

Abstract
While initial studies of Toll-like Receptor (TLR) signaling mainly focused on genetic analysis of signal transduction, recent work has highlighted the importance of understanding the basic cell biology underlying receptor function. Nowhere is this issue more important than in the study of the nucleic acid-sensing TLRs. These receptors face the unique challenge of distinguishing microbial nucleic acids from similar host-derived molecules. The physiological cost of not making this distinction can be readily observed in studies of autoimmunity, a cause of which is often inappropriate detection of self nucleic acids. In this review, we highlight recent research that has revealed myriad ways in which mammalian cells control the function of nucleic acid-sensing TLRs. A fundamental challenge we now face is to uncover unifying themes that can explain both the activation of TLR signaling and its control.
Keywords: Toll-like Receptors, Innate Immunity, nucleic acids, TLR9, autoimmunity, endosomal signal transduction
2. INTRODUCTION
The human body is equipped with a powerful set of tools for defending itself against various microorganisms, such as viruses, bacteria, and parasites. Although this repertoire of defense mechanisms evolved to recognize and neutralize pathogens, there are instances when the immune system goes awry and triggers an inappropriate response in the absence of an infection. In these instances, the body’s immune system recognizes self molecules and inappropriately attacks its own cells and tissues. This misdirected immune response is the cause of various autoimmune conditions, such as lupus, rheumatoid arthritis, and type I diabetes (1-3). Considering these severe consequences, it is not surprising that the immune system has evolved a way to discriminate self from non-self. This ability to differentiate foreign molecules from self molecules is mediated by several families of innate immune receptors, known as Pattern Recognition Receptors (PRRs) (4). These receptors recognize highly conserved components of pathogens, such as lipopolysaccharides, lipopeptides, and nucleic acids (5).
Of the nucleic acid-sensing PRRs, the Toll-like receptor (TLR) family is the best characterized group. Ten human TLRs and 13 murine TLRs have been identified; however, only TLR3, TLR7, TLR8, & TLR9 detect RNA or DNA in both species (5). TLR3 activates immune cells in response to double-stranded RNA, TLR9 detects unmethylated CpG motifs in bacterial and viral DNA, and TLR7 recognizes single-stranded RNA (6-9). TLR8 has also been reported to play a role in sensing single-stranded RNA in humans, but until recently, this receptor was thought to be nonfunctional in mice (6, 10). Now it is known that murine TLR8 is required for sensing A/T-rich DNA (11). It is worth noting that other sensors, such as AIM2 and RIGI, participate in the recognition of nucleic acids; however, discussion of these additional sensors is beyond the scope of this review (see (12, 13) for detailed reviews).
Binding of the various ligands to their respective receptors initiates a signal transduction cascade. Interestingly, not all of the nucleic acid-sensing TLRs utilize the same signaling pathway nor are all responses the same in different cell types (14). The cytoplasmic domains of TLRs contain a Toll-interleukin 1 receptor (IL-1R) homology domain (TIR domain) that serves as a platform to recruit TIR domain containing adaptor proteins, such as MyD88 and TRIF (14). The particular adaptor used determines which signaling pathway will be activated. In the case of TLR3, the adaptor, TRIF, is used to activate TBK1 (TANK-binding kinase 1) and IKK-alpha (IkappaB kinase-alpha, which phosphorylate and activate the transcription factor, IRF3 (14, 15). TRIF also interacts with TRAF6 and RIP1 to activate NF-kappaB and MAPKs in order to induce the transcription of proinflammatory cytokines (16, 17). These TLR3 signaling pathways appear to function in all cell types. In contrast to TLR3, TLR7, TLR8, and TLR9 signal through the adaptor molecule, MyD88 (18). MyD88 recruits the IRAK family of protein kinases and TRAF6 to initiate a signal transduction cascade that culminates in the activation of NF-kappaB to induce proinflammatory cytokines (18). In addition to the NF-kappaB-dependent pathway, another MyD88-dependent pathway exists in plasmacytoid dendritic cells (pDCs). In pDCs, MyD88 interacts directly with the transcription factor, IRF7, to induce type I interferons (19). Although there are differences in the mechanism of signal transduction, the outcome of TLR signaling is virtually the same for all nucleic acid-sensing TLRs. Ultimately, the activation of IRF and NF-kappaB transcription factors induces the transcription of various genes, leading to an inflammatory response.
