

Abstract
The nematode Caenorhabditis elegans secretes a family of water-soluble small molecules, known as the ascarosides, into its environment and uses these ascarosides in chemical communication. The ascarosides are derivatives of the 3,6-dideoxysugar ascarylose, modified with different fatty acid-derived side chains. C. elegans uses specific ascarosides, which are together known as the dauer pheromone, to trigger entry into the stress-resistant dauer larval stage. In addition, C. elegans uses specific ascarosides to control certain behaviors, including mating attraction, aggregation, and avoidance. Although in general the concentration of the ascarosides in the environment increases with population density, C. elegans can vary the types and amounts of ascarosides that it secretes depending on the culture conditions under which it has been grown and its developmental history. Here, we describe how to grow high-density worm cultures and the bacterial food for those cultures, as well as how to extract the culture medium to generate a crude pheromone extract. Then, we discuss how to analyze the types and amounts of ascarosides in that extract using mass spectrometry and NMR spectroscopy.
Keywords: Dauer, Ascarosides, Pheromone, C. elegans, Mass spectrometry, NMR spectroscopy, dqf-COSY
1 Introduction
C. elegans secretes ascarosides as chemical signals to coordinate its development and behavior. All ascarosides are derivatives of the 3,6-dideoxysugar ascarylose, with different fatty acid-derived side chains [1-9]. The ascarosides that are found in C. elegans culture medium can be divided into several structural classes (Table 1). In this chapter, we describe a general method for growing high-density worm cultures and extracting the culture medium in order to generate a crude pheromone extract that contains secreted ascarosides (Subheadings 3.1 and 3.2). This crude pheromone extract can be directly used in biological assays, such as the dauer formation assay (see Chapter 20), it can be fractionated chromatographically for bioactivity-guided purification of ascarosides [1-4 , 6], or the ascarosides in the extract can be analyzed using analytical techniques, such as liquid chromatography-mass spectrometry (LC-MS) (Subheading 3.3), LC-MS/MS (Subheading 3.4), and NMR spectroscopy (Subheading 3.5). Each of these techniques has its own advantages and disadvantages, as described below. The structural class of a specific ascaroside, as well as its concentration in the crude pheromone extract, can affect whether or not it can be detected in the extract using a specific analytical technique.
Table 1.
Summary of ascarosides found in wild-type (N2) C. elegans culture medium that have had their structures confirmed through chemical synthesisa
| Structure-based name | Other name(s) | Orig. ref. | Mol. formula | [M+Na]+ | [M−H]− | n | R | Structure |
|---|---|---|---|---|---|---|---|---|
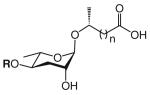
| Ascarosides with a saturated fatty acid side chain that is attached to the sugar at the penultimate (ω–1) carbon |
|
|||||||
| asc-C4 | ascr#11 | [7] | C10H18O6 | 257.1001 | 233.1025 | 1 | H | |
| asc-C5 | ascr#9 | [7,8] | C11H20O6 | 271.1158 | 247.1182 | 2 | H | |
| IC-asc-C5 | indolecarboxyl- ascaroside C5; ascaroside C5; icas#S |
[6] | C20H25NO7 | 414.1529 | 390.1553 | 2 | IC | |
| OS-asc-C5 | osas#9 | [9] | C23H33NO10 | 506.2002 | 482.2026 | 2 | OS | |
| asc-C7 | daumone 1; ascr#l; ascaroside C7 |
[1] | C13H24O6 | 299.1471 | 275.1495 | 4 | H | |
| IC-asc-C7 | icas#l | [7,8] | C22H29NO7 | 442.1842 | 418.1866 | 4 | IC | |
| asc-C9 | ascr#10 | [7,8] | C15H28O6 | 327.1784 | 303.1808 | 6 | H | |
|
| ||||||||
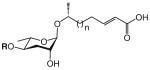
| Ascarosides with an α,β-unsaturated (Δ) fatty acid side chain that is attached to the sugar at the penultimate (ω–1) carbon | ||||||||
| asc-ΔC7 | ascr#7 | [5] | C13H22O6 | 297.1314 | 273.1339 | 2 | H |
|
| IC-asc-ΔC7 | icas #7 | [7,8] | C24H33NO7 | 440.1685 | 416.1709 | 2 | IC | |
| asc-ΔC9 | ascaroside C9; ascr#3; daumone 3 |
[2] | C15H26O6 | 325.1627 | 301.1651 | 4 | H | |
| IC-asc-ΔC9 | icas#3 | [7,8] | C24H31NO7 | 468.1998 | 444.2022 | 4 | IC | |
| HB-asc-ΔC9 | hbas#3 | [7] | C22H30O8 | 445.1838 | 421.1862 | 4 | HB | |
| MB-asc-ΔC9 | mbas#3 | [7] | C20H32O7 | 407.2046 | 383.2070 | 4 | MB | |
|
| ||||||||
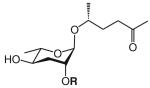
| Ascarosides with a side chain that is attached to the sugar at the penultimate (ω–1) carbon and terminates in a methyl ketone (MK)b | ||||||||
| asc-C6-MK | ascaroside C6; ascr#2; daumone 2 |
[2] | C12H22O5 | 269.1365 | n.a. | 2 | H |
|
| Glu-asc-C6-MK | ascr#4; daumone 4 | [4] | C18H32O10 | 431.1893 | n.a. | 2 | Glu | |
|
| ||||||||
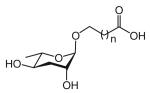
| Ascarosides with a saturated fatty acid side chain that is attached to the sugar at the terminal (ω) carbon | ||||||||
| asc-ωC3 | ascaroside C3; ascr#5; daumone 5 |
[3] | C9H16O6 | 243.0845 | 219.0869 | 1 | – |
|
| asc-ωC5 | oscr#9 | [7] | C11H20O6 | 271.1158 | 247.1182 | 3 | – | |
| asc-ωC9 | oscr#10 | [7] | C15H28O6 | 327.1784 | 303.1808 | 7 | – | |
|
| ||||||||
| Ascarosides with an α,β-unsaturated (Δ) side chain attached at the penultimate (ω–1) carbon and linked to p-aminobenzoic acid (PABA) | ||||||||
| asc-ΔC7-PABA | ascr#8 | [5] | C20H27NO7 | n.a. | 392.1709 | 2 | - |
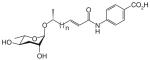
|
|
| ||||||||
| R group definitions: | ||||||||
