

Abstract
To efficiently deliver therapeutics into cancer cells, a number of strategies have been recently investigated. The toxicity associated with the administration of chemotherapeutic drugs due to their random interactions throughout the body necessitates the development of drug-encapsulating nanopreparations that significantly mask, or reduce, the toxic side effects of the drugs. In addition to reduced side effects associated with drug encapsulation, nanocarriers preferentially accumulate in tumors as a result of its abnormally leaky vasculature via the Enhanced Permeability and Retention (EPR) effect. However, simple passive nanocarrier delivery to the tumor site is unlikely to be enough to elicit a maximum therapeutic response as the drug-loaded carriers must reach the intracellular target sites. Therefore, efficient translocation of the nanocarrier through the cell membrane is necessary for cytosolic delivery of the cargo. However, Crossing the cell membrane barrier and reaching cytosol might still not be enough for achieving maximum therapeutic benefit, which necessitates the delivery of drugs directly to intracellular targets, such as bringing pro-apoptotic drugs to mitochondria, nucleic acid therapeutics to nuclei, and lysosomal enzymes to defective lysosomes. In this review, we discuss the strategies developed for tumor targeting, cytosolic delivery via cell membrane translocation, and finally organelle-specific targeting, which may be applied for developing highly efficacious, truly multifunctional, cancer-targeted nanopreparations.
Keywords: Nanopreparations, Intracellular, Organelle-specific, Drug delivery, Endocytosis, Cancer
1. Introduction
Based on the description of Hanahan and Weinberg, cancer can be characterized by six distinctive hallmarks, including limitless replication potential, insensitivity to growth inhibitory signals, evasion of apoptosis, sustained proliferative signals, induction of angiogenesis, invasion through capillary walls and basal membranes and metastasis to other locations [1]. Since cancer cells are mutated normal cells, chemotherapeutic agents with the potential to kill cancer cells also produce non-specific toxicity to normal tissues.
Nanotechnology has the potential to make a significant impact in cancer therapy by various approaches including an improved toxicity profile of the loaded chemotherapeutic drugs [2, 3]. Nanopreparations or nanoparticles (NP) are typically nano-sized uniformly dispersed particles, composed of biocompatible and biodegradable synthetic polymers, self-assembled lipids, or inorganic materials, capable of carrying therapeutic entities such as small molecule drugs, peptides, proteins, nucleic acids and delivering the pay-loads in a controlled manner to a desired site of action. Nanoparticles may carry drug molecules to the tumor site by encapsulation or as covalently linked drug molecules on the nanoparticles surface. Nanoparticular drug delivery systems offer unique advantages for cancer therapy over free drug administration since NPs
increase drug concentration in the tumor tissue through passive and active targeting, which reduces the drug concentration in normal tissues and reduces toxic side effects.
improve the solubility, pharmacokinetics and pharmacodynamic profiles of the drugs.
can decrease drug release during transit and increase it at the target site.
improve drug stability by reducing its degradation in the systemic circulation.
improve cellular internalization and organelle-specific delivery of the loaded drug that results from adoption of various surface functionalization strategies.
The unique advantages provided with the use of nanoparticles opened up an opportunity to re-evaluate potent drugs with poor pharmacokinetics that had been previously discarded [4]. Over the last two decades, a large number of nanoparticular drug delivery systems consisting of organic or inorganic materials have been evaluated for cancer therapy. Some have translated to the clinic or are being investigated in advanced pre-clinical stages of development [5]. Nanoparticulate drug delivery systems for anti-cancer applications include liposomes, polymeric nanoparticles, micelles, nanoshells, dendrimers, inorganic/metallic nanoparticles, magnetic and bacterial nanoparticles [2].
Owing to the abnormalities of the tumor vasculature, nanoparticles with a size range of 10–500 nm can eventually accumulate at the tumor site [6]. However, the nanoparticles have to unload their pay-loads at the site of disease for effective drug action, more specifically, the encapsulated drug has to successfully reach its sub-cellular target. Therefore, the drug loaded nanocarriers have to overcome systemic, extracellular and intracellular barriers to deliver the drug to the specific organelles for effective therapeutic benefit [7, 8]. More recently developed nanoparticles now being investigated for cancer therapy have been designed with multifunctional capabilities such as long systemic circulation, tumor targeting, cytosolic translocation, organelle-specific targeting for effective cytotoxic effect [2, 3, 8–11].
Functionalization of nanocarriers for organelle-specific targeting of bioactive molecules to specific intracellular compartments can sharply increase the efficiency of various treatment protocols [7]. Generally, nanocarriers are internalized by receptor-mediated endocytosis and reach lysosomal compartment where the nanocarriers and any released drug molecules encounter numerous degradative enzymes. Lysosomal degradation allows only a fraction of intracellular drugs or nanoparticles to become available to a site of action or specific organelles [7]. Bypass of the endocytic pathway or disruption of the endosomes could be powerful strategy for improved cytosolic delivery that enhances the therapeutic efficacy of the loaded drug. Therapeutic efficacy of drugs could further be improved if the nanocarriers were actively targeted to the specific organelles [7]. This is particularly important for anticancer pro-apoptotic drugs and therapeutic nucleic acids, whose the primary site of action is the mitochondrial membrane and the nuclear or mitochondrial genome, respectively. Additional evidence of the role of lysosomes in executing apoptosis has revealed that lysosomes should be explored as an intracellular target for anticancer therapeutics [12–14]. The membrane characteristics of organelles, including charge, composition or specific receptor expression can be exploited by surface modification of nanocarriers with organelle-targeted ligands for organelle-specific delivery. The review descrtibes various strategies that have been adopted for intracellular and organelle-specific delivery of nanocarriers with emphasis on cancer therapy.
2. Fundamentals of tumor targeting by nanopreparations
Several nanopreparations have emerged as potentially useful cancer therapeutics [15]. The physico-chemical properties, including size, surface charge, shape, and density of surface-associated targeting ligands can allow nanoparticles to avoid renal clearance, reach designated cellular destinations in sufficient amounts, undergo active cellular uptake and elicit a biological response with minimal non-specific interactions. The following is a brief discussion about the passive and active targeting of nano-sized drug delivery vehicles (Figure 1).
Figure 1.

Representation of (A) passive (via the EPR effect) and (B) active (receptor-mediated) targeting utilized for targeting nanopreparations to tumors.
2.1. Passive nanoparticle targeting
2.1.1. Role of size
Owing to pathophysiological differences of tumor and infarcted regions from normal tissues, macromolecules larger than 40 KDa and small particles ranging from 10–500 nm in size can leave the vascular capillary bed and accumulate in the interstitial space of such regions [6]. Due to the poor lymphatic drainage of interstitial fluid in tumor, nanoparticles eventually accumulate. The phenomenon of selective extravasation and retention of macromolecules or nanoparticles of specific size range in tumor or infarcted areas is known as the Enhanced Permeability and Retention (EPR) effect [6, 16–20]. The EPR effect, which relies on these unique physiologic features of the tumor microenvironment has revolutionized the anticancer therapy [19]. As EPR effect driven passive drug delivery occurs only in tumor, the drug loaded nanoparticles have limited penetration in normal tissues resulting in decreasing the side effects of chemotherapy [16–18, 20–22].
2.1.2. Role of surface properties
2.1.2.1. Charge
The surface property such as charge of the NPs can be modified for active tumor targeting and cytosolic drug delivery. Nanoparticles with their high surface-to-volume ratio enable surface characteristics to control the behavior of the nanoparticles in the circulation. In this regard, surface charge of the nanoparticles affects cellular association and penetration of the large tumors. Studies from animal models suggest that slightly positively charged nanoparticles with a size range of 50–100 nm can penetrate throughout the large tumors following systemic administration [23].
2.1.2.2. Steric stabilization
A polymer coating such as PEGylation of the NP’s surface sterically stabilizes and makes it long circulating [8]. Scavenging of NPs by circulating macrophages and subsequent clearance by reticulo-endothelial system (RES) increases with increasing surface charge. Polymer coating on the NP surface helps them to evade the engulfment by the RES and thereby, prolong circulation.
2.2. Active nanoparticle targeting
Targeting of the nanoparticles to a desired location, once referred to as the “magic bullet” concept of visionary Paul Ehrlich, is now commonly known as “active targeting” of the nanocarriers. Active targeting of nanopreparations to a tumor can be accomplished by several strategies. Typically, receptor-mediated targeting, in which nanopreparations are surface-functionalized with specific ligands for recognition of specific receptors/antigens in tumor, and stimuli-sensitive drug targeting, in which nanocarriers are engineered to respond to subtle environmental changes in the pathological area to trigger drug release, are the two most commonly applied targeting strategies [8, 9, 24, 25]. To achieve receptor-mediated active nanoparticle targeting, various targeting ligands including proteins, peptides, antibodies, aptamers, or small molecules specific for particular recognition mechanism are conjugated to the nanoparticle surface. The ligand-anchored nanoparticles bind to the tumor-specific or over-expressed antigens leading to accumulation in the tumor tissue.
There are two common approaches for the receptor -mediated NP targeting. The first approach is to target the tumor cell surface receptors for generate the higher tumor cell association. The second is to target the tumor microenvironment including the extracellular matrix or the surface receptors on the capillary endothelial cells for targeting immune response and angiogenesis. The active targeting of internalization-prone cell-surface receptors, over-expressed by cancer cells improves the cellular uptake of the nanocarriers. The increased intracellular delivery is responsible for the enhanced anti-tumor efficacy of the actively targeted nanocarriers. By contrary, NP targeting based on the EPR effect and active targeting to extracellular antigens cause accumulation in the interstitial spaces of the tumor that are eventually endocytosed by the cancer cells. The following is an overview of receptor-mediated active tumor-targeting strategies.
2.2.1. Targeting of cancer cell-surface receptors
Advances in cancer proteomics and bioinformatics have generated discovery of many protein markers, over-expressed in tumors. Targeting ligands that bind to internalization prone receptors are utilized for surface modification of the nanocarriers. Receptors currently utilized for active cancer cell targeting for receptor-mediated endocytosis are depicted below.
2.2.1.1. Transferrin receptor
Transferrin is a vital serum glycoprotein, which is involved in iron homeostasis and the regulation of cell growth. Iron loaded transferrin traverses the cell by binding to a transferrin receptor and is subsequently internalized by this receptor-mediated endocytosis. Transferrin receptors are over-expressed (approximately 100-fold higher than the normal cells) on the surface of various cancer cells, which makes this an attractive target for nanocarrier delivery to tumors [9, 26–29]. In a recent study, a dual targeted solid lipid nanoparticular (SLN) gene delivery system was developed as an anticancer therapy which was modified with an amphiphilic polymer transferrin-PEG-PE and a synthetic glucocorticoid dexamethasone for enhancing cellular and nuclear uptake of the genetic materials, respectively [30]. Pro-apoptotic ceramide loaded liposomes was targeted to cancer cells by anchoring transferrin to the liposomal surface [13].
2.2.1.2. Folate receptor
Folic acid is an essential vitamin for cell survival due to its role in the synthesis of nucleotide bases. Folic acid is taken up by the folate receptor expressed on the cell surface by receptor-mediated endocytosis. Folate receptors are over-expressed in many human malignancies particularly in ovarian cancer. Various attempts have been undertaken to actively target nanopreparations to tumor by conjugating folic acid (FA) to a NP-surface [31–34]. In a recent study, ovarian cancer cell targeted nanopreparation, a folate conjugated ternary copolymer, FA-Polyethylene glycol-polyethyleneimine-poly(ε-caprolactone) (FA-PEG-PEI-PCL) was developed for co-delivery of drug and siRNA [35]. In a related study, folate-polymer coated liposomes were developed for targeted chemotherapy using doxorubicin [36]. In this study, FA conjugated poly-L-lysine was used to coat doxorubicin-loaded anionic liposomes. The FA-polymer coated liposomes demonstrated increased cellular uptake of loaded doxorubicin by folate receptor over-expressing human nasopharyngeal carcinoma KB cells that resulted in two-fold higher cytotoxicity compared to PLL-coated liposomal Doxorubicin.