In the event of infection, this response is necessary to clear the host of any invading microorganism as well as to generate an adaptive immune response against the invader (20). However, in the absence of an invader, setting off these pathways through recognition of self nucleic acids can have deleterious effects on the host. As such, an immediate question that comes to mind is why such receptors would have evolved. The answer to this question probably lies within the challenge of detecting viruses (21). Most of the determinants recognized by TLRs are prokaryotic-specific molecules essential to the integrity, function, or replication of a particular class of microbes, but viruses generally lack uniquely foreign features that easily distinguish them from the host. For example, viral proteins have a high mutation frequency, making them poor targets for the innate immune system. Viruses also lack their own unique metabolic systems and are dependent on the host cells that they infect to reproduce. Since a virus marshals the host machinery to replicate, the viral products made do not have any distinguishing features that allow the host to discriminate the virus from itself. Although no unique viral product exists, the fact that viruses utilize the host cell machinery to replicate has become their “Achilles heel” since these pathogens cannot mutate structural features of their genomes in order to take advantage of the host’s replication system. As such, viral nucleic acids have evolved as targets of numerous innate immune receptors.
Because pathogen and host nucleic acids have very similar structures, it was initially proposed that unique aspects of sequence or nucleotide modifications allow for host/pathogen discrimination (22). However, increasing evidence indicates that these differences are insufficient to prevent activation of the nucleic acid-sensing TLRs in response to self nucleic acids (23-25). Thus, activation of nucleotide-sensing TLRs has to be tightly regulated to mount an appropriate defense against microorganisms without deleteriously responding to self-derived molecules. This review highlights recent work that has furthered our understanding of the key mechanisms that regulate the activation of nucleic acid-sensing TLRs and prevent the recognition of self-derived products by these receptors.
3. LOCATION, LOCATION, LOCATION: REGULATION OF RECEPTOR COMPARTMENTALIZATION
The distinct cell biology associated with the immunostimulatory effects of nucleic acids had been recognized even before knowing the receptors responsible for detecting these ligands. Hacker et al reported that using compounds that prevent endosomal maturation, such as Chloroquine or bafilomycin A, block the effects of CpG-DNA, indicating that cellular up-take via endocytosis and endosomal maturation are necessary for the activation of CpG-induced immune responses (26). These results strongly suggested that signaling via oligonucleotides takes place within the cell. Indeed, the nucleic acid-sensing TLRs were subsequently identified and were shown to localize within intracellular compartments (7).
Of the TLRs found within the cell, the localization of TLR9 has been studied the most extensively. Several groups have reported that TLR9 predominantly resides in the endoplasmic reticulum (ER) of resting cells, but upon activation of CpG, TLR9 is recruited to endolysosomes (27, 28). The endolysosomal compartment was demonstrated to be the site of ligand recognition and signal transduction, as both TLR9 and MyD88 co-localize with fluorescently-tagged CpG in this intracellular location (28, 29). For quite some time, it was assumed that TLR9 resided in the ER at steady state and that this receptor was only recruited to endolysosomes after cells were stimulated with CpG DNA. However, in light of recent data that revealed a cleaved version of this receptor (see below), it is now known that a pool of TLR9 constitutively resides in endolysosomes (30, 31). This endolysosomal pool of TLR9 is most likely poised to initiate a rapid response to endocytosed viral and/or bacterial DNA.
Similar to TLR9, TLR7 and TLR8 are also localized intracellularly (32, 33). TLR7 mediates ligand recognition and signaling initiation from endolysosomal compartments, as was shown for TLR9 (6). However, no data regarding the location of ligand recognition or signal initiation has been reported for TLR8. This lack of data for TLR8 results from the fact that TLR7 and TLR8 have been primarily studied as one entity. TLR7 and TLR8 share the highest degree of sequence similarity among the TLR family members involved in nucleic acid recognition (34). Furthermore, human TLR8 genetically complements TLR7-deficient mouse cells (6). As such, TLR8 had been assumed to be the functional equivalent of TLR7. New data, however, suggests that murine TLR8 plays its own role in viral recognition by recognizing A/T-rich DNA (11). More refined analysis of TLR8 will surely uncover whether TLR8 initiates signaling from its intracellular locale as has been reported for the other members of its TLR subfamily.
