

Abstract
With billions of years of evolution under its belt, Nature has been expanding and optimizing its biosynthetic capabilities. Chemically complex secondary metabolites continue to challenge and inspire today’s most talented synthetic chemists. A brief glance at these natural products, especially the substantial structural variation within a class of compounds, clearly demonstrates that Nature has long played the role of medicinal chemist. The recent explosion in genome sequencing has expanded our appreciation of natural product space and the vastness of uncharted territory that remains. One small corner of natural product chemical space is occupied by the recently dubbed thiazole/oxazole-modified microcins (TOMMs), which are ribosomally produced peptides with posttranslationally installed heterocycles derived from cysteine, serine and threonine residues. As with other classes of natural products, the genetic capacity to synthesize TOMMs has been widely disseminated among bacteria. Over the evolutionary timescale, Nature has tested countless random mutations and selected for gain of function in TOMM biosynthetic gene clusters, yielding several privileged molecular scaffolds. Today, this burgeoning class of natural products encompasses a structurally and functionally diverse set of molecules (i.e. microcin B17, cyanobactins, and thiopeptides). TOMMs presumably provide their producers with an ecological advantage. This advantage can include chemical weapons wielded in the battle for nutrients, disease-promoting virulence factors, or compounds presumably beneficial for symbiosis. Despite this plethora of functions, many TOMMs await experimental interrogation. This review will focus on the biosynthesis and natural combinatorial diversity of the TOMM family.
Introduction
One strategy that Nature employs in the production of biologically active secondary metabolites uses existing machinery to synthesize inactive precursor peptides. Upon posttranslational modification, these inactive precursors undergo structural rigidification and are endowed with function. The major advantages to this strategy are (i) producer organisms did not need to start from scratch as all organisms possess ribosomes, (ii) amino acid mutations, which provide chemical diversity, occur at the genomic level by simple changes in the codons, and (iii) conversion of inactive precursors to active products could be temporally and spatially controlled at the posttranslational level, allowing organisms to respond quickly to environmental changes. Two major classes of peptide-derived natural products follow the above strategy: the lantipeptides and the TOMMs. The lantipeptides contain (methyl)lanthionine crosslinks, which are installed through the Michael-type addition of a cysteine thiol to a dehydrated serine or threonine [1]. In the TOMM class of natural products, cysteine, serine, and threonine residues are heterocyclized, which conformationally restrains the flexibility of the peptide.
Indeed, thiazole and oxazole heterocycles are ubiquitous in bioactive molecules and show impressive functional versatility in TOMM, non-TOMM, and synthetic products. A few examples include thiostrepton (50S ribosome inhibitor) [2], trunkamide (anti-cancer compound) [3], microcin B17 (DNA gyrase inhibitor) [4], goadsporin (secondary metabolism inducer) [5], yersiniabactin (siderophore) [6], and Ritonavir (HIV-1 protease inhibitor) [7] (Figure 1b). Thus, it is not surprising that Nature has devised two enzymatic solutions to construct thiazol(in)es and oxazol(in)es, one of which operates on non-ribosomal peptides and the other on ribosomal peptides (TOMMs). In both systems, cyclodehydration converts amino acids with a beta-nucleophile (cysteine, serine, or threonine) into thiazoline or (methyl)oxazoline rings [8•,9]. Select azoline rings can then be oxidized to azoles, which in characterized cases is catalyzed by a flavin mononucleotide (FMN)-dependent dehydrogenase [8•,10]. The non-ribosomal peptide synthetase (NRPS) and TOMM enzymes that catalyze the cyclodehydration bear no amino acid similarity to each other and present an example of convergent evolution in biosynthetic processes.
Figure 1.

The biosynthesis and structure of thiazole/oxazole containing compounds. (a) Thiazoles and oxazoles are installed by peptide backbone cyclodehydration of a cysteine, serine, or threonine residue, resulting in a thiazoline or (methyl)oxazoline ring. A FMN-dependent oxidation then yields the thiazole or (methyl)oxazole ring. (b) Representative examples of biologically active thiazole/thiazoline/oxazole-containing compounds. Those that are TOMMs have a black carbon skeleton, while those that are non-TOMMs have a grey carbon skeleton. The latter are either products of a nonribosomal peptide synthetase or are of artificial synthetic origin. Azoles and azolines are colored in green and red, respectively, while other posttranslational modifications are blue. As specifically mentioned in the main text: Microcin B17, the 10-glycine spacer is underlined and bolded, while the first bisheterocycle site is boxed; Thiostrepton, alanines 2 and 4 are highlighted in orange and brown, respectively.
In addition to the cyclodehydratase and optional dehydrogenase, all characterized TOMM clusters contain a ‘docking protein’. The precise role of the docking protein has been debated, but the available data suggest that it plays a direct role in regulating cyclodehydratase activity and assembly of an active synthetic complex [11,12]. Despite this unclear role in TOMM biosynthesis, Nature has given us a hint as to the importance of the docking protein by fusing this gene to the C-terminus of the cyclodehydratase in approximately half of all known TOMM clusters. In the other cases, the cyclodehydratase is found as a separate open reading frame but is expected to form a complex with the docking protein, as demonstrated in microcin B17 biosynthesis [8•]. Beyond the heterocyclization machinery, TOMM clusters can also possess other posttranslational modification enzymes, a topic discussed later in this review.