2.2.1.3. The epidermal growth factor receptor (EGFR)
EGFR, over-expressed in a variety of solid tumors, plays a significant role in the progression of several human malignancies, since the activation of this receptor stimulates key tumor-promoting processes including cell proliferation, angiogenesis, invasion and metastasis. Human epidermal receptor-2 (HER-2) is over-expressed in a majority of patients with breast cancer [37–39]. Anti-EGFR or anti-HER-2 monoclonal antibody-grafted nanopreparations, mainly as immunoliposomes have been studied extensively as anticancer therapeutics [40–42]. Dox-loaded anti-HER-2 immunoliposomes have demonstrated increased therapeutic efficacy in breast cancer xenograft models when compared to unmodified PEGylated liposomes [40, 43]. Generally, the Fab’ portion of recombinant humanized anti-HER2 MAb, rhuMAb HER2 was covalently linked to the distal end of the PEG chain of sterically stabilized liposomes [40, 43].
2.2.2. Targeting the tumor microenvironment
Preventing tumors from recruiting new blood vessels and the destruction of existing endothelium inhibit tumor growth and promote subsequent tumor cell death [44, 45]. Targeting of the nanomedicines to the endothelial cells of the tumor blood vessels is an attractive strategy for inhibition of growth of the tumor, its blood supply, and capacity for metastasis [46]. The following are among the main targets on the tumor endothelial cells.
2.2.2.1. Vascular endothelial growth factor (VEGF)
VEGF is the key mediator of angiogenesis up-regulated by oncogene expression, a variety of growth factors and by hypoxia. VEGF binds to two VEGF receptors (VEGF receptor-1 and VEGF receptor-2) expressed on vascular endothelial cells [47, 48]. Upregulation of VEGF levels in the tumor cells results in upregulation of VEGFR-1 and VEGFR-2. This production of VEGF and other growth factors by the tumor and their interaction with receptors result in enhanced angiogenesis. Targeting VEGF or VEGFR inhibits neovascularization, resulting in tumor cell death due to lack of oxygen and nutrients. Two approaches have been developed to decrease angiogenesis (i) targeting VEGF to inhibit ligand binding to VEGFR, and (ii) targeting VEGFR to decrease VEGF binding to its receptor [44, 47, 49]. For targeting nanocarriers to the tumor, the human recombinant VEGF isoform VEGF121 was conjugated on a carrier surface [50]. Anti-VEGFR-2 mAb-labeled NPs were more effective in delaying tumor growth than Anti-VEGFR-2 mAb alone [51].
2.2.2.2. αvβ3 Integrin
αvβ3 integrin is an endothelial cell surface receptor for various extracellular matrix proteins with a specific amino acid sequence (Arginine-Glycine-Aspartic acid (RGD)) [52]. αvβ3 integrin is over-expressed in neovascular tumor endothelial cells and mediate migration of the endothelial cells. Targeting αvβ3 integrin is typically done by anchoring the RGD sequence on peptides and mimetic non-peptides [53]. An RGD-anchored cationic NP was reported to deliver genes selectively to angiogenic blood vessels that resulted in apoptosis leading to regression of primary and metastatic tumors [53].
2.2.2.3. Vascular cell adhesion molecules (VCAM)
VCAM-1, an immunoglobulin-like transmembrane glycoprotein that promotes cell-cell adhesion, is over-expressed on the surface of endothelial cells during inflammation. This signaling molecule is absent in the normal human vasculature, but readily inducible by inflammation and angiogenesis [54]. Active targeting of the nanopreparations to cancer cells was achieved by anchoring an anti-VCAM-1 monoclonal antibody (M/K-271) to the nanocarrier [55]. This VCAM-1 targeted nanopreparation actively targeted angiogenic tumor blood vessels. Therefore, VCAM-1 targeted nanocarriers can be further developed for the purpose of modifying the endothelial functions in tumor.
2.2.2.4. Matrix metalloproteinases (MMPs)
MMPs, a group of structurally related zinc-dependent endopeptidases are essential for angiogenesis, tumor invasion and metastasis [56]. Membrane type 1 MMP (MT1-MMP) plays a significant role in angiogenesis by extracellular matrix degradation, endothelial cell invasion, formation of capillary tubes, and recruitment of accessory cells [57]. MT1-MMP is expressed on endothelial and certain types of cancer cells including lung, gastric, colon, breast, cervical carcinomas, gliomas and melanomas [58–60]. Metalloproteinase aminopeptidase N (CD13) is another endothelial cell surface receptor that causes degradation of the extracellular matrix by unblocking the N-terminal segments of peptides and proteins [61]. A tumor homing-peptide NGR (Asn-Gly-Arg) has also been recognized that binds to aminopeptidase and inhibits angiogenesis [62]. Thus, targeting NPs to MMPs by surface functionation with MMP ligands or anti-MT1-MMP antibody may be an effective strategy for inhibiting angiogenesis in cancer therapy [63–65].
2.2.3. Targeting specific cancer cells using aptamers
Aptamers are readily synthesizable nucleic acid ligands that possess high affinity and specificity for target antigens. Aptamers could be utilized for dual purposes, as a drug candidate or for targeting nanopreparations towards tissues over-expressing target antigens [66]. Aptamers are single stranded DNA or RNA oligonucleotides folded into a stable conformation that recognize bio-macromolecules such as nucleic acids, proteins, phospholipids and sugars [67]. Aptamers specific for a target bio-macromolecule are identified from randomized nucleic acid libraries using high-throughput screening methodologies. In a recent study, a nuclease-resistant VEGF growth factor aptamer was conjugated to a lipid and the anchored into the liposomal lipid bilayer [68]. These VEGF-aptamer modified liposomes produced high affinity binding of liposomes to the VEGF. The lipid modification/liposomal incorporation improved the plasma residence time, the inhibitory activity of the aptamers towards VEGF-induced endothelial cell proliferation and angiogenesis in vitro and in vivo compared to the free aptamer. In another study, polymeric nanocarriers were surface-functionalized with A10 2′-fluoropyrimidine RNA aptamers that recognize the extracellular domain of prostate-specific membrane antigens, which is a well characterized antigen expressed on the surface of prostate cancer cells [69].
3. Nanopreparations for cancer therapy
Nanopreparations, clinically approved or at various stages of development for cancer therapy are listed in Tables 1 and 2, respectively. Liposomes loaded with small molecule chemotherapeutic drugs have been added to those approved for cancer therapy since mid-1990s. The first liposomal preparations approved for clinical use as formulation containing doxorubicin were Doxil® (PEG-coated) and Myocet® (uncoated). Other clinically approved liposomal preparations include DaunoXome®, encapsulating chemotherapeutic drug daunorubicin for the treatment of Kaposi sarcoma and Onco-TCS®, containing vincristine for non-Hodgkin lymphoma. Albraxane®, a solvent free, albumin-bound nanoparticle formulation of paclitaxel is currently approved for metastatic breast cancer. Abraxane has a better safety profile, higher response rate and improved pharmacokinetics compared to conventional paclitaxel.
Table 1.
Approved nanopreparations for cancer therapy.
| Nanopreparation | Drug | Trade Name | Treatment |
|---|---|---|---|
| Liposomes | Doxorubicin | Doxil®/Caelyx/Li podox | Ovarian, metastatic breast cancer, Kaposi Sarcoma |
| Liposomes | Doxorubicin | Myocet® | Breast cancer |
| Liposomes | Daunorubicin | DaunoXome® | Kaposi’s Sarcoma |
| Liposomes | Vincristine | Onco-TCS® | Non-Hodgkin’s lymphoma, |
| Liposomes | Cytarabine | DepoCyt® | Lymphomatus meningitis |
| Drug conjugate | Doxorubicin | Transdrug® | Hepatocarcinoma |
| Drug conjugate | Albumin-paclitaxel | Abraxen® | Non-small cell lung cancer, Metastatic breast cancer |
| Drug conjugate | PEG-L-asparaginase | Oncaspar | Acute lymphoblastic leukemia |
Table 2.
Example of nanopreparations undergoing clinical investigations for cancer therapy.
| Formulation | Drug | Name | Indication | Status |
|---|---|---|---|---|
| Liposomes | Doxorubicin | ThermoDox® | Various cancers | II |
| Liposomes | Doxorubicin | JNS002 | II | |
| Liposomes | Cisplatin | LiPlaCis | I | |
| Liposomes | Cisplatin | Lipoplatin | III | |
| Liposomes | Cisplatin | SPI-77 | II | |
| Liposomes | Cisplatin analog | L-NDDP/aroplatin | II | |
| Liposomes | Oxaliplatin | MBP426 | I | |
| Liposomes | Vincristine | Marqibo® | II | |
| Liposomes | Annamycin | Liposomal Annamycin | II | |
| Liposomes | Mitoxantrone | LEM | Pre-clinical | |
| Liposomes | Paclitaxel | PNU-93914 | II | |
| Liposomes | Paclitaxel | LEP-ETU | II | |
| Liposomes | CKD602 (Topoisomerase inhibitor) | S-CKD602 | Various cancers | I/II |
| Liposomes | ||||
| Polymeric Nanoparticles (Cyclodextrin) | Camptothecin | CRLX101 | Various cancers | II |
| Polymeric micelles (PEG-poly aspartate) | Paclitaxel | NK105 | Various cancers | II |
| Polymeric micelles (PEG-poly(D,L-lactide) | Paclitaxel | Genexol®-PM | Breast, lung, pancreatic cancer | II–III |
| Polymeric micelles (PEG-PLGA) | Docetaxel | BIND-014 | Various cancers | I |
| Polymeric micelles (PEG-poly(D,L-lactide) | Docetaxel | Nanoxel® | Advanced breast cancer | I |
| Polymeric micelles | Doxorubicin | NK911 | Various cancers | I |
| (PEG-poly aspartate | ||||
| Polymeric micelles (Glycoproteins) | Doxorubicin | SP1049C | Various cancers | II |
| Polymer-drug conjugate | Paclitaxel | Xyotax® | Breast, overian, advanced lung cancer | II |
| Polymer-drug conjugate | Paclitaxel | Taxoprexin® | Various cancers | II–III |
| Polymer-drug conjugate | Doxorubicin | PK1 | Breast, lung, colon cancers | II |
4. Intracellular delivery
A nanocarrier, once in the tumor has to cross the cell membrane barrier and translocate into the cytoplasm to exert its therapeutic action. The intracellular site of drug action can be cytoplasm, or specific organelles such as the mitochondrion, lysosome, or nucleus. Typically, gene and antisense therapy must be delivered to cell nuclei, pro-apoptotic drugs to mitochondria, lysosomal enzymes or apoptosis-inducers involving the lysosomal apoptotic pathway to lysosomes. In general, intracellular delivery of nanopreparations represents a challenge. Nanocarriers and other macromolecular therapeutics, unlike small molecules which cross cell membrane by random diffusion, requires energy-dependent endocytosis for cellular internalization. Molecules or nanoparticles that enter the cells by the endocytic pathway become entrapped in the endosomes, and eventually fuse with lysosomes, where active degradation of the nanoparticles and drugs takes place (Figure 2). As a result, only a small fraction of loaded drugs appear in the cytoplasm. To deliver nanocarriers effectively to the cytoplasm, a variety of strategies have been developed as described below.
Figure 2.

