

Abstract
The RIG-I-like receptors (RLR) RIG-I, MDA5 and LGP2 trigger innate immune responses against viral infections that serve to limit virus replication and to stimulate adaptive immunity. RLRs are cytosolic sensors for virus-derived RNA and thus responsible for intracellular immune surveillance against infection. RLR signaling requires the adapter protein MAVS to induce type I interferon, interferon-stimulated genes and proinflammatory cytokines. This review focusses on the molecular and cell biological requirements for RLR signal transduction.
General principles of the antiviral innate immune response
Viruses are obligate intracellular parasites and thus depend strictly on the biosynthetic machinery of the host in order to replicate and spread. As a result, the virus-driven exploitation of the host cell’s metabolic pathways and reprogramming of cellular processes often lead to cell death. The struggle for survival between virus and host is ancient and as a consequence both have evolved multiple strategies to antagonize each other. While mammalian hosts developed sophisticated mechanisms of antiviral immunity, viruses acquired strategies to evade the immune response. Therefore it is critical for the host to mount an effective innate and adaptive immune response immediately upon infection in order to successfully combat the pathogen.
The innate immune response constitutes the earliest phase of the host’s defense against pathogens and will stimulate and modulate the later onset adaptive response (Palm and Medzhitov, 2009). It operates through a set of germ line-encoded pattern recognition receptors (PRR) that recognize pathogen-associated molecular patterns (PAMP) of viruses, bacteria, fungi and protozoa. PAMPs are conserved within broad classes of pathogens. They are typically products of biosynthetic pathways that are essential for the survival of the pathogen and thus lack the potential for immune evasion through genetic variability (Medzhitov, 2007). Owing to the panel of PAMPs that is recognized by PRRs, the innate immune system achieves an impressively complete coverage of pathogens despite the genetically limited number of available receptors. Engagement of antiviral PRRs by their cognate PAMPs activates signaling pathways that lead to the production of defense factors such as pro-inflammatory cytokines, type I interferons (IFN-α and IFN-β), or interferon stimulated genes (ISG). ISGs induced by IFN secretion or cell-autonomously upon viral infection collectively establish an antiviral state that limits viral replication and prevents further spread of the infection (Katze, He and Gale, 2002).
Detection of viruses poses a particular challenge to the host as they lack features in line with the postulated characteristics of PAMPs, i. e. invariant structures required for survival. With few exceptions viral proteins are highly variable without being functionally compromised by mutation. Moreover, viruses are obligate parasites relying on the host metabolism for their replication. The evolutionary solution to this problem is to recognize viral nucleic acids, either virus genomes or replication intermediates. Undoubtedly, nucleic acid is not a PAMP that is unique to viruses and thus virus detection comes at the cost of the potential for autoimmunity (Barton and Kagan, 2009). Nucleic acid detection is accomplished by a growing list of PRRs, namely the cytosolic RIG-I-like receptors (RLR) RIG-I and MDA5 (Yoneyama et al., 2005; Yoneyama et al., 2004), the endosomal Toll-like receptors TLR3, TLR7/8, TLR9 and TLR13 (Kawai and Akira, 2010), the Ifi16/cGAS/STING axis (Ishikawa, Ma and Barber, 2009; Sun et al., 2012; Unterholzner et al., 2010; Wu et al., 2012), and the AIM2 inflammasome (Burckstummer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009; Roberts et al., 2009). This review will focus on virus-induced signaling by RIG-I like receptors; nucleic acid sensing by other receptor families is reviewed elsewhere (Barbalat et al., 2011).
II. RIG-I like receptors are RNA sensors
A. Common and distinct features of RIG-I like receptors and their signaling capabilities
RLRs detect RNA derived from RNA viruses and in some instances DNA viruses. In terms of specificity and signaling output, RLRs are most similar to TLR3, as both detect viral RNA and induce ISGs, type I IFN and proinflammatory cytokines (Alexopoulou et al., 2001; Matsumoto et al., 2003; Schulz et al., 2005). However, there is a fundamental conceptual difference in nucleic acid detection between TLRs and RLRs. The nucleic acid-specific endosomal TLRs TLR3, TLR7/8 and TLR9 recognize extracellular nucleic acids having reached the endosomes through endocytosis (Takeda and Akira, 2005), whereas RLRs are cytosolic receptors required for detection of intracellular viral RNA from actively replicating viruses (Kawai and Akira, 2006). As such, RLRs represent an indispensable means for determining if a given cell is infected or not. In line with this key role in antiviral immunity, RLR signaling operates in most cell types. In contrast, TLR expression is restricted to specialized immune cells such as macrophages and dendritic cells. Even though RLRs are expressed in plasmacytoid dendritic cells, TLRs but not RLRs are required for IFN-α production in this cell type (Kato et al., 2005).
Three highly related proteins constitute the family of RLRs: The founding member RIG-I, MDA5 and LGP2. They are characterized by a central ATPase containing DExD/H box helicase domain. RIG-I and MDA5 contain N-terminal tandem CARD domains that mediate downstream signaling, whereas LGP2 lacks a CARD (Yoneyama et al., 2005; Yoneyama et al., 2004). RIG-I and LGP2 also harbor a repressor domain (RD) in their C-terminal regulatory domains (CTD) (Figure 1). Due to the presence of the RD in RIG-I, its overexpression in the absence of an activating ligand does not result in signaling, whereas MDA5 overexpression is sufficient to activate the pathway. In accordance with their domain architecture RLRs lacking the CARDs have a dominant-negative phenotype. RIG-I devoid of the CTD or the N-terminal fragment comprising solely the CARDs signal constitutively (Cui et al., 2008; Saito et al., 2007; Takahasi et al., 2008). All RLRs are present at low levels in resting cells, but their expression is strongly induced by type I IFN creating a feed forward loop for a robust antiviral response (Kang et al., 2004; Yoneyama et al., 2005; Yoneyama et al., 2004).
Figure 1.