Akin to the other classes of ribosomal natural products, all characterized TOMM precursor peptides are bipartite. The N-terminal region of the precursor peptide, known as the leader peptide, contains key recognition motifs for the biosynthetic machinery [13••]. The C-terminal region of the precursor peptide, referred to as the core peptide, is rich in heterocyclizable residues and can be the site of numerous other posttranslational modifications [13••]. From the perspective of the producing organism, diversification of a TOMM is expected to be facile due to the relative ease of altering the composition of the core peptide [14]. As long as key binding sites are present within the leader peptide, the synthetase complex will carry out heterocyclization reactions on highly variable core peptides due to its promiscuous nature [15•,16,17•]. NRPS-derived natural products are expected to require a much more substantial genetic rearrangement to produce alternative compounds [18].
In this review, we use ‘TOMM’ as an umbrella term to refer to all ribosomal peptides posttranslationally modified by proteins with identifiable amino acid similarity to known cyclodehydratases and docking proteins. Such natural products have appeared in several recent reviews [1,2,14,19–27]. This review, however, will focus on the biosynthesis and natural combinatorial diversity of the TOMM family.
Microcin B17: the grandfather TOMM
The general biosynthetic route for the production of microcin B17 (Figure 1b), a TOMM from select strains of Escherichia coli, was first reported in 1996 [8•]. In this seminal paper, it was shown that a trimeric synthetase complex composed of McbB (cyclodehydratase), McbC (dehydrogenase), and McbD installed thiazole and oxazole moieties onto the McbA precursor peptide. This first enzymatic insight into the maturation of microcin B17 prompted numerous additional studies. In one such study, site-directed mutagenesis was used in conjunction with western blotting and mass spectrometry (MS) to biochemically dissect how the first bisheterocycle site was formed (Gly-Ser-Cys-Gly, boxed in Figure 1b). In vitro experiments suggested that the cysteine cyclized first, which then proceeded to the bisheterocycle. Alteration of the bisheterocycle site to Gly-Cys-Ser-Gly (Ser/Cys order reversed) did not mature to the bisheterocycle and predominantly formed only the one thiazole ring species [28]. This was a relevant observation since a naturally occurring Gly-Cys-Ser-Gly sequence resulted in a bisheterocycle elsewhere on the McbA peptide (Figure 1b). Clearly, heterocycle formation was not just dependent on the cyclizable residue, but also on the flanking residues. The glycine before the first bisheterocycle site proved to be crucial for heterocycle formation, as mutation of this residue to alanine precluded heterocycle formation [29]. Replacement of the C-terminal flanking glycine had a lesser effect. When mutated to alanine, valine, or asparagine, heterocycle formation was observed, although the rate was diminished by approximately two-fold in the valine and asparagine mutations [29]. Also, it was shown that cysteine residues cyclize 100-fold to 1000-fold faster than serine residues, depending on their location [30]. The increased rate was presumably due to the increased nucleophilicity of thiols, relative to alcohols. There also appeared to be a distance dependence on microcin B17 heterocycle formation. Upon shortening of the naturally occurring 10-glycine linker region between the leader sequence and the core domain (boldface/underlined in Figure 1b), a decrease in the rate of heterocyclization was observed. Likewise, a two-fold increase in rate was observed when the linker was lengthened to 11-glycines (Figure 1b) [29]. The order of heterocycle formation was also determined for microcin B17 via tandem MS. Overall, the installation of heterocycles was found to be directional, from the N-terminus to C-terminus. Furthermore, McbBCD released the McbA substrate after each heterocycle was formed, making microcin B17 biosynthesis a distributive process [31]. From all of these biochemical studies, it was evident that the microcin B17 biosynthetic machinery displayed regiospecific activity. However, this interpretation is somewhat convoluted due to the fact that in the N-terminal to C-terminal direction, four of the first five cyclizable residues encountered are cysteines, which are inherently more reactive (Figure 1b).
Cyanobacteria: marine medicinal chemists at work
The cyanobactins are a large family (>100) of macrocyclized TOMMs produced by cyanobacteria (e.g. trunkamide, Figure 1b) [17•,32,33]. A landmark discovery established that patellamides A and C were not being produced by the sea squirt (Lissoclinum patella), as was long suspected, but instead originated from its cyanobacterial symbiont (Prochloron didemni). Moreover, this report proved that these metabolites were of ribosomal origin [34••]. Since this initial discovery, the biosynthetic gene clusters for many cyanobactins have been identified [17•,32,35,36], some of which appear to have been distributed through horizontal gene transfer [37]. Additional studies with patellamide and trunkamide determined that the heterocycles were installed in a manner similar to microcin B17, supporting the classification of the heterocycle-containing cyanobactins as TOMMs. One key difference is that, unlike microcin B17, the cyanobactin hypervariable core peptides are flanked by highly conserved motifs. These motifs serve as proteolytic cleavage sites that ultimately lead to macrocyclization by an enzyme resembling subtilisin [38].
The cyanobactins provide a remarkable example of natural combinatorial biosynthesis, as illustrated by the variation at every position of the core peptide (Figure 2a). The least variance is observed at the C-terminus of the core peptide where a heterocycle is always found [33]. Interestingly, the penultimate position never contains a heterocyclizable residue. It remains unclear if the resulting molecules would be either biologically inactive or the biosynthetic machinery cannot carry out the transformation due to spatial considerations. A typical cyanobactin precursor peptide will contain two core peptides, allowing for the production of multiple cyanobactins from a single polypeptide [13••]. This natural library displays remarkable chemical diversity and supports the notion that the biosynthetic enzymes are permissive in their substrate tolerance. This inherent substrate flexibility allows for variation to emerge in the core domain and for new products with enhanced activity or novel modes of action. If the substrate tolerance and specificity of each cyanobactin modifying enzyme were known, it would allow the rational design of substrates to give rise to a desired product.
Figure 2.

