

Abstract
The steroid 17β-estradiol (E2) modulates energy homeostasis by reducing feeding behavior and increasing energy expenditure primarily through estrogen receptor α (ERα)-mediated mechanisms. Intact ERαKO female mice develop obesity as adults exhibiting decreased energy expenditure and increased fat deposition. However, intact transgenic female mice expressing a DNA-binding-deficient ERα (KIKO) are not obese and have similar energy expenditure, activity and fat deposition to wild type (WT) females, suggesting that non-Estrogen Response Element (ERE)-mediated signaling is important in E2 regulation of energy homeostasis. However, initial reports did not examine the effects of ovariectomy on energy homeostasis or E2’s attenuation of post-ovariectomy body weight gain. Therefore, we sought to determine if low physiological doses of E2 (250 ng QOD) known to suppress post-ovariectomy body weight gain in WT females, would suppress body weight gain in ovariectomized KIKO females. We observed that the post-ovariectomy increase in body weight was significantly greater in WT females than in KIKO females. Furthermore, E2 did not significantly attenuate the body weight gain in KIKO females as it did in WT females. E2 replacement suppressed food intake and fat accumulation while increasing nighttime oxygen consumption and activity only in WT females. E2 replacement also increased arcuate POMC gene expression in WT females only. These data suggest that in the intact female, ERE-independent mechanisms are sufficient to maintain normal energy homeostasis and to partially restore the normal response to ovariectomy. However, they are not sufficient for E2’s suppression of post-ovariectomy body weight gain and attenuation of decreases in metabolism and activity.
Keywords: 17β-estradiol, ovariectomy, energy homeostasis, ERα
Introduction
The reproductive steroid, 17β-estradiol (E2), regulates various aspects of energy homeostasis through both peripheral actions and central mechanisms. E2 suppresses feeding and fat accumulation and augments energy expenditure and activity through actions in the brain. The key brain regions mediating E2’s effects on energy homeostasis are the hypothalamus and the hindbrain [1–4]. A decrease in circulating estrogens due to ovariectomy or menopause is associated with positive weight gain in both rodent models and humans, which is attenuated by E2 replacement [5–7]. Furthermore, food intake varies during the menstrual cycle in humans and in intact animals with a luteal phase peak and a peri-ovulatory nadir in consumption illustrating E2’s effects [8–10].
E2 uses two classical nuclear steroid receptors, ERα and ERβ to control physiological and cellular functions and to regulate gene expression. ERα is highly expressed in the hypothalamus including the Arcuate nucleus and ERα-mediated actions are the primary mechanisms utilized by E2 to control energy homeostasis [11]. Furthermore, ERα knockouts (ERKO) exhibit an obesity phenotype with increased visceral adiposity and decreased energy expenditure [12,13]. ERα is also necessary for the attenuation of weight gain and food intake and is involved in the extrahypothalamic (NTS) control of food intake by cholecystokinin [3,13]. In the ventromedial hypothalamus, ERα knockdown by RNAi induces a phenotype defined by obesity, hyperphagia, glucose intolerance and reduces activity (energy expenditure), which is resistant to E2’s effect on activity [4].
Arcuate POMC and NPY neurons are involved in feeding and both express ERα. In POMC neurons, ERα is highly expressed (~75%) [14], with NPY neuronal ERα expression approximately 20% [15]. Indeed, specific deletion of ERα in POMC neurons significantly increased body weight, food intake and activity, while specific deletion of ERα in SF1 neurons of the ventromedial hypothalamus decreased energy expenditure and increased fat accumulation [16]. Furthermore, POMC and NPY neurons express a Gq-coupled membrane estrogen receptor that mediates E2’s effects on feeding, core body temperature and gene expression [17–21].
Both the classical ERs (ERα and ERβ) bind to DNA at estrogen response elements (ERE) to control gene expression. However, both receptors can control gene expression through ERE-independent signaling via protein-protein interactions with other transcription factors (Sp-1, Fos-Jun (AP-1) pCREB, STATs, and NFκB) and binding to other promoter sites [22]. ERs also activate membrane-initiated signaling cascades (MAPK, PLC, PI3K) that modulate cell physiology and control gene expression [18–20,23–25]. Recently, the restoration of ERE-independent signaling was reported to normalize energy balance in ERKO females [26]. These mice express an ERα (ERAA) that lacks the ability to bind to ERE due to mutations in the DNA-binding domain [27]. Females that only express the ERAA, also called ERα KIKO (KIKO), do not become obese like their ERKO counterparts.
In wild type females, ovariectomy induces weight gain that is attenuated by E2 replacement while in the ERKO female ovariectomy does not induce significant body weight gain. Ovariectomized ERKO females also do not respond to E2 replacement [13]. The lack of normal response to ovariectomy in the ERKO females is interpreted as an indicator of ERα’s role in mediating E2’s effect on energy homeostasis. Therefore, we sought to determine if the presence of ERE-independent signaling in KIKO females restores the post-ovariectomy body weight gain in females and if this positive state of energy balance is attenuated by E2 replacement. We assessed the effects of ovariectomy with or without E2 replacement in wild type, ERKO and KIKO littermate females by measuring body weight and food intake for four weeks followed by measurements of oxygen consumption, activity and body composition.
Experimental
Animal care
All animal treatments are in accordance with institutional guidelines based on National Institutes of Health standards, and were performed with Institutional Animal Care and Use Committee approval at Rutgers University. Female wild-type (WT), ERα KO (ERKO) and ERα KIKO (KIKO) transgenic mice (provided by Dr. Ken Korach, NIEHS) [28–30] were selectively bred in-house, and maintained under controlled temperature (25 °C) and photoperiod conditions (12/12 hr light/dark cycle) with food and water ad libitum. ERKO females were generated by breeding WT/KO heterozygotic males and females. KIKO females were generated by crossing the non-classical ER knock-in (NERKI) heterozygous males (WT/KI) with WT/KO heterozygotic females. Wild-type females were generated from both colonies and used with their KIKO and ERKO littermates. At weaning, females were tagged and ear clipped for genotyping. Genotype was determined by PCR of extracted DNA using previously published protocols [28–30].
Drugs
17β-estradiol benzoate (E2) was purchased from Steraloids (Newport, RI, USA) and dissolved in ethanol prior to dissolution in sesame oil (Sigma-Aldrich). Ketamine was purchased from Henry Schein (Melville, NY, USA) and used for sedation prior to killing.
Experimental design
At eight weeks of age, females were ovariectomized under isoflurane anesthesia using no touch sterile techniques and allowed to recover for 5 days prior to measurements of body composition using an EchoMRI 3-in-1 Body Composition Analyzer (Echo Medical Systems, Houston, TX, USA) followed by monitoring in a Comprehensive Lab Animal Monitoring System (CLAMS) (Columbus Instruments, Inc., Columbus, OH, USA) for 48 hrs. After monitoring, females were housed alone and allowed to recover for one day prior to injection of either sesame oil or 17β-estradiol benzoate (250 ng/dose) at which time body weight and food weight were measured. Females were injected and weighed every other day (QOD) for 4 weeks. At the end of four weeks, females were analyzed for body composition using the EchoMRI and metabolic parameters and activity using the CLAMS. Monitoring was followed by another day of recovery and an injection of oil or E2 24-hr prior to killing and brain and body dissections. See Figure 1 for a timeline of the experiment. A subset of females from each genotype were not ovariectomized and subjected to the same experimental paradigm.
Figure 1.
Experimental protocol for ovariectomized and intact females.