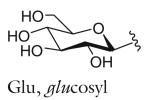
|
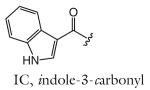
|
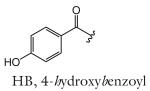
|
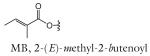
|
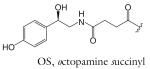
|
||||
The structure-based name given in the first column assumes that the canonical ascaroside structure is an ascaroside with a saturated fatty acid side chain that is attached to the sugar at the penultimate (ω–1) carbon. Deviations from the canonical structure are indicated such that each name conforms to the following: (modifications to the sugar)-asc-(deviations in the fatty acid)C#-(modifications at the end of the fatty acid)
Modifications to the sugar and their abbreviations are listed at the bottom of the table. Deviations in the fatty acid include attachment of the fatty acid to the sugar at the terminal (to) carbon, instead of penultimate (ω-1) carbon, as well as α-β (Δ) unsaturation of the fatty acid. Modifications at the end of the fatty acid include the methyl ketone (MK) moiety and addition of p-aminobenzoic acid (PABA)
Many other ascarosides, such as asc-C6 (ascr#12), asc-C8 (ascr#14), IC-asc-C9 (icas#10), asc-Cll (ascr#18), asc-ωCll (oscr#18), asc-C12 (ascr#20), OS-asc-C9 (osas#10), and OS-asc-C6-MK (osas#2), have also been detected in wild-type C. elegans culture medium, but their structures have not yet been fully confirmed through chemical synthesis [7-9]
Reduced versions of asc-C6-MK (ascr#6.1 and ascr#6.2) with a hydroxyl instead of a ketone have also been reported [5]
LC-MS analysis on a single quadrupole mass spectrometer (Subheading 3.3) is a rapid technique for analyzing the concentrations of specific known ascarosides in a sample [10 , 11]. In LC-ESI-MS, the molecules in the extract are separated on a column based on their polarity. As the molecules elute from the column, they are ionized by electrospray ionization (ESI), and the mass-to-charge (m/z) ratios of the ionized species are then detected. Most ascarosides will ionize most strongly as positively charged, sodium adducts. Thus, they can be detected by positive ion mode LC-MS, in which the ionized ascaroside is detected as [M+Na]+ , where M is the mass of the ascaroside (see Table 1 for which ascarosides ionize in positive ion mode). Many ascarosides, but not all, will also ionize as negatively charged, deprotonated species. Thus, they can also be detected by negative ion mode LC-MS, in which the ionized ascaroside is detected as [M−H]− (see Table 1 for which ascarosides ionize in negative ion mode). In order to use LC-MS to identify the presence of a specific ascaroside and to determine the concentration of that ascaroside in the extract, a synthetic standard is needed for the ascaroside of interest. The crude pheromone extract can contain non-ascaroside compounds with masses and retention times that are similar to the ascarosides of interest. For this reason, LC-MS-based identification of ascarosides is usually confirmed with additional techniques, including LC-MS/MS or NMR spectroscopy. LC-MS can be used to analyze the ascarosides in (1) the crude pheromone extract from the culture medium of a non-synchronized culture (generated by method given in Subheading 3.2), (2) the crude pheromone extract from the culture medium of a synchronized culture [11], (3) the crude extract from the worms themselves [10], or (4) the secretions from worms produced over a short period of time (“worm water”) [4]. LC-MS is not useful for characterizing the chemical structures of novel ascarosides, as this must be done using techniques that provide additional structural information, such as LC-MS/MS and NMR spectroscopy.
LC-MS/MS analysis on a triple quadrupole mass spectrometer (Subheading 3.4) allows the simultaneous identification of many of the ascarosides present in the crude pheromone extract [7]. LC-MS/MS is similar to LC-MS in that the molecules in the extract are separated on a column based on their polarity and ionized. The m/z ratios of the ionized species are then detected. However, in LC-MS/MS, the molecules are not only ionized to detect the ionized species (called the precursor ion), but also broken apart, resulting in product ions that provide additional structural information about the molecules. When LC-MS/MS is performed on a triple quadrupole mass spectrometer, a precursor ion scan can be used to detect only molecules that give rise to a specific product ion and report back the precursor ions that produced the specific product ion. This technique is particularly useful for the detection of classes of molecules with similar structures, such as the ascarosides, that all produce a common product ion. Those ascarosides that ionize in negative ion mode can be fragmented in LC-MS/MS to produce a common product ion of m/z 73 [C3H5O2]. Thus, this technique, which was developed by von Reuss and coworkers [7], can be used to detect all ascarosides in a crude pheromone extract that ionize as negative ions and report back the precursor ions (that is, [M−H]−) corresponding to those ascarosides (Subheading 3.4). Those ascarosides that ionize only in positive ion mode can be monitored by looking for a neutral loss of 130 amu [C6H10O3] that corresponds to the loss of the ascarylose sugar [7]. However, fragmentation of the ascarosides in positive ion mode is not as efficient as that in negative ion mode, leading to lower and less reliable signal, and will not be described in this review.