Phylogenetic analysis of the cyanobactins and thiopeptides. (a) A comparative analysis of the cyanobactins using the publicly available precursor peptide sequences. The core regions of the precursor peptides were used to generate a sequence logo for each clade. In the uncharacterized clades, sequence alignment was used to hypothesize the protease cleavage sites. The least variance is present in the last position of the core peptide, which is predominantly a cysteine that becomes a thiazol(in)e. (b) Same as above, except the thiopeptide precursor peptides were used for the analysis. The clades were classified to bind the 50S ribosomal subunit or elongation factor Tu based on characterized members. All phylogenetic analyses were conducted using MEGA4 [68] and all sequence logos were produced using WebLogo [69].
As the first steps towards the rational engineering of cyanobactins, preliminary biochemical work has been done on PatD and TruD. These heterocyclization enzymes are single polypeptide fusions of the cyclodehydratase and docking proteins (CD fusion) from the patellamide and trunkamide biosynthetic clusters, respectively [39,40]. These two proteins are 88% identical at the amino acid level, with essentially all of the differences occurring in the C-terminal docking protein domain [39]. Given their similarity, it is quite surprising that PatD and TruD have different regiospecificities [39]. After heterocyclization and prenylation, cyanobactins must be proteolytically cleaved from the precursor peptide and macrocyclized. In the patellamide biosynthetic cluster, PatA and PatG were shown to cleave the N-termini and C-termini of the core peptide, respectively [38]. PatG also catalyzed the final macrocyclization reaction through a proposed transamidation mechanism [38,41••]. PatG, which exhibits a broad substrate scope, catalyzed the proteolytic cleavage and macrocyclization of synthetic peptides of varying length and composition, even peptides containing nonproteinogenic residues [41••]. An early demonstration of this enzymatic promiscuity was illustrated by engineering the production of eptidemnamide, an entirely artificial substrate similar to the antiplatelet drug, eptifibatide [17•]. Again, the inherent substrate tolerance of the modifying enzymes permits for rapid alteration of the natural product at the genetic level to meet organismal demands upon encountering environmental challenges.
Streptolysin S-like toxins: disease-promoting TOMMs
For over a century, scientists have repeatedly sought to isolate and characterize the factor responsible for the classic, β-hemolytic phenotype exhibited by S. pyogenes [42•]. Early efforts were met with limited success due to the frustrating biophysical characteristics and non-antigenic nature of streptolysin S (SLS). A pioneering discovery in 1998 identified the first SLS-associated gene (sagA) and led to the discovery of the entire sag operon [43,44]. Further studies demonstrated that the biosynthetic gene cluster required for SLS production was composed of a dehydrogenase (SagB), a cyclodehydratase (SagC), and a docking protein (SagD), which act collectively to install thiazole and oxazole heterocycles onto an inactive precursor peptide (SagA) [42•]. After heterocycle installation by in vitro reconstitution, SagA was converted into a broadly active cytolysin, providing the first molecular level description of the factor responsible for the β-hemolytic phenotype of S. pyogenes [42•]. Given that the biosynthetic route to SLS was highly reminiscent of microcin B17, it was subsequently demonstrated that reacting McbA with SagBCD resulted in the formation of heterocycles on McbA, again highlighting the enzymatic promiscuity of the TOMM machinery [42•]. As with the early studies on SLS, the problematic amino acid composition of SagA thwarted numerous modern MS methods to identify sites of posttranslational modification after reaction with SagBCD. However, a Herculean effort, involving derivatization of SagBCD-treated SagA, followed by tandem MS, and extensive data analysis demonstrated that SagBCD installed heterocycles onto the SagA core peptide [45]. Similarity to the sag operon provided genetic evidence that SLS-like toxins may promote the virulence of non-Streptococcal human pathogens (Clostridium botulinum, Listeria monocytogenes, and Staphylococcus aureus), which have since been experimentally validated to be cytolytic using a combination of genetic deletions and in vitro reconstitution [16,45,46].
The promiscuity observed with cyanobactin biosynthesis is echoed in the cytolytic TOMMs. This was first illustrated by the ability of SagBCD to install heterocycles onto McbA and the SagBCD-dependent conversion of the precursor peptide from C. botulinum (ClosA) into a potent cytolysin [42•]. However, incubating the precursor peptides of S. aureus and L. monocytogenes (StaphA and ListA, respectively) with SagBCD or complementing their genes into a ΔsagA S. pyogenes strain did not result in cytolytic activity [16]. Upon further study, it was established that although the heterocycle-forming enzymes tolerate diverse substrates, specificity is achieved by the leader peptides having distinct binding motifs, which direct the precursor peptide to the biosynthetic machinery [13••,16,47]. The requirement of these binding motifs prevents the heterocyclization of all cellular peptides and proteins, which would presumably be lethal to the producing organism. Therefore, cytolytic activity was not observed with StaphA and ListA because their leader peptides lack important binding motifs recognized by SagBCD [13••,16]. Further studies utilizing chimeric precursor peptides containing the SagA leader peptide and the StaphA and ListA core peptides resulted in cytolytic activity after treatment with SagBCD [16]. The promiscuity of the SagBCD complex has also been further illustrated by the processing of artificial substrates into cytolysins [16].
Owing to the lack of structural information on SLS, inferences about regiospecificity and chemospecificity of the biosynthetic machinery must be based on cytolytic activity of site-directed mutants. As expected, some residues of SagA were critical for the cytolytic activity, as mutation to alanine abolished activity [16]. Replacement of a cysteine for a serine or vice versa at a critical location resulted in slightly reduced cytolytic activity in vitro and in vivo [16]. Intriguingly, prolines were also tolerated at such positions. The diminished activity suggested that heterocycles were likely present and played mostly a structural role, but given that the unnatural heterocycle decreased the cytolytic potency, electronic contributions cannot be ruled out.
