydrophobic clusters were shown to form cores of residual structures in the unfolded state. In the present study, however, the preferential loss of peaks volume of these hydrophobic residues is due to intermolecular interactions because the same group of peaks were preferentially diminished when HSQC spectra were taken of progressively higher concentrations of 0SS, 2–6 mg·ml–1, at 1 bar (data not shown) to induce intermolecular association of the sort shown in Fig. 1B. Taken together, these observations indicate that the hydrophobic clusters involved in intramolecular network in an unfolded monomer state can participate in intermolecular association as well. If there exists specificity in the interactions between hydrophobic clusters, then successive exchanges of an intramolecularly reacting cluster with the one from another molecule can produce, like the mechanism of domain swapping (41), an assemblage of some regular structures even starting from unfolded monomers.
Conclusion
The present result clearly demonstrates that, despite the general expectation that “partial” unfolding or “partial” folding of a globular protein is a prerequisite, an essentially unfolded conformer can attain amyloid fibrillogenesis. It is further disclosed that this reaction proceeds at least in two steps; a reversible association of the unfolded monomers followed by the irreversible formation of amyloid fibrils. Namely,
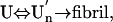 |
where two-way arrows represent reversible processes, one-way arrow represents an irreversible process, and U and U′n represent the unfolded monomeric conformer and the soluble assemblage of unfolded conformers with intermolecular β-structure, respectively. Furthermore, although the present experiment was performed on intrinsically denatured protein U, an equilibrium N ⇔ U is always present for a protein in solution where N represents the native conformer, and by various internal and external perturbations in living cells such as hereditary mutation, this equilibrium can be significantly shifted toward U. In such cases, the above mechanism could be general for any amyloidotic proteins for which folded conformers are normally dominant. We therefore present the following as a common mechanism for the formation of amyloid fibers, namely,
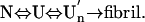 |
This mechanism may explain why many globular proteins form amyloidotic fiber of common morphology, despite the fact that they form quite different tertiary structures in the native state, because in U their conformational characteristics would be largely lost. This scheme may not appear to be consistent with the protein species specificity of amyloid fibrillation. However, the formation of highly ordered amyloid fibrils, which takes considerably long periods unlike the formation of nonspecific aggregates, will require the formation of intricate steric arrangement of side chains and favorable interactions between them in every successive subunit interface, which do not appear to be easily attainable with proteins of different primary structures. It should be noted that, although happening rarely, α-syn and tau, which normally adopt an unfolded conformation, copolymerize in amyloid fibrils (42).
Another important aspect of the present study is the finding that pressure effectively and reversibly dissociates the soluble assembly, thus preventing the irreversible fibril formation. Moreover, although pressure dissociation of protein aggregation has been reported in literatures, to our knowledge, this is the first case that thermodynamic analysis is carried out for the direct characterization of the early association events in amyloid-like fibrillogenesis. Pressure perturbation is uniquely suited to the thermodynamic analysis of highly associable systems because of a large volume change in the association reaction (27). Previous NMR studies of the structure of monomeric amyloidogenic intermediates (43–45) or amyloid fibrils (46) used hydrogen exchange labeling, titration with increasing concentrations of urea, or hydrogen exchange combined with dimethyl sulfoxide dissolution. As shown here, pressure perturbation combined with two-dimensional NMR provides a new route for the thermodynamic and structural analysis of the association events in amyloid-like fibrillogenesis.
Acknowledgments
We thank Y. Tsunashima, S. Okada, and M. Morita for sedimentation measurements and S. Segawa for fluorescence measurements. This work was supported by Grant-in-Aid for Scientific Research 13558082 from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and in part by a grant from the National Institute of Animal Health.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: 0SS, genetically engineered disulfide-deficient variant of hen lysozyme; HSQC, heteronuclear single quantum correlation.
