

Abstract
Objectives. Glucokinase encoded by GCK is a key enzyme that facilitates phosphorylation of glucose to glucose-6-phosphate. Variants of GCK gene were shown to be associated with type 2 diabetes (T2D) and coronary heart disease (CHD). The goal of this study was to investigate the contribution of GCK gene-body methylation to the risk of CHD. Design and Methods. 36 patients (18 males and 18 females) and 36 age- and sex-matched controls were collected for the current methylation research. DNA methylation level of the CpG island (CGI) region on the GCK gene-body was measured through the sodium bisulfite DNA conversion and pyrosequencing technology. Results. Our results indicated that CHD cases have a much lower methylation level (49.77 ± 6.43%) compared with controls (54.47 ± 7.65%, P = 0.018). In addition, GCK gene-body methylation was found to be positively associated with aging in controls (r = 0.443, P = 0.010). Conclusions. Our study indicated that the hypomethylation of GCK gene-body was significantly associated with the risk of CHD. Aging correlates with an elevation of GCK methylation in healthy controls.
1. Introduction
Diabetes and cardiovascular disease (CVD) are two common diseases with close relationship [1]. At least 65% of diabetic patients died from heart diseases or stroke. Even when glucose levels are under control, diabetes would greatly increase twofold of the risk of heart disease and stroke [2]. As a major type of CVD, coronary heart disease (CHD) has drawn a lot of attention in its pathogenic mechanism. Evidence has shown that variation in CHD incidence and mortality among different populations may be explained by the interaction between environmental and genetic factors [3]. Environmental factors contributing to the development of CHD include diabetes, hypertension, plasma lipid, smoking, and obesity [4, 5].
Aberrant epigenetic modifications may bridge the environmental and genetic factors and thus lead to pathological consequences such as diabetes and CVD [6]. Familial aggregation of CHD, diabetes, and obesity can reflect epigenetic processes that can be explained by common environmental factors such as diet habits [3, 7]. By introducing or removing CpG sites, some single nucleotide polymorphisms (SNPs) may impact gene expression through the alteration of DNA methylation level and contribute to the risk of disease [8].
DNA methylation is an important modification of epigenetics and often occurs at CpG dinucleotides in mammals [9]. Hypermethylation in promoter regions has been known to repress gene expression [10, 11]. However, DNA methylation in gene-body shows a bell-shaped correlation with gene expression [12, 13]. Specifically, genes with middle-level expression have higher levels of gene-body methylation in plants and invertebrates [13]. Positive correlation between expression and gene-body methylation is also presented in a variety of human tissue types [12, 14]. Adrian Bird proposed that gene-body methylation represented “orphan promoters” that might be used at the early stages of development [15]. Methylation of gene-body may activate the transcription of canonical gene, although the functions of the gene-body CGI remain largely unknown [11].
As a candidate gene of type 2 diabetes (T2D) [16], GCK encoded glucokinase that is a key enzyme of glucose phosphorylation [17, 18]. Studies have shown that the occurrence of cardiovascular morbidity and mortality is positively associated with the plasma glucose levels [19, 20]. Glucokinase is able to facilitate hepatic glucose uptake during hyperglycemia and determines the threshold for glucose-stimulated insulin secretion [17, 21–23]. The specific molecular mechanisms underlying the progression to hyperglycemic states remain uncertain. A GCK promoter variant (−30G>A, rs1799884) was shown to increase the risk of both T2D [24, 25] and CHD [26, 27]. Hypermethylation of hepatic GCK promoter in aging rats contributes to diabetogenic potential [28]. Human GCK CpG island (CGI) is located in the gene-body but not near promoter. Here, we performed an association study of GCK gene-body methylation with the risk of CHD in a well matched case-control cohort.
2. Materials and Methods
2.1. Samples and Clinical Data
36 CHD patients (18 males and 18 females) and 36 age- and sex-matched controls were collected from Ningbo Lihuili Hospital. The details of the inclusion criteria were presented in our previous publication [29]. All the collected individuals were Han Chinese from Ningbo city in eastern China. All of the experiments were approved by the Ethical Committee of Ningbo Lihuili Hospital, and written informed consent was obtained from all the subjects.