Schematic drawing of the cytosolic delivery and organelle-specific targeting of drug loaded nanoparticles via receptor-mediated endocytosis. After receptor mediated cell association with nanoparticles, the nanoparticles are engulfed in a vesicle known as an early endosome. Nanoparticles formulated with an endosome disrupting property disrupt the endosomes followed by cytoplasmic delivery. On other hand, if nanoparticles are captured in early endosomes, they may make their way to lysosomes as late endosomes where their degradation takes place. Only fraction of non-degraded drug released in the cytoplasm interacts with cellular organelles in a random fashion. However, cytosolic delivery of a fraction of organelle-targeted nanoparticles via endosomal escape or from lysosomes travel to the targeting organelles to deliver their therapeutic cargo.
4.1. Cell penetrating peptides (CPPs)
Over the last two decades, many short peptide sequences, commonly referred to as cell penetrating peptides have been identified that are capable of efficiently entering cells alone or when linked to bulky cargos such as peptides, proteins, oligonucleotides, pDNA, or liposomes [70–74]. The common characteristic of all CPPs is the net cationic charge due to the presence of the basic amino acids lysine and arginine. Among many CPPs, the peptide sequence of positions 48–60, referred to as Tat peptide (Tat-p), derived from the 86-mer trans-activating transcriptional activator (Tat) protein encoded by human immunodeficiency virus type-1 (HIV)-1 was the first discovered and most extensively utilized for intracellular delivery of biomacromolecules [75–79]. Other CPPs include pVEC derived from murine vascular endothelial cadherin, signal-sequence-based peptides and membrane translocating sequences (MTSs) [80, 81]. Model amphipathic peptides (MAP) or chimetic peptides, produced by the fusion of hydrophilic and hydrophobic domains from various sources can act as CPPs. Examples include transportan, a fusion of mastoparan and galanin, and MPG, a fusion of HIV-1 gp41 protein and peptide from the nuclear localization sequence of SV40 T-antigen [82, 83]. Synthetic oligo-arginines, most often octa-arginines (R8) have shown potent cell membrane translocation efficiency [71, 73, 84, 85]. Transportan and MAP have lysine, whereas Tat-p, penetratin, and pVEC have arginine as the primary cationic charge contributor. MAP has the highest cellular uptake and cargo delivery efficiency followed by transportan, Tat-p (48–60) and penetratin [86].
4.1.1. Tat-peptide
Tat-p (48–60), the most studied CPP consists of 13 amino acids including six arginine and two lysine residues (GRKKRRQRRRPPQ) that contribute to its basicity and hydrophilicity. The first reported modification of a nanopreparation with Tat-p was a biocompatible, dextran coated super-paramagnetic iron oxide nanoparticle derivatized with Tat-p [87]. In another study, the derivatized magnetic nanoparticles were internalized by lymphocytes over 100-fold more efficiently than unlabeled particles and the labeled cells were highly magnetic as detected by NMR imaging. Tat-p functionalization of the liposomal surface produced efficient intracellular delivery of liposomes, even at low temperature and in the presence of metabolic inhibitors, which indicates that the cargo delivery mediated by Tat-p is primarily a non-energy dependent process [88]. Another study reported the Tat-p modification of liposomes in ovarian carcinoma cells was by enhanced endocytosis rather than direct cytosolic delivery by plasma membrane translocation [89].
Tat-p has been utilized for intracellular delivery of nucleic acids [90–92]. Tat-p functionalized cationic liposomes, complexed with a model plasmid encoding for the enhanced-green fluorescence protein (pEGFP) demonstrated efficient transfection both in vitro and in vivo [90]. A dual targeted nanopreparation, a cationic liposome-plasmid DNA complex modified with tat-p and monoclonal antimyosin antibody 2G4 specific for cardiac myosin, was developed for gene delivery into the ischemic myocardium [92]. mAb 2G4-modified Tat-p lipoplexes demonstrated increased accumulation and enhanced transfection of hypoxic cardiomyocites both in vitro and in vivo.
4.1.2. Penetratin
Another cell penetrating peptide called penetratin, derived from the third helix of the homodomain of Drosophila antennapedia protein (positions 43–58) possesses the ability to be internalized even at 4 °C, suggesting an energy-independent mechanism of translocation rather than classical endocytosis [93]. Penetratin, which is rich in lysine residues, has been linked to a variety of cargos. The anti-androgen drug bicalutamide was linked to penetratin for intracellular and nuclear-targeted delivery [94]. Both CPPs, Tat-p and penetratin were conjugated to the chemotherapeutic doxorubicin to counteract the tumor cell resistance towards doxorubicin [95]. The strategy significantly enhanced the cellular uptake and cytotoxicity in a variety of tested sensitive- and drug-resistant cancer cell lines. The penetratin sequence linked in a P53-derived peptide (as the anticancer drug, PNC-28) induced tumor cell necrosis in cancer cells, but not in normal cells [96] Grb-7 peptide was conjugated to either Tat-p or penetratin. Both the peptide conjugates were able to inhibit the proliferation of breast cancer cells [97].
4.1.3. Octa-arginine (R8)
Oligo-arginine cell penetrating peptides have gained a great deal of attention as a cell membrane penetration enhancer [98, 99]. R8 has been successfully utilized for intracellular delivery of several nanoparticles [100] Releasable, R8-conjugated paclitaxel overcame multidrug resistance elicited by Taxol® [101]. Octa-arginine-linked nanocarriers were efficiently internalized and demonstrated improved in vivo efficacy [71–73, 102, 103]. A multifunctional envelope-type nanodevice (MEND), based on programmed packaging, has been developed that utilizes R8 for efficient intracellular delivery [10, 71]. Our group demonstrated that the bleomycin-loaded octa-arginine modified liposomes resulted in improved tumor growth inhibition [104]. Surface functionalization of doxorubicin-loaded liposomes, commercially available as Doxil® or Lipodox® with octa-arginine resulted enhanced anticancer activity in vitro and in vivo compared to the unmodified liposomes [103]. Figure 4 represents the schematic diagram for the modification of Lipodox® with R8-linked PEG-PE polymer. The confocal laser scanning micrograph in Figure 4 indicated that the R8-modified doxorubicin-loaded liposomes delivered the pay-load more efficiently to the cytosol, possibly via endosome disruption, that resulted in enhanced Dox accumulation in the nucleus.
Figure 4.
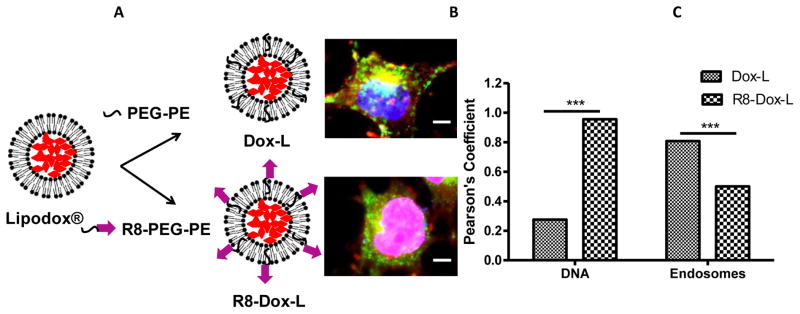
Modification of doxorubicin-loaded commercially available Lipodox® with cell-penetrating synthetic peptide, octa-arginine, to achieve improved anticancer activity. (A) Dox-L and R8-Dox-L were prepared by incorporation of hydrophilic co-polymer polyethyleneglycol-phosphatidylethanolamine (PEG-PE) and R8-PEG-PE into the liposomal lipid bilayer. (B) The confocal laser scanning micrograph shows murine mammary carcinoma cell (4T1) treated with Dox-L and R8-Dox-L at a Dox concentration 6 μg/mL. The cells were additionally stained with a DNA stain (blue) and an endosomal marker (green) for visualization of nuclei and endosomes. The merged picture indicated that R8-Dox-L delivered its pay-load to the nucleus more efficiently than Dox-L. (Scale bar. 15 nm.) (C) Co-localization of Dox signal with DNA and endosomes quantified by Image J software. The Pearson’s coefficient of colocalization of Dox-L or R8-Dox-L with DNA and endosomes indicates that R8-Dox-L had significantly higher co-localization with DNA than endosomes, indicating an endosome-disrupting property of R8-Dox-L which resulted in more cytosolic delivery of the loaded doxorubicin compared to Dox-L.
4.2. Endosomal escape
Various attempts have been undertaken to develop ‘SMART’ nanocarriers that allow endosomal disruption for release of drugs or drug-loaded nanocarriers to the cytoplasm before their passage to lysosomes.
4.2.1. Endosome disrupting peptides
High density R8-modified liposomes are reported to be internalized via macropinocytosis, which avoids the lysosomal fate of the endocytic pathway [72]. The high content of basic residues in CPPs allows interaction with the endosomal membrane at lowered pH, which causes endosomal disruption and pore formation [105]. Histidine-rich peptides can either fuse with endosomal membrane causing pore formation or buffer the proton pump causing membrane lysis [105]. A pH-sensitive fusogenic peptide GALA with amino acid sequence WEAALAEALAEALAEHLAEALAEALEALAA on the liposomal surface has produced efficient cytosolic delivery of loaded cargo [106, 107]. A pH-independent fusion inducer peptide, KALA (WEAKLAKALAKALAKHLAKALAKALKA) has been utilized for cytosolic delivery of a multi-layered nanoparticle as a gene carrier [108].
4.2.2. Fusogenic lipids
Various lipids used to formulate lipid-based nanocarriers such as liposomes and micelles have been identified with the ability to fuse with the endosomal membrane to destabilize of the nanocarriers and release of a drug pay-load. Typically, dioleoyl phosphatidylethanolamine (DOPE) is used extensively as a fusogenic lipid [109, 110]. DOPE exists in two conformations, lamellar and hexagonal phase. Transformation from lamellar to inverted hexagonal, which takes place at the low pH characteristic of the endosomal compartment promotes fusion of the nanocarrier with the endosomal membrane and resulted in the escape of the enclosed cargo [111] A combination of anionic lipids, cholesteryl hemisuccinate (CHEMS) and DOPE or phosphatidic acid have been successfully used as fusogenic lipid components [112].
4.3. Stimuli sensitivity
Nanopreparations, in this category are specially devised to be sensitive to externally applied energy such as ultrasound, light, heat, or internal stimuli such as change in pH, redox potential, and enzymes to trigger drug release. These nanoparticles maintain a hidden or stealth character during systemic circulation. However, they are transformed or degraded to trigger localized drug release and subsequent enhanced intracellular delivery in response to environmental or applied, focused stimuli [45]. Application of ultrasound for chemotherapeutic drug release from nanopreparations in solid tumors has been extensively studied by Rapoport et al [113–115]. In ultrasound-mediated micellar drug delivery, the mild hyperthermia induced may enhance micellar extravasation into tumor tissue, whereas the mechanical action of ultrasound triggers drug release from micelles and enhances intracellular uptake of the drug. In a recent study conducted by this group, perfluoro-15-crown-5-ether (PFCE) was used as a core forming compound in perfluorocarbon nanoemulsions, which manifested both ultrasound and fluorine ((19)F) MR contrast properties [113]. Paclitaxel-loaded PFCE nanodroplets had demonstrated excellent therapeutic properties as indicated by significant tumor regression and suppression of metastasis.
Light-triggered activation of nanocarriers composed of various photo-sensitive polymers has shown promising applications for various biomedical applications and cancer therapy [116–120]. Light-triggered activation of nanocarriers that liberate the pay-load is governed by various underlying mechanisms including photo-driven isomerization and activation, de-crosslinking, surface plasmon absorption and photochemical effects, hydrophobicity changes and polymer backbone fragmentation [121]. External magnetic field-driven iron oxide nanoparticles are being investigated extensively as diagnostics and to improve the efficacy of anticancer drugs [122–124]. In addition to the application of these external stimuli, various internal stimuli including altered pH, temperature, redox potential, over-expressed proteolytic enzymes of the tumor microenvironment potentiate activation of nanocarriers by deshielding specific moieties or coatings to expose useful functionalities [125, 126].
