

Abstract
Our previous work showed that chronic activation of the membrane-bound estrogen receptor GPR30/GPER significantly lowers blood pressure in ovariectomized hypertensive mRen2.Lewis female rats which may, in part, reflect direct vasodilatory actions. The current study assessed the hypothesis that cyclic adenosine monophosphate (cAMP) signaling contributes to GPER-mediated vasorelaxation. In mesenteric resistance arteries from intact Lewis females, relaxation to 17-β-estradiol (E2; 47±3% of phenylephrine contraction vs. vehicle 89±2%, P<0.001) or G-1 (44±8%, P<0.001) was blunted to a similar extent by denuding (P<0.001) or the nitric oxide synthase inhibitor L-NAME (P<0.001). In contrast, the cyclooxygenase inhibitor indomethacin did not alter vasodilation (P>0.05). The cAMP analogue Rp-cAMPS partially attenuated vasodilation (65±7%, P<0.001), while the combination of L-NAME and Rp-cAMPS exhibited additive effects to effectively abolish vasorelaxation (P>0.05 vs. vehicle). Pretreatment of endothelium-intact vessels with the adenylyl cyclase inhibitor SQ (63±6%) or the guanylyl cyclase inhibitor ODQ (62±9%) both partially inhibited the response to G-1 (P<0.01), while pretreatment with the both inhibitors completely abolished vasorelaxation (P>0.05 vs. vehicle). In denuded vessels only SQ reduced the response (88±3%, P<0.001). Moreover, G-1 significantly increased intracellular cAMP levels in cultured mesenteric smooth muscle cells (P<0.05). We conclude that GPER-dependent vasorelaxation apparently involves both endothelial release of nitric oxide which activates guanylyl cyclase and smooth muscle cell activation of adenylyl cyclase. Downstream production of cyclic nucleotides and stimulation of protein kinases may phosphorylate proteins to promote vascular smooth muscle cell relaxation. The ability of GPER to initiate these signaling pathways may contribute to the beneficial vascular effects of estrogen.
Keywords: GPER, GPR30, estrogen, cAMP, vasodilation, G proteins
Introduction
In ovariectomized hypertensive mRen2.Lewis rats, chronic administration of the selective GPER agonist G-1 significantly reduces blood pressure [1]. While this antihypertensive response may be partially due to local alterations in the renin-angiotensin system, GPER activation may also have direct effects on vascular tone. Indeed, we previously showed that acute administration of the GPER agonist G-1 induces vasodilation of mesenteric resistance arteries that is not different from the nonselective agonist 17-β-estradiol in hypertensive mRen2.Lewis females and normotensive Lewis controls [2]. The vasorelaxation induced by G-1 and 17-β-estradiol is partially dependent on endothelial nitric oxide production but also comprises an endothelium-independent component. Moreover, we demonstrated GPER immunoreactive staining on both intimal and medial aspects of the mesenteric vasculature.
In the initial characterization of its expression and function in breast cancer cell lines, GPER was found to activate acute signaling pathways including cAMP accumulation [3]. Despite the numerous studies verifying GPER expression in vascular smooth muscle [1, 4–8] and demonstrating endothelium-independent relaxation to G-1 [2, 7, 9], no studies as of yet have identified the intracellular signaling pathways that mediate GPER’s vasodilatory effects in smooth muscle. Therefore, the current study assessed the hypothesis that stimulation of cyclic AMP is an integral pathway in the vasorelaxant actions of GPER in vascular smooth muscle cells.
Experimental Methods
Animals
Lewis female rats (Harlan, Indianapolis, IN) were used for both myography and cell isolation at 15 weeks of age. Adequate measures were taken to minimize pain or discomfort, and all experiments were approved by the Tulane University Animal Care and Use Committee.