Tissue Dissections
At the end of the experiments, females were decapitated after sedation with ketamine (100 μl of 100 mg/ml, i.p.) 24 h after the final treatment injection. Trunk blood was collected and analyzed for triglyceride and glucose levels using a CardioChek (Polymer Technology Systems, Inc., Indianapolis, IN, USA) and prepared for serum analysis of 17β-estradiol levels using Mouse Calbiotech ELISA conducted by the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core [31]. The E2 assay had an intra-assay variability of 3.1% and a sensitivity of 0.3–300 pg/ml. Hypothalamic nuclei were micro-dissected for RNA extraction and gene expression analysis. The basal hypothalamus (BH) was cut using a brain slicer (Ted Pella, Inc., Redding, CA, USA), into one mm thick coronal rostral and caudal blocks corresponding to Plates 42 to 47 and Plates 48 to 53, respectively, from The Mouse Brain in Stereotaxic Coordinates (Paxinos & Franklin 2008, 3rd Edition) [32]. The BH blocks were transferred to RNAlater (Life Technologies, Inc., Grand Island, NE, USA) and stored overnight at 4 °C. The rostral and caudal parts of the Arcuate nucleus were dissected from slices using a dissecting microscope. Dissected tissue was stored at −80 °C. Total RNA was extracted from the combined nuclei (rostral and caudal arcuate) using Ambion RNAqueous-Micro Kits (Life Technologies, Inc.) according to the manufacturer’s protocol. Total RNA was also DNase I-treated, using the extraction kits, at 37 °C for 30 min to minimize any genomic DNA contamination. RNA quantity and quality were determined using a NanoDrop ND-2000 spectrophotometer (ThermoFisher, Inc., Waltham, MA, USA) and an Agilent 2100 Bioanalyzer and RNA Nano Chips (Agilent Technologies, Inc., Santa Clara, CA, USA).
Quantitative real-time PCR
cDNA was synthesized from 200 ng of total RNA using Superscript III reverse transcriptase (Life Technologies, Inc.), 4 μl 5x Buffer, 25 mM MgCl2, 10 mM dNTP (Clontech Laboratories, Inc., Mountain View, CA, USA), 100 ng random hexamer primers (Promega Corporation, Madison, WI, USA), 40 U/μl Rnasin (Promega) and 100 mM DTT in DEPC-treated water (Gene Mate, Bioexpress, Inc., Kaysville, UT, USA) in total volume of 20 μl. Reverse transcription was conducted using the following protocol: 5 min at 25 °C, 60 min at 50 °C, 15 min at 70 °C. The cDNA was diluted to 1:20 with Nuclease-free water (Gene Mate, Bioexpress) for a final cDNA concentration of 0.5 ng/μl and stored at −20 °C. BH test tissue RNA was used for positive and negative controls (no reverse transcriptase) and processed simultaneously with the experimental samples.
All primers were designed to span exon-exon junctions and synthesized by Life Technologies, Inc., using Clone Manager 5 software (Sci Ed Software, Cary, NC, USA). See Table 1 for a listing of all the primer sets used for quantitative real-time PCR (qPCR). For qPCR, 4 μl cDNA template (an equivalent of 2 ng total RNA) was amplified using either PowerSYBR Green master mix (Life Technologies) or Sso Advanced SYBR Green (BioRad, Inc., Hercules, CA, USA) on CFX-Connect Real-time PCR instrument (BioRad). Standard curves for each primer pair were prepared using serial dilutions of BH cDNA in triplicate to determine the efficiency [E=10(−1/m) − 1, m=slope] of each primer pair. All efficiencies expressed as percent efficiency were approximately equal (one doubling per cycle, 90–100%, Table 1). Therefore, the relative mRNA expression data was analyzed using the ΔΔCT method [33,34]. The amplification protocol for all the genes was as follows: initial denaturing −95 °C for 10 min (PowerSYBR) or 3 min (SsoAdvanced) followed by 40 cycles of amplification at 94 °C for 10 sec (denaturing), 60 °C for 45 sec (annealing), and completed with a dissociation step for melting point analysis with 60 cycles of 95 °C for 10 sec, 65 °C to 95 °C (in increments of 0.5 °C) for 5 sec and 95 °C for 5 sec. The reference genes used were Actb and/or Gapdh. Positive and negative controls were added to each amplification run including a water blank. Quantification values were generated only from samples showing a single product at the expected melting point.
Table 1.
Primers sequences used for qPCR.
| Name | Product Length | Primer Efficiency | Primer sequence | Base pair # | Accession # |
|---|---|---|---|---|---|
| AgRP | 146 | 105 | F: CTCCACTGAAGGGCATCAGAA | 287–307 | NM_007427.2 |
| R: ATCTAGCACCTCCGCCAAA | 414–432 | ||||
| β-actin | 63 | 100 | F: GCCCTGAGGCTCTTTTCCA | 849–867 | NM_007393.3 |
| R: TAGTTTCATGGATGCCACAGGA | 890–911 | ||||
| CART | 169 | 95 | F: GCTCAAGAGTAAACGCATTCC | 277–297 | NM_013732 |
| R: GTCCCTTCACAAGCACTTCAA | 425–445 | ||||
| Gapdh | 98 | 93 | F: TGACGTGCCGCCTGGAGAAA | 778–797 | NM_008084.2 |
| R: AGTGTAGCCCAAGATGCCCTTCAG | 852–875 | ||||
| NPY | 182 | 100 | F: ACTGACCCTCGCTCTATCTC | 106–125 | NM_023456 |
| R: TCTCAGGGCTGGATCTCTTG | 268–287 | ||||
| POMC | 200 | 103 | F: GGAAGATGCCGAGATTCTGC | 145–164 | NM_008895 |
| R: TCCGTTGCCAGGAAACAC | 327–344 |
primer is listed first with the antisense primer below. AgRP, agouti-related peptide; CART, cocaine-and-amphetamine regulated transcript; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NPY, neuropeptide Y; POMC, pro-opiomelanocortin.
Final relative quantitation was done using the comparative CT method [33,34] utilizing a calibrator of diluted (1:20) cDNA from the BH of an intact male. The data were reported as relative mRNA expression. To determine the CT for each transcript, the threshold was consistently set at the lowest point of the exponential curve where the slope of the curve was the steepest for all plates. The relative linear quantity of target molecules was calculated using the formula 2−ΔΔCT. All gene expression data were expressed as an n-fold difference relative to the calibrator. The n-fold difference was averaged for each treatment and analyzed statistically using a two-tailed Student’s t-test (p ≤ 0.05).
Data analysis
All data was expressed as mean ± SEM. All the data from the bi-daily body weight measurements and CLAMS analysis were analyzed using a two-way ANOVA followed by a post-hoc Bonferroni-Dunn multiple comparison tests. All data from the food intake studies, body composition determination, quantitative real-time PCR experiments and other measurements were analyzed using a one-way ANOVA followed by a post-hoc Bonferroni-Dunn multiple comparison tests and/or Student’s t-test (unpaired). All data was analyzed using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA) and in all cases, effects were considered significant at p ≤ 0.05.
Results
Serum E2 levels and uterine weights
Serum E2 levels were measured in all animals including intact females. Intact KIKO females had slightly higher serum E2 levels than intact WT females (WT: 3.3 ± 0.2 pg/ml (n=7) vs. KIKO: 4.7 ± 0.6 pg/ml (n=7); p<0.05) respectively (Figure 2A). Intact ERKO females had significantly higher serum E2 levels (22.3 ± 4.2 pg/ml (n=7), p<0.01) than both intact WT and KIKO females. In oil-treated, ovariectomized (ovx) WT, KIKO and ERKO females, the serum levels of E2 were not significantly different (3.9 ± 0.5 pg/ml (n=8); 8.5 ± 3.3 pg/ml (n=8); and 5.9 ± 1.3 pg/ml (n=8); respectively). In the E2-treated, ovx WT, KIKO and ERKO females, the serum levels of E2 were not significantly different (23.7 ± 3.5 pg/ml (n=8); 26.5 ± 4.8 pg/ml (n=8) and 33.3 ± 7.5 pg/ml (n=8); respectively) but were significantly higher than in the oil-treated counterpart females (p<0.0001, p<0.01, p<0.01, respectively).
Figure 2. Serum 17β-estradiol levels and uterine weights from intact and ovariectomized females.