5. Organelle-specific delivery
Delivering drug or a nanocarrier to the cytoplasm is often not enough to elicit a maximum therapeutic effect. Intracellular delivery can only result in random interaction of nanocarriers with cellular organelles. Organelle-specific delivery or delivery of the drug or drug-loaded nanocarriers directly to the site of drug action is required to achieve maximum therapeutic with a minimum of off-target effects.
5.1. Mitochondrial delivery
Besides being the powerhouse of cells, mitochondria are also considered a suicidal weapons store. Dozens of cell death-associated signal transduction pathways are activated through mitochondrion’s action. Mitochondria control the activation of programmed cell death by regulating the translocation of pro-apoptotic proteins from the mitochondrial intermediate space to the cytosol.
5.1.1. Role of mitochondria in cancer
Mitochondrial dysfunction has been linked to many of the hallmarks of cancer cells including their limitless proliferative potential, impaired apoptosis, insensitivity to anti-growth signals, enhanced anabolism and decreased autophagy [127, 128]. The microenvironment of rapidly proliferative cancer cells become hypoxic owing to the inability of the local vasculature to supply adequate oxygen [129]. This chronic hypoxic condition is lethal to normal cells. However, owing to the inability of mitochondria to provide enough ATP for cell survival under hypoxic conditions, malignant cells upregulate the glycolytic pathway as a alternate means of energy production by induction of hypoxia-inducible factor-1 (HIF-1) [130]. In cancer cells, down-regulation of mitochondrial respiration causes impairment of normal cellular functions. Kreb’s cycle substrates such as succinate accumulate as a result of inhibition or mutation of succinate dehydrogenase and signal stimulation of glycolysis and the activation of HIF-1α.
Tumor cells successfully escape hypoxia-mediated cell death by lowering expression or causing mutation of p53 [131]. p53, a tumor suppressor protein known as a central regulator of the cellular stress response. Mutation of p53 in tumors causes down-regulation of mitochondrial respiration and a shift of cellular energy metabolism towards glycolysis. Another important factor associated with mitochondrial dysfunction is reactive oxygen species (ROS) production. A dysfunctional mitochondrial respiratory chain produce excessive ROS, causing an imbalance between production of ROS and antioxidant defense, which is defined as oxidative stress. The ROS oxidize proteins, induce lipid peroxidation and damage mitochondrial DNA (mtDNA). The level of oxidatively modified bases such as 8-hydroxyguanosine is 10–20-fold higher than that found in nuclear DNA [129].
In cancer, the mitochondria-mediated intrinsic apoptotic pathway is suppressed, owing to the over-expression of anti-apoptotic proteins that protects mitochondrial membrane from permeabilization. Mitochondrial membrane permeabilization is the crucial step in the triggering of cell death via apoptosis. Over-expressed anti-apoptotic proteins such as Bcl-2, Bcl-XL, Mcl-1, and Bcl-w in cancer cells interact with pro-apoptotic proteins Bax and Bak to prevent their oligomerization necessary for mitochondrial membrane permeabilization.
5.1.2. Mitochondrial drug targeting
Since mitochondria play a major role in executing apoptosis-mediated cell death and cancer cells have suppressed apoptosis, mitochondria-directed drugs designed to trigger apoptosis are likely to be promising treatment strategy for curbing cancer. Curbing the mitochondrial ROS production by delivery of antioxidants to the mitochondria, activating mitochondrial membrane permeability, targeting Bcl-2 proteins to inhibit or down-regulate anti-apoptotic action, delivering therapeutic genes for the expression of pro-apoptotic proteins including Bax, Bak are a few of the effective mitochondrial targeted treatment strategies in cancer [132–135].
5.1.2.1. Mitochondrial- targeted ligands
Mitochondrial-targeted drug delivery systems are emerging as a prime pharmacological target in the treatment of cancer. Considerable efforts are being undertaken to discover molecules that target mitochondria [136]. Figure 3 summarizes various mitochondria-targeting strategies. Mitochondrial targeting is achieved by various delocalized lipophilic cations such as small molecule ligands, mitochondria-penetrating-peptides (MPP), cationic ‘bola-lipid’-based vesicles, mitochondrial protein import machineries and molecules targeting mitochondrial inner/outer membranes.
Figure 3.

Summary of current strategies for mitochondrial targeting. (A) attachment of lipophilic cations such as triphenylphosphonium to small molecules or nanocarriers for targeting to the mitochondria; (B) Mitochondrial targeting Szeto-Schiller (SS) peptides containing an aromatic-cationic sequence motif selectively partition into the inner mitochondrial membrane independent of the mitochondrial membrane potential; (C) A dicationic mitochondriotropic compound, dequalinium chloride, self-assembles and forms vesicle-like aggregates called DQAsomes. These vesicles are taken up by endocytosis, fuse with the mitochondrial outer membrane and enter to the mitochondrial matrix via the mitochondrial protein import machinery; (D) MITO-porter is a liposome-based carrier that fuses with the mitochondrial membrane and releases its cargo to the intra-mitochondrial compartment; (E) Mitochondrial targeted signal peptides attached to non-mitochondrial proteins create a chimeric protein taken up by the mitochondrial matrix via the mitochondrial protein import machinery. Cargo consisting of a drug or nucleic acid attached to this chimeric protein can be selectively transferred to mitochondria.
5.1.2.1.1. Lipophilic cations
Sufficient lipophilicity combined with delocalized positive charge are a pre-requisite for mitochondriotropic molecules. Various lipophilic cations including triphenylphosphonium (TPP), rhodamine 123, Flupirtine, MKT-077 and anthracyclins have demonstrated selective accumulation in the mitochondria upon intracellular delivery [137]. Mitochondria maintain a constant membrane potential of about −180 to −200 mV across their lipid bilayer by using channel pumps and oxidative phophorylation pathways [138]. This high negative membrane potential is not present in any other cellular organelle, which offers a unique chemical opportunity for selective accumulation of lipophilic cations to the mitochondria.
Owing to their possession of a delocalized cationic charge over a large surface area comprised of three phenyl rings, compounds with a triphenylphosphonium (TPP) moiety can easily pass through the hydrophobic barrier presented by the lipid bilayer [139]. Several biologically active molecules have been modified with a TPP moiety to selectively targeting them to the mitochondria [139–143]. Typically, antioxidants are targeted towards their site of action within the mitochondria [144]. The TPP group has been conjugated to the phenolic moiety of Vitamin E for its selective delivery to mitochondria. Results have demonstrated that increased antioxidant accumulation to the site of action protected mitochondria from oxidative damage more effectively than Vitamin E [141]. Mitochondrially-targeted antioxidant, MitoQ, a TPP-conjugated ubiquinone derivative has been used extensively to prevent oxidative damage in diseases associated with oxidative stress [145–151]. MitoQ accumulates several 100-fold within mitochondria and is significantly more potent than the non-targeted CoQ10 analogue cecycloubiquinone [152]. The TPP-conjugation also circumvents poor solubility problems associated with the naturally occurring antioxidant, coenzyme Q10 [153].
Drug-loaded liposomes have been surface-functionalized with TPP for effective delivery of therapeutic cargo to the mitochondria [154, 155]. The TPP cation has been conjugated to a lipophilic stearyl moiety to form stearyl triphenylphosphonium (STPP), which was easily incorporated into the liposomal lipid bilayer, anchoring the TPP group on the liposomal surface [154]. The STPP-modified mitochondriotropic liposomes were loaded with a sphingolipid signaling molecule, ceramide. An elevated ceramide level results in the formation of ceramide channel in the mitochondrial membrane, which leads to the release of cytochrome-c from the mitochondrial intermembrane space and initiation of apoptosis. Mitochondrial delivery of ceramide via STPP-modified liposomes enhanced its cytotoxicity both in vitro and in vivo in a murine mammary carcinoma model.
In our work, the toxicity associated with STPP was overcome by conjugating the TPP group to the amphiphilic poly(ethylene glycol)-phosphatidylethanolamine moiety [155]. The amphiphilic TPP-PEG-PE was incorporated into the liposomes, where the lipid group was embedded in the liposomal lipid bilayer and the TPP group, anchored at the distal end of the PEG chain was on the surface. These mitochondriotropic liposomes effectively delivered a pro-apoptotic chemotherapeutic paclitaxel to the mitochondria in comparison to the non-mitochondria targeted liposomes and enhanced the antitumor effect in vitro and in vivo (Figure 5).
Figure 5.

Evaluation of efficacy of mitochondria-targeted liposomes. The liposomal surface was modified with a co-polymer, triphenylphosphonium-PEG-PE conjugate, for mitochondrial targeting (TPP-L). (A) Confocal microscopy of HeLa cells treated with rhodamine-labeled unmodified liposomes (PL) and TPP-L. Yellow dots in the merged picture indicate co-localization of red (liposome) and green (mitotracker) signals. (B) Evaluation of the cell-killing efficacy of paclitaxel-loaded TPP-L (TPP-L-PTX) on HeLa cells. The targeted delivery of paclitaxel to mitochondria resulted in a further decrease in cell viability. (C) The in vivo evaluation of the efficacy of TPP-L-PTX in reducing tumor volume. The arrows indicate the day of intravenous administration of liposomes at a paclitaxel dose of 1 mg/Kg. The significantly higher reduction in tumor volumes (p<0.01) indicated the higher therapeutic efficacy of mitochondrial-targeted PTX liposomes.
In a related study, surface conjugation of TPP to a macromolecule dendrimer resulted in efficient mitochondrial targeting [156]. A N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer-based mitochondriotropic drug delivery system was developed by conjugating a TPP group and a photosensitizer mesochlorine e6 (Mce6) on the polymer backbone [157]. This mitochondrial targeted HPMA-copolymer-drug conjugate enhanced in vitro cytotoxicity compared to non-targeted HPMA-Mce6 conjugates.
Rhodamine 123 is another lipophilic cation utilized for drug delivery to the mitochondria. A complex of tetrachloroplatinate and rhodamine-123 was used to generate cytotoxic and antitumor effect. In our work, paclitaxel liposomes were modified with rhodamine 123-conjugated pEG-PE polymer. This drug delivery system produced efficient mitochondrial targeting and increased cytotoxicity of loaded paclitaxel compared to unmodified paclitaxel loaded liposomes [158].
5.1.2.1.2. Mitochondria-penetrating peptides
A class of mitochondrial targeting peptides, known as Szeto-Schiller (SS) peptides possess a structurally similar aromatic cationic motif in which the aromatic group, either tyrosine or a dimethyltyrosine, alternates with a basic amino acid. The SS-peptides are rapidly taken up by cells and target the mitochondria by an energy-independent mechanism [153, 159, 160] The SS-peptides localize to the inner mitochondrial membrane, where they serve as antioxidants, scavenge hydrogen peroxide, peroxynitrile and inhibit lipid peroxidation [161]. They have been shown to protect ischemia-reperfusion mediated cell injury [162]
5.1.2.2. Mitochondrial targeting via membrane fusion
5.1.2.2.1. MITO-Porter
A mitochondrial-targeting nanopreparation, MITO-Porter, developed by Harashima group holds promise as an efficacious system for the delivery of both large and small bioactive molecules into mitochondria [163]. MITO-Porter is a liposome based nanocarrier composed of DOPE/sphingmyelin/Strearyl-R8 (9:2:1) that delivers its macromolecular cargo to the mitochondrial interior via membrane fusion [163–165]. The liposomal surface-modification with high-density R8 was responsible for its efficient cytosolic delivery after cell uptake by macropinocytosis. Figure 5 represents a schematic diagram of the pathway followed by MITO-Porter for mitochondrial delivery. Upon cytosolic release from macropinosomes, MITO-Porters translocate into the mitochondrial membrane via electrostatic interaction between the MITO-Porters and mitochondria and induce fusion [164]. The lipid composition of the MITO-Porter promotes both fusion with the mitochondrial membrane and releases of its cargo to the intra-mitochondrial compartment. MITO-Porter encapsulating propidium iodide (PI), a fluorescent dye commonly used to stain nuclei, successfully delivered PI as cargo to the mitochondrial matrix [163]. A dual function MITO-Porter, a nanopreparation combining both cytoplasmic delivery and mitochondrial macromolecule delivery has been developed in which MITO-Porter liposomes were surface functionalized with high-density octa-arginine (R8) [166]. This dual function nanopreparation had efficient cytoplasmic delivery through the cell membrane due to R8-modification followed by mitochondrial delivery through the mitochondria-membrane via membrane fusion. A ligand of adenine nucleotide translocator, Bongkrekic acid (BKA) was encapsulated in the lipid envelope of the MITO-porter. The MITO-porter encapsulated BKA inhibited the mitochondrial permeability transition associated with apoptosis after its delivery to the mitochondria using MITO-Porter. The strong anti-apoptotic effect of MITO-Porter encapsulated BKA was observed compared to naked BKA in HeLa cells [167].