Myography
Second-order mesenteric vessels (~200 μm I.D.) were isolated and sectioned into 2 mm rings. Vessels were mounted in a DMT wire myograph containing Krebs-Henseleit solution (in mM): NaCl (118), KCl (4.8), CaCl2 (2.5), MgSO4 (1.2), NaHCO3 (25), KH2PO4 (1.2) and glucose (11) at 37°C and oxygenated with 95% O2 - 5% CO2. Internal circumference was normalized to 0.9·IC100, where IC100 is the internal circumference at a transmural pressure of 100 mmHg. Viability and endothelial integrity were tested with phenylephrine (PE, 10−5 M; MP Biomedicals, Solon, OH) followed by acetylcholine (Ach, 10−6 M; Enzo, Farmingdale, NY) and rings with > 50% relaxation to Ach were considered endothelium intact. Rings were washed and again constricted with PE before cumulative additions of vehicle (DMSO), 17-β-estradiol (E2; EMD Millipore, Billerica, MA), or G-1 (EMD Millipore). The following inhibitors were added to the bath 10 minutes prior to PE preconstriction for the concentration response curve: NG-Nitro-L-arginine-methyl ester (L-NAME, 10−4 M; Enzo), indomethacin (10−5 M; Sigma, St. Louis, MO), Rp-Diastereomer of adenosine-3′,5′-cyclic monophosphothioate (Rp-cAMPS, 10−5 M; Santa Cruz Biotechnology, Santa Cruz, CA), SQ 22536 (SQ, 10−4 M; Enzo), and ODQ (10−4 M; Enzo). All data are expressed as percent of the PE preconstriction, and two-way ANOVA was used to compare treatment groups and agonist concentrations.
cAMP measurements
Smooth muscle cells were isolated from the mesenteric arterial branch as previously described [10]. Cells were passaged a maximum of four times, as previous studies show that GPER expression is only stable for this time in culture [7]. Cells were cultured in Medium 199 containing 10% fetal bovine serum and penicillin/streptomycin, then switched to phenol red-free medium containing 5% charcoal-stripped serum two days before experiments. Cells were pretreated with the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX, 10−9 M; Sigma) for 20 minutes before the addition of vehicle (DMSO) or G-1. In some experiments, cells were also pretreated with the GPER antagonist G15 (10−8 M; EMD Millipore). Cells were incubated at 37 °C for 15 minutes, and cAMP was measured by EIA kit (Cayman Chemical, Ann Arbor, MI). One-way ANOVA was used to determine differences between groups.
Results
In isolated mesenteric resistance arteries from 15 week-old Lewis females, vasorelaxation to 17-β-estradiol (E2) and the GPER agonist G-1 was dose-dependent and became significant at 10−6 M. The maximum dilation reached was 47 ± 3% for E2 and 44 ± 8% for G-1 (P<0.001 vs. vehicle, Figure 1A–B). The cyclooxygenase inhibitor indomethacin had no effect on vasodilation to either E2 or G-1 (P>0.05 vs. agonist). Denuding or pretreatment with the nitric oxide synthase inhibitor L-NAME (10−4 M) similarly attenuated vasodilation to E2 and G-1 (P<0.001) as previously reported in mRen2.Lewis female vessels [7].
Figure 1.


(A) Estradiol (E2) and (B) G-1 significantly relaxed mesenteric arteries (*P<0.01). While the response to E2 was not altered in the presence of indomethacin (10−5 M, P>0.05 vs E2 or G-1), both L-NAME (10−4 μM) and endothelial denuding inhibited vasorelaxation to a similar extent (*P<0.001).
We next determined the signaling mechanisms involved in the endothelium-independent portion of the vasodilatory response. To ascertain whether the signaling molecule cyclic adenosine monophosphate (cAMP) was involved in the smooth muscle cell mechanism, endothelium-intact vessels were pretreated with the cell permeable cAMP analogue Rp-cAMPS, and the response to G-1 was partially inhibited (P<0.001; Figure 2). Pretreatment of intact vessels with a combination of L-NAME, to inhibit the endothelial component, and Rp-cAMPS completely abolished the response (P>0.05 vs. vehicle).
Figure 2.

The cAMP inhibitor Rp-cAMPS (10−5 M) partially reduced the response to G-1 (P<0.001), while pretreatment with Rp-cAMPS and L-NAME abolished the response (P>0.05 versus vehicle).