In the case of TLR3, reports regarding the localization of this receptor have varied greatly depending on the cell type. TLR3 has been reported to localize to the plasma membrane in human fibroblasts, while in human monocyte-derived iDCs and epithelial cells, TLR3 is primarily found in an intracellular site (35-38). This discrepancy in reported localization may reflect differences in cell-type; however, it may also be a reflection of the poor tools available to study the cell biology of TLRs. The most widely used antibodies against the various TLRs were generated against the ectodomain of these proteins where there is considerable sequence homology between the different TLRs. Thus, it is possible that another TLR was visualized in the experiments that reported TLR3 at the plasma membrane. Confirming the specificity of any TLR3 antibody in TLR3 KO cells will be necessary to provide a definitive answer to the question of TLR3 localization. Regardless of this technical issue, even if a small pool of TLR3 exists at the cell surface, the majority of this receptor is found within the cell, and this intracellular pool appears to be responsible for signal initiation (36). Interestingly, TLR3 has been shown to co-localize with TLR7 as well as TLR9, suggesting that nucleic acid recognition by different TLRs may occur within the same endocytic compartment (33, 39). Despite this co-localization with other intracellular TLRs, TLR3 does not unambiguously co-localize with any intracellular marker. Late endosome, lysosome, Golgi, endoplasmic reticulum, and mitochondria markers do not co-localize with TLR3 (35, 36). Some TLR3-positive compartments overlap with early endosomal markers as well as a marker for an acidic organelle (35, 36, 40), but because TLR3 localization does not definitively correspond to any intracellular marker, the specific TLR3-containing compartment has yet to be conclusively identified. Needless to say, further work needs to be done to determine the exact cellular compartment in which dsRNA is recognized by TLR3 and whether this site of recognition is shared with other nucleotide-sensing TLRs.
Regardless of whether these various intracellular TLRs signal from the same subcompartment, it is clear that this intracellular location is important for regulating the access of these receptors to their respective ligands. Microbial DNA is invisible for immune cells until pathogenic DNA is exposed inside the endolysosomal compartment during processes that affect the structure of the pathogen. By sequestering the nucleic acid-sensing TLRs within the cell, the host optimizes the chance of encountering foreign molecules. Another added benefit of this intracellular location is that this internal site safeguards the nucleic acid-sensing TLRs against contact with self-nucleic acids. The extracellular milieu is known to contain self-DNA and self-RNA; thus, by placing these nucleic acid-sensing TLRs inside the cell, the host has minimized the chance of encountering self molecules. The importance of this avoidance tactic was demonstrated by placing the ectodomain of TLR9 at the cell surface (41). When placed at this inappropriate site, the ectodomain of TLR9 was able to recognize mammalian DNA, indicating that there is nothing inherent about host DNA that prevents TLR9 from recognizing self DNA as a ligand (41). Thus, it seems that the intracellular location controls access of these receptors to different sources of nucleic acids.
Since these intracellular TLRs are capable of recognizing host nucleic acids, what prevents host molecules from entering TLR-containing endocytic vesicles? Secreted DNases, such as DNase I, appear to play an important role in degrading extracellular DNA (42), but what regulates the exclusion of self-RNA remains poorly defined. We speculate that secreted RNases may serve an analogous role to DNase I, but further work needs to be done to understand the exact mechanisms that normally prevent self ligands from reaching intracellular TLRs.
4. GOOD THINGS COME IN SMALL PACKAGES: REGULATION BY PROTEOLYTIC CLEAVAGE
Lysosomes contain a large variety of hydrolytic enzymes that degrade proteins and other substances (43). Due to their potent degradative properties, many lysosomal enzymes are synthesized in the ER as proproteins. Only upon delivery to their lysosomal compartment are these harsh digestive proproteins converted into their active form. By delaying the activation of these proproteins until they reach the lysosome, the cell prevents these destructive enzymes from functioning in inappropriate cellular locations. Similarly, intracellular TLRs are also synthesized as proproteins that are activated by a cleavage event in the endolysosomal compartment in order to prevent the activation of these receptors in inappropriate locations (31, 44). Considering the negative immunological consequences of activating a TLR inappropriately and the potential of recognizing host nucleic acids in other locations within the cell, this form of regulation makes sense. Despite being conceptually pleasing, the discovery of a cleaved form of any nucleic acid-sensing TLR was not made until very recently.
Biochemical analysis of TLR9 led to the identification of its processed form (31, 44). This form of TLR9 consists of approximately half of the ectodomain, the transmembrane domain, and the entire cytoplasmic domain of the full-length receptor. Although both full-length and processed forms of the receptor are capable of binding CpG oligonucleotides, it appears that only the cleaved form of the receptor is functional (31, 44). First, a retrovirus encoding the processed form is sufficient to complement cells mutant for TLR9 (44). Second, conditions that prevent receptor proteolysis, such as relocalization of full-length TLR9 to the cell surface or treatment of cells with protease inhibitors, interfere with the ability of this receptor to respond to CpG ligands (31, 44). Lastly, MyD88 selectively interacts with the truncated form following CpG stimulation (31). Taken together, these data indicate that cleavage of endosomal TLR9 is an additional regulatory event necessary for limiting signal transduction to the appropriate cellular compartment.