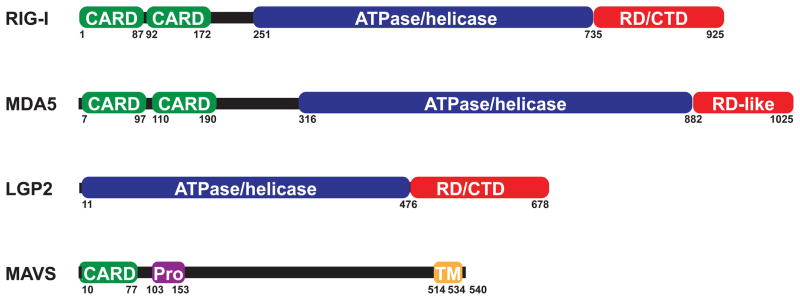
Despite different ligand specificities for viral RNA, both RIG-I and MDA5 rely on the same signaling cascade to trigger the expression of type I IFNs, ISGs and proinflammatory cytokines (Yoneyama et al., 2005). The adapter protein MAVS (also known as IPS-1, VISA and Cardif) (Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005) acts immediately downstream of the receptors and represents a node from which RLR signaling branches in several directions in order to promote the activation of NF-κB through the canonical IKKs IKK-α, IKK-β and IKK-γ, of ATF2/c-jun through MAPK activation and most importantly of members of the interferon regulatory factor (IRF) family of transcription factors (Kawai et al., 2005; Meylan et al., 2005; Mikkelsen et al., 2009; Poeck et al., 2010; Seth et al., 2005; Xu et al., 2005). IRF3 and IRF7 are the essential transcription factors for IFN-β gene transcription, as activation of NF-κB and ATF-2/c-Jun alone is not sufficient for IFN-β induction. Interestingly, in dendritic cells, IRF5 can also function to promote IFN-β expression (Lazear et al., 2013). They reside in the cytosol in their latent forms until viral infection activates the non-canonical IKKs TBK1 and IKK-i. Phosphorylation of IRF3 and IRF7 by these kinases causes hetero- or homodimerization and nuclear translocation. IRF3 and/or IRF7, NF-κB and ATF-2/c-Jun together with the transcriptional coactivator CBP/p300 and the architectural protein HMG I(Y) assemble in an enhanceosome to direct IFN-β transcription (Hiscott, 2007; Honda, Takaoka and Taniguchi, 2006) (Figure 2).
Figure 2.

Once IFN-β is secreted it binds to the IFN-α/β receptor (IFNAR) in an autocrine and paracrine manner resulting in JAK-STAT signaling and thus expression of several hundred ISGs by the ISGF3 transcription factor, which consists of STAT1, STAT2 and IRF9 (Platanias, 2005). However, despite their namesake, ISGs may also be induced independently of a preceding secretion of type I IFN (Collins, Noyce and Mossman, 2004; Mossman et al., 2001). Many ISGs function as direct antiviral effectors, acting to prevent viral genome replication, viral particle assembly, or virion release from infected cells. Others encode components of signaling pathways such as receptors for pathogen recognition or transcription factors resulting in a stronger IFN response and thereby creating a positive feedback loop.
The role of LGP2 in antiviral immunity is less clear. LGP2 lacks a CARD domain (Figure 1). Devoid of a signaling domain, LGP2 was proposed to be a negative regulator of RLR signaling. Overexpression of LGP2 does not activate IFN-β induction. On the contrary, reduced IRF3 activation was observed when LGP2 overexpressing cells were infected with Newcastle disease virus (NDV) (Rothenfusser et al., 2005; Yoneyama et al., 2005). In vivo experiments with different lines of LGP2 deficient mice strongly contradict the previous data generated by in vitro studies and implicate LGP2 as a positive regulator (Satoh et al., 2010; Venkataraman et al., 2007). In the absence of LGP2, both RIG-I and particularly MDA5-dependent responses to RNA virus infection are impaired, whereas responses to synthetic ligands of these RLRs are unaffected (Satoh et al., 2010). Presumably LGP2 facilitates binding of viral RNA – potentially in complex with protein – to its cognate receptor, whereas the affinity of RIG-I and MDA5 is sufficiently strong to bind to “naked” synthetic agonists. Structural analysis of the binding interface of RNA with the CTD of RIG-I supports this model, as it predicts weaker affinity of MDA5 than RIG-I to its ligand (Takahasi et al., 2009). In addition to confirming the role of LGP2 as a positive, yet non-essential regulator of RLR signaling, a recent report implicates LGP2 as a cell-intrinsic regulator of virus-specific CD8+ T cell survival and effector functions. CD8+T cells are crucial for controlling West Nile virus (WNV) pathology in the brain. LGP2 deficient mice displayed higher viral burden, and significantly lower WNV-specific CD8+ T cells in the brain leading to increased mortality as compared to wild-type animals (Suthar et al., 2012). Nonetheless further clarification is required to determine the role of LGP2 in RLR signaling.
B. Structural characteristics of synthetic RLR ligands
The two best characterized RLRs, RIG-I and MDA5, recognize structurally distinct RNA species that have reached the cytosol by infection or by means of transfection. Being cytosolic receptors, RIG-I and MDA5 do not respond to extracellular nucleic acid.
The RIG-I ligand comprises a RNA molecule with two features: i) a 5′-triphosphate (Hornung et al., 2006; Pichlmair et al., 2006) and ii) base pairing at the 5′-end due to secondary RNA structures such as hairpin or panhandle conformations (Schlee et al., 2009; Schmidt et al., 2009). Studies aimed at the characterization of molecular features of the RIG-I ligand largely rely on in vitro transcripts. In vitro transcribed RNA by all known RNA polymerases leaves a triphosphate at the 5′ end of the transcript (pppRNA) (Schlee and Hartmann, 2010). Transfection of pppRNA into monocytes resulted in robust IFN-α secretion, whereas RNA lacking a triphosphate did not (Hornung et al., 2006). Similarly, highly immunogenic RNA extracted from influenza-infected cells was rendered nonstimulatory after phosphatase treatment (Pichlmair et al., 2006). However, a 5′-triphosphate alone is not sufficient to mark a single-stranded (ss) RNA molecule as non-self and to render it immunogenic. In support of this notion, synthetic 5′-triphosphate-ssRNA did not activate RIG-I signaling. In contrast, when the same ssRNA molecule was generated by in vitro transcription, it was stimulatory. Reverse cloning and sequencing of the latter RNA species revealed the presence of sequences generated by self-coding intramolecular 3′-extension leading to blunt ended RNA with complementary 5′ and 3′-ends. Thus aberrant in vitro transcription products are responsible for the immunostimulatory properties of such preparations. The minimal length of the 5′-base paired region was found to be 19 bp. Furthermore, a 3′-overhang of 2 nt reduced the stimulatory activity by 70%, while no 5′-overhang was tolerated (Schlee et al., 2009). Alternatively to 5′-base pairing, sequence composition may contribute to the stimulatory potential of pppRNA. Hepatitis C virus (HCV) genomic ssRNA is characterized by polyuridine motifs with interspersed C nucleotides (referred to as poly-U/UC motifs) and a 5′-triphosphate. Deletion of the poly-U/UC motif abrogated the stimulatory activity of HCV genomic RNA (Saito et al., 2008; Uzri and Gehrke, 2009). Thus both panhandle structures and poly-U/UC may serve as a secondary PAMP for pppRNA. However, short synthetic double-stranded (ds) RNA without a 5′-triphosphate was reported to activate RIG-I as well (Kato et al., 2008; Takahasi et al., 2008). Notably, the antiviral protein RNaseL, can cleave ssRNA of virus or host origin and thereby generate short (200 nt) ligands devoid of a 5′-triphosphate for RIG-I and MDA5 (Malathi et al., 2007). The molecular features responsible for the immunogenicity of RNaseL-generated ligands have not been identified.