Nitric oxide activates guanylyl cyclase in vascular smooth muscle to increase the production of cyclic GMP, while adenylyl cyclase is responsible for the production of cAMP. To ascertain the role of these two enzymes in the GPER vasodilatory response, we utilized the soluble guanylyl cyclase inhibitor ODQ and the adenylyl cyclase inhibitor SQ. As shown in Figure 3A, both ODQ and SQ partially attenuated GPER-mediated vasodilation in endothelium-intact vessels (P<0.01), and the combination of the two inhibitors completely blocked relaxation (P>0.05 versus vehicle). Additional studies showed that ODQ was ineffective in the absence of the endothelium, while the adenylyl cyclase inhibitor SQ completely abolished relaxation in denuded vessels (P>0.05 versus vehicle; Figure 3B).
Figure 3.

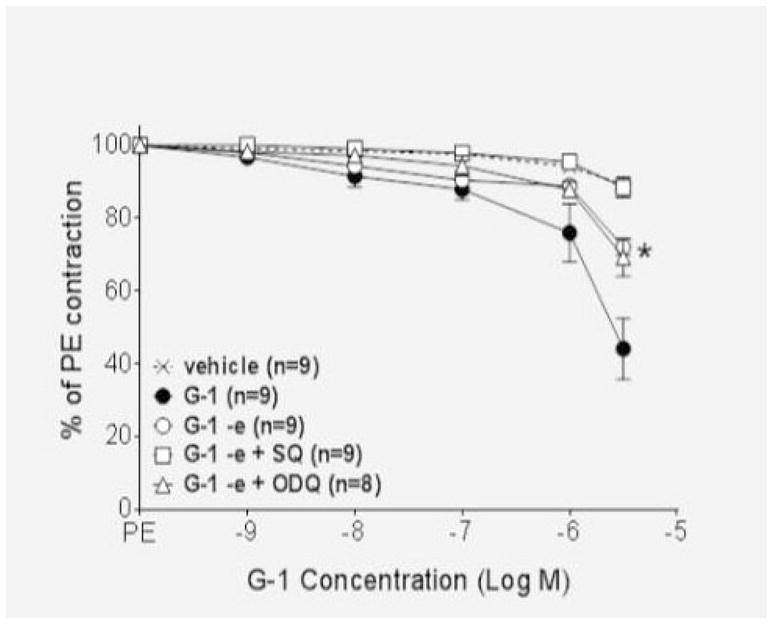
(A) The adenylyl cyclase inhibitor SQ (10−4 M) and the guanylyl cyclase inhibitor ODQ (10−4 M) both partially inhibited the response to G-1 in intact vessels (P<0.01). The combination of the two inhibitors abolished the response (P>0.05 versus vehicle). (B) In denuded vessels, ODQ did not alter the response to G-1 (P>0.05 vs. G-1) while SQ abolished endothelium-independent vasorelaxation (P>0.05 versus vehicle).
In order to confirm that GPER activation by G-1 increases cAMP in vascular smooth muscle, we isolated cells from the same arteries used in the myograph experiments. We previously showed that GPER expression in these cells that was stable in culture through passage four [7]. As shown in Figure 4, G-1 elicited a significant increase in cAMP production in mesenteric artery smooth muscle cells that was significant at nanomolar concentrations. Pretreatment with the GPER antagonist G15 abolished the cAMP response to G-1.
Figure 4.

In cultured cells, 15 minute treatment with G-1 (10−9 M to 10−5 M) increased the production of cAMP in comparison to DMSO (*P<0.05, n=4–13). The GPER antagonist G15 (10−8 M) abolished the response to 10−9 M G-1 (P>0.05 vs. DMSO, n=5).
Discussion
The present study demonstrates that GPER-dependent relaxation of female Lewis mesenteric arteries involves both endothelial and smooth muscle cell signaling pathways. In endothelial cells, GPER stimulates the release of nitric oxide which diffuses to smooth muscle and activates guanylyl cyclase. In smooth muscle cells, GPER activates adenylyl cyclase resulting in an increase in cAMP. The ability of G-1 to increase cAMP was confirmed in cultured vascular smooth muscle cells isolated from the mesenteric vessels. Because the G protein subunit Gαs is known to activate adenylyl cyclase and stimulate the production of cAMP, we propose that in vascular smooth muscle cells GPER is coupled to this alpha subunit and upon ligand binding utilizes this signaling mechanism to induce vasodilation (Figure 5).
Figure 5.