The exact lysosomal protease (s) involved in the cleavage events remain to be identified, but several lines of evidence point to the involvement of a cathepsin. First, cathepsin B and cathepsin L were functionally cloned as molecules required for TLR9 responses in a complementation screen using a cell line defective for TLR9 signaling (45). Overexpression of cathepsin S and cathepsin F were also able to confer CpG responsiveness on this same cell line (45). Second, pharmacological inhibition of various cathepsins has been reported to block TLR9 signaling in numerous cell types (44-46). Lastly, several recombinant cathepsins were shown to cleave full-length TLR9 to generate the processed form of the receptor in vitro (31, 44). Taken together, these data strongly implicate this class of lysosomal proteases as being responsible for cleaving TLR9; however, none of these individual proteins has been undeniably identified as the protease solely responsible for this processing event. It has been argued that multiple cathepsins are capable of processing TLR9 and the role of any individual protease in this cleavage event is redundant (31, 44). This line of reasoning is supported by the greater inhibitory potency of the broad spectrum inhibitors over specific inhibitors as well as the fact that mice deficient for individual cathepsins display varying phenotypes from having no defects at all to having partial impairment of TLR9 signaling (31, 44, 46). It is also possible that multiple cathepsins are actually involved in the processing of TLR9 since at least two cleavage events occur. As such, identifying which particular member (s) of the cathepsin family is required for TLR9 signaling poses a difficult challenge.
Despite there being conflicting reports (31, 44), TLR7 has also been shown to undergo proteolytic cleavage, suggesting that proteolysis is a general regulatory feature shared by nucleic-acid sensing TLRs (31). Although proteolytic degradation has been suggested to negatively regulate a number of TLRs (47), to date, no data has been reported regarding a cleavage event that activates TLR3 or TLR8. TLR7, TLR8, and TLR9 share high sequence homologies (34); thus, it is likely that TLR8 is regulated in a similar fashion as TLR9. TLR3, on the other hand, does not appear to be regulated in this manner. Along with structural studies, mutational analysis has demonstrated that residues within the N-terminal region of the ectodomain of TLR3 are necessary for ligand binding, suggesting that full-length TLR3 is the active form of this receptor (48, 49). The fact that all other nucleic acid-sensing TLRs appear to be cleaved raises the question as to why this level of regulation is not extended to TLR3. It is possible that because TLR3 recognizes dsRNA (a molecule that is rarely found within mammalian cells) this additional regulatory mechanism is not needed to prevent TLR3 from recognizing self ligands. In this sense, dsRNA should be viewed more like a prokaryotic PAMP in that it is unique to the microbe as compared to ssRNA or DNA, which are highly abundant in mammalian cells. However, it is also possible that TLR3 is cleaved, but since the ectodomain of the full-length receptor is much smaller than the other nucleic-sensing TLRs, this cleavage event has gone undetected. Consistent with this hypothesis, TLR9 appears to be cleaved in at least two sites, as a slightly larger “pre-C-terminal fragment” has been identified for TLR9 (31, 44). Drawing a parallel to TLR3, this “pre-C-terminal fragment” may correlate to the full-length receptor of TLR3, which when cleaved leads to the active form of the receptor. If such a cleavage event occurs for TLR3, one would expect the protease activity of cathepsins to be required for TLR3 responses. Indeed, cathepsin inhibitors suppress the immunological effects of polyI:C treatment in splenic B cells (45). This finding suggests that cathepsins are required for TLR3 signaling, but further work needs to be done to determine how these proteases affect TLR3 responses and if cleaved form of this receptor exists.
Why such a processing event would be evolutionarily necessary for TLR activation remains unclear. As stated above, it is possible that cells have evolved this compartmentalized cleavage in order to prevent receptors that leak out to the cell surface from erroneously responding to self nucleic acids. However, this control step would only account for newly synthesized receptors that were mislocalized to the plasma membrane during biosynthetic transport to endolysosomes. Once the receptor is cleaved in the endocytic compartments of the cell, it may also be delivered to the plasma membrane mistakenly. In this instance, the risk of self-recognition returns. This latter possibility diminishes the “fail-safe” means of preventing self nucleic acid recognition by compartmentalized cleavage events. Another possible reason for the need for this cleavage event is to prevent mistaken dimerization-induced signaling during glycosylation in the ER or Golgi compartment while en route to endolysosomes. However, this potential risk does not uniquely apply to nucleic acid sensing TLRs, and thus is unlikely to explain this unique level of regulation of these receptors. Future work that specifically addresses the functional “costs and benefits” of using TLRs as proproteins must be undertaken before an evolutionary basis for this intriguing level of regulation is explained.