NMR spectroscopy can be used to analyze the ascarosides in a crude pheromone extract or chromatography fractions derived from that extract (Subheading 3.5). One type of NMR experiment that is particularly informative is the double quantum-filtered correlation spectroscopy (dqf-COSY) experiment [5 , 10]. Like a standard COSY experiment, this experiment detects correlations between protons on neighboring carbons in a molecule, and these correlations appear as peaks off the diagonal of the spectrum. However, the dqf-COSY experiment is generally run in a phase-sensitive mode and gives a much cleaner spectrum near the diagonal than COSY, which is important for analysis of mixtures containing multiple components, such as crude pheromone extract, which may have many peaks near the diagonal. Each dqf-COSY correlation provides additional information about the protons involved in the correlation, including the coupling constant between those two protons (active coupling), as well as the coupling constants of those protons with other neighboring protons (passive coupling). The dqf-COSY spectrum of the crude pheromone extract contains peaks that are diagnostic for the ascarosides. Furthermore, dqf-COSY spectra have been acquired for many of the known ascarosides, and thus, the presence of a particular ascaroside in a crude pheromone extract or chromatography fraction can be substantiated by dqf-COSY. It should be noted, however, that the dqf-COSY spectrum of ascarosides from the same structural class, but with different side chain lengths, will be similar. One drawback of using NMR spectroscopy to analyze the ascarosides in the crude pheromone extract is that less abundant ascarosides cannot be detected because NMR spectroscopy is an inherently less sensitive technique than mass spectrometry. Further fractionation of the crude pheromone extract to give a less complex sample containing the specific ascaroside of interest is needed. However, chromatographic purification of individual ascarosides is dependent on the polarity of those ascarosides and done on an ad hoc basis, and thus, it is beyond the scope of this review.
2 Materials
2.1 25× Bacterial Stock
E. coli strain OP50 or HB101, grown on Luria-Bertani (LB)-agar.
LB broth.
Falcon tube (50 mL) for bacterial mini-culture.
Laminar flow hood.
2.8 L baffled flask for large bacterial culture.
Shaker for shaking bacterial culture at 37 °C.
Refrigerated table-top centrifuge.
Potassium phosphate (1 M, pH 6): In 0.8 L water, dissolve 136.1 g KH2PO4 . Add potassium hydroxide until the pH reaches 6. Then add water to a final volume of 1 L. Autoclave.
S basal: 0.1 M NaCl, 0.05 M potassium phosphate (pH 6). Autoclave.
Microfuge and 1.5 mL Eppendorf tubes.
2.2 Worm Culturing
C. elegans, wild-type (N2) or mutant strain.
Stereomicroscope and worm pick for manipulating worms.
Cholesterol (5 mg/mL in ethanol): Dissolve 50 mg of cholesterol in 10 mL of ethanol (200 proof). Sonicate for approximately 10 min or shake until the cholesterol dissolves.
25× bacterial stock from Subheading 3.1 for feeding worm cultures. 25 mL of 25× stock (corresponding to 625 mL-worth of bacterial culture) is needed for every 150 mL of worm culture that will be grown.
10 cm NGM-agar plates with bacterial lawn: In 500 mL water, add 1.5 g NaCl, 8.5 g granulated agar, and 1.25 g peptone. Autoclave, and then add: 0.5 mL 1 M CaCl2, 0.5 mL 1 M MgSO4, 12.5 mL potassium phosphate (pH 6), and 0.5 mL 5 mg/mL cholesterol. Pour into 10 cm plates, and let them dry on the benchtop overnight. Pipet 0.75 mL 25× bacterial stock onto each plate, spread on plate (but do not allow lawn to touch plate edge), and dry in laminar flow hood or on benchtop. Approximately one plate is needed for every 150 mL of worm culture that will be grown.
Potassium citrate (1 M, pH 6): In 0.8 L water, dissolve 210.14 g citric acid monohydrate. Add potassium hydroxide until the pH reaches 6. Then add water to a final volume of 1 L. Autoclave.
Trace metals solution: In 1 L water, dissolve 1.86 g disodium EDTA (5 mM), 0.69 g FeSO4 · 7H2O (2.5 mM), 0.20 g MnCl2 · 4H2O (1 mM), 0.29 g ZnSO4 · 7H2O (1 mM), 0.025 g CuSO4 · 5H2O (0.1 mM). Autoclave. Wrap bottle in foil, and store in the dark.
S medium: Autoclave 150 mL of S basal in a 500 mL Erlenmeyer flask, covering the top with foil. Just before use, add 1.5 mL 1 M potassium citrate (pH 6), 1.5 mL trace metals solution, 0.45 mL 1 M CaCl2 , 0.45 mL 1 M MgSO4 , and 150 μL cholesterol (5 mg/mL).
Optional: To help prevent contamination of the cultures, add the antifungal nystatin (100 U/mL final concentration) and the antibiotics penicillin–streptomycin (50 U/mL final concentration for penicillin and 50 μg/mL final concentration for streptomycin).
Refrigerated shaker for culturing worms in shaker flasks at 22.5 °C.
Graduated cylinder (250 mL) and ice bath.
Refrigerated table-top centrifuge and polypropylene centrifuge bottles with screw caps.
Lyophilizer and lyophilization flask (600 mL).
Mortar and pestle.
Erlenmeyer flask (500 mL) and funnel.
Ethanol (190 proof).
Tabletop shaker for performing extractions.
Filter paper (Whatman, No. 41), Buchner funnel, neoprene adaptor, and Erlenmeyer filtering flask (500 mL).
Rotary evaporator and round bottom flask (250 mL).
Short (5.75 in.) and long (9 in.) Pasteur pipets and bulbs.
Clean ball of cotton.
Bendable spatula.
Speedvac, rotor for 8 mL glass vials, and 8 mL glass vials (e.g., Fisher, cat. 03-340-60B) (see Note 1).
2.3 LC-MS
1. Methanol.
Sonicator.
Speedvac and rotor for 8 mL glass vials (see Note 1).
Microfuge and methanol-safe 1.5 mL Eppendorf tubes (e.g., Fisher, cat. 05-408-129).
Mass spectrometer: Agilent 6130 single quad mass spectrometer with API-ES source and operating in dual negative/positive scan mode.
LC: Agilent 1260 infinity binary pump with autosampler.
Column: Phenomenex 5 μm Luna C18 2, 100 Å (100 × 4.6 mm).
LCMS-grade water with 0.1 % formic acid (solvent A).
LCMS-grade acetonitrile with 0.1 % formic acid (solvent B).
Autosampler vials and glass inserts with springs.
2.4 LC-MS/MS
Methanol.
Sonicator.
Speedvac and rotor for 8 mL glass vials (see Note 1).