GPER activation causes increases in nitric oxide (NO) through an endothelium-dependent mechanism. In vascular smooth muscle cells, GPER may couple to the αs subunit to activate adenylyl cyclase (AC), increase cAMP, and trigger protein kinases (PKA, PKG) to phosphorylate proteins involved in vascular smooth muscle cell contractility. IP3R: inositol trisphosphate receptor, SERCA: sarcoplasmic reticulum Ca2+-ATPase, PLB: phospholamban.
Our results showing a contribution of both cell types to vasorelaxation are similar to those recently reported by Li and colleagues in the thoracic aorta of ovariectomized Sprague-Dawley rats [11]. However, others have shown that the GPER response is completely endothelium-dependent in epicardial porcine coronary arteries [12] and Sprague-Dawley carotid arteries [6]. Still others have shown a response that is completely endothelium-independent in female Sprague-Dawley inferior vena cava [13] and porcine coronary arteries [9]. The reason for these disparate results is unclear and may suggest differing levels of GPER expression and/or signaling in endothelial versus smooth muscle cells depending on the model and arterial bed investigated. For example, Reslan et al. showed greater vasodilation to G-1 in mesenteric and renal arteries versus carotid and pulmonary arteries [14]. Furthermore, we and others have demonstrated sex and age differences in the contribution of endothelium versus smooth muscle in GPER-mediated vasodilation [2, 15] and have also shown that disease conditions such as salt-loading influence this response [16].
The cAMP analogue Rp-cAMPs and the adenylyl cyclase inhibitor SQ partially inhibited the vasodilatory response in intact vessels. However, in denuded or L-NAME pretreated vessels these inhibitors were completely abolished vasorelaxation. Therefore, the signaling mechanism in endothelial cells is distinct from the activation of adenylyl cyclase. In endothelial cells, GPER may couple to a different alpha subunit or utilize a signaling pathway which does not include adenylyl cyclase. Additional studies using isolated endothelial cells may elucidate the mechanisms by which GPER influences nitric oxide synthase and release of nitric oxide.
In 1967, Szego and Davis showed that 17-β-estradiol stimulates uterine cAMP levels in less than one minute [17]. This acute cAMP response results from stimulation of adenylyl cyclase and is not dependent on RNA or protein synthesis, suggesting a non-genomic mechanism [18]. The estrogen-induced increase in cAMP is one possible mechanism for the acute vasodilatory effects of the hormone. In rat aorta, Rp-cAMPs inhibits the vasodilatory response to estrogen and diethylstilbestrol [19, 20]. In addition, cAMP and adenylyl cyclase are implicated in estradiol-induced relaxation of porcine coronary arteries [21]. We show here that the vasodilatory response to GPER activation also involves the adenylyl cyclase/cAMP signaling pathway in vascular smooth muscle.
Increases in cAMP can activate protein kinase A and can also cross-activate other protein kinases such as protein kinase G [22]. Subsequently, these protein kinases phosphorylate effector proteins that decrease the contractile state of smooth muscle cell. Possible targets include plasma membrane ion channels such as L-type calcium channels [23] and large-conductance potassium channels [24]. Alterations in sarcoplasmic reticulum proteins, such as the inositol trisphosphate receptor [25] and phospholamban [26], may increase intracellular calcium uptake. Alternative targets may include proteins which directly regulate the contractile apparatus, including myosin light chain kinase [27] and myosin phosphatase [28]. While the ability of GPER to alter many of these targets remains to be studied, Yu and colleagues recently found that G-1 alters large-conductance potassium channel activity in porcine coronary arteries [9]. In addition, Gros et al. showed that GPER increases myosin light chain phosphorylation in rat aortic smooth muscle cells [8]. Studies are in progress to identify the protein kinases and phosphorylation targets of GPER in mesenteric artery smooth muscle cells of the Lewis female rat.
Highlights.
Activation of the estrogen receptor GPER relaxes Lewis female mesenteric arteries.
Vasorelaxation is partially dependent on endothelial production of nitric oxide.
In vascular smooth muscle, vasorelaxation is dependent on cAMP production.
In isolated vascular smooth muscle cells, GPER activation increases cAMP.
These signaling pathways may promote vasorelaxation and oppose hypertension.