5. THE CELLULAR NAVIGATION SYSTEM: REGULATION BY RECEPTOR TRAFFICKING
The intracellular localization of nucleic acid-sensing TLRs is clearly important for recognition of viral DNA and RNA, but the mechanism by which these receptors are positioned within cells has only recently begun to be elucidated. Most research on the trafficking of intracellular TLRs has focused on TLR9 with the assumption that the other intracellular TLRs are subject to similar regulatory mechanisms. While this assumption awaits experimental verification, we will use TLR9 as a reference to explain the trafficking of all endosomal TLRs.
Although recognition of DNA by TLR9 occurs in endolysosomes, the majority of TLR9 is found in the endoplasmic reticulum at steady state (27, 28). Initial studies of the carbohydrates on the full length protein of TLR9 revealed that that this mature protein lacked modifications that are consistent with transit through the Golgi. Based on these findings, it was suggested that a special unconventional mechanism existed that bypassed the Golgi complex to transport TLR9 from the ER to early endosomes (28). Attempts to determine the mechanistic details by which this unusual transport took place led to the identification of a processed form of the receptor (see discussion above). Analysis of the carbohydrates on this processed form indicated that the cleaved receptor passed through the Golgi (31, 44). This discovery negated the hypothesis that an unconventional mode of transport exists and supported a model in which TLR9 traffics to the endolysosome via a conventional secretory pathway. Indeed, recent work has confirmed that full length TLR9 is sorted in the ER, traffics through the Golgi, and is then sent to the endolysosome where it is processed to initiate signaling (30, 31).
Even though recent work has debunked the hypothesis that a novel transport pathway exists for endosomal TLRs, there are unique components involved in the trafficking of these receptors. One such molecule is UNC93B1. This gene was first identified in a forward genetic screen that sought to find genes that disrupt various immune processes (50). The identified UNC93B1 mutant mice (termed ‘3d’) display defects in TLR3, TLR7, and TLR9 signaling and have increased susceptibility to infection by a variety of pathogens (50). Studies of UNC93B1 revealed that this 12-membrane-spanning protein specifically binds to TLR3, TLR7, TRL9, and TLR13 in the ER (51). Despite the initial claims that UNC93B1 did not control the transport of intracellular TLRs (50), the interaction of UNC93B1 with TLR7 and TLR9 is necessary for the delivery of these receptors to their signaling compartment as neither TLR7 nor TLR9 can leave the ER in cells of 3d mice (52). Further analysis of the role of UNC93B1 in TLR signaling revealed that UNC93B1 travels along with TLR7 and TLR9 to the endolysosome; however, this polytopic membrane protein is not required for ligand recognition nor is it needed for the initiation of signaling events by TLRs (52). The fact that UNC93B1 is dispensable for ligand recognition and signal transduction suggests that UNC93B1’s sole role in TLR signaling is to traffic these intracellular receptors to their site of activation. In support of this finding, aberrantly locating UNC93B1 to the cell surface also directs TLR9 to the plasma membrane (52). This key finding establishes that UNC93B1 is both necessary and sufficient to determine the intracellular location of nucleic acid-sensing TLRs.
Interestingly, the studies of UNC93B1 not only shed light on who the nucleic acid-sensing TLRs traffic to their intracellular location, but this work also provides clues about the most poorly characterized member of the Toll-like receptor family, TLR13. Based on its interaction with UNC93B1 (51), it is very likely that TLR13 is located within endosomes. Since other intracellular TLRs are nucleic acid sensors and are mainly involved in the recognition of viruses, it is also likely that TLR13 plays a similar role in virus detection. In support of this hypothesis, TLR13 is highly expressed in plasmacytoid dendritic cells, which are known to produce copious amounts of interferons in response to viral infection (53). Interferons have also been reported to increase the expression of TLR13, suggesting a positive feedback loop to maximally respond to a viral infection (53). It is likely that TLR13 recognizes viral nucleic acids, but the question regarding which one (s) remains to be addressed. The sensors of ssDNA (TLR9), dsDNA (TLR8), ssRNA (TLR7) and dsRNA (TLR3) have already been accounted for, so what could TLR13 detect? Of the known nucleic acid sensing TLRs, TLR3 is the most enigmatic. It was the first TLR identified that detects RNA, but its role in numerous RNA virus infections is minimal (54). Thus, perhaps another TLR detects dsRNA that complements the role of TLR3 in viral infections. We speculate that TLR13 may serve such a function.