Microfuge and methanol-safe 1.5 mL Eppendorf tubes (e.g., Fisher, cat. 05-408-129).
Mass spectrometer: Thermo TSQ quantum ultra triple quadrupole mass spectrometer using argon collision gas, operating in negative ion, heated (H)-ESI, precursor scanning mode, selecting for a product ion of 73.0.
LC: Accela UHPLC (max pressure of 1,000 bar) with autosampler.
Column: Kinetex 2.6 μm C18 100 Å (100 × 2.10 mm).
LCMS-grade water (solvent A).
LCMS-grade acetonitrile with 0.1 % acetic acid (solvent B).
Autosampler vials and glass inserts with springs.
2.5 NMR Spectroscopy
Lyophilizer.
Methanol-d4.
Sonicator.
Short (5.75 in.) and long (9 in.) Pasteur pipets and bulbs.
Clean ball of cotton.
5 mm methanol-matched Shigemi NMR tube (Shigemi, cat. MMS-005v) (see Note 2).
Parafilm.
NMR spectrometer: Varian 500 MHz NMR spectrometer.
NMR probe: 5 mm Varian indirect-detection, triple resonance probe (1H/13C/(31P-15N)PFG).
Pasteur pipets with extra long tips for removing samples from NMR tubes (Fisher, cat. 13-678-6).
Speedvac, rotor for 8 mL glass vials, and 8 mL glass vials (e.g., Fisher, cat. 03-340-60B).
3 Methods
3.1 Preparing 25× Bacterial Stock
This method prepares the bacterial food (25× bacterial stock) needed for growing the worm cultures in Subheading 3.2. Sterile technique is important in the preparation of the 25× bacterial stock. If the 25× bacterial stock is contaminated with bacteria or fungus, this contamination may overwhelm the worm cultures that are grown in Subheading 3.2 , regardless of whether antibiotics and antifungals are added to the worm culture medium.
Inoculate 5 mL LB with a colony of OP50 or HB101 bacteria. Let the mini-culture grow for 6–8 h in a shaker at 250 rpm at 37 °C (see Note 3).
Use 1 mL of this mini-culture to inoculate 1 L of LB broth in a 2.8 L baffled flask. Grow culture overnight in a shaker at 250 rpm at 37 °C.
Collect bacteria by centrifuging at 2,700 × g for 10 min. Discard the supernatant, and resuspend in enough S basal to make the final volume equal to 40 mL to give a 25× bacterial stock. Transfer to a 50 mL falcon tube, and store at 4 °C (see Notes 4-6).
3.2 Unsynchronized Worm Cultures and Generating Crude Pheromone Extract
This method describes how to grow unsynchronized worm cultures and how to extract the culture medium to generate crude pheromone extract. Production of many of the ascarosides is maximized by feeding the worm cultures regularly with sufficient bacterial food to allow reproduction and increase worm density. It is important not to feed the worms too much on any 1 day because they will suffocate, but it is also important not to deprive the worms of food. The exact culture conditions used will influence the amounts of the ascarosides produced.
Passage 20 N2 worms onto a 10 cm NGM-agar plate with a bacterial lawn.
Let worms grow for ~4 days at room temperature until just before the bacterial food supply runs out.
Wash the plate with 10 mL of S medium, and put in a 15 mL tube. Count the number of worms in 3 × 10 μL drops under the stereomicroscope to gauge the density of worms. Make sure that you invert the tube several times before taking out each 10 μL sample (see Note 7).
Add ~90,000 worms per 150 mL culture.
Feed the worms 3 mL of 25× bacterial stock (110–125 mg/mL). For our feeding schedule, this day is considered day 0.
Shake at 22.5 °C at 225 rpm for 9 days. Feed worms 1 mL 25× bacterial stock on day 1, and 3 mL 25× bacterial stock on days 2–8.
On day 9, harvest the culture by transferring the culture to a tall container, such as a 250 mL graduated cylinder, and place the container in an ice-bath for 30 min to 1 h in order to settle the worms. Transfer the supernatant to a centrifuge bottle until you cannot pipet any more of the supernatant without disturbing the settled worms. Transfer the remaining worms and culture medium from the bottle to a 50 mL falcon tube, centrifuge at 200 × g for 5 min. Pipet the remaining supernatant from the falcon tube into the centrifuge bottle together with the previously pipetted supernatant. Centrifuge the centrifuge bottle at 2,700 × g for 10 min to remove the bacteria from the supernatant. Then carefully transfer the supernatant to a 600 mL lyophilization flask, and freeze by placing it at −20 °C overnight.
Lyophilize the frozen culture medium to dryness (2–3 days) (see Note 8).
Scrape out solid material into a mortar, and grind with a pestle to a fine powder.
Place the powder in a 500 mL Erlenmeyer flask using a funnel, and extract by adding 150 mL ethanol (190 proof) and shaking in a table-top shaker at 250 rpm for 2 h.
Filter the extraction mixture through filter paper in a Buchner funnel.
Place the retentate back in the Erlenmeyer flask from step 10 , and extract a second time with 150 mL of ethanol (190 proof), shaking overnight (see Note 9).
Filter the extraction mixture through filter paper in a Buchner funnel, and combine the filtrate with the filtrate from step 11.
Rotovap the combined filtrates in a round bottom flask to a small volume (~3 mL).
Place a small amount of cotton in a short Pasteur pipet, and pack it down inside the Pasteur pipet using a long Pasteur pipet, such that the packed cotton plug is about 3 mm in length.
Wash the cotton in the Pasteur pipet with a small amount of ethanol to remove any impurities from the cotton. Use a bulb to push the ethanol through the Pasteur pipet and dry the cotton.
Filter the sample through the cotton plug in the Pasteur pipet into a 8 mL glass vial. Use a bulb to push the sample through the Pasteur pipet.
Wash the round bottom with ~3 mL of ethanol (190 proof). Scrape the sides of the round bottom with a metal spatula to make sure that as much of the solid is exposed to the ethanol as possible.