The molecular nature of the MDA5 ligand remains poorly characterized. The stereotypic MDA5 agonist is poly I:C (Gitlin et al., 2006; Kato et al., 2006), a synthetic RNA molecule lacking 5′-triphosphates that is generated by annealing of poly-inosine strands to poly-cytidine strands of various lengths. Thus polyI:C contains an ill-defined mix of ramified ds and ssRNA. Size fractionation of polyI:C revealed that MDA5 responds to high molecular weight (HMW) polyI:C, whereas polyI:C shorter than 1000 nucleotides acts as a RIG-I agonist (Kato et al., 2008). Size fractionation of total RNA isolated fom encephalomyocarditis virus (EMCV)-infected cells yielded a prominent dsRNA fraction of 11 kb and an even larger molecular weight RNA aggregate with variable ss and dsRNA content. Of note only the RNA aggregate, but not the dsRNA stimulated MDA5 activity. Furthermore, this fraction required its intact secondary and tertiary structure to remain fully active (Pichlmair et al., 2009). Thus MDA5 preferentially binds to high molecular weight dsRNA that presumably adopts a weblike conformation much like the synthetic RNA analog polyI:C.
C. Viruses
The structural features of viral RNA that are displayed by a given virus depend on its replication cycle. As a consequence, the different ligand specificities of RIG-I and MDA5 are reflected by the largely non-overlapping pattern of virus susceptibility of mice deficient in either of the two RLRs. RIG-I is required for innate responses to many ssRNA viruses. The best studied examples among these are the negative-stranded viruses of the orthomyxoviridae e.g. influenza A and B virus, paramyxoviridae e. g. Newcastle disease virus (NDV), Sendai virus (SeV), respiratory syncytial virus (RSV) and measles virus and rhabdoviridae e. g. vesicular stomatitis virus (VSV) and rabies virus (Hornung et al., 2006; Kato et al., 2006; Loo et al., 2008; Plumet et al., 2007). Moreover, detection of positive-stranded flaviviruses including hepatitis C virus (HCV) and Japanese encephalitis virus (JEV) is RIG-I-dependent (Kato et al., 2006; Saito et al., 2007; Sumpter et al., 2005). In addition, recognition of cytoplasmic DNA can also feed into the RIG-I pathway. RIG-I does not detect DNA directly, but can do so after RNA polymerase III-mediated transcription of AT-rich DNA. IFN induction in response to infection with DNA viruses such as adenovirus, herpes simplex Virus-1 (HSV-1) and Epstein-Barr virus (EBV) relies on this pathway (Ablasser et al., 2009; Chiu, Macmillan and Chen, 2009; Samanta et al., 2006). MDA5 is required for protection against picornaviruses such as encephalomyocarditis virus (EMCV), Theiler’s virus, mengovirus, murine norovirus and murine hepatitis virus (Gitlin et al., 2006; Kato et al., 2006; McCartney et al., 2008; Roth-Cross, Bender and Weiss, 2008). Similarly to RIG-I, MDA5 has also been implicated in DNA virus detection. Vaccinia virus, a dsDNA virus of the poxvirus family, activates MDA5 via a yet to be characterized mechanism (Pichlmair et al., 2009). Some viruses like West Nile virus (WNV), Dengue virus, reovirus and lymphocytic choriomeningitis virus (LCMV) (Fredericksen et al., 2008; Loo et al., 2008; Zhou et al., 2010) trigger both RIG-I- and MDA5-dependent innate immune responses. RLR-dependence of the aforementioned viruses was determined by infection of different RLR-deficient cell types or mice with purified virions and is summarized in table 1.
Table 1.
RIG-I and MDA5 detect different sets of viruses
| Viruses detected by RIG-I | ||
|
| ||
| Orthomyxoviridae (−) ssRNA, NS | Influenza A virus | (Kato et al., 2006) |
| Influenza B virus | (Loo et al., 2008) | |
|
| ||
| Paramyxoviridae (−) ssRNA, NS | Sendai virus | (Kato et al., 2006) |
| Newcastle disease virus | (Kato et al., 2006) | |
| Respiratory syncytial virus | (Loo et al., 2008) | |
| Measles virus | (Plumet et al., 2007) | |
|
| ||
| Rhabdoviridae (−) ssRNA, NS | Vesicular stomatitis virus | (Kato et al., 2006) |
| Rabies virus | (Hornung et al., 2006) | |
| Flaviviridae (+) ssRNA NS | Hepatitis C virus | (Saito et al., 2007; Sumpter et al., 2005) |
| Japanese encephalitis virus | (Kato et al., 2006) | |
|
| ||
| dsDNA-viruses | Epstein-Barr virus | (Ablasser et al., 2009; Chiu, Macmillan and Chen, 2009; Samanta et al., 2006) |
| Herpes simplex virus-1 | (Chiu, Macmillan and Chen, 2009) | |
| Adenovirus | (Chiu, Macmillan and Chen, 2009) | |
|
| ||
| Viruses detected by MDA5 | ||
|
| ||
| Picornaviridae (+) ssRNA, NS | Encephalomyocarditis virus | (Gitlin et al., 2006; Kato et al., 2006) |
| Theiler’s virus | (Kato et al., 2006) | |
| Mengovirus | (Kato et al., 2006) | |
|
| ||
| Caliciviridae (+) ssRNA, NS | Murine norovirus-1 | (McCartney et al., 2008) |
|
| ||
| Coronaviridae (+) ssRNA NS | Murine hepatitis virus | (Roth-Cross, Bender and Weiss, 2008) |
|
| ||
| Viruses detected by RIG-I and MDA5 | ||
|
| ||
| Flaviviridae (+) ssRNA, NS | Dengue virus | (Loo et al., 2008) |
| West Nile virus | (Fredericksen et al., 2008; Loo et al., 2008) | |
|
| ||
| Reoviridae dsRNA S | Reovirus | (Loo et al., 2008) |
|
| ||
| Arenaviridae (−) ssRNA, S | Lymphocytic choriomeningitis virus | (Zhou et al., 2010) |
D. Bacteria
Various bacteria including Francisella tularensis, Mycobacteria tuberculosis, Brucella abortis, group B streptococcus (GBS), Listeria monocytogenes and Legionella pneumophila have been shown to induce type I IFN in a TLR-independent manner (Charrel-Dennis et al., 2008; Henry et al., 2007; O’Riordan et al., 2002; Opitz et al., 2006; Roux et al., 2007; Stanley et al., 2007; Stetson and Medzhitov, 2006). While it is well appreciated that viral replication is inhibited by type I IFN, the role of IFN in bacterial infections is less clear, e. g. IFN has a protective effect during GBS infection (Mancuso et al., 2007), whereas it is disadvantageous during Listeria infection (Auerbuch et al., 2004; Carrero, Calderon and Unanue, 2004; O’Connell et al., 2004). Even less clear is which bacterial ligands and host receptors trigger IFN secretion.