5.1.2.2.2. Colloidal dequalinium vesicles
Dequalinium is a dicationic amphiphilic compound that self-assembles and forms vesicle-like aggregates referred to as dequalinium liposomes or DQAsomes [168–171]. This vesicles translocate into cells via endocytosis and fuse with the mitochondrial outer membrane. DQAsomes have been evaluated to determine their potential to serve as a mitochondriotropic, non-viral transfection vector for delivery of mitochondrial DNA to the mitochondrial membrane [172–174]. In the first study, to deliver plasmid DNA (pDNA) more effectively to the mitochondria via DQAsomes, the mitochondrial DNA was conjugated to a mitochondrial leader sequence peptide (MLS) and condensed in the DQAsomes. After accumulation of mitochondrial DNA in the mitochondrial membrane by DQAsomes, it was taken in to the mitochondrial matrix by the mitochondrial protein import machinery [172]. These results demonstrate that DQAsomes are able to transport the pDNA to the mitochondria by escaping from endosomes without losing their pDNA load.
5.1.2.3. Mitochondrial protein import machinery
Mitochondrial targeting for protein delivery to the mitochondria employs creating a chimeric protein linked with a mitochondrial signal peptide, an N-terminal specific amino acid sequence to the proteins to be delivered [140]. These chimeric proteins are taken up into the mitochondrial matrix via the protein import machinery [175]. Delivery of a oligonucleotide or DNA to the mitochondria has also been achieved by covalently linking a mitochondrial precursor protein at the C-terminus or a mitochondrial protein targeting peptide [176, 177]. In another study, a mitochondrially targeted DNA delivery vector was developed by linking an N-terminal mitochondrial targeting peptide to a polyamide nucleic acid (PNA), a moiety in which the deoxyribose phosphate backbone of DNA was replaced with an achiral polyamide backbone [178]. This mitochondrial targeting peptide-PNA conjugate was annealed to an oligonucleotide with a selected sequence and transfected into the cytosol via polycations or transient membrane channels. The nanopreparation efficiently delivered labeled oligonucleotides into the mitochondrial matrix.
Proteins located in the mitochondrial inner or outer membrane can be targeted for mitochondrial delivery of therapeutics. A ligand of multimeric translocator protein, located in the outer mitochondrial membrane, commonly referred to as peripheral benzodiazepine receptor was conjugated to a dendrimer imaging agent for mitochondrial delivery [179]. Even though significant effort has been undertaken to develop the mitochondria-targeted nanopreparations, the full understanding of the process of intracellular trafficking of nanopreparation, pay-load delivery and time-dependent localization have yet to be achieved.
5.2. Lysosomal delivery
Lysosomes are the main digestive organelles of the cells and interconnect the extracellular environment with the inside of the cell via endocytosis, phagocytosis and exocytosis. Lysosomes are the final destination of endocytic cargos and macromolecules for degradation. The notion of lysosomes as merely “garbage disposal units” has changed dramatically due to recent discoveries of other roles of lysosomes and their contents, the most importantly in cell-death mechanisms in cancer [12, 180, 181]. The lysosomal enzyme, cathepsins perform more specific functions, including a role in bone remodeling, antigen presentation, pro-hormone processing, apoptosis, angiogenesis and cancer cell invasion [182, 183].
5.2.1. Lysosomal storage diseases (LSD)
Lysosomal storage disease is a set of more than 40 inherited disorders associated with the deficiency of certain lysosomal enzymes [184]. The main approach for the treatment of LSD has been enzyme replacement therapy with supplemental exogenous enzymes. However, poor delivery and pharmacokinetics of the enzymes limit the efficacy of this approach. The use of nanopreparations loaded with therapeutic cargo for delivery of agents, including enzymes and other small molecules to the lysosomes has been suggested [7]. In an attempt to prepare a lysosome-targeted nanocarrier, Koshkaryev, et al, surface-modified liposomes with the lysosomotropic ligand octadecyl-rhodamine B (RhB) [14]. The modified liposomes successfully delivered the loaded model cargo, fluorescein isothiocyanate (FITC)-dextran (FD) to the lysosomes of HeLa cells. In the confocal microscopy study, RhB-modified liposomes demonstrated enhanced co-localization of the liposomal RhB and a specific lysosomal marker, Lamp-2 (tracked with anti-Lamp2 antibodies) compared to plain RhB-unmodified liposomes. The fluorescence of the lysosome-enriched fractions of the cells, treated with FD-loaded, RhB-modified liposomes was at least 2-fold higher compared with cells, treated with unmodified liposomes. Flow cytometry analysis revealed that the treatment of cells with same amount of FD, loaded in unmodified and RhB-liposomes, led to the same level of FITC fluorescence indicating that the enhanced fluorescence associated with lysosomal fractions of cells treated with RhB-liposomes was due to the enhanced lysosomal delivery mediated by lysosomotropic RhB-modified liposomes. These lysosome-targeted octadecyl-RhB liposomes were also utilized for the lysosomal delivery of the enzyme glucocerebrosidase into the Gaucher’s fibroblasts in vitro [185]. The lysosomotropic liposomes improved the lysosomal accumulation of liposomal enzymes both in nonphagocytic Gaucher’s fibroblasts and phagocytic monocyte-derived macrophages.
5.2.2. Lysosomes as target for cancer therapy
Cancer cells can activate or inactivate a number of pathways to inhibit caspase activation to evade apoptosis. However, recent findings on the role of lysosomes in apoptosis showed that the cancer cell death could be triggered by the release of lysosomal enzymes. Various molecules of an endogenous or synthetic nature have been identified which disrupt the lysosomal membranes, causing lysosomal membrane permeabilization (LMP) [186, 187]. This LMP can be induced by classic apoptotic stimuli including endogenous reactive oxygen species, detergents such as sphingosine and other lysosomotropic toxins [188, 189]. Other LMP inducers include various lysosomotropic compounds that become trapped and accumulate in the lysosomes after protonation. When the concentration of a lysosomotropic compound reaches a critical level, the compound stimulates LMP and the release of lysosomal contents in the cytosol. Released lysosomal proteolytic cathepsins cause the cleavage of Bid, degrade the anti-apoptotic Bcl-2 protein, triggers caspase, Bax activation, recruit mitochondrial pathway for executing apoptosis by causing mitochondrial outer membrane permeabilization [190–193]. Lysosomal cell death pathways are turned on not only by lysosomal disrupters, but also by classic endogenous apoptotic stimuli such as the death receptor and p53 activation [194, 195]. The lysosome-mediated cell death pathway can be activated by many conventional chemotherapeutics through LMP [196]. Ceramide, a precursor of sphingosine produced by the lysosomal enzyme, acid ceramidase, is a promising molecule for the induction of LMP [197]. Intracellular delivery of ceramides via transferrin-functionalized liposomes induced increased apoptosis in vitro in HeLa and in vivo in A2780-ovarian carcinoma xenograft mouse model [13].
In cancer cells, lysosomes have increased expression of lysosomal enzymes that participate in tissue invasion and tumor growth. In addition, cancer cells over-express endogenous molecules that protect lysosomal membranes against permeabilization including heat-shock protein 70 (HSP-70), lysosomes-associated membrane proteins 1 and 2 (LAMP-1 and 2) as a defense mechanism of cancer cells against apoptosis [198]. Depletion or down-regulation or inhibition of HSP-70 can activate the lysosomal cell death pathway in a tumor and induce a therapeutic cytotoxic effect [199].
5.3. Nuclear delivery
Generally, the transport of biomolecules to the nucleus’ interior occurs through the nuclear pore complexes (NPCs) [105]. The diameter of NPCs is only ~9 nm, which limits the diffusion of macromolecules in the nucleus. However, many nuclear localization signal (NLS) peptides have been discovered that target the DNA to the nucleus and allow entry through NPCs via an active transport process [200, 201]. NLS peptides containing basic amino acid residues are recognized by cytoplasmic receptors such as importins. These NLS peptides bind to NPC followed by translocation through the pore [202]. NLS bio-conjugates have been successfully used to target therapeutic genes to the nucleus [203] The most well-known NLS for nuclear gene delivery is SV40, derived from a tumor antigen of simian virus 40 [204, 205].
The nuclear membrane represents a major barrier to nuclear delivery. The nuclear membrane allows pDNA to enter the nucleus only during M-phase of the dividing cells. For gene delivery to the nucleus, a tetralamellar multifunctional envelope-type nanodevice (T-MEND) has been developed by Harashima group [10]. This group proposed a nuclear pore complex-independent nuclear transport approach in which T-MEND is fused with the nuclear membrane. T-MEND is a multi-layered nanopreparation possessing a pDNA-polycation condensed core, coated with two envelopes, including an inner envelope composed of a mixture of cardiolipin and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) for fusion with the nuclear membrane plus an outer envelope composed of phosphatidic acid and DOPE to disrupt the endosomes [206]. The nanopreparation was made positively charged by incorporation of stearyl octa-arginine (R8). Transfection of dendritic JAWS II cells using T-MEND as the non-viral gene delivery vector increased by several hundred-fold compared to another MEND-nanopreparation developed by this group. The T-MEND system was further upgraded with a pH-independent fusogenic peptide, KALA, in the outer layer and an increased charge ratio of the core [108]. KALA-peptide improved the membrane fusion process with endosome- and nuclear membranes. This upgraded nanopreparation had a 20-fold higher transgene expression compared to the original R8-T-MEND in JAWS II cells.
Jing Yao et al simultaneously targeted delivery of all-trans-retinoic acid (ATRA) and paclitaxel to tumors with a hyaluronic acid-based (HRA) multifunctional nanocarrier to provide a synergistic combination chemotherapy. HRA-nanoparticles had both efficient cytoplasmic followed by nuclear delivery via a HA-receptor-mediated endocytosis and ATRA-mediated nuclear translocation, respectively. ATRA translocated into the nucleus by binding to specific cytosolic proteins [207].
5.4. Delivery to the endoplasmic reticulum (ER)
The ER plays an essential role in protein synthesis, folding, protein trafficking and dynamic calcium homeostasis. In addition, the activation of caspase-8-mediated apoptotic pathway occurs in response to the ER stress [208]. The ER lumens represent a major site of stored intracellular Ca2+, the release of which promotes cleavage of caspase-8 and initiates ER stress- induced apoptosis. In addition, the ER is a major site of peptide loading and promotes cross presentation in antigen presenting cells. Delivery of antigenic peptides to the ER induces for inducing cross presentation in dendritic cells that leads to antitumor immunity. PLGA nanoparticles were targeted to the ER of dendritic cells for delivery of antigenic peptides (SIINFEKL) with the goal to prepare an anticancer vaccine [209]. Nanocarriers were targeted to the ER by surface modification with a peptide containing a specific ER targeting sequence. The in vitro studies demonstrated that the surface modification with this ER signal peptide enhanced endocytosis, intracellular trafficking to the ER, and induced prolonged cross presentation of the loaded antigenic peptides. Analysis of intracellular trafficking confirmed that poly(γ-glutamic acid)-based nanoparticles (γ-PGA NP), a vaccine carrier, accumulated in both the ER and endosomal compartments within 2 h [210]. Further analysis strongly suggested that the γ-PGA NP enhanced ER-endosome fusion for antigen cross presentation.