Filter this ethanol wash through the cotton plug in the Pasteur pipet into the 8 mL glass vial from step 17 . Use a small amount of ethanol to wash the plug, and add this last wash to the glass vial as well.
Speedvac the sample in the glass vial to dryness. Store the sample at −20 °C until use.
3.3 LC-MS Quantitation of Ascarosides
This method can be used to quantify the concentrations of the ascarosides in a particular sample, such as in the crude pheromone extract (Fig. 1) or in worm secretions produced over a short period of time (“worm water”) [4]. The mass spectrometer must be well maintained with low background signal. It should be noted that this method can underestimate the amount of an ascaroside in a sample, especially if the sample is complex, if ion suppression occurs.
Fig. 1.

LC-MS-based determination of the concentrations of asc-ΔC9, asc-C6-MK, asc-ωC3, and IC-asc-C5 in crude pheromone extract. A calibration curve of synthetic ascarosides was generated by injecting 5 μL of several concentrations of the ascarosides and fi tting a quadratic curve to the data. 5 μL of crude pheromone extract was also injected, and the relevant ion for each ascaroside was extracted from the positive total ion count trace. The area under the peak for each ascaroside was used to estimate the number of pmol in 5 μL of crude pheromone extract. This number then was used to estimate the concentration of each ascaroside in the conditioned medium that was used to generate the crude pheromone extract. ( a) The concentration of asc-ΔC9 in the conditioned medium is ~273.7 nM. ( b) The concentration of asc-C6-MK in the conditioned medium is ~204.9 nM. ( c) The concentration of asc-ωC3 in the conditioned medium is ~144.5 nM. ( d) The concentration of IC-asc-C5 in the conditioned medium is ~77.4 nM
Resuspend the sample from Subheading 3.2 , step 20 , by adding 1 mL methanol. Sonicate for 20 min, vortex briefly to dislodge any remaining solids on the sides of the vial, and sonicate again for 20 min. Centrifuge the vial (with the cap closed) in the speedvac for 2 min to pellet most of the particulates.
Pipet 100 μL of the supernatant into a 1.5 mL Eppendorf tube, and centrifuge at 17,600 × g for 1 min to pellet any remaining particulates. Pipet 50 μL from the top of the sample to an autosampler vial. Pipet the remaining 50 μL of the sample from the tube back into vial (see Note 10).
Make 0.2, 0.4, 2, 4, 8, 20, 40, 60, 80, and 100 μM stocks of each synthetic standard in methanol. Transfer 20 μL of each stock to an autosampler vial (see Note 11).
For an Agilent 6130 single quadrupole mass spectrometer, use the following parameters for the spray chamber of the ion source: Drying gas flow of 12 L/min, nebulizer pressure of 35 psig, capillary voltage of 3,000 V, and drying gas temperature of 350 °C. Acquire signals in dual positive–negative scan mode with a 50 % cycle time between the two modes, peak width of 0.1 min, and cycle time of 1.08 s/cycle. Use a mass range of 100–1,000, fragmentor voltage of 125 V, gain of 1.00, threshold of 150, and stepsize of 0.10 (see Note 12).
Set the column temperature to 25 °C.
Set up the following chromatography method, where the solvent A is water with 0.1 % formic acid and solvent B is acetonitrile with 0.1 % formic acid: (1) ramp from 5 % B to 100 % B over 20 min, (2) hold at 100 % B for 2 min, (3) ramp from 100 % B to 5 % B over 1 min, and (4) hold at 5 % B for 2 min. Set the flow rate at 0.7 mL/min (see Note 13).
In order to clean the column, wash the column with 100 % acetonitrile for 10 min at 0.7 mL/min.
In order to equilibrate the column, wash the column with 5 % B for 10 min at 0.7 mL/min.
Use the autosampler to inject 5 μL of methanol into the LC-MS. This blank run will allow you to assess the background level of signal (see Note 14).
Use the autosampler to inject 5 μL of sample into the LC-MS.
Extract the relevant ion for the ascaroside of interest. That is, extract [M+Na]+ for the ascaroside of interest from the positive total ion count trace, or extract [M-H]− for the ascaroside of interest from the negative total ion count trace. Obtain the area under the peak for the extracted ion.
Use the autosampler to inject 5 μL of each concentration of the synthetic standards in turn into the LC-MS.
For each concentration of synthetic standard injected into the LC-MS, extract the relevant ion from the total ion count trace, and use the area under the peak to generate a standard curve.
Use the standard curve generated to estimate the concentration of the ascaroside of interest in the sample (see Fig. 1).
In order to clean and store the column, wash the column with 100 % acetonitrile for 10 min.
3.4 LC-MS/MS Analysis of Ascarosides
This method can be used to analyze the ascarosides in the crude pheromone extract that ionize in negative ion mode (Fig. 2). This method assumes that the user has a basic knowledge of LC-MS/MS and is assisted by an experienced mass spectrometry facility manager. The mass spectrometer must be well maintained with low background signal.
Fig. 2.

LC-MS/MS trace in precursor ion scanning mode (selecting for m/z 73.0 product ion) for crude pheromone extract. Asterisks indicate signals from unidentified compounds or non-ascarosides
Resuspend the sample from Subheading 3.2 , step 20 , by adding 1 mL methanol. Sonicate for 20 min, vortex briefly to dislodge any remaining solids on the sides of the vial, and sonicate again for 20 min. Centrifuge the vial (with the cap closed) in the speedvac for 2 min to pellet most of the particulates.
Pipet 100 μL of the supernatant into a 1.5 mL Eppendorf tube, and centrifuge at 13,700 rpm for 1 min to pellet any remaining particulates. Pipet 50 μL from the top of the sample to an autosampler vial with glass insert with spring. Pipet the remaining 50 μL of the sample from the tube back into vial (see Note 10).
Set the mass spectrometer to operate in negative ion, heated (H)-ESI, precursor scanning mode, selecting for a product ion of 73.0. If you are using a Thermo TSQ quantum ultra mass spectrometer, use the following parameters for the mass spectrometer: spray voltage of 3,000 V, vaporizer temperature of 300 °C, sheath gas pressure of 40 arb, auxillary gas pressure of 20 arb, capillary temperature of 350 °C, skimmer offset of 5 V, collision pressure of 0.8 mTorr, collision energy of 25 eV, and scan time 0.4 s. We usually set the mass range to m/v 200–500 (see Notes 15 and 16).