In addition to UNC93B1, gp96 and PRAT4A have also been reported to regulate the trafficking of TLRs (55, 56). However, unlike UNC93B1, which appears to be specific for the trafficking of only intracellular TLRs, gp96 and PRAT4A appear to be master regulators for the trafficking of all TLRs. Gp96 is an ER paralog of the Hsp90 family that acts as an ER chaperone protein for multiple protein substrates, including immunoglobulin chains, some integrins, and TLRs (56). Macrophages lacking gp96 are hyporesponsive to ligands for all TLRs, indicating gp96 is essential for signaling via TLRs found at both the plasma membrane and within endosomes (56). Similarly, PRAT4A deficient cells have impaired cytokine production in response to ligands from all TLRs, with the exception of TLR3 (55). Because PRAT4A is involved in the trafficking of all other TLRs, it is a bit confusing that only TLR3 would not require this molecule for its localization. It is possible that the observed response in PRAT4A deficient cells following polyI:C treatment is due to the activation of the cytosolic dsRNA sensor MDA-5, and not TLR3. TLR3 and MDA-5 have been implicated in the recognition of polyI:C, but each of these receptors contribute to the induction of different antiviral genes following treatment with this particular ligand (57, 58). Perhaps analyzing responses that are unique to TLR3 will help distinguish PRAT4A’s role in the trafficking of this particular TLR. It would also be interesting to clarify the exact contribution of each trafficking factor in the localization of the various TLRs. The overlapping requirements of gp96, PRAT4A, and UNC93B1 in TLR trafficking suggest that these proteins may function together to deliver these receptors to their particular signaling location. How this targeting event occurs and what the relationships between the various trafficking factors remain to be determined.
6. FUTURE DIRECTIONS/CONCLUSIONS
In the last five years, we have witnessed an explosion of interest in the trafficking and function of nucleic acid sensing TLRs (Figure 1). We now know that these receptors are delivered to endosomes in complex with UNC93B1, by a conventional biosynthetic transport route. Once in endosomes, at least two of these TLRs are cleaved by cathepsins to form signaling-competent receptors that detect viral, bacterial, and in rare instances, self nucleic acids. However, what happens upon receptor activation by nucleic acids remains an important “black box” in the field. For example, recent work has highlighted the role of the small GTPase Rab7 (59, 60) and the E3 ubiquitin ligase TRIAD3a (61) in regulating the stability of some TLRs, including TLR9, after they have encountered their ligands. However, like the situation described above for gp96, PRAT4A, and UNC93B1, the relationship between Rab7 and TRAID3a in controlling TLR stability remains obscure. In addition, the emerging field of small RNA biology has reached the TLR field as well, with the discovery that miR-21 functions to control the signaling functions of some TLRs (62, 63). These findings raise the possibility that other small RNAs function to indirectly control the transport and/or signaling functions of TLRs. Finally, we still do not understand why some endosomal TLRs utilize a MyD88-dependent pathway to promote signal transduction (e.g. TLR9) whereas others utilize a TRIF-dependent pathway (e.g. TLR3). Based on the reference of TLR4, which uses both of these adaptors but from different compartments of the cell, it is possible that TLR3 and TLR9 use different adaptors because they also signal from different locations. What would be the evolutionary advantage to this? A fundamental challenge we now face is in defining the precise subcellular sites of TLR signaling, how the cytosolic machinery is recruited to these sites, and why a common site of signaling by endosomal TLRs does not appear to exist. Detailed cell biological analysis of these receptors is essential to answer these questions, and the recent emergence of these approaches to study innate immunity (64) should yield many exciting discoveries in the years to come.
FIGURE 1. MULTIPLE LEVELS OF ENDOSOMAL TLR REGULATION.