2.2. Biochemical Analyses
Nucleic acid extraction analyzer (Lab-Aid 820, Xiamen, China) was used to extract genomic DNA from peripheral blood samples. The concentrations of extracted DNA were measured by the ultramicro nucleic acid ultraviolet tester (NANODROP 1000, Wilmington, USA). Plasma levels of biochemical factors (including TG, TC, HDL, LDL, ApoA 1, ApoB, ApoE, Lp(a), hs-CRP, ALB, GLB, ALT, AST, ALP and GGT) were measured using the methods described in our previous study [30]. DNA methylation was measured using the sodium bisulphite DNA conversion coupled with pyrosequencing [30]. Genomic DNA was chemically modified by sodium bisulfite (EpiTech Bisulfite Kits; Qiagen) to convert all unmethylated cytosines to uracils while the methylated cytosines unchanged. The bisulfite converted DNA and the polymerase chain reaction (PCR) primers which were designed by PyroMark Assay Design software were mixed and performed with PCR (Pyromark PCR Kit; Qiagen). The PCR products were degenerated and released to single strand products for pyrosequencing and the sequencing was conducted by Q24 machine and reagents (Pyromark Gold Q24 Reagents; Qiagen). The forward primer sequence was 5′-TGGATGGTTTAGTGTATAAGTTGTATT-3′. The reverse primer sequence was 5′-Biotin-CACCTCATCCTCCACATTCAT-3′, and the sequencing primer sequence was 5′-AAGTGGGGTTTAAAAAG-3′.
2.3. Statistical Analyses
Statistical analyses were performed using the SPSS package (version 16.0) to investigate the association of GCK methylation with CHD and various biochemical factors. Values of biochemical indicators deviated from normality were corrected via a logarithmic transformation. A more conservative nonparametric approach was used for data which were unable to be normalized. The correlations of DNA methylation with age and biochemical indicators were performed using SPSS package and R statistical software. All the P values were adjusted for the history of age, smoking, diabetes, and hypertension. Methylation levels were presented as means ± SD. The adjusted calculation was analyzed by SPSS package with binary logistic regression. A two-tailed P < 0.05 was considered to be significant.
3. Results
Two CGIs of GCK gene (hg19, chr7: 44183870-44229022) were located in the gene-body. One of the CGIs was too short (223 bp) to design the primers for bisulfite pyrosequencing. Therefore, the other CGI was used for the methylation assay. As shown in Figure 1, this CGI spans exon 9 and part of exon 10 (hg19, chr7: 44184771-44185695). DNA methylation percentages of four CpG sites were obtained using the bisulfite pyrosequencing assay on a 210 bp fragment of intron 9 and exon 9 (hg19, chr7: 44184929-44185138). Significant correlation of the DNA methylation levels was observed among the four CpGs (Figure 1, r = 0.6–0.8, P < 0.0001). The methylation levels of the four CpG sites on GCK gene-body were measured using the Pyromark Q24 instrument (Figure 2). In our study, the raw data of methylation levels of the four CpG sites on GCK gene-body were presented in Supplemental Table 1 (See Supplementary Material available at http://dx.doi.org/10.1155/2014/151723). Mean methylation level and each of the four CpG sites were used to compare the differences between cases and controls, between males and females, and the relationship between GCK methylation and biochemical indicators.
Figure 1.
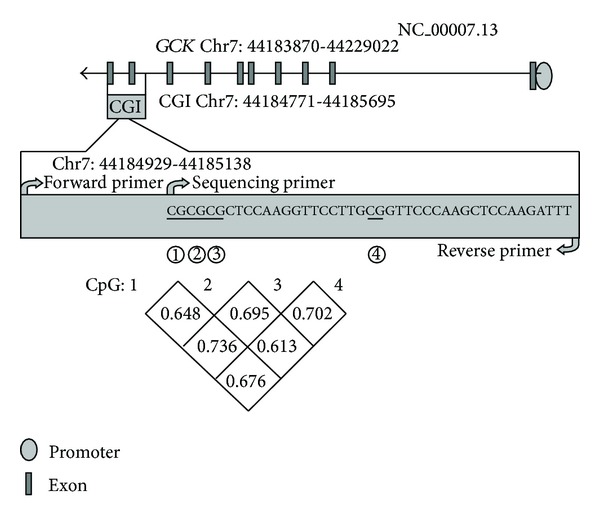
The four tested CpG sites in GCK gene.