Thiopeptides: posttranslational modification taken to the extreme
The thiopeptides, defined by a central (tetrahydro)pyridine ring and at least one thiazole substituent (e.g. thiostrepton, Figure 1b) [2], were first discovered in 1948 [48]. Although the thiopeptides have a diverse array of functions, the most well known activity is to inhibit protein synthesis by interacting with the 50S ribosomal subunit or elongation factor Tu [2]. Early feeding experiments using labeled amino acids demonstrated that these highly modified natural products are amino acid derived [2,49]. However, for over half a century, the biosynthetic pathway to this class of natural products remained elusive until four independent research labs reported the biosynthetic genes responsible for several thiopeptides within the span of a few months [50–53]. These initial studies have led to a number of other discoveries for various thiopeptide biosynthetic gene clusters [54–58]. Thiopeptides feature an extraordinary array of posttranslational modifications. Thiostrepton, for example, contains four thiazoles, a thiazoline, a dehydrobutyrine, three dehydroalanines, a quinaldic acid moiety, a tetrahydropyridine ring, a dihydroxylated isoleucine, and a carboxy amide (Figure 1b) [53].
It was hypothesized that two dehydroalanines underwent a [4+2] cycloaddition to produce the central 6-membered ring found in all thiopeptides, which was later supported by feeding experiments [49,59]. The ‘hetero-Diels-Alderase’ responsible for this remarkable transformation was recently identified (TclM in thiocillin biosynthesis), which confirmed the original dehydroalanine hypothesis [60••]. Other enzymes involved in the ancillary posttranslational modifications have also been elucidated through genetic and biochemical studies [50,54,61].
Although thiopeptide biosynthesis is a fascinating example of chemical and genetic complexity, the use of these molecules in clinical medicine has been plagued by poor water solubility [27]. In an effort to increase water solubility, and thus bioavailability, initial studies to determine the structure-activity relationship have been conducted using the thiostrepton and thiocillin biosynthetic frameworks [62,63•,64]. Introduction of a fosmid that contained the thiostrepton biosynthetic cluster into a precursor peptide deficient strain of Streptomyces laurentii (thiostrepton producer) allowed genetic manipulation of the precursor peptide [62]. Alanines 2 and 4 (numbered N-terminus to C-terminus, Figure 1b) in thiostrepton were permissive toward mutation [62]. A parallel study with the thiocillin core peptide demonstrated that some positions were tolerant to substitution, while mutation to other positions either attenuated or abolished activity [63•,64]. Notably, threonine-3 is the only unmodified residue in thiocillin, and mutation of this position to serine resulted in a moderate loss of antibiotic activity (~50%) [64]. Attempts to increase the water solubility by incorporating charged residues into the more tolerant positions dramatically decreased antibiotic activity [63•]. Thus, the quest to engineer more bioavailable thiopeptide analogs continues.
Emerging TOMMs
The aforementioned TOMMs only cover about half of the currently known landscape occupied by this natural product class, as illustrated in Figure 3. Some light was recently shed on a subset of the uncharacterized TOMMs with the discovery of two new families of precursor peptides that share two traits: uncharacteristically long leader peptides that are homologous to known proteins and hypervariable C-terminal core regions [15•]. One family of these precursor peptides shares homology with the alpha subunit of nitrile hydratase [15•]. These nitrile hydratase-related leader peptides (NHLPs) are present in a diverse set of organisms, with a distinct lineage being present in the Burkholderia order. These peptides have been subjected to natural combinatorial biosynthesis, as demonstrated by paralogous duplication within many genomes and peptides with hypervariable C-termini [13••,15•]. Thus, multiple NHLPs are predicted to be biosynthesized by a single organism. This is exemplified by the 12 NHLPs found in Pelotomaculum thermopropionicum SI [15•]. The second family of recently discovered precursor peptides has homology to the Nif11 protein family, which are found in nitrogen-fixing bacteria but have an unknown function. In many cases, the TOMM biosynthetic machinery is not present in the genomes with Nif11-related precursor peptides and is replaced with lanthionine biosynthetic machinery. Numerous Nif11-like peptides from Prochlorococcus marinus have been experimentally validated to produce mature lantipeptides. In this non-TOMM system, one bifunctional LanM enzyme was shown to install (methyl)lanthionine rings on 17 different substrates but likely modifies all 29 genetically encoded substrates [65]. Catalytic promiscuity allows bacteria with compact genomes to produce an array of secondary metabolites with minimal increase in genome size [65].
Figure 3.

TOMM’s Family Tree. A phylogenetic analysis of the global TOMM family constructed using the amino acid sequence of the docking protein. For docking proteins that are naturally fused to the cyclodehydratase, only the predicted docking protein domain was used. The phyla represented are listed for each half of the tree. Cladal annotations for reported families are given and color-coded. The data for the phylogenetic analysis were collected and analyzed using the Biopython library and MEGA4 software, respectively [68,70].
Another recently characterized TOMM, plantazolicin, from the plant growth-promoting bacterium, Bacillus amyloliquefaciens FZB42, exhibits antibacterial properties towards select gram-positive bacteria through an unknown mechanism [66]. The structure of this natural product has yet to be established, but deletion studies have indicated that the compound is extensively posttranslationally modified [66].