Set the column temperature to 30 °C.
Set up the following chromatography method, where solvent A is water and solvent B is acetonitrile with 0.1 % acetic acid: (1) hold at 2 % B for 1 min, (2) ramp from 2 % B to 50 % B over 34 min, (3) ramp from 50 % B to 100 % B over 1 min, (4) hold at 100 % B for 4 min, (5) ramp from 100 % B to 2 % B over 1 min, (6) hold at 2 % B for 4 min. Set the flow rate at 0.25 mL/min (see Note 17).
In order to clean the column before use, wash the column with 100 % acetonitrile for 10 min at 0.25 mL/min.
In order to equilibrate the column, wash the column with 2 % solvent B for 10 min at 0.25 mL/min.
Use the autosampler to inject 5 μL of methanol into the LC-MS/MS. This blank run will allow you to assess the background level of signal (see Note 14).
Use the autosampler to inject 5 μL of sample into the LC-MS/MS.
After the run is complete, smooth the trace using a Boxcar or Gaussian function, with 5–7 smoothing points (see Note 18).
Obtain the areas under the peaks for each ascaroside by extracting each specific precursor ion from the total ion count trace (see Note 19).
In order to validate that a given peak is a given ascaroside, you will need to acquire a synthetic standard. Dissolve the standard(s) in ethanol (200 proof) at 270 μM. Transfer some of the sample to an autosampler vial, and use the autosampler to inject 5 μL from the autosampler vial into the LC-MS/MS.
After the run is complete, verify that the synthetic standard has the same retention time as the tentatively assigned peak from step 11.
In order to clean and store the column, wash the column with 100 % acetonitrile for 10 min.
3.5 NMR Spectroscopy-Based Analysis of Ascarosides Using the dqf-COSY Experiment
This method can be used to analyze the ascarosides in the crude pheromone extract (Fig. 3) or chromatography fractions generated from the crude pheromone extract. The ascarosides must be present at sufficient concentrations in the crude pheromone extract for detection by NMR spectroscopy. Subheading 3.5 assumes that the user has a basic knowledge of NMR and is assisted by an experienced NMR facility manager. The NMR spectrometer must be well maintained with an updated shim set. Although we use a Varian NMR spectrometer in our own experiments, we employ terminology in this method that should be interpretable by users familiar with either Agilent (Varian) or Bruker NMR spectrometers. In order to have the necessary chemical shift separation in the complex spectrum obtained from crude pheromone extract, an NMR spectrometer with a magnetic field of 500 MHz or higher should be used. The number of points in both dimensions should provide for a resolution of 1–2 Hz per point. The concentrations of the ascarosides in the crude pheromone extract are relatively low compared to the concentrations of other more abundant metabolites. Therefore, it is critical to maximize the signal-to-noise ratio for the sample, and thus, an indirect detection probe or a cold probe is recommended.
Fig. 3.

dqf-COSY spectrum of crude pheromone extract, highlighting specific correlations for bolded protons for specifi c ascarosides (asc-ΔC9, asc-C6-MK, and asc-ωC3) that are visible in the spectrum. ( a and b) Highlight the below diagonal and above diagonal correlations between the bolded proton at 5.81 ppm and the bolded proton at 6.92 ppm in asc-ΔC9. The active coupling can be measured in ( a) and ( b) to give the coupling constant between the two bolded protons (15.6 Hz). The passive coupling can be measured in ( b) to give the coupling constant between the bolded proton at 6.92 ppm and the other protons to which it is coupled (6.8 Hz). ( c) Highlights the above diagonal correlations between the bolded protons at 2.61 ppm and the two nonequivalent bolded protons at 1.71 ppm and 1.77 ppm in asc-C6-MK. The active coupling between the bolded protons can be measured in ( c). The measured value of 14.8 Hz must be divided in two (to give coupling constants of approximately 7.4 Hz) because the bolded proton at 2.61 ppm is a triplet and oppositely phased peaks in the center of the triplet cancel each other. The below diagonal correlation is not highlighted as it is too faint. ( d) Highlights the below diagonal correlation between the two bolded protons at 2.56 ppm and the bolded proton at 3.94 ppm in asc-ωC3. The above diagonal correlation is not highlighted as it is obscured by a vertical stripe
Resuspend the sample from Subheading 3.2, step 20 , by adding 1 mL methanol-d4 . Sonicate for 20 min, vortex briefly to dislodge any remaining solids on the sides of the vial, and sonicate again for 20 min. Centrifuge in the speedvac (with the cap closed) for 2 min to pellet any particulates that do not dissolve.
Transfer the supernatant to a new vial, and speedvac or rotovap the sample to dryness (see Note 20).
Place the vial of dry sample, a short Pasteur pipet, and an empty 5 mm methanol-matched Shigemi tube on the lyophilizer for 1–2 h. This drying step will remove any trace amounts of methanol and water in the sample that would otherwise interfere with the signal of the sample in the NMR spectrum.
Resuspend your sample by adding 300 μL of methanol-d4 and sonicating for 5 min.
Place a small amount of cotton in the short Pasteur pipet, and pack it down inside the Pasteur pipet using a long Pasteur pipet, such that the packed cotton plug is about 3 mm in length.
Wash the cotton in the Pasteur pipet with a small amount of methanol-d4 to remove any impurities from the cotton. Use a bulb to push the methanol-d4 through the Pasteur pipet and dry the cotton.
Filter the sample through the cotton plug in the Pasteur pipet, directly into the Shigemi tube. Use a bulb to push the sample through the Pasteur pipet. Use a few drops of methanol-d4 to wash the cotton plug, allowing the wash to drip directly into the Shigemi tube (see Note 21).
Insert the solvent-matched Shigemi tube insert into the Shigemi tube, taking care to avoid any bubbles between the sample and the insert. Use a small strip of parafilm (about 0.5 × 2 cm in length) to wrap around the top of the tube to prevent any evaporation of the sample while it is in the NMR spectrometer (see Notes 22 and 23).