The intracellular gram-negative bacterium Legionella pneumophila infects macrophages and causes Legionnaires‘ disease. IFN-β induction in lung epithelial cells and macrophages depends on MAVS (Monroe, McWhirter and Vance, 2009; Opitz et al., 2006). However, the signaling events upstream of MAVS activation are a matter of debate. Chiu et al. propose that AT-rich DNA reaches the host cytosol and is transcribed into an RNA-ligand for RIG-I in a RNA polymerase III-dependent manner (Chiu, Macmillan and Chen, 2009). In contrast, Monroe et al. argue that the IFN response to Legionella genomic DNA does not require MAVS in mouse macrophages as MAVS-deficient and wild-type macrophages display comparable levels of IFN. Instead their data supports a model where Legionella RNA is directly detected by both RIG-I and MDA5 as macrophages deficient in either receptor display a partial phenotype (Monroe, McWhirter and Vance, 2009).
Shigella flexneri, the causative agent of bacillary dysentery, infects macrophages of the colonic epithelium and rapidly induces cell death by pyroptosis. Escaping bacteria invade colonic epithelial cells where they replicate in the cytosol. Type II IFN-γ is critical for inhibiting S. flexneri cytosolic growth. It is at this stage that IFN-γ exerts its antimicrobial effect through RIG-I signaling in non-myeloid cells. Both RIG-I-deficient and MAVS-deficient mouse embryonic fibroblasts failed to restrict IFN-γ dependent S. flexneri replication. Inhibition of RNA polymerase III also reduced the antimicrobial effect of IFN-γ suggesting that RIG-I signaling is triggered by RNA polymerase III-generated RNA mediates. Interestingly, type I IFN induction is not required for this effect as IFNAR-deficient mouse embryonic fibroblasts (MEF) that are completely unresponsive to type I IFNs do not impair IFN-γ-mediated growth inhibition of S. flexneri. In contrast, in primary macrophages, RIG-I signaling is dispensable for IFN-γ mediated growth arrest (Jehl et al., 2012). These findings underscore the importance of the interplay of distinct innate immunity pathways in order to successfully combat pathogens.
III. RIG-I activation and receptor proximal signal propagation
RLR activation is a multi-stage process that requires a well coordinated interplay of receptor, ligand, and several accessory proteins. In contrast to RIG-I, the specific requirements for efficient MDA5 activation are unclea, but it stands to reason that both proinflammatory RLRs follow a similar mechanism. As exemplified by RIG-I our current understanding of this process involves the following sequence of events: 1. In resting cells RIG-I adopts a closed conformation resulting in an autoinhibited (non-signaling) state. 2. pppRNA binding to RIG-I induces conformational changes that lead to dimerization and exposure of CARDs in the open conformation. 3. Dephosphorylation of RIG-I and TRIM25-dependent ubiquitination events fully activate the signaling capability of RIG-I, 4. RIG-I associates with MAVS in a CARD-dependent manner. 5. MAVS accumulates in signaling aggregates by a prion-like mechanism.
In the absence of infection RIG-I is kept in an autoinhibited state by intramolecular interactions between the CARDs and the helicase domain, which sterically hinders RNA binding to the helicase domain and prevents the CARDs from signaling (Kowalinski et al., 2011; Saito et al., 2007). Accordingly, the N-terminus of RIG-I comprising the 2 CARDs has a constitutively active phenotype when overexpressed (Yoneyama et al., 2004). Furthermore, phosphorylation of threonine 170 (and serine 8 in primate orthologs) by PKC-α and PKC-β suppresses RIG-I activity at steady state (Gack et al., 2010; Maharaj et al., 2012; Nistal-Villan et al., 2010).
Only upon ligand binding does the closed conformation open up to facilitate downstream signaling by the CARDs. Biochemical studies have identified the CTD as the sensor for pppRNA. Receptor-ligand interactions were examined by measuring ATPase activity of purified deletion mutants of RIG-I lacking either the CARDs (ΔCARD), the CTD (ΔCTD) or both (helicase) in response to treatment with a panel of RNA ligands derived from the rabies virus leader (RVL) sequence, i.e. pppRNA (pppRVL), non-phosphorylated ssRNA (ssRVL) as well as dsRNA (dsRVL). ssRVL did not activate ATPase activity in any of the RIG-I variants. pppRVL strongly stimulated ATPase activity of wild-type RIG-I. Deletion of the CARDs did not interfere with pppRVL-stimulated ATPase activity. Neither the helicase domain alone nor RIG-I lacking the CTD displayed ATPase activity in response to pppRVL. dsRNA weakly stimulated wild-type RIG-I and the isolated helicase domain. Of note, dsRNA activated ΔCARD more efficiently than pppRVL achieving ATPase activity levels comparable to wild-type RIG-I in complex with pppRVL. These findings suggest that the CARDs inhibit dsRNA binding in an inactive conformation while CTD promotes pppRVL binding in an active conformation. Further binding studies clearly demonstrated that the pppRNA binding site resides within the CTD. X-ray crystallography of the CTD revealed 2 features that are required for pppRNA binding: 1. a zinc coordination site comprised of four highly conserved cystidine residues (C810, C813, C864, C869). These cystidines are conserved in a paralogous and orthologous manner within the family of RLRs. 2. a conserved groove with a positively charged patch at the center of which a RIG-I invariant lysine is located (K888) (Cui et al., 2008).
Chrystallographic structures of RIG-I give detailed insight into the conformational changes triggered by ligand binding and required for signal initiation. The structural data suggests a model where in the autorepressed state the CTD is devoid of intramolecular interactions and thus can freely engage in pppRNA binding. This initial event increases the local RNA concentration and leads to cooperative binding of RNA and ATP to the helicase domain resulting in dramatic rearrangements within the helicase domain that are orchestrated by the pincher domain that connects the helicase domain with the CTD. The helicase domain and the CTD completely surround the RNA clasping onto the helix by numerous intermolecular interactions. This channel covers 9–10 bp along the RNA. Longer RNA molecules allow the binding of two RIG-I monomers simultaneously. However, this apparent dimerization is devoid of a a protein-protein interface but much rather reflects an RNA guided oligomerization (Kowalinski et al., 2011; Luo et al., 2011). In line with the structural data of RNA bound RIG-I full-length RIG-I but not the ΔCTD mutant or MDA5 eluted as dimers after gelfiltration when incubated with pppRNA (Cui et al., 2008).