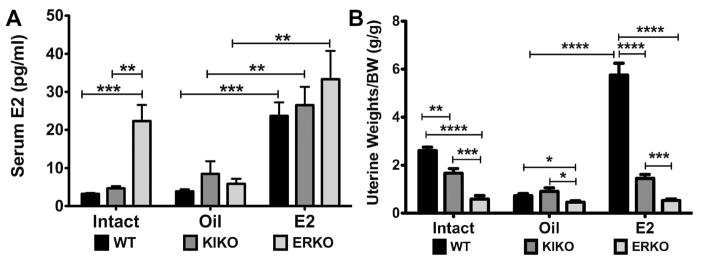
A. Serum E2 (pg/ml) were measured using a Mouse Calbiotech ELISA in WT (black bars), KIKO (dark gray bars) and ERKO (light gray bars) in intact females as well as oil-treated and E2-treated ovx females. Each intact genotype group had 7 females and each ovx group had 8 females. B. Uterine weights normalized to body weight (g/g) in the same animals. Data was analyzed by a one-way ANOVA with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001).
We measured uterine weights at the end of the experiment to confirm the presence of E2 in the WT ovx females (Figure 2B). In WT females, E2 replacement is known to increase uterine weights through an ERα-mediated mechanism [35]. Intact WT females had significantly higher uterine weights/body weight than either intact KIKO or ERKO females (WT=2.61 ± 0.4 g/g; KIKO=1.7 ± 0.5 g/g; ERKO=0.6 ± 0.4 g/g; WT vs. KIKO: p<0.01; WT vs. ERKO: p<0.0001).
In WT females, the oil-treated, ovx females had a uterine weight/body weight (g/g) that was significantly less than the E2-treated, ovx WT females (0.7 ± 0.3 g/g vs. 5.8 ± 0.5 g/g, p<0.0001). E2 also increased the uterine weight/body weight in ovx KIKO females, although the difference was not as great between oil- and E2-treated groups (0.9 ± 0.1 g/g vs. 1.5 ± 0.2 g/g, p<0.5). E2 replacement had no effect on ERKO uterine weights (0.5 ± 0.1 g/g vs. 0.5 ± 0.1 g/g). Furthermore, E2-treated WT females had significantly higher uterine weights than both E2-treated KIKO and ERKO females (p<0.0001 for both) and E2-treated KIKO females had significantly higher uterine weights than ERKO females (p<0.001).
Post-ovariectomy body weight gain and E2 attenuation
Ovariectomy in WT female mice results in significant body weight gain. Conversely, ovariectomy in ERKO females does not cause a significant increase in body weight [13]. Prior to ovariectomy, there was no significant difference in body weights across or within the genotypes. WT females weighed 18.4 ± 0.3 g (oil) and 18.7 ± 0.4 g (E2); KIKO females weighed 17.7 ± 0.6 g (oil) and 17.9 ± 0.3 g (E2); and ERKO females weighed 18.9 ± 0.5 g (oil) and 19.3 ± 0.6 g (E2).
In this study, oil-treated, ovx WT females gained more weight than both the KIKO and ERKO oil-treated, ovx females (Figure 3A). Significant differences appeared between the WT, KIKO and ERKO ovx females on day 15 and day 9, respectively (WT vs. KIKO: p<0.01, df=1, F=9.08; WT vs. ERKO: p<0.001, df=1, F=18.09). The weight gain in the ovx KIKO, as seen in Figure 3A, is intermediary between the ovx WT and ERKO females. Weight gain between the oil-treated, ovx KIKO and ERKO females was not significantly different (p=0.076) although the weight gain in the KIKO females was observably higher. This was due, in part, to a large amount of variation in the response to ovariectomy by ERKO females with three animals gaining weight and the remaining either not gaining weight or losing weight over the 27 days of the experiment.
Figure 3. Post-ovariectomy body weight gain and the attenuation by E2.

A. Bi-daily (QOD) cumulative body weight gain in oil-treated, ovariectomized WT (black circles), KIKO (dark gray triangles) and ERKO (light gray squares). Each ovariectomized group had 8 females. Data was analyzed by a two-way ANOVA (genotype: p<0.01, df=2, F=9.84) with Bonferroni-Dunn multiple comparison tests (a = p<0.05; b = p<0.01; c = p<0.001; d = p<0.0001, compared to WT). B–D. Cumulative body weight gain in oil-treated, ovariectomized (black circles); E2-treated, ovariectomized (gray squares); and intact (dark gray triangles) females (B=WT, C=KIKO; D=ERKO). Ovariectomized groups had 8 females and the intact groups had 7 females each. Data was analyzed by a two-way ANOVA (WT: p<0.001, df=1, F=20.68; KIKO & ERKO: not significant (ns)) with Bonferroni-Dunn multiple comparison tests (a = p<0.05; b = p<0.01; c = p<0.001; d = p<0.0001, oil vs. E2 comparisons only).
Furthermore, post-ovariectomy body weight gain in WT was significantly greater than intact WT females (cumulative weight gain (day 27): intact WT: 1.4 ± 0.3 g vs. ovx WT: 3.8 ± 0.2 g; p<0.0001; Figure 3B). Intact KIKO females gained 1.4 ± 0.3 g over the 27 days while oil-treated, ovx females gain an average of 2.1 ± 0.3 g (p=0.11; Figure 3C). Intact ERKO females gained an average of 1.4 ± 0.9 g during the experiment while oil-treated, ovx ERKO females gained 0.5 ± 0.7 g (Figure 3D). However, ovx KIKO females did temporarily have greater cumulative weight gain at day 9 through day 13 compared to intact KIKO females (p<0.01) indicating a significant, initial post-ovariectomy weight gain that was not sustained like in the ovx WT females.
Only in ovx WT females did E2 replacement attenuate the post-ovariectomy body weight gain (Figure 3B; ANOVA: p<0.001, df=1, F=20.68). In KIKO females, E2 did not significantly reduce body weight gain although there was a clear separation in body weights between oil- and E2-treated KIKO females (Figure 3C). As previously reported, there is no effect of E2 on body weight gain in ovx ERKO females (Figure 3D). Similar to the response to ovariectomy, the response of the body weight in KIKO females to E2 replacement was intermediate to WT and ERKO females suggesting that ERE-independent signaling may be involved, but not sufficient for E2’s attenuation of post-ovariectomy body weight gain.
E2 effects on feeding behavior
During the 4 weeks of the experiment, food intake was measured every other day (QOD) for both intact and ovx females. The average 48 hr food intake was significantly higher in intact WT females (6.7 ± 0.3 g) than both intact KIKO (5.6 ± 0.2 g, p<0.01) and intact ERKO (5.7 ± 0.3 g, p<0.05) (Figure 4A). This pattern continued for both oil-treated and E2-treated ovx females. WT ovx females, regardless of steroid treatment consumed more food than both the KIKO and ERKO ovx females (two-way ANOVA: p<0.0001, df=2, F=39.1). Furthermore, oil-treated WT females consumed more food than their E2-treated counterparts (oil: 7.6 ± 0.2 g vs. E2: 6.9 ± 0.2 g, p<0.05) and their intact WT counterparts (p<0.05) (one-way ANOVA: p<0.05, df=2, F=4.8). Intact KIKO females ate significantly less food than either oil-treated or E2-treated KIKO females (oil: 6.2 ± 0.2 g; E2: 6.1 ± 0.1 g, p<0.05). There was no effect of E2 treatment on either KIKO or ERKO 48 hr food intake nor was there any difference between either intact ERKO food intake or oil-treated or E2-treated ERKO females.
Figure 4. Average 48 hr food intake and food chewed for both intact and ovariectomized females.
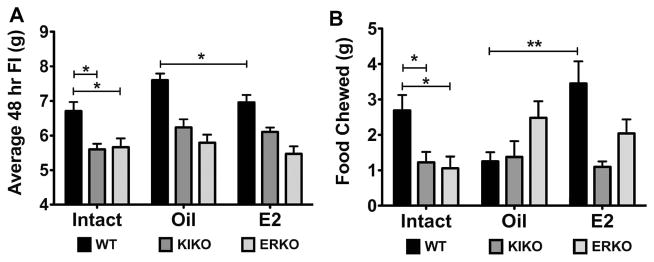
A. Average 48 hr food intake (g) over the course of the 4 weeks measured for each animal from each group. WT (black bars), KIKO (dark gray bars) and ERKO (light gray bars) in intact and oil-treated and E2-treated ovariectomized females. Data was analyzed by a two-way ANOVA (genotype: p<0.0001, df=2, F=39.1; steroid: p<0.01; df=2, F=5.25) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05; ** p<0.01). B. The average 48 hr amount of food chewed but not consumed by each animal within each group. Data was analyzed by a one-way ANOVA (Intact: p<0.01, df=2; F=6.28; E2: p<0.01, df=2, F=6.37; oil: ns) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05; ** p<0.01).