References
- 1.Dobson, C. M. (1999) Trends Biochem. Sci. 24, 329–332. [DOI] [PubMed] [Google Scholar]
- 2.Konno, T., Murata, K. & Nagayama, K. (1999) FEBS Lett. 454, 122–126. [DOI] [PubMed] [Google Scholar]
- 3.Fändrich, M., Fletcher, M. A. & Dobson, C. M. (2001) Nature 410, 165–166. [DOI] [PubMed] [Google Scholar]
- 4.Fink, A. L. (1998) Fold. Des. 3, R9–R23. [DOI] [PubMed] [Google Scholar]
- 5.Kelly, J. W. (1998) Curr. Opin. Struct. Biol. 8, 101–106. [DOI] [PubMed] [Google Scholar]
- 6.Lansbury, P. T., Jr. (1999) Proc. Natl. Acad. Sci. USA 96, 3342–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Booth, D. R., Sunde, M., Belloti, V., Robinson, C. V., Hutchinson, W. L., Fraser, P. E., Hawkins, P. N., Dobson, C. M., Radford, S. E., Blake, C. C., et al. (1997) Nature 385, 787–793. [DOI] [PubMed] [Google Scholar]
- 8.Morozova-Roche, L. A., Zurdo, J., Spencer, A., Noppe, W., Receveur, V., Archer, D. B., Joniau, M. & Dobson, C. M. (2000) J. Struct. Biol. 130, 339–351. [DOI] [PubMed] [Google Scholar]
- 9.Krebs, M. R. H., Wilkins, D. K., Chung, E. W., Pitkeathly, M. C., Chamberlain, A. K., Zurdo, J., Robinson, C. V. & Dobson, C. M. (2000) J. Mol. Biol. 300, 541–549. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg, M. E., Rudolph, R. & Jaenicke, R. (1991) Biochemistry 30, 2790–2797. [DOI] [PubMed] [Google Scholar]
- 11.Tachibana, H. (2000) FEBS Lett. 480, 175–178. [DOI] [PubMed] [Google Scholar]
- 12.Silva, J. L., Foguel, D. & Royer, C. A. (2001) Trends Biochem. Sci. 26, 612–618. [DOI] [PubMed] [Google Scholar]
- 13.Randolph, T. W., Seefeldt, M. & Carpenter, J. F. (2002) Biochim. Biophys. Acta 1595, 224–234. [DOI] [PubMed] [Google Scholar]
- 14.Ferrão-Gonzales, A. D., Souto, S. O., Silva, J. L. & Foguel, D. (2000) Proc. Natl. Acad. Sci. USA 97, 6445–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niraula, T. N., Haraoka, K., Ando, Y., Li, H., Yamada, H. & Akasaka, K. (2002) J. Mol. Biol. 320, 333–342. [DOI] [PubMed] [Google Scholar]
- 16.Dubois, J., Ismail, A. A., Chan, S. L. & Ali-Khan, Z. (1999) Scand. J. Immunol. 49, 376–380. [DOI] [PubMed] [Google Scholar]
- 17.Torrent, J., Alvarez-Martinez, M. T., Heitz, F., Liautard, J.-P., Balny, C. & Lange, R. (2003) Biochemistry 42, 1318–1325. [DOI] [PubMed] [Google Scholar]
- 18.Foguel, D., Suarez, M. C., Ferrão-Gonzales, A. D., Porto, T. C., Palmieri, L., Einsiedler, C. M., Andrade, L. R., Lashuel, H. A., Lansbury, P. T., Kelly, J. W. & Silva, J. L. (2003) Proc. Natl. Acad. Sci. USA 100, 9831–9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akasaka, K., Tezuka, T. & Yamada, H. (1997) J. Mol. Biol. 271, 671–678. [DOI] [PubMed] [Google Scholar]
- 20.Kamatari, Y. O., Yamada, H., Akasaka, K., Jones, J. A., Dobson, C. M. & Smith, L. J. (2001) Eur. J. Biochem. 268, 1782–1793. [PubMed] [Google Scholar]
- 21.Akasaka, K. & Yamada, H. (2001) Methods Enzymol. 338, 134–158. [DOI] [PubMed] [Google Scholar]
- 22.Tachibana, H., Oka, T. & Akasaka, K. (2001) J. Mol. Biol. 314, 311–320. [DOI] [PubMed] [Google Scholar]
- 23.Noda, Y., Yokota, A., Horii, D., Tominaga, T., Tanisaka, Y., Tachibana, H. & Segawa, S. (2002) Biochemistry 41, 2130–2139. [DOI] [PubMed] [Google Scholar]
- 24.Prakash, V., Loucheux, C., Scheufele, S., Gorbunoff, M. J. & Timasheff, S. N. (1981) Arch. Biochem. Biophys. 210, 455–464. [DOI] [PubMed] [Google Scholar]
- 25.Schwalbe, H., Fiebig, K. M., Buck, M., Jones, J. A., Grimshaw, S. B., Spencer, A., Glaser, S. J., Smith, L. J. & Dobson, C. M. (1997) Biochemistry 36, 8977–8991. [DOI] [PubMed] [Google Scholar]
- 26.Niraula, T. N. (2002) Ph.D. thesis (Kobe Univ., Kobe, Japan).