Evolutionary aspects of TOMMs
TOMMs can be diversified using multiple mechanisms (Figure 4). In regard to precursor diversification, the bipartite character of TOMM precursor peptides allows the TOMM biosynthetic enzymes to be both specific for substrate binding yet promiscuous in terms of installing posttranslational modifications. This allows an organism to assess the functional ramification of core peptide mutations, of which some may yield increased target potency or a new target altogether (Figure 4). Nature has iteratively performed this operation over the eons to arrive at numerous privileged scaffolds. Much like medicinal chemists would diversify a promising lead compound, TOMM producers can obtain/evolve genes encoding enzymes that further elaborate existing TOMMs. An example of this is the ‘hetero-Diels-Alderase’ of the thiopeptides. Here, Nature took a thiopeptide ancestor, likely resembling goadsporin (Figure 1b) [67], and developed an enzyme that could join two dehydroalanines into a tetrahydropyridine ring (Figure 4). This quantum leap in biosynthetic capacity transformed the mode of action of the resultant molecule into a potent inhibitor of protein synthesis. Thus, a powerful weapon for niche competition was born, which is now disseminated in many genera of soil bacteria [2]. It is probable that the scope of functionality for this natural product family has not yet been fully realized.
Figure 4.
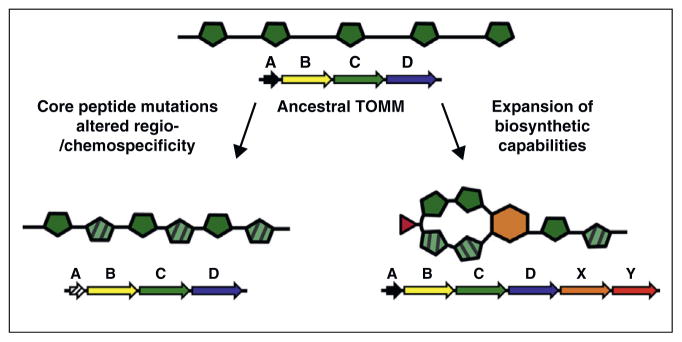
Genetically encoded medicinal chemistry. Shown is an ancestral TOMM produced by a basic biosynthetic cluster consisting of a precursor peptide (A, black arrow), dehydrogenase (B, yellow arrow), cyclodehydratase (C, green arrow), and docking protein (D, blue arrow). Core peptide mutations (black and white striped arrow) and/or altered regiospecificity/chemospecificity of the biosynthetic machinery provide one route to diversify the ancestral TOMM. This not only changes the number and position of cyclized residues, but also can alter the physicochemical properties of the final product. Another method to structurally diversify the TOMM involves the acquisition of ancillary enzymes (X and Y) that carryout additional posttranslational modifications (orange hexagon and red triangle) to diversify the TOMM scaffold. This can result in an entirely new function, relative to the ancestral TOMM.
Summary and outlook
By sifting through publicly available genome databases, bioinformatics-based approaches have identified over 300 TOMM biosynthetic gene clusters (Figure 3). The function of many of these can be inferred based on close phylogenetic relationships to characterized TOMMs. We refer to this process as bioinformatics-guided chemotyping. However, there is a vast area of TOMM chemical/genetic space that awaits characterization, and will continue to be an area of growth. This unexplored area has been growing linearly with efforts in microbial genome sequencing, suggesting that new TOMM subfamilies will continue to be uncovered.
Future studies, from both chemical and biological perspectives, will provide a more complete knowledge of the enzymes responsible for the peptide modifications, as well as the order and specificity in which they occur. Additionally, the role of the docking protein in orchestrating TOMM biosynthesis remains unclear. Future efforts should focus on the docking protein’s role in heterocycle formation. The biochemical knowledge of critical and ancillary enzymes will allow for the rational design and production of artificial TOMMs. Other future studies will focus on the production and screening of combinatorial libraries for increased potency and novel modes of action. Overall, the TOMM natural product family is a prolific source of chemical and functional diversity, and represents one strategy Nature adopted to synthesize complex natural products from a ribosomal template.
Acknowledgments
We would like to thank members of the Mitchell lab for critical reading of the manuscript. We also acknowledge the generous support from the Department of Chemistry and the Institute for Genomic Biology at the University of Illinois at Urbana-Champaign.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Velasquez JE, van der Donk WA. Genome mining for ribosomally synthesized natural products. Curr Opin Chem Biol. 2010 doi: 10.1016/j.cbpa.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagley MC, Dale JW, Merritt EA, Xiong A. Thiopeptide antibiotics. Chem Rev. 2005;105:685–714. doi: 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- 3.Salvatella X, Caba JM, Albericio F, Giralt E. Solution structure of the antitumor candidate trunkamide A by 2D NMR and restrained simulated annealing methods. J Org Chem. 2003;68:211–215. doi: 10.1021/jo026464s. [DOI] [PubMed] [Google Scholar]
- 4.Vizan JL, Hernandez-Chico C, del Castillo I, Moreno F. The peptide antibiotic microcin B17 induces double-strand cleavage of DNA mediated by E. coli DNA gyrase. EMBO J. 1991;10:467–476. doi: 10.1002/j.1460-2075.1991.tb07969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Igarashi Y, Kan Y, Fujii K, Fujita T, Harada K, Naoki H, Tabata H, Onaka H, Furumai T. Goadsporin, a chemical substance which promotes secondary metabolism and morphogenesis in streptomycetes. II. Structure determination. J Antibiot (Tokyo) 2001;54:1045–1053. doi: 10.7164/antibiotics.54.1045. [DOI] [PubMed] [Google Scholar]