During the 4 weeks of food intake measurements, we also observed a significant amount of food in the bottom of the cage. Therefore, we decided to measure the amount of food left in the cage every 48 hr and called it ‘food chewed’. The amount of food chewed correlated with the average 48 hr food intake in the intact females. Intact WT females chewed significantly more food than both the KIKO and ERKO intact females (WT: 2.7 ± 0.4 g vs. KIKO: 1.2 ± 0.3 g vs. ERKO: 1.1 ± 0.3 g, p<0.05) (Figure 4B). In the oil-treated, ovx females, the amount of food chewed was 1.3 ± 0.3 g for the WT females; 1.4 ± 0.4 g for the KIKO and 2.5 ± 0.5 g for the ERKO females (no significant effect of genotype). However, ovariectomy did decrease the amount of food chewed compared to intact females in the WT (p<0.05). In the E2-treated, ovx females, WT females (3.4 ± 0.6 g) chewed more food than KIKO females (1.1 ± 0.2 g; p<0.01), but not ERKO females (2.0 ± 0.4 g). E2-treated WT females also chewed significantly more food than oil-treated WT females (p<0.01) and E2-treated ERKO females chewed more food than E2-treated KIKO (E2: p<0.05). Food chewing may be an indication of greater activity, anxiety/boredom or pica, non-nutritive food intake [36].
The effect of ovariectomy and E2 on body composition and metabolic chemistry
To determine the change in lean mass and fat accumulation over the course of the 4-week treatment, we measured body composition 5 days after ovariectomy and after 4 weeks using an EchoMRI Body Composition analyzer. The change in body fat was positive for both WT (22.3 ± 7.1%) and KIKO (13.3 ± 4.8%) females over the 4 weeks (Figure 5A). ERKO females did not have a significant change in body fat (−9.1 ± 13.7), and there were no significant differences between the genotypes. The change in body fat in intact WT and KIKO females correlates with the small increase in body weight over the 27 days of the experimental period. ERKO females did not increase in body fat significantly but had larger fat deposits at the start of the experiment than then WT and KIKO females (data not shown).
Figure 5. E2 replacement suppressed body fat accumulation and increases blood triglycerides levels.

A. Average percent change in body fat/body weight during the 4 weeks averaged for each group. WT (black bars), KIKO (dark gray bars) and ERKO (light gray bars) in intact and oil-treated and E2-treated ovariectomized females. Data was analyzed by a one-way ANOVA (WT: p<0.001, df=2, F=13.6; KIKO: p<0.01, df=2, F=7.26; ERKO: p<0.05, df=2, F=4.26) with Bonferroni-Dunn multiple comparison tests across and within genotypes (** p<0.01, *** p<0.001). B. The average percent change in lean mass/body weight averaged for each group. Data was analyzed by a one-way ANOVA (E2: p<0.05, df=2; F=3.65; oil: p<0.01, df=2, F=7.65; oil: ns) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05; ** p<0.01). C. Blood triglycerides levels (mg/dl) averaged for each group. Data was analyzed by a one-way ANOVA with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05). D. Blood glucose levels (mg/dl) averaged for each group. Data was analyzed by a one-way ANOVA with Bonferroni-Dunn multiple comparison tests across and within genotypes.
Amongst the oil-treated, ovx animals, the change in percent body fat was greatest in the WT compared to both the other oil-treated genotype groups and to the E2-treated WT females. Amongst the oil-treated females, the change in percent body fat increased by 78.0 ± 10.2% in the WT, 49.2 ± 6.6% in the KIKO and 25.4 ± 12.3% in the ERKO (WT vs. KIKO: p<0.05; WT vs. ERKO: p<0.01). In the E2-treated WT females, the change in percent body fat was 49.3 ± 4.1% (WT oil vs. WT E2: p<0.01). The changes in percent body fat for KIKO (38.9 ± 7.9%) and ERKO (36.3 ± 8.1%) E2-treated females were not significantly different than the WT. There were no significant effects of steroid treatment or genotype on percent change in lean mass (Figure 5B) except for a significant difference between both oil-treated and E2-treated WT and their ERKO counterparts (WT oil vs. ERKO oil: p<0.01; WT E2 vs. ERKO E2: p<0.05).
We also measured non-fasting, whole blood triglyceride and glucose levels from collected trunk blood. We did not fast the animals prior to killing due to the effects of fasting on arcuate neuropeptide gene expression [37]. E2 treatment increased the triglyceride levels in WT females (WT oil: 61.5 ± 3.0 mg/dl vs. WT E2: 74.3 ± 4.1 mg/dl, p<0.5). However, blood triglycerides were not significantly affected by genotype in the intact females or the oil-treated and E2-treated ovx females (Figure 5C). Blood glucose levels were highly variable and not significant different across the genotypes for intact or ovx females (Figure 5D).
The effect of ovariectomy and E2 on energy expenditure and locomotor activity
To measure changes in energy expenditure, we used a Comprehensive Lab Animal Monitoring System to measure oxygen consumption (V.O2), CO2 production (V.CO2), respiratory exchange ratio (RER), heat and activity (X and Z). Each female was analyzed for these parameters 5 days after ovariectomy for 48 hr and after 4 weeks of steroid treatment for another 48 hr. All data from the last 24 hr of the CLAMS run was used in the analysis and was further separated into day (7 am to 7 pm) and night (7 pm to 7 am). Comparisons were made between the pre-treatment and the post-treatment CLAMS results within genotype and across steroid treatment (intact, oil, E2).
In both day (data not shown) and night (Figure 6A), V.O2 was significantly lower in the animals post-treatment compared to pre-treatment for WT oil (p<0.001), WT E2 (p<0.01), KIKO oil (p<0.05) and KIKO E2 (p<0.05). There was no difference in V.O2 in either ERKO oil or ERKO E2 during the day or night between pre-treatment and post-treatment (Two-way ANOVA: day-night: p<0.0001, df= 1, F= 36.97). Furthermore, WT-oil, WT-E2, KIKO-oil and KIKO-E2 pre-treatment nighttime V.O2 was significantly higher than the ERKO-oil or ERKO-E2 pre-treatment nighttime V.O2 (WT oil: p<0.01; WT E2: p<0.01; KIKO oil: p<0.05; KIKO E2: p<0.05). These data indicate that the suppressive effects of ovariectomy on energy expenditure increase over time in both WT and KIKO females but not in ERKO females. These data also corroborates earlier research reporting that ERKO females have a lower energy expenditure compared to both WT and KIKO females [26]
Figure 6. Oxygen consumption (V.O2) and carbon dioxide production (V.CO2) decreases after ovariectomy, which is only attenuated by E2 in WT females.

A. Average nightime V.O2 (ml/min/kg) for each group before and after treatment. Unhatched bar = pre-treatment and hatched bars = post-treatment. Data was analyzed by a two-way ANOVA (genotype X steroid: p<0.001, df=5, F=4.09; pre-post: p<0.0001, df=1, F=36.97) with Bonferroni-Dunn multiple comparison tests across and within genotypes. B. Average daytime and nighttime V.O2 (ml/min/kg) for each group post-treatment. Data was analyzed by a two-way ANOVA (day-night: p<0.0001, df=1; F=36.74; genotype X steroid: ns (p=0.06)) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05, ** p<0.01, *** p<0.001, compared to nighttime group). C. Average nighttime V.CO2 (ml/min/kg) for each group before and after treatment. Unhatched bar = pre-treatment and hatched bars = post-treatment. Data was analyzed by a two-way ANOVA (genotype X steroid: ns; pre-post: p<0.0001, df=1, F=30.85) with Bonferroni-Dunn multiple comparison tests across and within genotypes. D. Average daytime and nighttime V.CO2 (ml/min/kg) for each group post-treatment. Data was analyzed by a two-way ANOVA (day-night: p<0.0001, df=1; F=77.71; genotype X steroid: p<0.01, df=5, F=3.63) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, compared to nighttime group).