The ER and mitochondria were simultaneously targeted by photodynamic therapy using a small molecule hexaminolevulinate to induce apoptosis in human lymphoma cells [211]. Phototherapy damage of the ER calcium pump protein SERCA2 released Ca2+ that initiated caspase-8 cleavage.
Investigations by Kabanov and co-workers have revealed that the block copolymer Pluronic P85 can be internalized by caveoli-mediated endocytosis and delivered directly to the ER [212]. The hydrophobic poly(propylene oxide) (PPO) block in the P85 was suggested to be the moiety promoting the affinity for ER accumulation. In another study, silicon quantum dots (Si-QDs) were coated with a block copolymer Pluronic F127 (PEO-PPO-PEO) with PPO as a hydrophobic moiety for selective labeling of the ER of live HUVEC cells [213]. Pollock et al studied a number of liposomes of various phospholipid composition and reported that the liposomal system composed of phosphatidyl ethanolamine: phosphatidyl choline: phosphatidyl inositol: phosphatidyl serine (1.5:1.5:1:1) trafficked directly to the ER, fused with the ER membrane, to deliver hydrophobic drugs or markers [214].
5.5. Delivery to other organelles
Drug delivery to the other intracellular organelles has been relatively unexplored. Targeting nanocarriers to caveolae to promote clathrin-dependent endocytosis is an approach being studied for cytosolic delivery of nanocarriers [215, 216]. Clathrin-mediated endocytosis by-passes fusion with lysosomes, and provides an intracellular delivery route with an unique advantage by avoiding lysosome- mediated degradation of the therapeutic cargos. Various small molecules or macromolecular complexes interact with caveolar receptors of the cell surface and are internalized after docking onto caveolar receptors. Internalized caveolae fuse with caveosomes and deliver their content in non-lysosomal subcellular organelles. Caveolar invaginations are found only on the surface of cells expressing caveolin I proteins. Additionally, “lipid rafts” constitute a caveolar-equivalent plasma membrane domain, assembled from the same lipid constituents and proteins as caveolae, but flat in structure, are found in cells without caveolae. Lipid-mediated drug delivery prevents lysosomal uptake as well [217].
6. Conclusion
Considerable efforts have been undertaken to improve the therapeutic efficacy of bioactive molecules including cancer therapeutics. The highly complex disease pathology of cancer and the toxic effects associated with chemotherapeutics necessitates the development of multifunctional nanomedicines with high efficacy and minimized adverse effects. Besides encapsulating the drugs in nanopreparations and targeting the drug-loaded nanocarriers to the disease site to generate so called “magic bullets”, a significant effort is being undertaken to design and develop strategies to improve the cellular uptake of the therapeutic molecules and their delivery to an organelle of interest. Although organelle-specific delivery is challenging, when achieved, it can dramatically increase the interaction of the bioactive molecules with target organelles. The targeted delivery has been shown to reduce the toxicity and increase the therapeutic efficacy many-fold. Therefore, this strategy remains attractive for delivering pro-apoptotic compounds to the mitochondria; molecules that alter lysosomal function to lysosomes; gene silencers or nucleic acid therapeutics to the nucleus. Still, further in-depth studies to provide detailed understanding of the intracellular trafficking of organelle-targeted nanopreparations as well as the loaded drugs, together with their time-dependent fate and drug release inside the organelles are much needed. Shedding a light in that direction would rationalize the application of organelle-specific drug delivery in cancer.
Figure 6.

Schematic diagram of mitochondrial delivery of MITO-Porter encapsulated drugs. MITO-Porters enter cells via macropinocytosis. Disruption of macropinosomes liberates MITO-Porter, which is translocated to the mitochondria via electrostatic interaction of mitochondria with the MITO-Porter membrane component, R8. The liposomal cargo is delivered to the mitochondria via mitochondrial membrane fusion.
Acknowledgments
The work was supported in part by NIH grants RO1 CA121838 and RO1 CA128486 to Vladimir P. Torchilin. We thank Dr. William Hartner for his help in preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Alexis F, Pridgen EM, Langer R, Farokhzad OC. Nanoparticle technologies for cancer therapy. Handb Exp Pharmacol. 2010:55–86. doi: 10.1007/978-3-642-00477-3_2. [DOI] [PubMed] [Google Scholar]
- 3.Tiwari M. Nano cancer therapy strategies. J Cancer Res Ther. 2012;8:19–22. doi: 10.4103/0973-1482.95168. [DOI] [PubMed] [Google Scholar]
- 4.Langer R. Drug delivery and targeting. Nature. 1998;392:5–10. [PubMed] [Google Scholar]
- 5.Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3:16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 6.Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63:131–135. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Torchilin VP. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu Rev Biomed Eng. 2006;8:343–375. doi: 10.1146/annurev.bioeng.8.061505.095735. [DOI] [PubMed] [Google Scholar]
- 8.Torchilin VP. Multifunctional nanocarriers. Advanced Drug Delivery Reviews. 2006;58:1532–1555. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura T, Akita H, Yamada Y, Hatakeyama H, Harashima H. A multifunctional envelope-type nanodevice for use in nanomedicine: concept and applications. Acc Chem Res. 2012;45:1113–1121. doi: 10.1021/ar200254s. [DOI] [PubMed] [Google Scholar]
- 11.Taratula O, Garbuzenko O, Savla R, Wang YA, He H, Minko T. Multifunctional nanomedicine platform for cancer specific delivery of siRNA by superparamagnetic iron oxide nanoparticles-dendrimer complexes. Curr Drug Deliv. 2011;8:59–69. doi: 10.2174/156720111793663642. [DOI] [PubMed] [Google Scholar]
- 12.Kirkegaard T, Jaattela M. Lysosomal involvement in cell death and cancer. Biochim Biophys Acta. 2009;1793:746–754. doi: 10.1016/j.bbamcr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Koshkaryev A, Piroyan A, Torchilin VP. Increased apoptosis in cancer cells in vitro and in vivo by ceramides in transferrin-modified liposomes. Cancer Biol Ther. 2012;13:50–60. doi: 10.4161/cbt.13.1.18871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koshkaryev A, Thekkedath R, Pagano C, Meerovich I, Torchilin VP. Targeting of lysosomes by liposomes modified with octadecyl-rhodamine B. J Drug Target. 2011;19:606–614. doi: 10.3109/1061186X.2010.550921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 16.Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Advanced Drug Delivery Reviews. 2011;63:136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Maeda H. Advances in Enzyme Regulation. Elsevier Science Ltd; Oxford: 2001. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting; pp. 189–207. [DOI] [PubMed] [Google Scholar]
- 18.Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71:409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Maeda H, Greish K, Fang J. The EPR Effect and Polymeric Drugs: A Paradigm Shift for Cancer Chemotherapy in the 21st Century. In: Satchi-Fainaro R, Duncan R, editors. Polymer Therapeutics II. Springer; Berlin Heidelberg: 2006. pp. 103–121. [Google Scholar]
- 20.Seki T, Fang J, Maeda H. Tumor-Targeted Macromolecular Drug Delivery Based on the Enhanced Permeability and Retention Effect in Solid Tumor. In: Lu Y, Mahato RI, editors. Pharmaceutical Perspectives of Cancer Therapeutics. Springer; US: 2009. pp. 93–120. [Google Scholar]
- 21.Maeda H. SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy. Adv Drug Deliv Rev. 2001;46:169–185. doi: 10.1016/s0169-409x(00)00134-4. [DOI] [PubMed] [Google Scholar]
- 22.Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11:812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res. 2005;65:8984–8992. doi: 10.1158/0008-5472.CAN-05-0565. [DOI] [PubMed] [Google Scholar]
- 24.Arias JL. Drug targeting strategies in cancer treatment: an overview. Mini Rev Med Chem. 2011;11:1–17. doi: 10.2174/138955711793564024. [DOI] [PubMed] [Google Scholar]
- 25.Couvreur P, Vauthier C. Nanotechnology: intelligent design to treat complex disease. Pharm Res. 2006;23:1417–1450. doi: 10.1007/s11095-006-0284-8. [DOI] [PubMed] [Google Scholar]
- 26.Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121:159–176. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Derycke AS, Kamuhabwa A, Gijsens A, Roskams T, De Vos D, Kasran A, Huwyler J, Missiaen L, de Witte PA. Transferrin-conjugated liposome targeting of photosensitizer AlPcS4 to rat bladder carcinoma cells. J Natl Cancer Inst. 2004;96:1620–1630. doi: 10.1093/jnci/djh314. [DOI] [PubMed] [Google Scholar]
- 28.Sahoo SK, Ma W, Labhasetwar V. Efficacy of transferrin-conjugated paclitaxel-loaded nanoparticles in a murine model of prostate cancer. Int J Cancer. 2004;112:335–340. doi: 10.1002/ijc.20405. [DOI] [PubMed] [Google Scholar]
- 29.Sahoo SK, Labhasetwar V. Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharm. 2005;2:373–383. doi: 10.1021/mp050032z. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Zhou F, Ge L, Liu X, Kong F. Transferrin-PEG-PE modified dexamethasone conjugated cationic lipid carrier mediated gene delivery system for tumor-targeted transfection. Int J Nanomedicine. 2012;7:2513–2522. doi: 10.2147/IJN.S31915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SH, Jeong JH, Chun KW, Park TG. Target-specific cellular uptake of PLGA nanoparticles coated with poly(L-lysine)-poly(ethylene glycol)-folate conjugate. Langmuir. 2005;21:8852–8857. doi: 10.1021/la0502084. [DOI] [PubMed] [Google Scholar]
- 32.Esmaeili F, Ghahremani MH, Ostad SN, Atyabi F, Seyedabadi M, Malekshahi MR, Amini M, Dinarvand R. Folate-receptor-targeted delivery of docetaxel nanoparticles prepared by PLGA-PEG-folate conjugate. J Drug Target. 2008;16:415–423. doi: 10.1080/10611860802088630. [DOI] [PubMed] [Google Scholar]
- 33.Stella B, Arpicco S, Peracchia MT, Desmaele D, Hoebeke J, Renoir M, D’Angelo J, Cattel L, Couvreur P. Design of folic acid-conjugated nanoparticles for drug targeting. J Pharm Sci. 2000;89:1452–1464. doi: 10.1002/1520-6017(200011)89:11<1452::aid-jps8>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 34.Liang B, He ML, Chan CY, Chen YC, Li XP, Li Y, Zheng D, Lin MC, Kung HF, Shuai XT, Peng Y. The use of folate-PEG-grafted-hybranched-PEI nonviral vector for the inhibition of glioma growth in the rat. Biomaterials. 2009;30:4014–4020. doi: 10.1016/j.biomaterials.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 35.Zou S, Cao N, Cheng D, Zheng R, Wang J, Zhu K, Shuai X. Enhanced apoptosis of ovarian cancer cells via nanocarrier-mediated codelivery of siRNA and doxorubicin. Int J Nanomedicine. 2012;7:3823–3835. doi: 10.2147/IJN.S29328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe K, Kaneko M, Maitani Y. Functional coating of liposomes using a folate- polymer conjugate to target folate receptors. Int J Nanomedicine. 2012;7:3679–3688. doi: 10.2147/IJN.S32853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 38.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 39.Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, Sklarin NT, Seidman AD, Hudis CA, Moore J, Rosen PP, Twaddell T, Henderson IC, Norton L. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14:737–744. doi: 10.1200/JCO.1996.14.3.737. [DOI] [PubMed] [Google Scholar]