- 27.Royer, C. A. (1995) Methods Enzymol. 259, 357–377. [DOI] [PubMed] [Google Scholar]
- 28.Imoto, T., Johnson, L. N., North, A. C. T., Phillips, D. C. & Rupley, J. A. (1972) in The Enzyme, ed. Boyer, P. D. (Academic, New York), Vol. 7, pp. 665–868. [Google Scholar]
- 29.Isaacs, N. S. (1981) Liquid Phase High Pressure Chemistry (Wiley, Chichester, U.K.).
- 30.Cantor, C. R. & Schimmel, P. R. (1980) Biophysical Chemistry (Freeman, New York), Parts 2 and 3.
- 31.Tanford, C. (1961) Physical Chemistry of Macromolecules (Wiley, New York).
- 32.Josephs, R. & Harrington, W. F. (1967) Proc. Natl. Acad. Sci. USA 58, 1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LeVine, H., III. (1993) Protein Sci. 2, 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walsh, D. M., Hartley, D. M., Kusumoto, Y., Fezoui, Y., Condron, M. M., Lomakin, A., Benedek, G. B., Selkoe, D. J. & Teplow, D. B. (1999) J. Biol. Chem. 274, 25945–25952. [DOI] [PubMed] [Google Scholar]
- 35.Harper, J. D., Wong, S. S., Lieber, C. M. & Lansbury, P. T., Jr. (1999) Biochemistry 38, 8972–8980. [DOI] [PubMed] [Google Scholar]
- 36.Silva, J. L. & Weber, G. (1993) Annu. Rev. Phys. Chem. 44, 89–113. [DOI] [PubMed] [Google Scholar]
- 37.Silva, J. L. & Weber, G. (1988) J. Mol. Biol. 199, 149–159. [DOI] [PubMed] [Google Scholar]
- 38.Silva, J. L., Villas-Boas, M., Bonafe, C. F. S. & Meirelles, N. C. (1989) J. Biol. Chem. 264, 15863–15868. [PubMed] [Google Scholar]
- 39.Klein-Seetharaman, J., Oikawa, M., Grimshaw, S. B., Wirmer, J., Duchardt, E., Ueda, T., Imoto, T., Smith, L. J., Dobson, C. M. & Schwalbe, H. (2002) Science 295, 1719–1722. [DOI] [PubMed] [Google Scholar]
- 40.Baldwin, R. L. (2002) Science 295, 1657–1658. [DOI] [PubMed] [Google Scholar]
- 41.Liu, Y., Gotte, G., Libonati, M. & Eisenberg, D. (2001) Nat. Struct. Biol. 8, 211–214. [DOI] [PubMed] [Google Scholar]
- 42.Giasson, B. I., Forman, M. S., Higuchi, M., Golbe, L. I., Graves, C. L., Kotzbauer, P. T., Trojanowski, J. Q. & Lee, V. M.-Y. (2003) Science 300, 636–640. [DOI] [PubMed] [Google Scholar]
- 43.Liu, K., Cho, H. S., Lashuel, H. A., Kelly, J. W. & Wemmer, D. E. (2000) Nat. Struct. Biol. 7, 754–757. [DOI] [PubMed] [Google Scholar]
- 44.Canet, D., Last, A. M., Tito, P., Sunde, M., Spencer, A., Archer, D. B., Redfield, C., Robinson, C. V. & Dobson, C. M. (2002) Nat. Struct. Biol. 9, 308–315. [DOI] [PubMed] [Google Scholar]
- 45.McParland, V. J., Kalverda, A. P., Homans, S. W. & Radford, S. E. (2002) Nat. Struct. Biol. 9, 326–331. [DOI] [PubMed] [Google Scholar]
- 46.Hoshino, M., Katou, H., Hagihara, Y., Hasegawa, K., Naiki, H. & Goto, Y. (2002) Nat. Struct. Biol. 9, 332–336. [DOI] [PubMed] [Google Scholar]