Insert your sample into the spectrometer. Setup parameters for a 1H spectrum in methanol (see Note 24).
Lock the sample, and optimize the lock phase.
Gradient shim the sample.
Tune the 1H channel of the probe.
Turn off the spinner (see Note 25).
Acquire a 1H spectrum with 16 scans (and with the spinner off) (see Notes 26 and 27).
Set the sweep width to the region of the 1H spectrum that you are interested in for the 2D experiment. Usually you will select the region where there are peaks, plus 0.5 ppm on either side. In general, for the crude pheromone extract, the sweep width will extend from 0.5 to 9.5 ppm.
Determine the 90° pulse width. Make sure that the pulse width is equal to this new optimized value (see Note 28).
Acquire another 1H spectrum (see Note 29).
Move the 1H spectrum into a new experiment in order to move all optimized parameters, including sweep width, 90° pulse width, and gain, into the new experiment. Setup up a dqf-COSY experiment with the default parameters.
Change the default parameters in the following manner: the number of scans to 16, the number of iterations to 1,024, the number of points to 4,096, and the relaxation delay to 1 (see Notes 30 and 31).
Start the experiment, and once finished, process the data (see Note 32).
Acknowledgments
Funding for this work was provided by the National Institutes of Health (GM87533 to R.A.B.) and the Human Frontiers Science Program (RGY0042/2010 subcontract to R.A.B.). Our work on the triple quadrupole mass spectrometer was supported in part by the National Institutes of Health and National Center for Research Resources CTSA grant 1UL 1RR029890. We would like to thank Dr. Tim Garrett (University of Florida) for helpful suggestions on the LC-MS/MS parameters and Dr. Ion Ghiviriga (University of Florida) for helpful suggestions on the NMR parameters. We would like to thank Prof. Justin R. Ragains (Louisiana State University) for providing the synthetic ascaroside standards that are used in our work.
Footnotes
If a speedvac with a rotor for 8 mL glass vials is not available, the material can be dried on a rotovap with a 24/40 glass adaptor (e.g., Chemglass cat. CG-1318-10) and connector (e.g., Chemglass cat. CG-1318-21) for the 8 mL glass vials.
This tube is magnetic susceptibility-matched to methanol. The catalog number given is for a tube that is designed for use with a 5 mm Varian NMR probe.
Because sterile technique is important, we perform as many of the steps as possible in a laminar flow hood.
To gauge the sterility of the 25× bacterial stock, streak it out on an LB-agar plate and allow colonies to grow. There should be only one colony type.
The 25× bacterial stock can be stored at 4 °C for up to 1 month.
Typically, if HB101 or OP50 is grown in LB broth as described, 110–125 mg/mL of bacteria will be obtained for the 25× stock. Calculate the stock concentration by weighing an empty 1.5 mL Eppendorf tube and then adding 1 mL of 25× stock to it. Centrifuge the tube in the microfuge at 13,700 rpm for 1 min, and remove the supernatant by pipetting. Centrifuge again, and remove any remaining supernatant by pipetting. Reweigh the tube with the pellet in it. The weight of the pellet gives the stock concentration in mg/mL.
Generally, for N2, ~90,000 worms are obtained from a single 10 cm plate, but for some mutants, especially for those that like to dig into the agar, this number can be lower.
It is important that there are no chunks of frozen material remaining, since it will interfere with the extraction process. After 2 days, it is helpful to use a spatula to break apart any remaining material, and place the flask back on the lyophilizer.
The second extraction significantly increases the pheromone yield.
If the sample will be used again within a week, store at −20 °C. If the sample will not be used again within a week, speedvac it to dryness, and store at −20 °C.
If a standard curve will be generated for more than one synthetic ascaroside, it is convenient to put all synthetic standards together at a given concentration for simultaneous injection.
These parameters are instrument dependent. The fragmentor voltage was optimized on our instrument using a synthetic ascaroside standard (20 μM asc-ΔC9 in ethanol). If a mass spectrometer other than an Agilent 6130 single quadrupole mass spectrometer will be used, these parameters may need to be re-optimized.
This chromatography protocol was optimized for monitoring the concentrations of specific ascarosides (asc-ωC3, asc-C6-MK, asc-ΔC9, IC-asc-C5, as shown in Fig. 1). It should be noted that this gradient is a short one, and so two or more ascarosides in the crude pheromone extract may elute at the same time. For example, two ascarosides with similar polarities and the same mass (e.g., asc-C5 and asc-ωC5) may elute from the column at the same time, which would be problematic if the objective was to analyze their concentrations. To prevent co-elution of two or more ascarosides, a longer gradient may be required such as the gradient used in Subheading 3.4.
If there is a high background level of signal, it most likely indicates that the column is dirty and must be cleaned more thoroughly before use.
These parameters are instrument dependent. They were optimized on our instrument based on direct infusion of a synthetic ascaroside standard (4 μM asc-ΔC9 in ethanol). If a mass spectrometer other than a Thermo TSQ quantum mass spectrometer will be used, these parameters will need to be re-optimized.
This mass range should be shifted if you are interested in analyzing ascarosides with larger masses, such as the long-chain ascarosides.
This chromatography protocol was optimized for the separation and analysis of more polar, short-chain ascarosides, but not for the separation and analysis of less polar, long-chain ascarosides. If you are interested in separating and analyzing long-chain ascarosides, you will need to extend the ramp from 50 % B to 100 % B over a longer period of time.
Using greater than seven smoothing points may create artificial peaks.
Different ascarosides ionize and fragment with different efficiencies, so the area of a given peak is not a very good indication of its relative abundance.
This drying step will remove trace amounts of methanol and water in your sample that would otherwise interfere with the signal of the sample the NMR spectrum.
The sample should extend exactly 2 cm in the Shigemi tube. This volume of sample will allow the sample to extend the full length of the receiver coil when inserted into the NMR spectrometer. When washing the cotton filter with a few drops of methanol-d4 , take great care not to add too much volume, since the sample will then extend longer than 2 cm in the Shigemi tube and will be more dilute. A more dilute sample will give lower signal in the NMR spectrum.