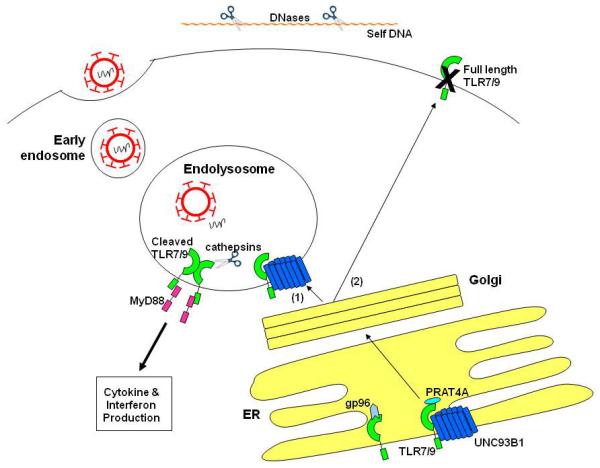
Nucleotides are sensed by TLRs in endolysosomes in order to allow for the efficient discrimination between pathogen-associated and self-derived nucleic acids. Viral genomes are protected by their capsids until they are released within the endolysosomal compartment, whereas extracellular self-derived nucleic acids are degraded by DNases before reaching endolysosomes. Endosomal TLR responses are also limited by controlling their intracellular trafficking. Endosomal TRLs associate with gp96, PRAT4A, and UNC93B1 in the ER. These chaperone proteins facilitate the transport of intracellular TLRs through the Golgi via the conventional secretory pathway to the endolysosome (denoted by (1)). In some instances, however, full length TLRs may be trafficked to the plasma membrane (denoted by (2)). These mislocalized TLRs cannot signal since they are not processed to their active form at the plasma membrane like they are in the endolysosomal compartment by proteases, such as cathepsins.
REFERENCES
- 1.Swiecki M, Colonna M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol Rev. 234(1):142–62. doi: 10.1111/j.0105-2896.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lehuen A, et al. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 10(7):501–13. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- 3.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 140(5):619–30. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R, Janeway CA., Jr. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296(5566):298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 5.Barton GM, Medzhitov R. Toll-like receptors and their ligands. Curr Top Microbiol Immunol. 2002;270:81–92. doi: 10.1007/978-3-642-59430-4_5. [DOI] [PubMed] [Google Scholar]
- 6.Heil F, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 7.Hemmi H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 8.Alexopoulou L, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 9.Diebold SS, et al. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–31. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 10.Jurk M, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3(6):499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 11.Martinez J, Huang X, Yang Y. Toll-like receptor 8-mediated activation of murine plasmacytoid dendritic cells by vaccinia viral DNA. Proc Natl Acad Sci U S A. 107(14):6442–7. doi: 10.1073/pnas.0913291107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakhaei P, et al. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin Immunol. 2009;21(4):215–22. doi: 10.1016/j.smim.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol. 22(1):28–33. doi: 10.1016/j.coi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 15.Fitzgerald KA, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4(5):491–6. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 16.Sato S, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol. 2003;171(8):4304–10. doi: 10.4049/jimmunol.171.8.4304. [DOI] [PubMed] [Google Scholar]
- 17.Meylan E, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5(5):503–7. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 18.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–64. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5(10):1061–8. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 20.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 21.Barton GM. Viral recognition by Toll-like receptors. Semin Immunol. 2007;19(1):33–40. doi: 10.1016/j.smim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 23.Avalos AM, Busconi L, Marshak-Rothstein A. Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity. 43(1):76–83. doi: 10.3109/08916930903374618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okuya K, et al. Spatiotemporal regulation of heat shock protein 90-chaperoned self-DNA and CpG-oligodeoxynucleotide for type I IFN induction via targeting to static early endosome. J Immunol. 184(12):7092–9. doi: 10.4049/jimmunol.1000490. [DOI] [PubMed] [Google Scholar]
- 25.Ganguly D, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206(9):1983–94. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hacker H, et al. CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. Embo J. 1998;17(21):6230–40. doi: 10.1093/emboj/17.21.6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leifer CA, et al. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol. 2004;173(2):1179–83. doi: 10.4049/jimmunol.173.2.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Latz E, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5(2):190–8. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 29.Ahmad-Nejad P, et al. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur J Immunol. 2002;32(7):1958–68. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 30.Chockalingam A, et al. TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol Cell Biol. 2009;87(3):209–17. doi: 10.1038/icb.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ewald SE, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456(7222):658–62. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishiya T, DeFranco AL. Ligand-regulated chimeric receptor approach reveals distinctive subcellular localization and signaling properties of the Toll-like receptors. J Biol Chem. 2004;279(18):19008–17. doi: 10.1074/jbc.M311618200. [DOI] [PubMed] [Google Scholar]