After the 4 weeks of treatment, V.O2 is significantly lower during the daytime compared to the nighttime for all genotype-steroid treatments including ERKO-oil and ERKO-E2 (Two-way ANOVA: day-night: p<0.0001, df= 1, F= 36.74; Figure 6B). The greatest difference was between WT-E2 day and WT-E2 night V.O2 (day: 3726 ± 130 ml/min/kg vs. night: 4657 ± 143 ml/min/kg, p<0.001). E2 only had a significant effect on V.O2 in the WT females during the nighttime hours (WT-oil: 4234 ± 168 ml/min/kg vs. WT-E2: 4657 ± 143 ml/min/kg, p<0.05). These data suggest that the attenuation of body weight gain in the WT females by E2 is due, in part, to a small but significant increase in nighttime V.O2.
Similar results were observed between the pre-treatment and post-treatment nighttime V.CO2 (Figure 6C). V.CO2 was significantly higher in the pre-treatment females compared to post-treatment females in WT-oil (p<0.001), WT-E2 (p<0.05), KIKO-oil (p<0.05), and KIKO-E2 (p<0.05) but not in the ERKO-oil or ERKO-E2 females (Two-way ANOVA: day-night: p<0.0001, df= 1, F= 30.85). V.CO2 in WT females prior to treatment was also significantly higher than in the ERKO females (p<0.05). Pre-treatment KIKO females were not significantly different from either WT or ERKO females. After 4 weeks of treatment, V.CO2 was significantly lower during the daytime compared to the nighttime for all genotype-steroid treatments (Two-way ANOVA: day-night: p<0.0001, df= 1, F= 77.71; Figure 6D). Although E2 had no significant effect on V.CO2 between genotypes either in the daytime or nighttime, the greatest difference between time periods was WT-E2 day and WT-E2 night V.O2 (day: 3501 ± 118 ml/min/kg vs. night: 4496 ± 151 ml/min/kg, p<0.001).
There was no effect of E2 on the respiratory exchange ratio (V.CO2/V.O2) or RER within the genotypes nor was there a significant effect of genotype on RER either during the daytime or nighttime (Figure 7A). However, there was a significant decrease in RER during the daytime compared to the nighttime between all genotype-treatments (Two-way ANOVA: day-night: p<0.0001, df= 1, F= 79.23) with greatest difference between ERKO-E2 day and night (day: 0.87 0.01 vs. night: 0.99 0.02, p<0.0001). Because RER is an estimation of the respiratory quotient, which indicates the type of fuel (fat or carbohydrates) being metabolized [38], we can infer that during the day the females are utilizing a mixture of fat and carbohydrates with more carbohydrates being utilized during the night, the primary feeding hours. We also measured body heat (CV x V.O2 or 3.815 + 1.232 x RER) using the CLAMS units (Figure 7B). As with RER, there was no significant effect of E2 on heat within genotypes during the daytime or nighttime but there was a significant decrease in heat between the daytime and nighttime between all genotype-treatments (Two-way ANOVA: day-night: p<0.0001, df= 1, F= 31.89).
Figure 7. Respiratory Exchange Ratio (RER), heat production and activity.

A. Average daytime and nighttime RER for each group post-treatment. Data was analyzed by a two-way ANOVA (day-night: p<0.0001, df=1; F=79.23; genotype X steroid: ns) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, compared to nighttime group). B. Average daytime and nighttime heat production for each group post-treatment. Data was analyzed by a two-way ANOVA (day-night: p<0.0001, df=1; F=31.89; genotype X steroid: p<0.0001, df=5; F=11.97) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05, ** p<0.01, compared to nighttime group). C. Average nighttime X-plane activity (counts) for each group before and after treatment. Unhatched bar = pre-treatment and hatched bars = post-treatment. Data was analyzed by a two-way ANOVA (genotype X steroid: p<0.0001, df=5, F=5.84; pre-post: ns) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05). D. Average daytime and nighttime X-plane activity for each group post-treatment. Data was analyzed by a two-way ANOVA (day-night: p<0.0001, df=1; F=140.2; genotype X steroid: p<0.0001, df=5, F=6.69) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, compared to nighttime group).
Locomotor activity was also measured using the CLAMS units. Dissimilar to V.O2 and V.CO2, there was no significant change between the nighttime pre-treatment and post-treatment total X-plane activity, although there was a negative trend in the WT-oil females compared to the other treatments (Figure 7C). The decrease in nighttime WT-oil activity between pretreatment and post-treatment was not observed in the WT-E2 females. Indeed, the difference in the X-plane beam breaks between the post-treatment WT-oil (347 ± 62 counts) and WT-E2 (535 ± 68 counts) females was the only significant effect of E2 on activity (p<0.05) (Figure 7C & 7D). There was also a significant decrease in locomotor activity between the daytime and nighttime between all genotype-treatments (Two-way ANOVA: day-night: p<0.0001, df= 1, F= 140; Figure 7D) with the greatest difference between the WT-E2 day activity (167 ± 19 counts) and the WT-E2 night activity (534 ± 68 counts), (p<0.0001). Similar results were observed for Z-plane activity (data not shown). These data indicate that the attenuation of body weight gain in the WT females by E2 is due, in part, to a small but significant increase in nighttime activity, which correlates to the increase in V.O2 (Figure 6B).
Neuropeptide genes regulated by E2 and across genotypes
To determine if there are any genotype or steroid effects on the expression of arcuate neuropeptide genes known to be involved in energy homeostasis, arcuate nuclei were dissected and processed for quantitative real-time PCR from all intact and ovx females (n are smaller than in the physiological experiments due to a loss of tissue samples during RNA extraction). We analyzed two anorexigenic genes, proopiomelanocortin (POMC) and cocaine-and-amphetamine-regulated transcript (CART), and two orexigenic genes, neuropeptide Y (NPY) and agouti-related peptide (AgRP). In the arcuate, POMC and NPY are co-expressed in the same neurons and NPY and AgRP are co-expressed.
In intact females, we observed no significant difference in gene expression between the genotypes (WT, ERKO, KIKO) for CART, NPY and AgRP (Figure 8A). For POMC, ERKO POMC expression was significantly lower than both WT and KIKO females (WT: 1.04 ± 0.14 (n=6); KIKO: 0.96 ± 0.11 (n=6); ERKO: 0.52 ± 0.17 (n=5); p<0.05 for both comparisons; genes were normalized to WT females for all genes). In the ovx females, POMC gene expression was significantly different within genotype between the oil-treated and E2-treated females in the WT (WT-oil: 1.09 ± 0.09 (n=8) vs. 1.52 ± 0.08 (n=8), p<0.01) (Figure 8B). In comparing gene expression across the genotypes when normalized to the WT-oil females, there was a significant effect of genotype-treatment (One-way ANOVA: p<0.0001, df=5, F=14.7). Indeed, the KIKO-E2, ERKO-oil and ERKO-E2 were all significantly lower than the WT-oil (KIKO-E2: p<0.05; ERKO-oil: p<0.05; ERKO-E2: p<0.001). KIKO-oil females were almost significantly less than the WT-oil (p=0.051).
Figure 8. Genotype and steroid treatment affect relative expression of Arcuate neuropeptide genes.

A. Average relative mRNA expression for POMC, CART, NPY and AgRP neuropeptides in the intact females. Data was analyzed by a one-way ANOVA (POMC: p<0.05, df=2; F=3.85) with Bonferroni-Dunn multiple comparison tests across and within genotypes (* p<0.05). B–E. Average relative mRNA expression in oil-treated and E2-treated, ovariectomized females for each neuropeptide. Data was normalized to WT-oil samples and analyzed by a one-way ANOVA (POMC: p<0.0001, df=5, F=14.7; CART: p<0.05, df=5, F=3.22; NPY: p<0.001, df=5, F=5.8; AgRP: p<0.05, df=5, F=3.44) with Bonferroni-Dunn multiple comparison tests across and within genotypes (a = p<0.05, b = p<0.01, c = p<0.001, d = p<0.0001).