- 40.Park JW, Hong K, Kirpotin DB, Colbern G, Shalaby R, Baselga J, Shao Y, Nielsen UB, Marks JD, Moore D, Papahadjopoulos D, Benz CC. Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin Cancer Res. 2002;8:1172–1181. [PubMed] [Google Scholar]
- 41.Mamot C, Drummond DC, Noble CO, Kallab V, Guo Z, Hong K, Kirpotin DB, Park JW. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005;65:11631–11638. doi: 10.1158/0008-5472.CAN-05-1093. [DOI] [PubMed] [Google Scholar]
- 42.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89:4285–4289. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park JW, Kirpotin DB, Hong K, Shalaby R, Shao Y, Nielsen UB, Marks JD, Papahadjopoulos D, Benz CC. Tumor targeting using anti-her2 immunoliposomes. J Control Release. 2001;74:95–113. doi: 10.1016/s0168-3659(01)00315-7. [DOI] [PubMed] [Google Scholar]
- 44.Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615–1626. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 45.Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 46.Lammers T, Hennink WE, Storm G. Tumour-targeted nanomedicines: principles and practice. Br J Cancer. 2008;99:392–397. doi: 10.1038/sj.bjc.6604483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl 3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 48.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 49.Veikkola T, Karkkainen M, Claesson-Welsh L, Alitalo K. Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res. 2000;60:203–212. [PubMed] [Google Scholar]
- 50.Backer MV, Gaynutdinov TI, Patel V, Bandyopadhyaya AK, Thirumamagal BT, Tjarks W, Barth RF, Claffey K, Backer JM. Vascular endothelial growth factor selectively targets boronated dendrimers to tumor vasculature. Mol Cancer Ther. 2005;4:1423–1429. doi: 10.1158/1535-7163.MCT-05-0161. [DOI] [PubMed] [Google Scholar]
- 51.Chen J, Wu H, Han D, Xie C. Using anti-VEGF McAb and magnetic nanoparticles as double-targeting vector for the radioimmunotherapy of liver cancer. Cancer Lett. 2006;231:169–175. doi: 10.1016/j.canlet.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 52.Li L, Wartchow CA, Danthi SN, Shen Z, Dechene N, Pease J, Choi HS, Doede T, Chu P, Ning S, Lee DY, Bednarski MD, Knox SJ. A novel antiangiogenesis therapy using an integrin antagonist or anti-Flk-1 antibody coated 90Y-labeled nanoparticles. Int J Radiat Oncol Biol Phys. 2004;58:1215–1227. doi: 10.1016/j.ijrobp.2003.10.057. [DOI] [PubMed] [Google Scholar]
- 53.Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, Cheresh DA. Tumor regression by targeted gene delivery to the neovasculature. Science. 2002;296:2404–2407. doi: 10.1126/science.1070200. [DOI] [PubMed] [Google Scholar]
- 54.Cook-Mills JM, Marchese ME, Abdala-Valencia H. Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants. Antioxid Redox Signal. 2011;15:1607–1638. doi: 10.1089/ars.2010.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gosk S, Moos T, Gottstein C, Bendas G. VCAM-1 directed immunoliposomes selectively target tumor vasculature in vivo. Biochim Biophys Acta. 2008;1778:854–863. doi: 10.1016/j.bbamem.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 56.Vihinen P, Ala-aho R, Kahari VM. Matrix metalloproteinases as therapeutic targets in cancer. Curr Cancer Drug Targets. 2005;5:203–220. doi: 10.2174/1568009053765799. [DOI] [PubMed] [Google Scholar]
- 57.Genis L, Galvez BG, Gonzalo P, Arroyo AG. MT1-MMP: universal or particular player in angiogenesis? Cancer Metastasis Rev. 2006;25:77–86. doi: 10.1007/s10555-006-7891-z. [DOI] [PubMed] [Google Scholar]
- 58.Jiang A, Lehti K, Wang X, Weiss SJ, Keski-Oja J, Pei D. Regulation of membrane-type matrix metalloproteinase 1 activity by dynamin-mediated endocytosis. Proc Natl Acad Sci U S A. 2001;98:13693–13698. doi: 10.1073/pnas.241293698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sato H, Takino T, Miyamori H. Roles of membrane-type matrix metalloproteinase-1 in tumor invasion and metastasis. Cancer Sci. 2005;96:212–217. doi: 10.1111/j.1349-7006.2005.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yana I, Seiki M. MT-MMPs play pivotal roles in cancer dissemination. Clin Exp Metastasis. 2002;19:209–215. doi: 10.1023/a:1015527220537. [DOI] [PubMed] [Google Scholar]
- 61.Saiki I, Fujii H, Yoneda J, Abe F, Nakajima M, Tsuruo T, Azuma I. Role of aminopeptidase N (CD13) in tumor-cell invasion and extracellular matrix degradation. Int J Cancer. 1993;54:137–143. doi: 10.1002/ijc.2910540122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, Ashmun RA, Shapiro LH, Arap W, Ruoslahti E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60:722–727. [PMC free article] [PubMed] [Google Scholar]
- 63.Kondo M, Asai T, Katanasaka Y, Sadzuka Y, Tsukada H, Ogino K, Taki T, Baba K, Oku N. Anti-neovascular therapy by liposomal drug targeted to membrane type-1 matrix metalloproteinase. Int J Cancer. 2004;108:301–306. doi: 10.1002/ijc.11526. [DOI] [PubMed] [Google Scholar]
- 64.Hatakeyama H, Akita H, Ishida E, Hashimoto K, Kobayashi H, Aoki T, Yasuda J, Obata K, Kikuchi H, Ishida T, Kiwada H, Harashima H. Tumor targeting of doxorubicin by anti-MT1-MMP antibody-modified PEG liposomes. Int J Pharm. 2007;342:194–200. doi: 10.1016/j.ijpharm.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 65.Pastorino F, Brignole C, Marimpietri D, Cilli M, Gambini C, Ribatti D, Longhi R, Allen TM, Corti A, Ponzoni M. Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res. 2003;63:7400–7409. [PubMed] [Google Scholar]
- 66.Levy-Nissenbaum E, Radovic-Moreno AF, Wang AZ, Langer R, Farokhzad OC. Nanotechnology and aptamers: applications in drug delivery. Trends Biotechnol. 2008;26:442–449. doi: 10.1016/j.tibtech.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 67.Wang KY, McCurdy S, Shea RG, Swaminathan S, Bolton PH. A DNA aptamer which binds to and inhibits thrombin exhibits a new structural motif for DNA. Biochemistry. 1993;32:1899–1904. doi: 10.1021/bi00059a003. [DOI] [PubMed] [Google Scholar]
- 68.Willis MC, Collins BD, Zhang T, Green LS, Sebesta DP, Bell C, Kellogg E, Gill SC, Magallanez A, Knauer S, Bendele RA, Gill PS, Janjic N. Liposome-anchored vascular endothelial growth factor aptamers. Bioconjug Chem. 1998;9:573–582. doi: 10.1021/bc980002x. [DOI] [PubMed] [Google Scholar]
- 69.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mae M, Langel U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr Opin Pharmacol. 2006;6:509–514. doi: 10.1016/j.coph.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 71.Khalil IA, Kogure K, Futaki S, Hama S, Akita H, Ueno M, Kishida H, Kudoh M, Mishina Y, Kataoka K, Yamada M, Harashima H. Octaarginine-modified multifunctional envelope-type nanoparticles for gene delivery. Gene Ther. 2007;14:682–689. doi: 10.1038/sj.gt.3302910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khalil IA, Kogure K, Futaki S, Harashima H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J Biol Chem. 2006;281:3544–3551. doi: 10.1074/jbc.M503202200. [DOI] [PubMed] [Google Scholar]
- 73.Khalil IA, Kogure K, Futaki S, Harashima H. Octaarginine-modified liposomes: enhanced cellular uptake and controlled intracellular trafficking. Int J Pharm. 2008;354:39–48. doi: 10.1016/j.ijpharm.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 74.Lindgren M, Hallbrink M, Prochiantz A, Langel U. Cell-penetrating peptides. Trends Pharmacol Sci. 2000;21:99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 75.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 76.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci U S A. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 78.Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med. 1998;4:1449–1452. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- 79.Jeang KT, Xiao H, Rich EA. Multifaceted activities of the HIV-1 transactivator of transcription. Tat, J Biol Chem. 1999;274:28837–28840. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
- 80.Elmquist A, Lindgren M, Bartfai T, Langel U. VE-cadherin-derived cell-penetrating peptide, pVEC, with carrier functions. Exp Cell Res. 2001;269:237–244. doi: 10.1006/excr.2001.5316. [DOI] [PubMed] [Google Scholar]
- 81.Rojas M, Donahue JP, Tan Z, Lin YZ. Genetic engineering of proteins with cell membrane permeability. Nat Biotechnol. 1998;16:370–375. doi: 10.1038/nbt0498-370. [DOI] [PubMed] [Google Scholar]
- 82.Pooga M, Hallbrink M, Zorko M, Langel U. Cell penetration by transportan. FASEB J. 1998;12:67–77. doi: 10.1096/fasebj.12.1.67. [DOI] [PubMed] [Google Scholar]
- 83.Morris MC, Vidal P, Chaloin L, Heitz F, Divita G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997;25:2730–2736. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fretz MM, Penning NA, Al-Taei S, Futaki S, Takeuchi T, Nakase I, Storm G, Jones AT. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem J. 2007;403:335–342. doi: 10.1042/BJ20061808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vazquez O, Blanco-Canosa JB, Vazquez ME, Martinez-Costas J, Castedo L, Mascarenas JL. Efficient DNA binding and nuclear uptake by distamycin derivatives conjugated to octa-arginine sequences. Chembiochem. 2008;9:2822–2829. doi: 10.1002/cbic.200800345. [DOI] [PubMed] [Google Scholar]
- 86.Hallbrink M, Floren A, Elmquist A, Pooga M, Bartfai T, Langel U. Cargo delivery kinetics of cell-penetrating peptides. Biochim Biophys Acta. 2001;1515:101–109. doi: 10.1016/s0005-2736(01)00398-4. [DOI] [PubMed] [Google Scholar]
- 87.Josephson L, Tung CH, Moore A, Weissleder R. High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates. Bioconjug Chem. 1999;10:186–191. doi: 10.1021/bc980125h. [DOI] [PubMed] [Google Scholar]
- 88.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci U S A. 2001;98:8786–8791. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fretz MM, Koning GA, Mastrobattista E, Jiskoot W, Storm G. OVCAR-3 cells internalize TAT-peptide modified liposomes by endocytosis. Biochim Biophys Acta. 2004;1665:48–56. doi: 10.1016/j.bbamem.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 90.Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D’Souza GG. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc Natl Acad Sci U S A. 2003;100:1972–1977. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pappalardo JS, Quattrocchi V, Langellotti C, Di Giacomo S, Gnazzo V, Olivera V, Calamante G, Zamorano PI, Levchenko TS, Torchilin VP. Improved transfection of spleen-derived antigen-presenting cells in culture using TATp-liposomes. J Control Release. 2009;134:41–46. doi: 10.1016/j.jconrel.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 92.Ko YT, Hartner WC, Kale A, Torchilin VP. Gene delivery into ischemic myocardium by double-targeted lipoplexes with anti-myosin antibody and TAT peptide. Gene Ther. 2009;16:52–59. doi: 10.1038/gt.2008.135. [DOI] [PubMed] [Google Scholar]