Figure 2.
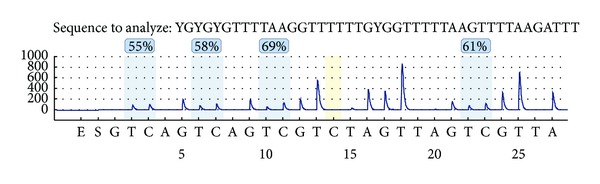
The methylation levels of four CpG sites on GCK gene-body of one sample.
As shown in Table 1 and Supplemental Figure 1, significant difference was observed between CHD cases and healthy controls. CHD cases have a significantly lower methylation level (49.77 ± 6.43%) compared with controls (54.47 ± 7.65%, P = 0.018). The similar trend was observed in three CpGs (Table 1 and Supplemental Figure 2: CpG2: 43.42 ± 7.93% versus 49.42 ± 8.26%, P = 0.005; CpG3: 56.81 ± 8.74% versus 62.06 ± 9.58%, P = 0.036; CpG4: 48.86 ± 7.44% versus 53.67 ± 7.11%, P = 0.031). There was no difference of GCK methylation level between males and females and no significant interaction between gender and disease (Table 1, P > 0.05). A significant difference of the CpG2 methylation level with CHD was observed in males (Table 2, P = 0.030). Mean GCK methylation was associated with aging in controls (r = 0.443, P = 0.010; Figure 3). No gender-specific difference was found for the association between GCK methylation and age (data not shown).
Table 1.
Comparison of GCK gene DNA methylation levels within subgroups and gender separately.
| DNA methylation (%) | Case | Control | Subgroup P | Male | Female | Gender P | Gender ∗ subgroup Interaction P |
|---|---|---|---|---|---|---|---|
| CpG1 | 50.00 ± 8.34 | 52.72 ± 8.25 | 0.232 | 52.11 ± 8.01 | 50.61 ± 8.73 | 0.955 | 0.556 |
| CpG2 | 43.42 ± 7.93 | 49.42 ± 8.26 | 0.005* | 46.14 ± 9.16 | 46.69 ± 8.10 | 0.932 | 0.668 |
| CpG3 | 56.81 ± 8.74 | 62.06 ± 9.58 | 0.036* | 59.92 ± 9.04 | 58.94 ± 10.00 | 0.887 | 0.909 |
| CpG4 | 48.86 ± 7.44 | 53.67 ± 7.11 | 0.031* | 50.83 ± 7.48 | 51.69 ± 7.83 | 0.258 | 0.667 |
| Mean | 49.77 ± 6.43 | 54.47 ± 7.65 | 0.018* | 52.25 ± 7.23 | 51.99 ± 7.67 | 0.842 | 0.658 |
The P values were adjusted by age, history of smoking, diabetes, and hypertension.
*P < 0.05.
Table 2.
Comparison of GCK gene DNA methylation levels between cases and controls in male and female separately.
| DNA methylation (%) | Male | Female | ||||
|---|---|---|---|---|---|---|
| Case | Control | P | Case | Control | P | |
| CpG1 | 50.17 ± 8.50 | 54.06 ± 7.19 | 0.321 | 49.83 ± 8.42 | 51.39 ± 9.20 | 0.674 |
| CpG2 | 42.72 ± 9.52 | 49.56 ± 7.57 | 0.030* | 44.11 ± 6.15 | 49.28 ± 9.12 | 0.067 |
| CpG3 | 57.17 ± 8.99 | 62.67 ± 8.46 | 0.155 | 56.44 ± 8.73 | 61.44 ± 10.79 | 0.138 |
| CpG4 | 48.06 ± 7.01 | 53.61 ± 7.06 | 0.085 | 49.67 ± 7.96 | 53.72 ± 7.36 | 0.228 |
| Mean | 49.53 ± 6.58 | 54.97 ± 6.98 | 0.063 | 50.01 ± 6.45 | 53.96 ± 8.44 | 0.168 |
The P values were adjusted by age, history of smoking, diabetes, and hypertension.