Amongst the other three arcuate genes, CART expression in the WT females was the only pair to have significant change due to E2 treatment (Figure 8C). CART expression in WT-E2 (0.55 ± 0.1) was significantly lower than in WT-oil (1.08 ± 0.15; p<0.05). Across genotypes, there was a significant effect of genotype (One-way ANOVA: p<0.05, df=5, F=3.2) and both WT-oil and KIKO-oil (1.05 ± 0.19) was significantly higher than ERKO-E2 (0.43 ± 0.16; p<0.05). There was no effect of E2 treatment within a genotype for either of the two orexigenic genes, NPY and AgRP (Figure 8D & 8E). However, in both genes there was a significant effect of genotype (One-way ANOVA: NPY - p<0.001, df=5, F=5.8; AgRP - p<0.05, df=5, F=3.4). In NPY, ERKO gene expression was higher than in the WT and KIKO females (WT vs. ERKO-oil (p<0.05) and ERKO-E2 (p<0.001); KIKO vs. ERKO-oil (p<0.05) and ERKO-E2 (p<0.01)). In AgRP, ERKO-E2 was significantly higher than WT-oil (p<0.001). These data collectively suggest that there are genotype differences in the gene expression of these neuropeptides that may be involved in the changes we measured in energy homeostasis.
Discussion
Previously, it has been shown that ovariectomy in wild-type female mice caused significant body weight gain, which is abrogated in ovx ERα KO (ERKO) female mice [13]. Furthermore, intact, adult ERKO mice developed obesity and the knockin of ERα ERE-independent signaling in ERKO females (ERα KIKO) restored normal energy homeostasis in intact females [13, 26]. We demonstrated here that ERE-independent signaling is not sufficient to either fully restore the positive weight gain observed in ovx WT females or fully reestablish the effects of E2 in attenuating post-ovariectomy body weight gain. In every measure of energy homeostasis conducted, the effects of E2 were only observed in WT females, suggesting that ERE-dependent signaling was necessary for effects of E2 replacement on feeding, oxygen consumption, activity, body fat accumulation and gene expression of arcuate neuropeptides in ovx females.
Vehicle-treated, ovx WT females were shown to gain significantly more weight than their E2-treated counterparts on regular and high-fat diets, respectively [13,39–41]. E2 replacement was associated with a suppression of food intake [13,40] and an increase in oxygen consumption [41]. Our data supported these findings in WT females. E2 replacement in ovx WT females increased oxygen consumption and activity and suppressed food intake compared to oil-treated WT females. Together, these effects of E2 on energy homeostasis attenuated the post-ovariectomy body weight gain in WT females.
Unexpectedly, none of these effects were observed in the E2-treated KIKO females although there was an observable, but non-significant, attenuation of the body weight gain. As in the WT females, ovariectomy did significantly suppress oxygen consumption in KIKO females. However, E2 did not increase oxygen consumption in the KIKO females suggesting that ERE-independent signaling was not sufficient to restore E2’s augmentation of oxygen consumption. While food intake did slightly increase due to ovariectomy in KIKO females, this increase was significantly less than in the WT females regardless of treatment. The smaller increase in the post-ovariectomy food intake in KIKO females is the most plausible explanation for the lower amount of weight gain in the oil-treated, ovx KIKO females. Therefore, the increase in body weight due to ovariectomy in KIKO females is primarily through a decrease in metabolism, independent of activity, and through a small but significant increase in food intake.
Modulation of arcuate POMC and NPY neurons is known to have a role in E2’s suppression of food intake [42,43,16]. For example, deletion of ERα in arcuate POMC neurons produced a hyperphagic response in intact female mice leading to an increase in body weight [16]. In our study, an E2-induced increase in POMC gene expression was only observed in WT females, indicating that the suppressive effects E2 on food intake in this study was mediated, in part, by anorexigenic POMC neurons. Gene expression of the anorectic (POMC/CART) and orexigenic (NPY/AgRP) arcuate neuropeptides was significantly affected by genotype. ERKO females expressed more orexigenic peptides and less anorexigenic peptides than the WT counterparts. However, the differences in neuropeptide gene expression did not correlate with lower amount of food intake in ERKO females. This apparent discrepancy between gene expression and feeding behavior suggests a dysregulation of the intrinsic control of feeding behavior by arcuate neurons in ERKO females.
E2’s effects on metabolism are mediated by its action on neurons of the ventromedial nucleus of the hypothalamus (VMH). Deletion of ERα in the SF-1 neurons of the VMH [16] or silencing of ERα by RNAi administered in the VMH [4] produced a phenotype of decreased energy expenditure. Likewise, total knockout of ERα caused a decrease in energy expenditure compared to WT females, which is one of the primary causes of obesity in ERKO mice [12,26]. In the current study, neither ovariectomy nor E2 replacement had an effect on energy expenditure in the ERKO females. Furthermore, intact WT and KIKO females had similar rates of oxygen consumption [26], as did WT and KIKO females within one week of ovariectomy. Long-term (12 weeks) ovariectomy in WT females caused a significant decrease in oxygen consumption leading to weight gain [44]. Similar to WT females, long-term (5 week) ovariectomy significantly decreased energy expenditure in KIKO females. However, E2 attenuated post-ovariectomy body weight gain only in WT females, which was partially due to an increase in nighttime oxygen consumption. These data suggest that ERE-independent ERα signaling, possibly in the VMH, controls energy expenditure in intact females. This type of ERα signaling also has a role in the post-ovariectomy decrease in energy expenditure, but was not sufficient for E2’s augmentation of energy expenditure.
During the experiment, we also observed a difference in the amount of food chewed, but not fully consumed, between genotypes. In wild-type females, there was a significant decrease in the amount of food chewed after ovariectomy that was not seen in the other genotypes. E2 replacement in the WT females increased the amount of food chewed to similar levels observed in the intact. This behavior correlated with the higher levels of activity observed in the intact (data not shown) and E2-treated WT females. Another potential causes is compulsive or anxiety-associated feeding behaviors or “pica”, [36] some of which may be augmented by E2 in mice [45], or increased anxiety due to the stress of single housing during the experiment [46].
The partial restoration of the post-ovariectomy body weight gain in KIKO females compared to WT females suggests a role for ERE-independent signaling. This role in energy homeostasis may function through the organizational effects of ERα signaling occurring during neurodevelopment. Thus, a potential cause behind the lack of body weight gain in ERKO females is the loss of ERα signaling during neurogenesis in the hypothalamus and other regions of the brain involved in homeostatic functions [47–49]. While no such direct role of organizational ERα signaling in energy homeostasis has been demonstrated, the partial restoration of the response in body weight to ovariectomy in KIKO females indicates that a portion of these mechanisms are ERE-independent. This hypothesis is supported by the normal weight phenotype of the intact KIKO females [26]. Therefore, we can hypothesize that ERα-mediated developmental programming and control of neural circuitry is necessary for the post-ovariectomy body weight gain. The development of a conditional ERα KO mice strain would be key in addressing this hypothesis.
Furthermore, it is unknown if the response to ovariectomy (body weight gain) is primarily mediated by central or peripheral organizational and activational mechanisms. A recently produced brain-specific ERKO strain exhibit higher body weight compared to wild-type littermates in both females and males [16]. These mice exhibit higher food intake and visceral fat accumulation, lower heat production and decreased locomotor activity indicating the central effects of E2 and ERα signaling are necessary for normal energy homeostasis. However, the response to ovariectomy has not been characterized in these mice. If there is a neurodevelopmental effect of ER signaling involved in the post-ovariectomy body weight gain, these mice should respond similar to the total ERKO females used in this study and others [13].
Peripheral expression of estrogen receptors includes organs and tissues involved in energy balance, metabolism and glucose homeostasis [50]. Thus, the actions of ERα are involved in a range of metabolic processes in intact females, which are altered by ovariectomy [51,52]. It is largely unknown if these peripheral effects of E2 and ERα are mediated by ERE-independent signaling. In liver-specific ERα knockout females, E2’s effects on insulin sensitivity and liver lipid deposition are impaired, without affecting E2’s suppression of adiposity on a high-fat diet [53]. In intact KIKO females, glucose homeostasis and the response to high-fat diet are normalized compared to WT females [26], thus indicating that ERα ERE-independent signaling is involved in peripheral metabolic processes. Indeed, membrane-initiated ERα signaling is implicated in the regulation of gene expression and lipid synthesis in the liver through an AMPK-mediated pathway [54]. Therefore, future experiments will examine the effects of high fat diets, liver function, glucose homeostasis and ovariectomy with or without E2 replacement in KIKO females [44,51,52].