- 6.Pelludat C, Rakin A, Jacobi CA, Schubert S, Heesemann J. The yersiniabactin biosynthetic gene cluster of Yersinia enterocolitica: organization and siderophore-dependent regulation. J Bacteriol. 1998;180:538–546. doi: 10.1128/jb.180.3.538-546.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 8•.Li YM, Milne JC, Madison LL, Kolter R, Walsh CT. From peptide precursors to oxazole and thiazole-containing peptide antibiotics: microcin B17 synthase. Science. 1996;274:1188–1193. doi: 10.1126/science.274.5290.1188. This paper characterized the enzymatic machinery involved in thiazole/oxazole biosynthesis, determined that the McbBCD proteins form a trimeric complex, and highlighted the importance of the leader peptide. [DOI] [PubMed] [Google Scholar]
- 9.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 10.Schneider TL, Shen B, Walsh CT. Oxidase domains in epothilone and bleomycin biosynthesis: thiazoline to thiazole oxidation during chain elongation. Biochemistry. 2003;42:9722–9730. doi: 10.1021/bi034792w. [DOI] [PubMed] [Google Scholar]
- 11.Milne JC, Eliot AC, Kelleher NL, Walsh CT. ATP/GTP hydrolysis is required for oxazole and thiazole biosynthesis in the peptide antibiotic microcin B17. Biochemistry. 1998;37:13250–13261. doi: 10.1021/bi980996e. [DOI] [PubMed] [Google Scholar]
- 12.Milne JC, Roy RS, Eliot AC, Kelleher NL, Wokhlu A, Nickels B, Walsh CT. Cofactor requirements and reconstitution of microcin B17 synthetase: a multienzyme complex that catalyzes the formation of oxazoles and thiazoles in the antibiotic microcin B17. Biochemistry. 1999;38:4768–4781. doi: 10.1021/bi982975q. [DOI] [PubMed] [Google Scholar]
- 13••.Oman TJ, van der Donk WA. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat Chem Biol. 2010;6:9–18. doi: 10.1038/nchembio.286. An informative review of leader peptide function for ribosomally templated natural products. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt EW. Trading molecules and tracking targets in symbiotic interactions. Nat Chem Biol. 2008;4:466–473. doi: 10.1038/nchembio.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Haft DH, Basu MK, Mitchell DA. Expansion of ribosomally produced natural products: a nitrile hydratase- and Nif11-related precursor family. BMC Biol. 2010;8:70. doi: 10.1186/1741-7007-8-70. Expanded the TOMM family to include two new precursor peptide families whose leader sequences share homology to other proteins. Many times, these precursor peptides are present in multiple variant copies in one genome, providing another example of the natural combinatorial biosynthesis of the TOMM class of natural products. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitchell DA, Lee SW, Pence MA, Markley AL, Limm JD, Nizet V, Dixon JE. Structural and functional dissection of the heterocyclic peptide cytotoxin streptolysin S. J Biol Chem. 2009;284:13004–13012. doi: 10.1074/jbc.M900802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17•.Donia MS, Hathaway BJ, Sudek S, Haygood MG, Rosovitz MJ, Ravel J, Schmidt EW. Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat Chem Biol. 2006;2:729–735. doi: 10.1038/nchembio829. The first paper to rationally design a substrate to be processed by the cyanobactin machinery and result in an artifical ‘natural’ product. This report also highlights the combinatorial biosynthetic capabilities of the cyanobactin biosynthetic pathway. [DOI] [PubMed] [Google Scholar]
- 18.Weissman KJ, Leadlay PF. Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol. 2005;3:925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- 19.Arndt HD, Schoof S, Lu JY. Thiopeptide antibiotic biosynthesis. Angew Chem Int Ed Engl. 2009;48:6770–6773. doi: 10.1002/anie.200901808. [DOI] [PubMed] [Google Scholar]
- 20.Li C, Kelly WL. Recent advances in thiopeptide antibiotic biosynthesis. Nat Prod Rep. 2010;27:153–164. doi: 10.1039/b922434c. [DOI] [PubMed] [Google Scholar]
- 21.Houssen WE, Jaspars M. Azole-based cyclic peptides from the sea squirt lissoclinum patella: old scaffolds, new avenues. Chembiochem. 2010;11:1803–1815. doi: 10.1002/cbic.201000230. [DOI] [PubMed] [Google Scholar]
- 22.Severinov K, Semenova E, Kazakov A, Kazakov T, Gelfand MS. Low-molecular-weight post-translationally modified microcins. Mol Microbiol. 2007;65:1380–1394. doi: 10.1111/j.1365-2958.2007.05874.x. [DOI] [PubMed] [Google Scholar]
- 23.Jones AC, Gu L, Sorrels CM, Sherman DH, Gerwick WH. New tricks from ancient algae: natural products biosynthesis in marine cyanobacteria. Curr Opin Chem Biol. 2009;13:216–223. doi: 10.1016/j.cbpa.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McIntosh JA, Donia MS, Schmidt EW. Ribosomal peptide natural products: bridging the ribosomal and nonribosomal worlds. Nat Prod Rep. 2009;26:537–559. doi: 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nolan EM, Walsh CT. How nature morphs peptide scaffolds into antibiotics. Chembiochem. 2009;10:34–53. doi: 10.1002/cbic.200800438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sivonen K, Leikoski N, Fewer DP, Jokela J. Cyanobactins-ribosomal cyclic peptides produced by cyanobacteria. Appl Microbiol Biotechnol. 2010;86:1213–1225. doi: 10.1007/s00253-010-2482-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walsh CT, Acker MG, Bowers AA. Thiazolyl peptide antibiotic biosynthesis: a cascade of post-translational modifications on ribosomal nascent proteins. J Biol Chem. 2010;285:27525–27531. doi: 10.1074/jbc.R110.135970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelleher NL, Belshaw PJ, Walsh CT. Regioselectivity and chemoselectivity analysis of oxazole and thiazole ring formation by the peptide-heterocyclizing microcin B17 synthetase using high-resolution MS/MS. J Am Chem Soc. 1998;120:9716–9717. [Google Scholar]