Acknowledgments
NIH (HL56973, HL51952, HL103974) and the American Heart Association (25515E, 3080005)
Abbreviations
- GPER
G protein-coupled estrogen receptor
- cAMP
cyclic adenosine monophosphate
- L-NAME
Nitro-L-arginine-methyl ester
- PE
phenylephrine
- Ach
acetylcholine
- E2
estrogen
- G-1
GPER agonist
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology. 2009;150:3753–8. doi: 10.1210/en.2008-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindsey SH, da Silva AS, Silva MS, Chappell MC. Reduced vasorelaxation to estradiol and G-1 in aged female and adult male rats is associated with GPR30 downregulation. American journal of physiology Endocrinology and metabolism. 2013;305:E113–8. doi: 10.1152/ajpendo.00649.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Molecular endocrinology (Baltimore, Md) 2002;16:70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 4.Isensee J, Meoli L, Zazzu V, Nabzdyk C, Witt H, Soewarto D, et al. Expression pattern of G protein-coupled receptor 30 in LacZ reporter mice. Endocrinology. 2009;150:1722–30. doi: 10.1210/en.2008-1488. [DOI] [PubMed] [Google Scholar]
- 5.Ma Y, Qiao X, Falone AE, Reslan OM, Sheppard SJ, Khalil RA. Gender-specific reduction in contraction is associated with increased estrogen receptor expression in single vascular smooth muscle cells of female rat. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2010;26:457–70. doi: 10.1159/000320569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broughton BR, Miller AA, Sobey CG. Endothelium-dependent relaxation by G protein-coupled receptor 30 agonists in rat carotid arteries. American journal of physiology Heart and circulatory physiology. 2010;298:H1055–61. doi: 10.1152/ajpheart.00878.2009. [DOI] [PubMed] [Google Scholar]
- 7.Lindsey SH, Carver KA, Prossnitz ER, Chappell MC. Vasodilation in response to the GPR30 agonist G-1 is not different from estradiol in the mRen2.Lewis female rat. Journal of cardiovascular pharmacology. 2011;57:598–603. doi: 10.1097/FJC.0b013e3182135f1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gros R, Ding Q, Davis M, Shaikh R, Liu B, Chorazyczewski J, et al. Delineating the receptor mechanisms underlying the rapid vascular contractile effects of aldosterone and estradiol. Canadian journal of physiology and pharmacology. 2011;89:655–63. doi: 10.1139/y11-062. [DOI] [PubMed] [Google Scholar]
- 9.Yu X, Ma H, Barman SA, Liu AT, Sellers M, Stallone JN, et al. Activation of G protein-coupled estrogen receptor induces endothelium-independent relaxation of coronary artery smooth muscle. American journal of physiology Endocrinology and metabolism. 2011;301:E882–8. doi: 10.1152/ajpendo.00037.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindsey SH, Songu-Mize E. Stretch-induced TRPC4 downregulation is accompanied by reduced capacitative Ca2+ entry in WKY but not SHR mesenteric smooth muscle cells. Clinical and experimental hypertension. 2010;32:288–92. doi: 10.3109/10641960903443525. [DOI] [PubMed] [Google Scholar]
- 11.Li ZL, Liu JC, Liu SB, Li XQ, Yi DH, Zhao MG. Improvement of vascular function by acute and chronic treatment with the GPR30 agonist G1 in experimental diabetes mellitus. PloS one. 2012;7:e38787. doi: 10.1371/journal.pone.0038787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer MR, Baretella O, Prossnitz ER, Barton M. Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology. 2010;86:58–64. doi: 10.1159/000315497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raffetto JD, Qiao X, Beauregard KG, Khalil RA. Estrogen receptor-mediated enhancement of venous relaxation in female rat: implications in sex-related differences in varicose veins. Journal of vascular surgery : official publication, the Society for Vascular Surgery [and] International Society for Cardiovascular Surgery, North American Chapter. 2010;51:972–81. doi: 10.1016/j.jvs.2009.11.