Downstream signaling by ligand-activated RIG-I is achieved by the N-terminal tandem CARDs. Deletion of the CARDs results in a dominant negative phenotype of RIG-I (Yoneyama et al., 2004). Huh7.5 cells, a subpopulation of the hepatocyte cell line Huh7 that is characterized by a threonine to isoleucine mutation at position 55 (T55I) in the first CARD of RIG-I, fail to respond to HCV infection. As a consequence the absence of a functional antiviral response creates conditions permissive for HCV replication in Huh7.5 (Sumpter et al., 2005). The T55I mutant interferes with binding of the TRIM25 E3 ubiqitin ligase that is required for activation of RIG-I signaling. Gack et al demonstrated that TRIM25 binds to the first CARD domain via its SPRY domain. Prerequisite for TRIM25 binding is dephosphorylation of RIG-I at T170 (and S8 in primates) by an unidentified phosphatase. The phosphomimetic mutation T170E abrogated binding of TRIM25 to RIG-I and interfered with downstream signaling events and antiviral actvity of RIG-I (Gack et al., 2010). TRIM25 transfers K63-linked ubiquitin moieties to the lysine 172 residue (K172) within the second CARD using its RING domain. Oligomerization of RIG-I with the adapter protein MAVS critically depends on this modification. Accordingly, TRIM25-deficient MEFs do not secrete IFN-β after SeV infection. The absence of antiviral defenses is reflected by markedly higher viral titers upon VSV infection (Gack et al., 2007). Although TRIM25 does not attach ubiquitin moieties to MDA5, polyubiquitin binding by MDA5 is required for its signaling functions (Jiang et al., 2012).
The requirement for ubiqitination of RIG-I for initiation of downstream signaling was challenged by a study using a cell-free system to identify the minimal components for RIG-I signal transduction. The RIG-I pathway was reconstituted in a mixture containing affinity-purified RIG-I, crude mitochondria and peroxisomes (containing the adapter MAVS), cytosolic extracts (containing TBK1), in vitro synthesized transcription factor IRF3 and ATP. RIG-I activation was quantified by measurement of dimerization of IRF3, a read-out for its activation. With this in vitro assay in place, the authors recapitulated key aspects of RIG-I signaling and revealed new regulatory mechanisms. IRF3 activation required MAVS and TRIM25 as depletion of these proteins by RNAi interfered with IRF3 dimerization. RIG-I needed to be isolated from virus-infected cells, to be activated by RNA ligand in vitro or be present as an N-terminal CARD fragment for IRF3 activation to occur. The ubiquitination machinery responsible for RIG-I activation was shown to be comprised of E1, the E2 Ubc5 and Ubc13 and the E3 TRIM25 as the mitochondrial fraction of virus-infected cells depleted from Ubc5 (isoform b and c) and Ubc13 no longer elicited IRF3 dimerization. In line with the notion that Ubc13 is specific for synthesis of lysine 63 (K63)-linked ubiquitin and previous findings on the importance of K63-linked polyubiquitin for RIG-I activation, ubiquitin proteins with a sole lysine residue at position 63 were capable to activate the pathway in the cell-free in vitro system (Zeng et al., 2010).
Thus a requirement for both TRIM25 and K63-linked ubiquitin for IFN-β induction by RIG-I were confirmed in this experimental setup. The major discrepancy between the studies by Gack et al. and Zeng et al. is the attachment of polyubiquitin. While in the former study covalent linkage to the K172 residue of RIG-I was proposed, the latter study suggests that unanchored polyubiquitin chains serve as essential cofactors for RIG-I activation. Two major lines of evidence support this proposition: 1. RIG-I CARDs isolated from E. coli that lack an ubiquitination system activated IRF3 when ubiquitin polymers were added to the cell-free system. 2. Endogenous polyubiquitin was co-precipitated with RIG-I CARDs from mammalian cells and subsequently recovered from the complex by selective heat denaturation. This preparation promoted IRF3 dimerization, but lost its activity when treated with the deubiquitination enzyme IsoT. Even though the K172 residue is not required as an acceptor for ubiquitination in this situation, its relevance for RIG-I signaling remains undisputed as it is critical for the binding affinity to polyubiquitin (Zeng et al., 2010).
Both RIG-I and MDA5 signaling depends on the adapter protein MAVS to link receptor activity to the downstream kinases TBK1 and IKK-i (Figure 2). MAVS is a 540 aa protein comprised of a N-terminal CARD domain, a central proline-rich region (Pro) and a C-terminal transmembrane domain (TM) (Seth et al., 2005) (Figure 1). While the transmembrane domain targets the adapter to its proper subcellular locations (mitochondria, peroxisomes and mitochondria-associated membranes (MAM), see section IV.B.), the CARD domain is required for signaling (Dixit et al., 2010; Horner et al., 2011; Seth et al., 2005). When MAVS was initially characterized as an RLR signaling adapter, the authors noted that viral infection results in formation of detergent resistant aggregates (Seth et al., 2005). Recent studies by the same group defined these aggregates as highly organized, self-propagating prion-like fibrils. Using the cell-free system for in vitro reconstitution of RLR signaling as described earlier, complexes of MAVS larger than the 26S proteasome were detected 9 hours after SeV infection which coincided with IRF3 dimerization. These complexes displayed several features characteristic for prions: 1. The MAVS CARD is necessary and sufficient for formation of fiber-like structures as determined by electron microscopy. 2. These fibrils are resistant to protease K treatment and detergent solubilisation. 3. Protease resistant fibrils convert MAVS on mitochondria that were extracted from uninfected cells into functional aggregates leading to IRF3 activation. Interestingly however, these MAVS aggregated did not stain with Congo Red, a dye that typically stains “classic” prion structures (chen prion paper). Conversely, mitochondria depleted of MAVS by RNAi prior to extraction did not result in IRF3 dimerization. Importantly, MAVS aggregates form within minutes upon activation of RLR signaling in the cell-fee reconstitution assay indicating that prion-like MAVS fibrils are a bona fide determinant of the MAVS activation status (Hou et al., 2011).
IV. Regulatory mechanisms of RIG-I signaling
A. Regulators of RLR signaling
Several proteins regulate RLR signaling along the pathway in order to tailor the response. Various E3 ubiquitin ligases regulate RIG-I activity. TRIM25 as discussed in section III. and Riplet (also known as RNF135 or REUL) positively regulate RIG-I activity through K63-linked ubiquitination at its N- or C- terminus, respectively (Gack et al., 2007; Gao et al., 2009; Oshiumi et al., 2009; Oshiumi et al., 2010). In contrast RNF125 mediates K48-linked ubiqitination that targets RIG-I for degradation and thus acts as a negative regulator (Arimoto et al., 2007). Recently, ZAPS was identified as a cofactor for RIG-I signaling. ZAPS is a member of the poly (ADP-ribose) polymerase (PARP) family, but lacks the PARP-like domain present in ZAPS due to alternative splicing. ZAPS was shown to directly associate with RIG-I in a ligand-dependent manner and to amplify downstream signaling events such as activation of the transcription factors IRF3 and NF-κB and induction of type I IFN. As a result ZAPS inhibited viral replication after infection with RIG-I-dependent viruses like influenza virus or NDV (Hayakawa et al., 2011). While a continuously growing number of accessory proteins that modify RIG-I signaling activity emerges, the interplay between these proteins, the order in which they act upon RIG-I and their relative significance for signaling output remain elusive until further systematic studies are done to address these questions.