- 29.Sinha Roy R, Belshaw PJ, Walsh CT. Mutational analysis of posttranslational heterocycle biosynthesis in the gyrase inhibitor microcin B17: distance dependence from propeptide and tolerance for substitution in a GSCG cyclizable sequence. Biochemistry. 1998;37:4125–4136. doi: 10.1021/bi9728250. [DOI] [PubMed] [Google Scholar]
- 30.Belshaw PJ, Roy RS, Kelleher NL, Walsh CT. Kinetics and regioselectivity of peptide-to-heterocycle conversions by microcin B17 synthetase. Chem Biol. 1998;5:373–384. doi: 10.1016/s1074-5521(98)90071-0. [DOI] [PubMed] [Google Scholar]
- 31.Kelleher NL, Hendrickson CL, Walsh CT. Posttranslational heterocyclization of cysteine and serine residues in the antibiotic microcin B17: distributivity and directionality. Biochemistry. 1999;38:15623–15630. doi: 10.1021/bi9913698. [DOI] [PubMed] [Google Scholar]
- 32.Donia MS, Ravel J, Schmidt EW. A global assembly line for cyanobactins. Nat Chem Biol. 2008;4:341–343. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donia MS, Schmidt EW. Cyanobactins-ubiquitous cyanobacterial ribosomal peptide metabolites. In: Mander L, Liu H-W, editors. Comprehensive Natural Products II. Elsevier Ltd; 2010. [Google Scholar]
- 34••.Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, Ravel J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc Natl Acad Sci U S A. 2005;102:7315–7320. doi: 10.1073/pnas.0501424102. Landmark paper that demonstrated the heterocyclized cyclic peptides isolated from sea squirts were not only produced by the symbiotic bacteria (Prochloron sp.) but were of ribosomal origin. The authors also hypothesized, based on sequence similarity, that the patellamide heterocycles were installed in a similar manner to microcin B17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziemert N, Ishida K, Quillardet P, Bouchier C, Hertweck C, de Marsac NT, Dittmann E. Microcyclamide biosynthesis in two strains of Microcystis aeruginosa: from structure to genes and vice versa. Appl Environ Microbiol. 2008;74:1791–1797. doi: 10.1128/AEM.02392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sudek S, Haygood MG, Youssef DTA, Schmidt EW. Structure of trichamide, a cyclic peptide from the bloom-forming cyanobacterium Trichodesmium erythraeum, predicted from the genome sequence. Appl Environ Microbiol. 2006;72:4382–4387. doi: 10.1128/AEM.00380-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leikoski N, Fewer DP, Sivonen K. Widespread occurrence and lateral transfer of the cyanobactin biosynthesis gene cluster in cyanobacteria. Appl Environ Microbiol. 2009;75:853–857. doi: 10.1128/AEM.02134-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee J, McIntosh J, Hathaway BJ, Schmidt EW. Using marine natural products to discover a protease that catalyzes peptide macrocyclization of diverse substrates. J Am Chem Soc. 2009;131:2122. doi: 10.1021/ja8092168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McIntosh JA, Donia MS, Schmidt EW. Insights into heterocyclization from two highly similar enzymes. J Am Chem Soc. 2010;132:4089–4091. doi: 10.1021/ja9107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McIntosh JA, Schmidt EW. Marine molecular machines: heterocyclization in cyanobactin biosynthesis. Chembiochem. 2010;11:1413–1421. doi: 10.1002/cbic.201000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41••.McIntosh JA, Robertson CR, Agarwal V, Nair SK, Bulaj GW, Schmidt EW. Circular logic: nonribosomal peptide-like macrocyclization with a ribosomal peptide catalyst. J Am Chem Soc. 2010;132:15499–15501. doi: 10.1021/ja1067806. This paper demonstrated the substrate promiscuity of the enzyme involved in cyanobactin macrocyclization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.Lee SW, Mitchell DA, Markley AL, Hensler ME, Gonzalez D, Wohlrab A, Dorrestein PC, Nizet V, Dixon JE. Discovery of a widely distributed toxin biosynthetic gene cluster. Proc Natl Acad Sci U S A. 2008;105:5879–5884. doi: 10.1073/pnas.0801338105. This paper provided structural insight into streptolysin S and also demonstrated the widespread nature of the TOMM biosynthetic gene cluster among bacteria and archaea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Betschel SD, Borgia SM, Barg NL, Low DE, De Azavedo JC. Reduced virulence of group A streptococcal Tn916 mutants that do not produce streptolysin S. Infect Immun. 1998;66:1671–1679. doi: 10.1128/iai.66.4.1671-1679.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nizet V, Beall B, Bast DJ, Datta V, Kilburn L, Low DE, De Azavedo JC. Genetic locus for streptolysin S production by group A streptococcus. Infect Immun. 2000;68:4245–4254. doi: 10.1128/iai.68.7.4245-4254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez DJ, Lee SW, Hensler ME, Markley AL, Dahesh S, Mitchell DA, Bandeira N, Nizet V, Dixon JE, Dorrestein PC. Clostridiolysin S, a post-translationally modified biotoxin from Clostridium botulinum. J Biol Chem. 2010;285:28220–28228. doi: 10.1074/jbc.M110.118554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cotter PD, Draper LA, Lawton EM, Daly KM, Groeger DS, Casey PG, Ross RP, Hill C. Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS Pathog. 2008;4:e1000144. doi: 10.1371/journal.ppat.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy RS, Kim S, Baleja JD, Walsh CT. Role of the microcin B17 propeptide in substrate recognition: solution structure and mutational analysis of McbA1-26. Chem Biol. 1998;5:217–228. doi: 10.1016/s1074-5521(98)90635-4. [DOI] [PubMed] [Google Scholar]