*P < 0.05.
Figure 3.
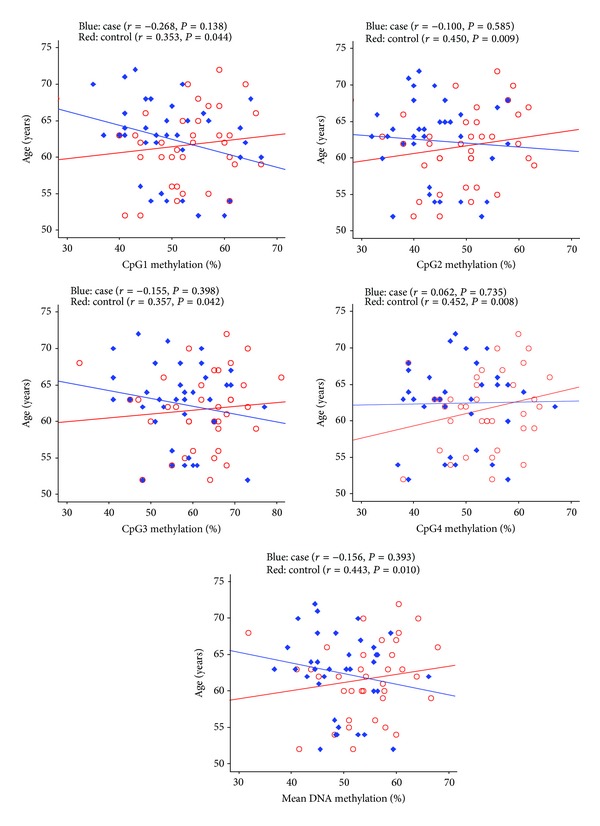
Correlation between GCK gene DNA methylation and age. The P values were adjusted by age, history of smoking, diabetes, and hypertension.
We explored the correlation of these biochemical indicators and DNA methylation. As shown in Supplemental Table 2, significant associations were observed for triglyceride (TG, CpG2: P = 0.044; CpG3: P = 0.019), low-density lipoprotein (LDL, CpG3: P = 0.029), apolipoprotein B (ApoB, CpG2: P = 0.048), alanine amiotransferase (ALT, CpG2: P = 0.044), and γ-glutamyl transpeptidase (GGT, CpG1: P = 0.020; CpG2: P = 0.039, CpG3: P = 0.006, CpG2: P = 0.033).
4. Discussion
Type 2 diabetes (T2D) patients often have high risk of developing cardiovascular diseases including CHD [31]. We hypothesize that GCK gene as a candidate diabetic gene may also contribute to the risk of CHD. Our results revealed that CHD cases have a hypomethylation level in GCK gene-body compared with controls. In addition, average GCK methylation level elevated along with increasing age in controls.
The methylation levels of both promoter and gene-body are highly dynamic [32]. DNA methylation causes gene silencing through blocking the binding of transcription factors or methyl-binding proteins [33]. Genome-wide study of DNA methylation has identified that aberrant methylation of gene bodies was able to contribute to the risk of heart failure [34]. DNA methylation marks on gene bodies were shown to regulate gene expression [11]. Positive correlation between gene-body methylation and gene expression was presented in a variety of human tissue types [12, 14]. Gene-body methylation was shown to play an important role in regulating cell context-specific alternative promoters in human and mouse tissues [35].
In the present study, we found that there was no specific CGI in the human GCK promoter. Evidence has shown that transcribed regions tend to have higher levels of methylation than intergenic or promoter regions [13, 36]. Hypermethylation of gene-body methylation often indicates a higher level of gene expression in human tissue and cell types [13, 35]. It can slow down the transcription elongation and suppress the initiation of abnormal transcription [37]. Gene-body methylation represented “orphan promoters” that might be used at the early stages of development [15]. DNA methylation can cause chromatin structure changes of the corresponding region in the genome and change the restriction endonuclease sites [38]. Thereby we speculate that the decrease of GCK methylation in CHD cases may influence the transcription and gene expression.