NLRX1 (also known as Nod9) was proposed to control RLR signal transduction at the level of MAVS, however its role is a matter of debate. NLRX1 was reported to reside at the outer mitochondrial membrane from where it physically disrupts the virus-induced RLR-MAVS interaction (Moore et al., 2008) (Figure 2). Alternatively, NLRX1 was found to be localized within the mitochondrial matrix which deems impossible the proposed function as a direct interactor of MAVS to modulate its activity. Rather it was shown that NLRX1 promotes the generation of reactive oxygen species (ROS) (Arnoult et al., 2009; Tattoli et al., 2008). Interestingly, several lines of evidence implicate ROS as modulators of RLR signaling. Cells deficient in autophagy accumulate dysfunctional mitochondria which entails increased ROS levels and display enhanced RLR signaling. Treatment with antioxidant reverses the effect (Tal et al., 2009). Conversely, mitochondrial uncoupling – a process by which ROS generation is decreased – reduced RLR signaling (Koshiba et al., 2011). Additional research is required to delineate the mechanism by which ROS regulate RLR-dependent antiviral responses.
STING (also known as MITA, MPYS or ERIS) (Ishikawa and Barber, 2008; Jin et al., 2011; Sun et al., 2009; Zhong et al., 2008) was originally identified as a regulator of RIG-I signaling owing to its ability to directly bind to RIG-I, MAVS and TBK1 and to and its knockout phenotype. Overexpression of the constitutively active fragment of RIG-I failed to induce IFN in STING-deficient MEFs. Moreover, VSV infection of STING-deficient mice resulted in significantly poorer survival rates and lower type I IFN serum levels relative to control littermates. It is of note that the response to transfected polyI:C remained unchanged in the absence of STING (Ishikawa, Ma and Barber, 2009). While STING was shown to play an undisputed role in the IFN response to cytosolic DNA from viruses or synthetic agonists, its implication in RLR signaling may not be essential.
B. Regulation of RLR signal transduction by subcellular compartmentalization
All three receptors of the RLR family are cytosolic proteins, and they have not been found to be associated with any subcellular structure at steady state. However, several signaling components downstream of the receptors are membrane proteins whose functional domains project into the cytosol from the surface of the respective organelles. More importantly, proper localization of these proteins is a prerequisite for their biological activity. The best characterized example is the adapter protein MAVS. MAVS resides on the outer mitochondrial membrane (Seth et al., 2005), peroxisomes (Dixit et al., 2010) and mitochondria-associated membranes (MAM) (Horner et al., 2011), a specialized subdomain of the ER that connects mitochondria and peroxisomes (Hayashi et al., 2009; Vance, 1990). Peroxisomal and mitochondrial MAVS both signal to induce ISG expression in MEFs. While mitochondrial MAVS induces type I IFN and as a consequence ISG expression in response to reovirus and influenza virus infection, peroxisomal MAVS directly induces ISG expression which creates a transient yet functional antiviral state. The lack of type I IFN induction by peroxisomal MAVS was also observed in macrophages. Unlike MEFs, macrophages upregulate not only expression of ISGs but also proinflammatory cytokines after reovirus infection (Dixit et al., 2010). A different study confirms the localization of MAVS on mitochondria and peroxisomes, and adds MAMs to the list of subcellular pools of MAVS. Moreover, the authors propose the MAM as an innate immune synapse for antiviral responses that coordinates MAVS-dependent signaling from mitochondria and peroxisomes (Horner et al., 2011). HCV-infected Huh7 hepatocytes are unable to induce IFN expression due to MAVS cleavage by the viral protease NS3/4A (Loo et al., 2006; Meylan et al., 2005). We and others have shown that cytosolic MAVS is unable to signal (Dixit et al., 2010; Seth et al., 2005). Given that NS3/4A cleaves MAM-localized MAVS, but not mitochondrial MAVS the authors conclude that-at least for HCV infections-mitochondrial MAVS is dispensable for RIG-I signaling. This notion is further supported by the finding that RIG-I is recruited specifically to MAM-resident MAVS upon HCV infection (Horner et al., 2011). In fact, a ternary complex consisting of active open-conformation RIG-I, TRIM25 and the chaperone 14-3-3ε is redistributed to MAMs upon infection (Liu et al., 2012). MFN2 tethers the ER to mitochondria and thus maintains the MAM mitochondrial contacts (de Brito and Scorrano, 2008). Depletion of MFN2 by RNAi destabilizes the antiviral synapse, which shifts MAVS to peroxisomes and thereby increases RIG-I-mediated signaling in response to SeV, VSV and HCV (at early timepoints before MAVS cleavage by NS3/4A) infection (Horner et al., 2011). It would be interesting to test the effect of MFN2 on the organelle-specific outcome of RLR signaling using cells with organelle restricted MAVS expression.
In addition to MFN2, MFN1 has been implicated in regulation of RIG-I signaling as well. Activation of RLRs by infection with SeV, NDV, influenza virus, VSV, Sindbis virus or EMCV and by transfection with pppRNA resulted in redistribution of mitochondrial MAVS. While some mitochondria accumulate MAVS, others become devoid of it during a process that depends on MFN1. RIG-I is evenly distributed throughout the cytosol in uninfected cells, but is concentrated in foci upon infection. However, no co-localization between RIG-I and MAVS was observed. On the contrary, RIG-I co-localized with viral nucleocapsid. As a consequence type I IFN induction after NDV infection was completely abolished in MFN1-deficient MEFs. These findings led the authors to propose a model where RIG-I is recruited to virus factories to maximize the chances of receptor-ligand interaction. Mitochondria serve as vehicles that position MAVS. Some mitochondria enrich MAVS through repeated fission and fusion events and surround the foci of active viral replication in order to enable IFN induction (Onoguchi et al., 2010). While this model outlines how mitochondrial signaling is optimized to perpetuate IFN induction for the duration of infection and to establish a sustained antiviral immune response, it leaves two important questions unanswered. First, what are the kinetics of this process? The earliest time point presented in the study is 9 h post infection. Second, what triggers mitochondrial remodeling and accumulation of MAVS? Regardless if activation of RLR signaling or a different stimulus initiates the rearrangement, this model does not explain RNA detection at the very first instant of virus encounter. Much rather it demands additional and disparate means of RLR signaling that ensure an immediate antiviral response until MAVS-enriched mitochondria are recruited to the periphery of virus factories.