- 48.Su TL. Micrococcin, an antibacterial substance formed by a strain of Micrococcus. Br J Exp Pathol. 1948;29:473–481. [PMC free article] [PubMed] [Google Scholar]
- 49.Mocek U, Zeng ZP, Ohagan D, Zhou P, Fan LDG, Beale JM, Floss HG. Biosynthesis of the modified peptide antibiotic thiostrepton in Streptomyces azureus and Streptomyces laurentii. J Am Chem Soc. 1993;115:7992–8001. [Google Scholar]
- 50.Kelly WL, Pan L, Li C. Thiostrepton biosynthesis: prototype for a new family of bacteriocins. J Am Chem Soc. 2009;131:4327–4334. doi: 10.1021/ja807890a. [DOI] [PubMed] [Google Scholar]
- 51.Liao R, Duan L, Lei C, Pan H, Ding Y, Zhang Q, Chen D, Shen B, Yu Y, Liu W. Thiopeptide biosynthesis featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications. Chem Biol. 2009;16:141–147. doi: 10.1016/j.chembiol.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morris RP, Leeds JA, Naegeli HU, Oberer L, Memmert K, Weber E, LaMarche MJ, Parker CN, Burrer N, Esterow S, et al. Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J Am Chem Soc. 2009;131:5946–5955. doi: 10.1021/ja900488a. [DOI] [PubMed] [Google Scholar]
- 53.Wieland Brown LC, Acker MG, Clardy J, Walsh CT, Fischbach MA. Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc Natl Acad Sci U S A. 2009;106:2549–2553. doi: 10.1073/pnas.0900008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu Y, Duan L, Zhang Q, Liao R, Ding Y, Pan H, Wendt-Pienkowski E, Tang G, Shen B, Liu W. Nosiheptide biosynthesis featuring a unique indole side ring formation on the characteristic thiopeptide framework. ACS Chem Biol. 2009;4:855–864. doi: 10.1021/cb900133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei M, Deng J, Wang S, Liu N, Chen Y. A simple reverse genetics approach to elucidating the biosynthetic pathway of nocathiacin. Biotechnol Lett. 2011;33:585–591. doi: 10.1007/s10529-010-0460-0. [DOI] [PubMed] [Google Scholar]
- 56.Ding Y, Yu Y, Pan H, Guo H, Li Y, Liu W. Moving posttranslational modifications forward to biosynthesize the glycosylated thiopeptide nocathiacin I in Nocardia sp. ATCC202099. Mol Biosyst. 2010;6:1180–1185. doi: 10.1039/c005121g. [DOI] [PubMed] [Google Scholar]
- 57.Engelhardt K, Degnes KF, Zotchev SB. Isolation and characterization of the biosynthetic gene cluster for thiopeptide antibiotic TP-1161. Appl Environ Microbiol. 2010;76:4969–4976. doi: 10.1128/AEM.01442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Yu Y, Tang K, Liu W, He X, Huang X, Deng Z. Identification and analysis of the biosynthetic gene cluster encoding the thiopeptide antibiotic cyclothiazomycin in Streptomyces hygroscopicus 10–22. Appl Environ Microbiol. 2010;76:2335–2344. doi: 10.1128/AEM.01790-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bycroft BW, Gowland MS. Structures of highly modified peptide antibiotics micrococcin P1 and P2. J Chem Soc-Chem Commun. 1978:256–258. [Google Scholar]
- 60••.Bowers AA, Walsh CT, Acker MG. Genetic interception and structural characterization of thiopeptide cyclization precursors from Bacillus cereus. J Am Chem Soc. 2010;132:12182–12184. doi: 10.1021/ja104524q. This is the first paper to characterize the ‘hetero-Diels-Alderase’ involved in thiopeptide (tetrahydro)pyridine ring formation. It also suggested that other posttranslational modifications seen in thiocillin precede cycloaddition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu Y, Guo H, Zhang Q, Duan L, Ding Y, Liao R, Lei C, Shen B, Liu W. NosA catalyzing carboxyl-terminal amide formation in nosiheptide maturation via an enamine dealkylation on the serine-extended precursor peptide. J Am Chem Soc. 2010 doi: 10.1021/ja106571g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li C, Zhang F, Kelly WL. Heterologous production of thiostrepton A and biosynthetic engineering of thiostrepton analogs. Mol Biosyst. 2010 doi: 10.1039/c0mb00129e. [DOI] [PubMed] [Google Scholar]
- 63•.Acker MG, Bowers AA, Walsh CT. Generation of thiocillin variants by prepeptide gene replacement and in vivo processing by Bacillus cereus. J Am Chem Soc. 2009;131:17563–17565. doi: 10.1021/ja908777t. The first paper demonstrating that variants of thiocillin can be produced by installing site directed mutations to the precursor peptide, followed by replacement of the naturally occurring precursor peptides. This has opened the door for engineering a more pharmaceutically tractable thiopeptide. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bowers AA, Acker MG, Koglin A, Walsh CT. Manipulation of thiocillin variants by prepeptide gene replacement: structure, conformation, and activity of heterocycle substitution mutants. J Am Chem Soc. 2010;132:7519–7527. doi: 10.1021/ja102339q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW, van der Donk WA. Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc Natl Acad Sci U S A. 2010;107:10430–10435. doi: 10.1073/pnas.0913677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scholz R, Molohon KJ, Nachtigall J, Vater J, Markley AL, Sussmuth RD, Mitchell DA, Borriss R. Plantazolicin, a novel microcin B17/streptolysin S-like natural product from Bacillus amyloliquefaciens FZB42. J Bacteriol. 2011;193:215–224. doi: 10.1128/JB.00784-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Onaka H, Nakaho M, Hayashi K, Igarashi Y, Furumai T. Cloning and characterization of the goadsporin biosynthetic gene cluster from Streptomyces sp. TP-A0584. Microbiology. 2005;151:3923–3933. doi: 10.1099/mic.0.28420-0. [DOI] [PubMed] [Google Scholar]
- 68.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4. 0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 69.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]