The rat study has shown that GCK gene methylation is important in the early development [39]. Reduced GCK expression was shown in aged rats through the elevation of GCK gene promoter methylation [28, 40]. Rat GCK mRNA in hepatocytes increased 4-fold upon treatment with 5-Aza-CdR, a cytosine methylation inhibitor [28]. Downregulation of GCK expression was reversed by the 5-Aza-CdR in the cell model of steatosis [40].
Differences of age-dependent methylation have been observed in multiple independent studies [41–43]. Aging induces global and complex changes of DNA methylation in humans [42]. Global methylation levels become more variable between monozygotic twins during their lifetime [44]. Age-dependent gene hypermethylation was also observed in human skin tissue [45]. In the present study, we also found a significant association between GCK gene-body hypermethylation and aging in healthy individuals.
Our study confirmed that human GCK gene-body methylation level was positively correlated with ageing in the healthy controls. We speculated that GCK hypermethylation might play an important role to protect people from cardiovascular diseases, although the exact mechanisms need to be clarified in the future study.
There are some limitations of our results to be taken with caution. Firstly, we explored only a part of CGI. Our findings of these four CpGs may not represent the whole CGI of GCK gene. Secondly, the sample size of our study is small. Validations of our findings are needed in larger samples and in other ethnic populations. Last but not least, we cannot exclude a chance that variations in the administrated drugs and diets may affect our findings on GCK methylation, although our results have been employed with a strict adjustment by multiple factors including hypertension, smoking habit, and diabetes.
5. Conclusion
In conclusion, our results found that lower methylation level of GCK gene-body was associated with the risk of CHD in Chinese. Our results also confirmed that GCK methylation level had a positive correlation with aging in humans.
Supplementary Material
Supplemental Table 1: The raw data of methylation levels of the four CpG sites on GCK gene body.
Supplemental Table 2: Correlation between GCK gene DNA methylation and biochemical indicators in cases.
Supplemental Figure 1:Comparison of GCK gene mean DNA methylation levels within subgroups and gender separately.
Supplemental Figure 2: Four CpG sites DNA methlation levels in cases and controls.
Acknowledgments
The research was supported by the Grants from National Natural Science Foundation of China (31100919 and 81371469), Natural Science Foundation of Zhejiang Province (LR13H020003), K. C. Wong Magna Fund in Ningbo University, Natural Science Foundation of Ningbo City (2011A610037), Ningbo Social Development Research Projects (2012C50032), Graduate Student Research Innovation Fund in Ningbo University (G13037), Advanced key Scientific and Technological Programs of Ningbo (2012C5017), Zhejiang Provincial Natural Science Foundation (LY12H16002), Science and Technology Innovation Team of Ningbo (2011B82015), and Advanced Key Scientific and Technological Programs of Ningbo (2011C51001).
Abbreviations
- GCK:
Glucokinase
- CHD:
Coronary heart disease
- CVD:
Cardiovascular disease
- GWAS:
Genome-wide association studies
- T2D:
Type 2 diabetes
- CGI:
CpG island
- LDL:
Low density lipoprotein
- TG:
Triglyceride
- ApoB:
Apolipoprotein B
- ALT:
Alanine amiotransferase
- GGT:
γ-Glutamyl transpeptidase.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Limin Xu, Dawei Zheng, and Lingyan Wang are co-first authors of this work.