V. Conclusions and future directions
RLR signaling is a crucial pathway for detection of intracellular viruses and mounting protective antiviral defenses. Since the identification of RIG-I and its related proteins MDA5 and LGP2 tremendous progress has been made in terms of the core components of this pathway and the regulatory mechanisms. Still many open questions remain on the pathogen as well as the host side. What are the biological ligands that arise during a given viral infection? Viral genomes, viral transcripts or replication intermediates are likely candidates. Do these naturally occurring ligands match the postulated structural features that were identified in vitro? Baum et al. sought to characterize such ligands by immunoprecipitation of endogenous RIG-I/RNA complexes from SeV and influenza virus-infected cells and subsequent deep sequencing. Copy-back defective interfering (DI) particles were identified as the natural ligand of both SeV and influenza virus. RIG-I also bound to (preferentially short segments of) genomic RNA of influenza virus. This study confirms the requirement for both a 5′ triphosphate and a panhandle structure for RIG-I activation during SeV and influenza virus infection (Baum, Sachidanandam and Garcia-Sastre, 2010). How accessible are these ligands during infection? In the light of co-evolution of virus and host, it stands to reason that viral PAMPs are spatially segregated from the respective PRRs. Is RLR-mediated virus detection merely possible by accidental escape of PAMPs or are mechanisms in place that actively sample sites of viral replication?
Regarding the host factors required for an effective antiviral response, our understanding of the spatiotemporal control of this pathway is very limited. Despite the designation of RLRs as cytosolic receptors, the signal transduction cascade initiated upon ligand engagement is certainly not cytosolic, but strictly dependent on proper subcellular localization of many components of this pathway. The adaptor protein MAVS resides on and signals distinctively from peroxisomes, MAM and mitochondria (Dixit et al., 2010; Horner et al., 2011; Seth et al., 2005). The negative regulator NLRX1 is also localized on mitochondria (Moore et al., 2008). In the course of infection mitochondria are rearranged to surround sites of viral replication in a MFN1-dependent manner. Failure to do so severely abrogates an antiviral response (Onoguchi et al., 2010). What is the benefit for the host of such an elaborate subcellular arrangement of a signal transduction pathway? Perhaps recruitment of molecules concentrated on an organelle might be faster and more energy efficient than recruiting every single molecule independently. Considering the different responses mediated by peroxisomal and mitochondrial MAVS, distribution of this pathway on two organelles might facilitate targeting of factors specifically required for each of the responses. A similar situation can be found with TLR4, the receptor for the prototypical PAMP lipopolysaccharide. Perhaps a positive regulator of direct ISG induction is only targeted to peroxisomes or an inhibitor of such a signaling pathway is located on mitochondria. The TLR4 pathway exemplifies how the spatial distribution of signaling components governs the signaling output. While plasma membrane bound TLR4 induces cytokine expression in a MyD88-dependent manner (Medzhitov, Preston-Hurlburt and Janeway, 1997; Medzhitov et al., 1998), endocytosis of TLR4 induces type I IFN induction in a TRIF dependent manner (Kagan et al., 2008; Yamamoto et al., 2002). For TLR4 signaling TRAF3 was proposed to be limited in its mobility. The inability of TRAF3 to be recruited to TLR4 at the plasma membrane necessitates TLR4 to be endocytosed. It is at the endosome that the TRAM-TRIF adaptor pair is recruited to engage TRAF 3 and to enable type I IFN signaling (Kagan et al., 2008). Similarly an essential factor for direct ISG induction may be available exclusively at peroxisomes. Experimental evidence for the organelle-specific presence of regulators of RLR signaling comes from NLRX1. Overexpression of NLRX1 inhibits signaling mediated by mitochondrial MAVS, but not by peroxisomal MAVS (Dixit et al., 2010). The spatial regulation may also be indicative of RLR signaling being a multistage process, wherein in an initial wave a nascent infection is sensed, in a later phase the process is optimized for a robust response during infection and finally is turned off. In order to address this possibility kinetic studies rather than late endpoints after infection would be helpful.
Supplementary Material
Acknowledgments
E. D. is supported by the Erwin Schrödinger Fellowship (J3295-B22) of the Austrian Science Fund (FWF). The National Institutes of Health grants AI093589 and P30 DK34854 support the work performed in the laboratory of J. Kagan. Dr. Kagan holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
References
- Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007;104:7500. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci. 2009;122:3161. doi: 10.1242/jcs.051193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9:535. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A, Sachidanandam R, Garcia-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A. 2010;107:16303. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. 2004;200:535. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrel-Dennis M, Latz E, Halmen KA, Trieu-Cuot P, Fitzgerald KA, Kasper DL, Golenbock DT. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe. 2008;4:543. doi: 10.1016/j.chom.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SE, Noyce RS, Mossman KL. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol. 2004;78:1706. doi: 10.1128/JVI.78.4.1706-1717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell. 2008;29:169. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M., Jr Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J Virol. 2008;82:609. doi: 10.1128/JVI.01305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Nistal-Villan E, Inn KS, Garcia-Sastre A, Jung JU. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J Virol. 2010;84:3220. doi: 10.1128/JVI.02241-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Gao D, Yang YK, Wang RP, Zhou X, Diao FC, Li MD, Zhai ZH, Jiang ZF, Chen DY. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS One. 2009;4:e5760. doi: 10.1371/journal.pone.0005760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103:8459. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa S, Shiratori S, Yamato H, Kameyama T, Kitatsuji C, Kashigi F, Goto S, Kameoka S, Fujikura D, Yamada T, Mizutani T, Kazumata M, Sato M, Tanaka J, Asaka M, Ohba Y, Miyazaki T, Imamura M, Takaoka A. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat Immunol. 2011;12:37. doi: 10.1038/ni.1963. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med. 2007;204:987. doi: 10.1084/jem.20062665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J. Convergence of the NF-kappaB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 2007;18:483. doi: 10.1016/j.cytogfr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011;108:14590. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehl SP, Nogueira CV, Zhang X, Starnbach MN. IFNgamma inhibits the cytosolic replication of Shigella flexneri via the cytoplasmic RNA sensor RIG-I. PLoS Pathog. 2012;8:e1002809. doi: 10.1371/journal.ppat.1002809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, Chen ZJ. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36:959. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B, Perraud AL, Cambier JC, Lenz LL. MPYS Is Required for IFN Response Factor 3 Activation and Type I IFN Production in the Response of Cultured Phagocytes to Bacterial Second Messengers Cyclic-di-AMP and Cyclic-di-GMP. J Immunol. 2011;187:2595. doi: 10.4049/jimmunol.1100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Lin L, Randolph A, Valerie K, Pestka S, Fisher PB. Expression analysis and genomic characterization of human melanoma differentiation associated gene-5, mda-5: a novel type I interferon-responsive apoptosis-inducing gene. Oncogene. 2004;23:1789. doi: 10.1038/sj.onc.1207300. [DOI] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Katze MG, He Y, Gale M., Jr Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal. 2011;4:ra7. doi: 10.1126/scisignal.2001147. [DOI] [PubMed] [Google Scholar]
- Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW, Nikolich-Zugich J, Moses AV, Gale M, Jr, Früh K, Diamond MS. IRF-3, IRF-5, and IRF-7 Coordinately Regulate the Type I IFN Response in Myeloid Dendritic Cells Downstream of MAVS Signaling. PLoS Pathog. 2013;9:e1000607. doi: 10.1371/journal.ppat.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HM, Loo YM, Horner SM, Zornetzer GA, Katze MG, Gale M., Jr The mitochondrial targeting chaperone 14-3-3 epsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe. 2012;11:528. doi: 10.1016/j.chom.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, Carney DS, Wang T, Ishida H, Yoneyama M, Fujita T, Saito T, Lee WM, Hagedorn CH, Lau DT, Weinman SA, Lemon SM, Gale M., Jr Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM. Structural insights into RNA recognition by RIG-I. Cell. 2011;147:409. doi: 10.1016/j.cell.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharaj NP, Wies E, Stoll A, Gack MU. Conventional protein kinase C-alpha (PKC-alpha) and PKC-beta negatively regulate RIG-I antiviral signal transduction. J Virol. 2012;86:1358. doi: 10.1128/JVI.06543-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M, Jr, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, Leanderson T, Teti G. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, Yamamoto A, Seya T. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol. 2003;171:3154. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- McCartney SA, Thackray LB, Gitlin L, Gilfillan S, Virgin HW, Colonna M. MDA-5 recognition of a murine norovirus. PLoS Pathog. 2008;4:e1000108. doi: 10.1371/journal.ppat.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Mikkelsen SS, Jensen SB, Chiliveru S, Melchjorsen J, Julkunen I, Gaestel M, Arthur JS, Flavell RA, Ghosh S, Paludan SR. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: dependence on TRAF2 and TAK1. J Biol Chem. 2009;284:10774. doi: 10.1074/jbc.M807272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe KM, McWhirter SM, Vance RE. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 2009;5:e1000665. doi: 10.1371/journal.ppat.1000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, Lich JD, Heise MT, Chen Z, Ting JP. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- Mossman KL, Macgregor PF, Rozmus JJ, Goryachev AB, Edwards AM, Smiley JR. Herpes simplex virus triggers and then disarms a host antiviral response. J Virol. 2001;75:750. doi: 10.1128/JVI.75.2.750-758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistal-Villan E, Gack MU, Martinez-Delgado G, Maharaj NP, Inn KS, Yang H, Wang R, Aggarwal AK, Jung JU, Garcia-Sastre A. Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-beta production. J Biol Chem. 2010;285:20252. doi: 10.1074/jbc.M109.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci U S A. 2002;99:13861. doi: 10.1073/pnas.202476699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoguchi K, Onomoto K, Takamatsu S, Jogi M, Takemura A, Morimoto S, Julkunen I, Namiki H, Yoneyama M, Fujita T. Virus-infection or 5′ ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog. 2010;6:e1001012. doi: 10.1371/journal.ppat.1001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz B, Vinzing M, van Laak V, Schmeck B, Heine G, Gunther S, Preissner R, Slevogt H, N’Guessan PD, Eitel J, Goldmann T, Flieger A, Suttorp N, Hippenstiel S. Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J Biol Chem. 2006;281:36173. doi: 10.1074/jbc.M604638200. [DOI] [PubMed] [Google Scholar]
- Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem. 2009;284:807. doi: 10.1074/jbc.M804259200. [DOI] [PubMed] [Google Scholar]
- Oshiumi H, Miyashita M, Inoue N, Okabe M, Matsumoto M, Seya T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe. 2010;8:496. doi: 10.1016/j.chom.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227:221. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol. 2009;83:10761. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Plumet S, Herschke F, Bourhis JM, Valentin H, Longhi S, Gerlier D. Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS One. 2007;2:e279. doi: 10.1371/journal.pone.0000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschlager N, Schlee M, Rothenfusser S, Barchet W, Kato H, Akira S, Inoue S, Endres S, Peschel C, Hartmann G, Hornung V, Ruland J. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, Hume DA, Stacey KJ. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- Roth-Cross JK, Bender SJ, Weiss SR. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol. 2008;82:9829. doi: 10.1128/JVI.01199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- Roux CM, Rolan HG, Santos RL, Beremand PD, Thomas TL, Adams LG, Tsolis RM. Brucella requires a functional Type IV secretion system to elicit innate immune responses in mice. Cell Microbiol. 2007;9:1851. doi: 10.1111/j.1462-5822.2007.00922.x. [DOI] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M., Jr Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007;104:582. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta M, Iwakiri D, Kanda T, Imaizumi T, Takada K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006;25:4207. doi: 10.1038/sj.emboj.7601314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A. 2010;107:1512. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M, Hartmann G. The chase for the RIG-I ligand--recent advances. Mol Ther. 2010;18:1254. doi: 10.1038/mt.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, Juranek S, Kato H, Kawai T, Poeck H, Fitzgerald KA, Takeuchi O, Akira S, Tuschl T, Latz E, Ludwig J, Hartmann G. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31:25. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, Hoffmann FS, Michallet MC, Besch R, Hopfner KP, Endres S, Rothenfusser S. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci U S A. 2009;106:12067. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljestrom P, Reis e Sousa C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol. 2007;178:3143. doi: 10.4049/jimmunol.178.5.3143. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science. 2012 doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. 2009;106:8653. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar MS, Ramos HJ, Brassil MM, Netland J, Chappell CP, Blahnik G, McMillan A, Diamond MS, Clark EA, Bevan MJ, Gale M., Jr The RIG-I-like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity. 2012;37:235. doi: 10.1016/j.immuni.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Kumeta H, Tsuduki N, Narita R, Shigemoto T, Hirai R, Yoneyama M, Horiuchi M, Ogura K, Fujita T, Inagaki F. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: identification of the RNA recognition loop in RIG-I-like receptors. J Biol Chem. 2009;284:17465. doi: 10.1074/jbc.M109.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M, Jr, Inagaki F, Fujita T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell. 2008;29:428. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106:2770. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, Arnoult D, Girardin SE. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008;9:293. doi: 10.1038/sj.embor.7401161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzri D, Gehrke L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J Virol. 2009;83:4174. doi: 10.1128/JVI.02449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265:7248. [PubMed] [Google Scholar]
- Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, Barber GN. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007;178:6444. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science. 2012 doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Zhou S, Cerny AM, Zacharia A, Fitzgerald KA, Kurt-Jones EA, Finberg RW. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J Virol. 2010;84:9452. doi: 10.1128/JVI.00155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.