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reslan OM, Yin Z, do Nascimento GR, Khalil RA. Subtype-specific estrogen receptor-mediated vasodilator activity in the cephalic, thoracic, and abdominal vasculature of female rat. Journal of cardiovascular pharmacology. 2013;62:26–40. doi: 10.1097/FJC.0b013e31828bc88a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murata T, Dietrich HH, Xiang C, Dacey RG., Jr G protein-coupled estrogen receptor agonist improves cerebral microvascular function after hypoxia/reoxygenation injury in male and female rats. Stroke; a journal of cerebral circulation. 2013;44:779–85. doi: 10.1161/STROKEAHA.112.678177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindsey SH, Yamaleyeva LM, Brosnihan KB, Gallagher PE, Chappell MC. Estrogen receptor GPR30 reduces oxidative stress and proteinuria in the salt-sensitive female mRen2.Lewis rat. Hypertension. 2011;58:665–71. doi: 10.1161/HYPERTENSIONAHA.111.175174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szego CM, Davis JS. Adenosine 3′,5′-monophosphate in rat uterus: acute elevation by estrogen. Proceedings of the National Academy of Sciences of the United States of America. 1967;58:1711–8. doi: 10.1073/pnas.58.4.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aronica SM, Kraus WL, Katzenellenbogen BS. Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:8517–21. doi: 10.1073/pnas.91.18.8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez J, Garcia de Boto MJ, Hidalgo A. Mechanisms involved in the relaxant effect of estrogens on rat aorta strips. Life sciences. 1996;58:607–15. doi: 10.1016/0024-3205(95)02330-5. [DOI] [PubMed] [Google Scholar]
- 20.Martinez C, Sanchez M, Hidalgo A, de Boto MJ. Mechanisms of diethylstilbestrol-induced relaxation in rat aorta smooth muscle. Vascular pharmacology. 2003;40:197–204. doi: 10.1016/j.vph.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Keung W, Vanhoutte PM, Man RY. Acute impairment of contractile responses by 17beta-estradiol is cAMP and protein kinase G dependent in vascular smooth muscle cells of the porcine coronary arteries. Br J Pharmacol. 2005;144:71–9. doi: 10.1038/sj.bjp.0706018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lincoln TM, Cornwell TL, Taylor AE. cGMP-dependent protein kinase mediates the reduction of Ca2+ by cAMP in vascular smooth muscle cells. American Journal of Physiology - Cell Physiology. 1990;258:C399–C407. doi: 10.1152/ajpcell.1990.258.3.C399. [DOI] [PubMed] [Google Scholar]
- 23.Xiong Z, Sperelakis N. Regulation of L-type calcium channels of vascular smooth muscle cells. Journal of molecular and cellular cardiology. 1995;27:75–91. doi: 10.1016/s0022-2828(08)80009-0. [DOI] [PubMed] [Google Scholar]
- 24.White RE, Kryman JP, El-Mowafy AM, Han G, Carrier GO. cAMP-dependent vasodilators crossactivate the cGMP-dependent protein kinase to stimulate BK(Ca) channel activity in coronary artery smooth muscle cells. Circulation research. 2000;86:897–905. doi: 10.1161/01.res.86.8.897. [DOI] [PubMed] [Google Scholar]
- 25.Komalavilas P, Lincoln TM. Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic GMP-dependent protein kinase. J Biol Chem. 1994;269:8701–7. [PubMed] [Google Scholar]
- 26.Cornwell TL, Pryzwansky KB, Wyatt TA, Lincoln TM. Regulation of sarcoplasmic reticulum protein phosphorylation by localized cyclic GMP-dependent protein kinase in vascular smooth muscle cells. Molecular pharmacology. 1991;40:923–31. [PubMed] [Google Scholar]
- 27.Hathaway DR, Konicki MV, Coolican SA. Phosphorylation of myosin light chain kinase from vascular smooth muscle by cAMP- and cGMP-dependent protein kinases. Journal of molecular and cellular cardiology. 1985;17:841–50. doi: 10.1016/s0022-2828(85)80098-5. [DOI] [PubMed] [Google Scholar]
- 28.Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, Ito M, et al. Regulation of Myosin Phosphatase by a Specific Interaction with cGMP-Dependent Protein Kinase Iα. Science (New York, NY) 1999;286:1583–7. doi: 10.1126/science.286.5444.1583. [DOI] [PubMed] [Google Scholar]