References
- 1.Currie CJ, Morgan CL, Peters JR. Patterns and costs of hospital care for coronary heart disease related and not related to diabetes. Heart. 1997;78(6):544–549. doi: 10.1136/hrt.78.6.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eriksson M, Carlberg B, Eliasson M. The disparity in long-term survival after a first stroke in patients with and without diabetes persists: the northern sweden monica study. Cerebrovascular Diseases. 2012;34:153–160. doi: 10.1159/000339763. [DOI] [PubMed] [Google Scholar]
- 3.Neufeld HN, Goldbourt U. Coronary heart disease: genetic aspects. Circulation. 1983;67(5):943–954. doi: 10.1161/01.cir.67.5.943. [DOI] [PubMed] [Google Scholar]
- 4.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hara M, Sakata Y, Sato H. [genetic factors in myocardial infarction] The Japanese Journal of Clinical Pathology. 2013;61:176–183. [PubMed] [Google Scholar]
- 6.Pan MH, Lai CS, Wu JC, Ho CT. Epigenetic and diseases targets by polyphenols. Current Pharmaceutical Design. 2013;19:6156–6185. doi: 10.2174/1381612811319340010. [DOI] [PubMed] [Google Scholar]
- 7.Drong AW, Lindgren CM, McCarthy MI. The genetic and epigenetic basis of type 2 diabetes and obesity. Clinical Pharmacology and Therapeutics. 2012;92:707–715. doi: 10.1038/clpt.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dayeh TA, Olsson AH, Volkov P, Almgren P, Ronn T, Ling C. Identification of cpg-snps associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia. 2013;56:1036–1046. doi: 10.1007/s00125-012-2815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. Journal of Molecular Biology. 1987;196(2):261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 10.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends in Biochemical Sciences. 2006;31(2):89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 12.Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing-related and gene body-specific methylation of active human genes. Human Molecular Genetics. 2011;20(4):670–680. doi: 10.1093/hmg/ddq513. [DOI] [PubMed] [Google Scholar]
- 13.Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–474. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nature Biotechnology. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Illingworth RS, Gruenewald-Schneider U, Webb S, et al. Orphan CpG Islands Identify numerous conserved promoters in the mammalian genome. PLoS Genetics. 2010;6(9) doi: 10.1371/journal.pgen.1001134.e1001134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iynedjian PB. Molecular physiology of mammalian glucokinase. Cellular and Molecular Life Sciences. 2009;66(1):27–42. doi: 10.1007/s00018-008-8322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heredia VV, Thomson J, Nettleton D, Sun S. Glucose-induced conformational changes in glucokinase mediate allosteric regulation: transient kinetic analysis. Biochemistry. 2006;45(24):7553–7562. doi: 10.1021/bi060253q. [DOI] [PubMed] [Google Scholar]
- 18.Kamata K, Mitsuya M, Nishimura T, Eiki J-I, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure. 2004;12(3):429–438. doi: 10.1016/j.str.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Coutinho M, Gerstein HC, Wang Y, Yusuf S. The relationship between glucose and incident cardiovascular events: A metaregression analysis of published data from 20 studies of 95,783 individuals followed for 12.4 years. Diabetes Care. 1999;22(2):233–240. doi: 10.2337/diacare.22.2.233. [DOI] [PubMed] [Google Scholar]
- 20.Rose CS, Ek J, Urhammer SA, et al. A -30G>A polymorphism of the β-cell-specific glucokinase promoter associates with hyperglycemia in the general population of whites. Diabetes. 2005;54(10):3026–3031. doi: 10.2337/diabetes.54.10.3026. [DOI] [PubMed] [Google Scholar]
- 21.Fu D, Cong X, Ma Y, et al. Genetic polymorphism of glucokinase on the risk of type 2 diabetes and impaired glucose regulation: evidence based on 298, 468 subjects. PloS ONE. 2013;8 doi: 10.1371/journal.pone.0055727.e55727 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Van Schaftingen E, Detheux M, da Cunha MV. Short-term control of glucokinase activity: role of a regulatory protein. The FASEB Journal. 1994;8(6):414–419. doi: 10.1096/fasebj.8.6.8168691. [DOI] [PubMed] [Google Scholar]
- 23.Matschinsky FM, Glaser B, Magnuson MA. Pancreatic β-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes. 1998;47(3):307–315. doi: 10.2337/diabetes.47.3.307. [DOI] [PubMed] [Google Scholar]