If bubbles appear, they must be removed since they may interfere with optimal shimming. They can be removed by slightly pushing the insert 1–2 mm further into the tube with a quick turning motion.
Shigemi tubes have a smaller sample window than regular 5 mm NMR tubes because both the bottom of the tube and the insert that fits into the tube are made of solvent-matched glass. Thus, the volume of solvent required to dissolve the sample and fill the tube is smaller, thereby increasing the concentration of the sample and the signal-to-noise ratio in the NMR spectrum.
Be sure to align the Shigemi tube in the spinner using the depth gauge so that once the tube is inserted into the NMR spectrometer, the deuterated solvent will be centered within the receiver coil and will extend the full length of the receiver coil. If the deuterated solvent does not extend the full length of the receiver coil, it will cause issues with shimming.
If the lock level drops significantly (>10 %) when the spinner is turned off, it indicates poor shimming. Adjust the non-spinning (x , y) shims accordingly.
The number of scans determines the signal-to-noise ratio of the spectrum. For concentrated samples such as the crude pheromone extract, 16 scans is generally good, but for more dilute samples, such as chromatography fractions from the crude pheromone extract, the number of scans should be set higher (e.g., 32). In general, if a sample is more dilute by a factor of 2, then the number of scans must be increased by a factor of 4 in order to obtain a spectrum with a similar signal-to-noise ratio. The number of scans in a dqf-COSY should be a multiple of 8 due to phase-cycling.
Make sure that the peak shapes are sharp and that the methanol peak at 3.31 ppm is a fully defined quintet. If the peak shape is not sharp or is skewed, it indicates poor shimming. Spinning the tube improves peak shape, but since spinning will be turned off during the 2D dqf-COSY experiment, it is important to assess peak shape in a 1H spectrum in which the spinner has been turned off.
Because the crude pheromone extract has a high concentration of salt, the 90° pulse width can be much higher than for more dilute samples. Optimizing the 90° pulse width will improve the signal-to-noise ratio in the 2D dqf-COSY experiment.
Acquiring this second 1H spectrum with an optimized 90° pulse width will give you a lower gain value than that obtained in the original 1H spectrum. Use this new lower gain value in the dqf-COSY experiment.
The number of iterations determines the resolution in the indirect (f1) dimension and should be at least 512 for a dqf-COSY experiment performed on a complex mixture. If the experiment time (which is proportional to the number of iterations) is not an issue, then the number of iterations should be increased to 1,024.
The resolution in the direct (f2) dimension depends on the number of points. Since increasing the number of points does not significantly increase the experiment time, there is no disadvantage to setting the number of points to as high as 4,096 (except that it will increase the file size). The number of points should be equal to a power of two since the Fourier transform uses 2n points. Given a sweep width of approximately 9 ppm, setting the number of points to 4,096 will give an acquisition time of approximately 0.5.
The desktop processing software, MestreNova, is a convenient way to process NMR data. In the f2 dimension, use a sine-bell window function with a 90° shift (to obtain high resolution) or a squared sine-bell window function with a 55° shift (if high noise is present). In the f1 dimension, use a sine-bell window function with a 90° shift. For zero-filling, the size of the Fourier transform should be 4,096 in both dimensions. The spectrum will also need to be phased.
References
- 1.Jeong PY, Jung M, Yim YH, et al. Chemical structure and biological activity of the Caenorhabditis elegans dauer-inducing pheromone. Nature. 2005;433:541–545. doi: 10.1038/nature03201. [DOI] [PubMed] [Google Scholar]
- 2.Butcher RA, Fujita M, Schroeder FC, et al. Small-molecule pheromones that control dauer development in Caenorhabditis elegans. Nat Chem Biol. 2007;3:420–422. doi: 10.1038/nchembio.2007.3. [DOI] [PubMed] [Google Scholar]
- 3.Butcher RA, Ragains JR, Kim E, et al. A potent dauer pheromone component in C. elegans that acts synergistically with other components. Proc Natl Acad Sci USA. 2008;105:14288–14292. doi: 10.1073/pnas.0806676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srinivasan J, Kaplan F, Ajredini R, et al. A blend of small molecules regulates both mating and development in Caenorhabditis elegans. Nature. 2008;454:1115–1118. doi: 10.1038/nature07168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pungaliya C, Srinivasan J, Fox BW, et al. A shortcut to identifying small molecule signals that regulate behavior and development in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2009;106(19):7708–7713. doi: 10.1073/pnas.0811918106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butcher RA, Ragains JR, Clardy J. An indole-containing dauer pheromone component with unusual dauer inhibitory activity at higher concentrations. Org Lett. 2009;11:3100–3103. doi: 10.1021/ol901011c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Reuss SH, Bose N, Srinivasan J, et al. Comparative metabolomics reveals biogenesis of ascarosides, a modular library of small-molecule signals in C. elegans. J Am Chem Soc. 2012;134:1817–1824. doi: 10.1021/ja210202y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srinivasan J, von Reuss SH, Bose N, et al. A modular library of small molecule signals regulates social behaviors in Caenorhabditis elegans. PLoS Biol. 2012;10:e1001237. doi: 10.1371/journal.pbio.1001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Artyukhin AB, Yim JJ, Srinivasan J, et al. Succinylated octopamine ascarosides and a new pathway of biogenic amine metabolism in C. elegans. J Biol Chem. 2013;288:18778–18783. doi: 10.1074/jbc.C113.477000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butcher RA, Ragains JR, Li W, et al. Biosynthesis of the Caenorhabditis elegans dauer pheromone. Proc Natl Acad Sci USA. 2009;106:1875–1879. doi: 10.1073/pnas.0810338106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplan F, Srinivasan J, Mahanti P, et al. Ascaroside expression in Caenorhabditis elegans is strongly dependent on diet and developmental stage. PLoS One. 2011;6:e17804. doi: 10.1371/journal.pone.0017804. [DOI] [PMC free article] [PubMed] [Google Scholar]