One potential qualification to the current study is the genotype of the ERKO backbone to the ERα KIKO mice. The ERKO and KIKO females used in this study were genotypically different than those used in either Geary et al. (2001) or Park et al. (2010) studies, respectively. In the current study, Dr. Ken Korach recently produced the ERKO backbone by global deletion of exon 3 of the ERα gene [30] while the KIKO mice characterized in Park et al. (2010) were produced with the ERKO mouse produced by Dr. Pierre Chambon and colleagues [55]. Furthermore, the ERKO mice originally described as not gaining significant weight post-ovariectomy in Geary et al. (2001) were a different ERKO strain produced by Dr. Ken Korach and colleagues [56]. Potential differences between these three strains of ERα knockouts may account for the differences reported in the results between earlier studies [12,13], the Park et al., (2010) study and this current study.
In conclusion, ERα is the primary receptor in E2’s effects on energy homeostasis, although other ERs (Gq-mER, ERβ) have been recently implicated in mediating E2’s control of energy homeostasis either by suppressing feeding in ovx rodents [18–20] or by attenuating adiposity and maintaining normal glucose homeostasis [57,58]. The multiple receptor-mediated mechanisms initiated by ERα are not fully understood yet previous studies [26,41,52] and the current study suggest that these mechanisms involve both ERE-dependent and -independent signaling pathways. Future studies must focus on the contributions of these two types of steroid signaling and their respective roles in both organizational and activational effects of E2 on energy homeostasis. Such studies may provide multiple therapeutic targets for selective hormone replacement therapy with limited deleterious side effects.
Highlights.
We compared the effects of ovariectomy and E2 on wild type, ERα KO and KIKO females.
Ovariectomized WT females had greater weight gain than ERα KO and KIKO females.
E2 replacement only had significant effects on female WT energy balance.
ERE-independent signaling is not sufficient for E2 to control energy balance.
Acknowledgments
The authors wish to thank Dr. Judy Storch for the use of the CLAMS unit and the EchoMRI Body Composition Analyzer. The University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core is supported by the Eunice Kennedy Shriver NICHD/NIH (SCCPIR) Grant U54-HD28934. This research is supported by National Institutes of Health Grant R00DK083457 (T.A.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Santollo J, Torregrossa A-M, Eckel LA. Estradiol acts in the medial preoptic area, arcuate nucleus, and dorsal raphe nucleus to reduce food intake in ovariectomized rats. Horm Behav. 2011;60:86–93. doi: 10.1016/j.yhbeh.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asarian L, Geary N. Estradiol enhances cholecystokinin-dependent lipid-induced satiation and activates estrogen receptor-α-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology. 2007;148:5656–5666. doi: 10.1210/en.2007-0341. [DOI] [PubMed] [Google Scholar]
- 3.Thammacharoen S, Lutz TA, Geary N, Asarian L. Hindbrain administration of estradiol inhibits feeding and activates estrogen receptor-α-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology. 2008;149:1609–1617. doi: 10.1210/en.2007-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Musatov S, Chen W, Pfaff DW, Mobbs CV, Yang X-J, Clegg DJ, et al. Silencing of estrogen receptor α in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci USA. 2007;104:2501–2506. doi: 10.1073/pnas.0610787104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asarian L, Geary N. Cyclic estradiol treatment normalizes body weight and restores physiological patterns of spontaneous feeding and sexual receptivity in ovariectomized rats. Horm Behav. 2002;42:461–471. doi: 10.1006/hbeh.2002.1835. [DOI] [PubMed] [Google Scholar]
- 6.Abel TW, Rance NE. Proopiomelanocortin gene expression is decreased in the infundibular nucleus of postmenopausal women. Mol Brain Res. 1999;69:202–208. doi: 10.1016/s0169-328x(99)00111-4. [DOI] [PubMed] [Google Scholar]
- 7.Reid RL, Quigley ME, Yen SS. The disappearance of opioidergic regulation of gonadotropin secretion in postmenopausal women. J Clin Endocrinol Metab. 1983;57:1107–1110. doi: 10.1210/jcem-57-6-1107. [DOI] [PubMed] [Google Scholar]
- 8.Buffenstein R, Poppitt SD, McDevitt RM, Prentice AM. Food intake and the menstrual cycle: a retrospective analysis, with implications for appetite research. Physiol Behav. 1995;58:1067–1077. doi: 10.1016/0031-9384(95)02003-9. [DOI] [PubMed] [Google Scholar]
- 9.Dye L, Blundell JE. Menstrual cycle and appetite control: implications for weight regulation. Hum Reprod. 1997;12:1142–1151. doi: 10.1093/humrep/12.6.1142. [DOI] [PubMed] [Google Scholar]
- 10.Paolisso G, Rizzo MR, Mazziotti G, Rotondi M, Tagliamonte MR, Varricchio G, et al. Lack of association between changes in plasma leptin concentration and in food intake during the menstrual cycle. Eur J Clin Invest. 1999;29:490–495. doi: 10.1046/j.1365-2362.1999.00488.x. [DOI] [PubMed] [Google Scholar]
- 11.Roepke TA. Oestrogen modulates hypothalamic control of energy homeostasis through multiple mechanisms. J Neuroendocrinol. 2009;21:141–150. doi: 10.1111/j.1365-2826.2008.01814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci USA. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S. Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-alpha null mice. Endocrinology. 2001;142:4751–4757. doi: 10.1210/endo.142.11.8504. [DOI] [PubMed] [Google Scholar]
- 14.Roepke TA, Malyala A, Bosch MA, Kelly MJ, Rønnekleiv OK. Estrogen regulation of genes important for K+ channel signaling in the arcuate nucleus. Endocrinology. 2007;148:4937–4951. doi: 10.1210/en.2007-0605. [DOI] [PubMed] [Google Scholar]
- 15.Roepke TA, Qui J, Smith AW, Ronnekleiv OK, Kelly MJ. Fasting and 17β-estradiol differentially modulate the M-current in neuropeptide Y neurons. J Neurosci. 2011;31:11825–11835. doi: 10.1523/JNEUROSCI.1395-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Y, Nedungadi TP, Zhu L, Sobhani N, Irani BG, Davis KE, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 2011;14:453–465. doi: 10.1016/j.cmet.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Rønnekleiv OK, Kelly MJ. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci. 2003;23:9529–9540. doi: 10.1523/JNEUROSCI.23-29-09529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu J, Bosch MA, Tobias SC, Krust A, Graham SM, Murphy SJ, et al. A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J Neurosci. 2006;26:5649–5655. doi: 10.1523/JNEUROSCI.0327-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roepke TA, Xue C, Bosch MA, Scanlan TS, Kelly MJ, Rønnekleiv OK. Genes associated with membrane-initiated signaling of estrogen and energy homeostasis. Endocrinology. 2008;149:6113–6124. doi: 10.1210/en.2008-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roepke TA, Bosch MA, Rick EA, Lee B, Wagner EJ, Seidlová-Wuttke D, et al. Contribution of a membrane estrogen receptor to the estrogenic regulation of body temperature and energy homeostasis. Endocrinology. 2010;151:4926–4937. doi: 10.1210/en.2010-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith AW, Bosch MA, Wagner EJ, Rønnekleiv OK, Kelly MJ. The membrane estrogen receptor ligand STX rapidly enhances GABAergic signaling in NPY/AgRP neurons: Role in mediating the anorexigenic effects of 17β-estradiol. Am J Physiol Endocrinol Metab. 2013;305:e632–e640. doi: 10.1152/ajpendo.00281.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammes SR, Levin ER. Extra-nuclear steroid receptors: nature and actions. Endocr Rev. 2007;28:726–741. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- 23.Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol. 2005;19:1951–1959. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vasudevan N, Pfaff DW. Membrane-initiated actions of estrogens in neuroendocrinology: emerging principles. Endocr Rev. 2007;28:1–19. doi: 10.1210/er.2005-0021. [DOI] [PubMed] [Google Scholar]