- 24.Cauchi S, Ezzidi I, El Achhab Y, et al. European genetic variants associated with type 2 diabetes in North African Arabs. Diabetes & Metabolism. 2012;38(4):316–323. doi: 10.1016/j.diabet.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Sotos-Prieto M, Guillen M, Vicente Sorli J, et al. Relevant associations of the glucokinase regulatory protein/glucokinase gene variation with tag concentrations in a high-cardiovascular risk population: modulation by the mediterranean diet. The British Journal of Nutrition. 2013;109:193–201. doi: 10.1017/S0007114512000918. [DOI] [PubMed] [Google Scholar]
- 26.Webster RJ, Warrington NM, Weedon MN, et al. The association of common genetic variants in the APOA5, LPL and GCK genes with longitudinal changes in metabolic and cardiovascular traits. Diabetologia. 2009;52(1):106–114. doi: 10.1007/s00125-008-1175-9. [DOI] [PubMed] [Google Scholar]
- 27.März W, Nauck M, Hoffmann MM, et al. G(-30)A polymorphism in the pancreatic promoter of the glucokinase gene associated with angiographic coronary artery disease and type 2 diabetes mellitus. Circulation. 2004;109(23):2844–2849. doi: 10.1161/01.CIR.0000129306.44085.C4. [DOI] [PubMed] [Google Scholar]
- 28.Jiang MH, Fei J, Lan MS, et al. Hypermethylation of hepatic Gck promoter in ageing rats contributes to diabetogenic potential. Diabetologia. 2008;51(8):1525–1533. doi: 10.1007/s00125-008-1034-8. [DOI] [PubMed] [Google Scholar]
- 29.Zhou J, Huang Y, Huang RS, et al. A case-control study provides evidence of association for a common snp rs974819 in pdgfd to coronary heart disease and suggests a sex-dependent effect. Thrombosis Research. 2012;130:602–606. doi: 10.1016/j.thromres.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 30.Jiang D, Zheng D, Wang L, et al. Elevated pla2g7 gene promoter methylation as a gender-specific marker of aging increases the risk of coronary heart disease in females. PloS ONE. 2013;8 doi: 10.1371/journal.pone.0059752.e59752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. The New England Journal of Medicine. 1998;339(4):229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature Reviews Genetics. 2008;9(6):465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 33.Papait R, Greco C, Kunderfranco P, Latronico MV, Condorelli G. Epigenetics: a new mechanism of regulation of heart failure? Basic Research in Cardiology. 2013;108:p. 361. doi: 10.1007/s00395-013-0361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Movassagh M, Choy M-K, Knowles DA, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124(22):2411–2422. doi: 10.1161/CIRCULATIONAHA.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Yazaki J, Sundaresan A, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126(6):1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nature Genetics. 2007;39(1):61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 38.Hoekenga OA, Muszynski MG, Cone KC. Developmental patterns of chromatin structure and DNA methylation responsible for epigenetic expression of a maize regulatory gene. Genetics. 2000;155(4):1889–1902. doi: 10.1093/genetics/155.4.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bogdarina I, Murphy HC, Burns SP, Clark AJL. Investigation of the role of epigenetic modification of the rat glucokinase gene in fetal programming. Life Sciences. 2004;74(11):1407–1415. doi: 10.1016/j.lfs.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 40.Jiang M, Zhang Y, Liu M, et al. Hypermethylation of hepatic glucokinase and L-type pyruvate kinase promoters in high-fat diet-induced obese rats. Endocrinology. 2011;152(4):1284–1289. doi: 10.1210/en.2010-1162. [DOI] [PubMed] [Google Scholar]
- 41.Raddatz G, Hagemann S, Aran D, et al. Aging is associated with highly defined epigenetic changes in the human epidermis. Epigenetics & Chromatin. 2013;6, article 36 doi: 10.1186/1756-8935-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christensen BC, Houseman EA, Marsit CJ, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CPG island context. PLoS Genetics. 2009;5(8) doi: 10.1371/journal.pgen.1000602.e1000602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winnefeld M, Lyko F. The aging epigenome: DNA methylation from the cradle to the grave. Genome Biology. 2012;13:p. 165. doi: 10.1186/gb-2012-13-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grönniger E, Weber B, Heil O, et al. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genetics. 2010;6(5) doi: 10.1371/journal.pgen.1000971.e1000971 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: The raw data of methylation levels of the four CpG sites on GCK gene body.
Supplemental Table 2: Correlation between GCK gene DNA methylation and biochemical indicators in cases.
Supplemental Figure 1:Comparison of GCK gene mean DNA methylation levels within subgroups and gender separately.
Supplemental Figure 2: Four CpG sites DNA methlation levels in cases and controls.