- 33.Nishiya T, et al. TLR3 and TLR7 are targeted to the same intracellular compartments by distinct regulatory elements. J Biol Chem. 2005;280(44):37107–17. doi: 10.1074/jbc.M504951200. [DOI] [PubMed] [Google Scholar]
- 34.Wagner H. The immunobiology of the TLR9 subfamily. Trends Immunol. 2004;25(7):381–6. doi: 10.1016/j.it.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 35.Funami K, et al. Spatiotemporal mobilization of Toll/IL-1 receptor domain-containing adaptor molecule-1 in response to dsRNA. J Immunol. 2007;179(10):6867–72. doi: 10.4049/jimmunol.179.10.6867. [DOI] [PubMed] [Google Scholar]
- 36.Funami K, et al. The cytoplasmic ‘linker region’ in Toll-like receptor 3 controls receptor localization and signaling. Int Immunol. 2004;16(8):1143–54. doi: 10.1093/intimm/dxh115. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto M, et al. Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem Biophys Res Commun. 2002;293(5):1364–9. doi: 10.1016/S0006-291X(02)00380-7. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto M, et al. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol. 2003;171(6):3154–62. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- 39.Kajita E, Nishiya T, Miwa S. The transmembrane domain directs TLR9 to intracellular compartments that contain TLR3. Biochem Biophys Res Commun. 2006;343(2):578–84. doi: 10.1016/j.bbrc.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 40.Johnsen IB, et al. Toll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling. Embo J. 2006;25(14):3335–46. doi: 10.1038/sj.emboj.7601222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7(1):49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 42.Napirei M, et al. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25(2):177–81. doi: 10.1038/76032. [DOI] [PubMed] [Google Scholar]
- 43.Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- 44.Park B, et al. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol. 2008;9(12):1407–14. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsumoto F, et al. Cathepsins are required for Toll-like receptor 9 responses. Biochem Biophys Res Commun. 2008;367(3):693–9. doi: 10.1016/j.bbrc.2007.12.130. [DOI] [PubMed] [Google Scholar]
- 46.Asagiri M, et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science. 2008;319(5863):624–7. doi: 10.1126/science.1150110. [DOI] [PubMed] [Google Scholar]
- 47.Choi YJ, et al. TRIF modulates TLR5-dependent responses by inducing proteolytic degradation of TLR5. J Biol Chem. 285(28):21382–90. doi: 10.1074/jbc.M110.115022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ranjith-Kumar CT, et al. Biochemical and functional analyses of the human Toll-like receptor 3 ectodomain. J Biol Chem. 2007;282(10):7668–78. doi: 10.1074/jbc.M610946200. [DOI] [PubMed] [Google Scholar]
- 49.Bell JK, et al. The dsRNA binding site of human Toll-like receptor 3. Proc Natl Acad Sci U S A. 2006;103(23):8792–7. doi: 10.1073/pnas.0603245103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabeta K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7(2):156–64. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 51.Brinkmann MM, et al. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol. 2007;177(2):265–75. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim YM, et al. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452(7184):234–8. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- 53.Shi Z, et al. Transcriptional regulation of the novel Toll-like receptor Tlr13. J Biol Chem. 2009;284(31):20540–7. doi: 10.1074/jbc.M109.022541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schroder M, Bowie AG. TLR3 in antiviral immunity: key player or bystander? Trends Immunol. 2005;26(9):462–8. doi: 10.1016/j.it.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 55.Takahashi K, et al. A protein associated with Toll-like receptor (TLR) 4 (PRAT4A) is required for TLR-dependent immune responses. J Exp Med. 2007;204(12):2963–76. doi: 10.1084/jem.20071132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Y, et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26(2):215–26. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gitlin L, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103(22):8459–64. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, et al. Lysosome-associated small Rab GTPase Rab7b negatively regulates TLR4 signaling in macrophages by promoting lysosomal degradation of TLR4. Blood. 2007;110(3):962–71. doi: 10.1182/blood-2007-01-066027. [DOI] [PubMed] [Google Scholar]
- 60.Yao M, et al. Late endosome/lysosome-localized Rab7b suppresses TLR9-initiated proinflammatory cytokine and type I IFN production in macrophages. J Immunol. 2009;183(3):1751–8. doi: 10.4049/jimmunol.0900249. [DOI] [PubMed] [Google Scholar]
- 61.Chuang TH, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol. 2004;5(5):495–502. doi: 10.1038/ni1066. [DOI] [PubMed] [Google Scholar]
- 62.Sheedy FJ, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 11(2):141–7. doi: 10.1038/ni.1828. [DOI] [PubMed] [Google Scholar]
- 63.Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 9(4):293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 64.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9(8):535–42. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]