- 25.Rønnekleiv OK, Malyala A, Kelly MJ. Membrane-initiated signaling of estrogen in the brain. Semin Reprod Med. 2007;25:165–176. doi: 10.1055/s-2007-973429. [DOI] [PubMed] [Google Scholar]
- 26.Park CJ, Zhao Z, Glidewell-Kenney C, Lazic M, Chambon P, Krust A, et al. Genetic rescue of nonclassical ERα signalling normalizes energy balance in obese ERα-null mutant mice. J Clin Invest. 2011;121:604–612. doi: 10.1172/JCI41702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakacka M, Ito M, Martinson F, Ishikawa T, Lee EJ, Jameson JL. An estrogen receptor (ER)α Deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathyway signaling in vivo. Mol Endocrinol. 2002;16:2188–2201. doi: 10.1210/me.2001-0174. [DOI] [PubMed] [Google Scholar]
- 28.Hewitt SC, Korach KS. Estrogenic Activity of bisphenol A and 2,2-bis(p-Hydroxyphenyl)-1,1,1-trichloroethane (HPTE) demonstrated in mouse uterine gene profiles. Environ Health Perspect. 2011;119:63–70. doi: 10.1289/ehp.1002347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hewitt SC, O’Brien JE, Jameson JL, Kissling GE, Korach KS. Selective disruption of ERα DNA-Binding activity alters uterine responsiveness to estradiol. Mol Endocrinol. 2009;23:2111–2116. doi: 10.1210/me.2009-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hewitt SJ, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS. Biological and biochemical consequences of global deletion of exon 3 from the ERα gene. FASEB J. 2010;24:4660–4667. doi: 10.1096/fj.10-163428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haisenleder DJ, Schoenfelder AH, Marcinko ES, Geddis LM, Mashall JC. Estimation of estradiol in mouse serum samples: evaluation of commercial estradiol immunoassays. Endocrinology. 2011;152:1–5. doi: 10.1210/en.2011-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paxinos G, Franklin SO. The coronal plates and diagrams. 3. Amsterdam: Elsevier Academic Press; 2008. The Mouse Brain in Stereotaxic Coordinates, Compact. [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurita T, Lee K-J, Saunders PTK, Cooke PS, Taylor JA, Lubahn DB, et al. Regulation of progesterone recepors and decidualization in uterine stroma of the estrogen receptor-α knockout mouse. Biol Repro. 2000;64:272–283. doi: 10.1095/biolreprod64.1.272. [DOI] [PubMed] [Google Scholar]
- 36.Chou-Green JM, Holscher TD, Dallman MF, Akana SF. Compulsive behavior in the 5-HT2c receptor knockout mouse. Physiol Behav. 2003;78:641–649. doi: 10.1016/s0031-9384(03)00047-7. [DOI] [PubMed] [Google Scholar]
- 37.Yoo SB, Ryu V, Park EY, Kim B-T, Kang D-W, Lee J-H, Jahng JW. The arcuate NPY, POMC, and CART expressions responding to food deprivation and exaggerated in young female rats that eperienced neonatal maternal speration. Neuropeptides. 2011;45:343–349. doi: 10.1016/j.npep.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Jamieson PM, Cleasby ME, Kuperman Y, Morton NM, Kelly PAT, Brownstein DG, et al. Urocortin 3 transgenic mice exhibit a metabolically favourable phenotype resisting obesity and hyperglycaemia on a high-fat diet. Diabetologia. 2011;54:2392–2403. doi: 10.1007/s00125-011-2205-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stubbins RE, Holcomb VB, Hong J, Núñez NP. Estrogen modulates abdominal adiposity and protects female mice from obesity and impaired glucose tolerance. Euro J Nutr. 2011;51:861–870. doi: 10.1007/s00394-011-0266-4. [DOI] [PubMed] [Google Scholar]
- 40.Zhu L, Yang Y, Xu P, Zou F, Yan X, Liao L, et al. Steroid receptor coactivator-1 mediates estrogenic actions to prevent body weight gain in female mice. Endocrinology. 2013;154:150–158. doi: 10.1210/en.2012-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Handgraaf S, Riant E, Fabre A, Waget A, Burcelin R, Lière P, et al. Prevention of obesity and insulin resistance by estrogens requires ERα activation function-2 (ERαAF-2), whereas ERαAF-1 is dispensable. Diabetes. 2013 doi: 10.2337/db13-0282. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao Q, Horvath TL. Neurobiology of feeding and energy expenditure. Ann Rev Neurosci. 2007;30:367–398. doi: 10.1146/annurev.neuro.30.051606.094324. [DOI] [PubMed] [Google Scholar]
- 43.Hirosawa M, Minata M, Harada KH, Hitomi T, Krust A, Koizumi A. Ablation of estrogen receptor alpha (ERα) prevents upregulation of POMC by leptin and insulin. Biochem Biophys Res Comm. 2008;371:320–323. doi: 10.1016/j.bbrc.2008.04.073. [DOI] [PubMed] [Google Scholar]
- 44.Rogers NH, Perfield JW, Strissel KJ, Obin MS, Greenberg AS. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinlogy. 2009;150:2161–2168. doi: 10.1210/en.2008-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kastenberger I, Lutsch C, Schwarzer C. Activation of the g-protein-coupled receptor GPR30 induces anxiogenic efects in mice, similar to oestradiol. Psychopharmacology. 2012;221:527–535. doi: 10.1007/s00213-011-2599-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nonogaki K, Nozue K, Oka Y. Social Isolation affects the development of obesity and type 2 diabetes in mice. Endocrinology. 2007;148:4658–4666. doi: 10.1210/en.2007-0296. [DOI] [PubMed] [Google Scholar]
- 47.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 48.Küppers E, Krust A, Chambon P, Beyer C. Functional alterations of the nigrostriatal dopamine system in estrogen receptor-α knockout (ERKO) mice. Psychoneuroendocrinology. 2008;33:832–838. doi: 10.1016/j.psyneuen.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Semaan SJ, Kauffman AS. Sexual differentiation and development of forebrain reproductive circuits. Neurobiology. 2010;20:424–431. doi: 10.1016/j.conb.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barros RPA, Gustafsson J-Å. Estrogen receptor and the metabolic network. Cell Metab. 2011:289–299. doi: 10.1016/j.cmet.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 51.Riant E, Waget A, Cogo H, Arnal J-F, Burcelin R, Gourdy P. Estrogens protect agains high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. 2009;150:2109–2117. doi: 10.1210/en.2008-0971. [DOI] [PubMed] [Google Scholar]
- 52.Camporez JP, Jornavaz FR, Lee HY, Kanda S, Guigni BA, Kahn M, et al. Cellular mechanism by which estradiol protects female ovarietomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology. 2013;154:1021–1028. doi: 10.1210/en.2012-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu L, Brown WC, Cai Q, Krust A, Chambon P, McGuinness OW, Stafford JM. Estrogen treatment after ovariectomy protects against fatty liver and may improve pathway-selective insulin resistance. Diabetes. 2013;62:424–434. doi: 10.2337/db11-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pedram A, Razandi M, O’Mahony FO, Harvey H, Harvey BJ, Levin ER. Estrogen reduces lipid content in the liver eclusively rom membrane receptor signaling. Physiology. 2013;6:1–12. doi: 10.1126/scisignal.2004013. [DOI] [PubMed] [Google Scholar]
- 55.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development. 2000;127:4277–4291. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]
- 56.Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA. 1993;90:11162–11166. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yepuru M, Eswaraka J, Kearbey JD, Barrett CM, Raghow S, Veverka KA, et al. Estrogen receptor-β-selective ligands alleviate high-fat diet- and overiectomy-induced obesity in mice. J Biol Chem. 2010;285:31292–31303. doi: 10.1074/jbc.M110.147850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alonso-Magdalena P, Ropero AB, Garcia-Arévalo M, Soiano S, Quesada I, Muhammed SJ, et al. Antidiabetic actions of an estrogen receptor β selective agonist. Diabetes. 2013;62:2015–2025. doi: 10.2337/db12-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]