- 93.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 94.Hodoniczky J, Sims CG, Best WM, Bentel JM, Wilce JA. The intracellular and nuclear-targeted delivery of an antiandrogen drug by carrier peptides. Biopolymers. 2008;90:595–603. doi: 10.1002/bip.20986. [DOI] [PubMed] [Google Scholar]
- 95.Aroui S, Brahim S, Waard MD, Kenani A. Cytotoxicity, intracellular distribution and uptake of doxorubicin and doxorubicin coupled to cell-penetrating peptides in different cell lines: a comparative study. Biochem Biophys Res Commun. 2010;391:419–425. doi: 10.1016/j.bbrc.2009.11.073. [DOI] [PubMed] [Google Scholar]
- 96.Bowne WB, Sookraj KA, Vishnevetsky M, Adler V, Sarafraz-Yazdi E, Lou S, Koenke J, Shteyler V, Ikram K, Harding M, Bluth MH, Ng M, Brandt-Rauf PW, Hannan R, Bradu S, Zenilman ME, Michl J, Pincus MR. The penetratin sequence in the anticancer PNC-28 peptide causes tumor cell necrosis rather than apoptosis of human pancreatic cancer cells. Ann Surg Oncol. 2008;15:3588–3600. doi: 10.1245/s10434-008-0147-0. [DOI] [PubMed] [Google Scholar]
- 97.Pero SC, Shukla GS, Cookson MM, Flemer S, Jr, Krag DN. Combination treatment with Grb7 peptide and Doxorubicin or Trastuzumab (Herceptin) results in cooperative cell growth inhibition in breast cancer cells. Br J Cancer. 2007;96:1520–1525. doi: 10.1038/sj.bjc.6603732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J Biol Chem. 2002;277:2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- 99.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 100.Futaki S, Niwa M, Nakase I, Tadokoro A, Zhang Y, Nagaoka M, Wakako N, Sugiura Y. Arginine carrier peptide bearing Ni(II) chelator to promote cellular uptake of histidine-tagged proteins. Bioconjug Chem. 2004;15:475–481. doi: 10.1021/bc034181g. [DOI] [PubMed] [Google Scholar]
- 101.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc Natl Acad Sci U S A. 2008;105:12128–12133. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koshkaryev A, Piroyan A, Torchilin VP. Bleomycin in octaarginine-modified fusogenic liposomes results in improved tumor growth inhibition. Cancer Letters. doi: 10.1016/j.canlet.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Biswas S, Dodwadkar NS, Deshpande PP, Parab S, Torchilin VP. Surface functionalization of doxorubicin-loaded liposomes with octa-arginine for enhanced anticancer activity. European Journal of Pharmaceutics and Biopharmaceutics. doi: 10.1016/j.ejpb.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koshkaryev A, Piroyan A, Torchilin VP. Bleomycin in octaarginine-modified fusogenic liposomes results in improved tumor growth inhibition. Cancer Lett. 2013;334:293–301. doi: 10.1016/j.canlet.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bolhassani A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim Biophys Acta. 2011;1816:232–246. doi: 10.1016/j.bbcan.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 106.Kakudo T, Chaki S, Futaki S, Nakase I, Akaji K, Kawakami T, Maruyama K, Kamiya H, Harashima H. Transferrin-modified liposomes equipped with a pH-sensitive fusogenic peptide: an artificial viral-like delivery system. Biochemistry. 2004;43:5618–5628. doi: 10.1021/bi035802w. [DOI] [PubMed] [Google Scholar]
- 107.Khalil IA, Hayashi Y, Mizuno R, Harashima H. Octaarginine- and pH sensitive fusogenic peptide-modified nanoparticles for liver gene delivery. Journal of Controlled Release. 2011;156:374–380. doi: 10.1016/j.jconrel.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 108.Shaheen SM, Akita H, Nakamura T, Takayama S, Futaki S, Yamashita A, Katoono R, Yui N, Harashima H. KALA-modified multi-layered nanoparticles as gene carriers for MHC class-I mediated antigen presentation for a DNA vaccine. Biomaterials. 2011;32:6342–6350. doi: 10.1016/j.biomaterials.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 109.Farhood H, Serbina N, Huang L. The role of dioleoyl phosphatidylethanolamine in cationic liposome mediated gene transfer. Biochim Biophys Acta. 1995;1235:289–295. doi: 10.1016/0005-2736(95)80016-9. [DOI] [PubMed] [Google Scholar]
- 110.Kogure K, Akita H, Harashima H. Multifunctional envelope-type nano device for non-viral gene delivery: concept and application of Programmed Packaging. J Control Release. 2007;122:246–251. doi: 10.1016/j.jconrel.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 111.Hafez IM, Cullis PR. Roles of lipid polymorphism in intracellular delivery. Adv Drug Deliv Rev. 2001;47:139–148. doi: 10.1016/s0169-409x(01)00103-x. [DOI] [PubMed] [Google Scholar]
- 112.Kogure K, Moriguchi R, Sasaki K, Ueno M, Futaki S, Harashima H. Development of a non-viral multifunctional envelope-type nano device by a novel lipid film hydration method. J Control Release. 2004;98:317–323. doi: 10.1016/j.jconrel.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 113.Rapoport N, Nam KH, Gupta R, Gao Z, Mohan P, Payne A, Todd N, Liu X, Kim T, Shea J, Scaife C, Parker DL, Jeong EK, Kennedy AM. Ultrasound-mediated tumor imaging and nanotherapy using drug loaded, block copolymer stabilized perfluorocarbon nanoemulsions. J Control Release. 2011;153:4–15. doi: 10.1016/j.jconrel.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rapoport N, Kennedy AM, Shea JE, Scaife CL, Nam KH. Ultrasonic nanotherapy of pancreatic cancer: lessons from ultrasound imaging. Mol Pharm. 2010;7:22–31. doi: 10.1021/mp900128x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rapoport NY, Kennedy AM, Shea JE, Scaife CL, Nam KH. Controlled and targeted tumor chemotherapy by ultrasound-activated nanoemulsions/microbubbles. J Control Release. 2009;138:268–276. doi: 10.1016/j.jconrel.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bisby RH, Mead C, Morgan CG. Photosensitive liposomes as ‘cages’ for laser-triggered solute delivery: the effect of bilayer cholesterol on kinetics of solute release. FEBS Lett. 1999;463:165–168. doi: 10.1016/s0014-5793(99)01612-9. [DOI] [PubMed] [Google Scholar]
- 117.Jochum FD, Theato P. Thermo- and light responsive micellation of azobenzene containing block copolymers. Chem Commun (Camb) 2010;46:6717–6719. doi: 10.1039/c0cc01288b. [DOI] [PubMed] [Google Scholar]
- 118.Wang Y, Han P, Xu H, Wang Z, Zhang X, Kabanov AV. Photocontrolled self-assembly and disassembly of block ionomer complex vesicles: a facile approach toward supramolecular polymer nanocontainers. Langmuir. 2010;26:709–715. doi: 10.1021/la9023844. [DOI] [PubMed] [Google Scholar]
- 119.You J, Zhang G, Li C. Exceptionally high payload of doxorubicin in hollow gold nanospheres for near-infrared light-triggered drug release. ACS Nano. 2010;4:1033–1041. doi: 10.1021/nn901181c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen J, Wang D, Xi J, Au L, Siekkinen A, Warsen A, Li ZY, Zhang H, Xia Y, Li X. Immuno gold nanocages with tailored optical properties for targeted photothermal destruction of cancer cells. Nano Lett. 2007;7:1318–1322. doi: 10.1021/nl070345g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fomina N, Sankaranarayanan J, Almutairi A. Photochemical mechanisms of light-triggered release from nanocarriers. Adv Drug Deliv Rev. 2012;64:1005–1020. doi: 10.1016/j.addr.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang G, Chen H, Dong Y, Luo X, Yu H, Moore Z, Bey EA, Boothman DA, Gao J. Superparamagnetic Iron Oxide Nanoparticles: Amplifying ROS Stress to Improve Anticancer Drug Efficacy. Theranostics. 2013;3:116–126. doi: 10.7150/thno.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.El-Dakdouki MH, Zhu DC, El-Boubbou K, Kamat M, Chen J, Li W, Huang X. Development of multifunctional hyaluronan-coated nanoparticles for imaging and drug delivery to cancer cells. Biomacromolecules. 2012;13:1144–1151. doi: 10.1021/bm300046h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kaaki K, Herve-Aubert K, Chiper M, Shkilnyy A, Souce M, Benoit R, Paillard A, Dubois P, Saboungi ML, Chourpa I. Magnetic nanocarriers of doxorubicin coated with poly(ethylene glycol) and folic acid: relation between coating structure, surface properties, colloidal stability, and cancer cell targeting. Langmuir. 2012;28:1496–1505. doi: 10.1021/la2037845. [DOI] [PubMed] [Google Scholar]
- 125.Fleige E, Quadir MA, Haag R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: concepts and applications. Adv Drug Deliv Rev. 2012;64:866–884. doi: 10.1016/j.addr.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 126.Sawant RM, Hurley JP, Salmaso S, Kale A, Tolcheva E, Levchenko TS, Torchilin VP. “SMART” drug delivery systems: double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjug Chem. 2006;17:943–949. doi: 10.1021/bc060080h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Modica-Napolitano JS, Singh KK. Mitochondrial dysfunction in cancer. Mitochondrion. 2004;4:755–762. doi: 10.1016/j.mito.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 128.Galluzzi L, Morselli E, Kepp O, Vitale I, Rigoni A, Vacchelli E, Michaud M, Zischka H, Castedo M, Kroemer G. Mitochondrial gateways to cancer. Mol Aspects Med. 2010;31:1–20. doi: 10.1016/j.mam.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 129.Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 2008;18:165–173. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 130.Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90:4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Moll UM, Schramm LM. p53--an acrobat in tumorigenesis. Crit Rev Oral Biol Med. 1998;9:23–37. doi: 10.1177/10454411980090010101. [DOI] [PubMed] [Google Scholar]
- 132.Ackermann EJ, Taylor JK, Narayana R, Bennett CF. The role of antiapoptotic Bcl-2 family members in endothelial apoptosis elucidated with antisense oligonucleotides. J Biol Chem. 1999;274:11245–11252. doi: 10.1074/jbc.274.16.11245. [DOI] [PubMed] [Google Scholar]
- 133.Marcucci G, Stock W, Dai G, Klisovic RB, Liu S, Klisovic MI, Blum W, Kefauver C, Sher DA, Green M, Moran M, Maharry K, Novick S, Bloomfield CD, Zwiebel JA, Larson RA, Grever MR, Chan KK, Byrd JC. Phase I study of oblimersen sodium, an antisense to Bcl-2, in untreated older patients with acute myeloid leukemia: pharmacokinetics, pharmacodynamics, and clinical activity. J Clin Oncol. 2005;23:3404–3411. doi: 10.1200/JCO.2005.09.118. [DOI] [PubMed] [Google Scholar]
- 134.Li X, Marani M, Yu J, Nan B, Roth JA, Kagawa S, Fang B, Denner L, Marcelli M. Adenovirus-mediated Bax overexpression for the induction of therapeutic apoptosis in prostate cancer. Cancer Res. 2001;61:186–191. [PubMed] [Google Scholar]
- 135.Costantini P, Belzacq AS, Vieira HL, Larochette N, de Pablo MA, Zamzami N, Susin SA, Brenner C, Kroemer G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene. 2000;19:307–314. doi: 10.1038/sj.onc.1203299. [DOI] [PubMed] [Google Scholar]
- 136.Weissig V, Boddapati SV, Souza GGM, Cheng SM. Targeting of Low-Molecular Weight Drugs to Mammalian Mitochondria. Drug Design Reviews-Online. 2004;1:15–28. [Google Scholar]
- 137.Ross MF, Kelso GF, Blaikie FH, James AM, Cocheme HM, Filipovska A, Da Ros T, Hurd TR, Smith RA, Murphy MP. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry (Mosc) 2005;70:222–230. doi: 10.1007/s10541-005-0104-5. [DOI] [PubMed] [Google Scholar]
- 138.Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- 139.Burns RJ, Smith RA, Murphy MP. Synthesis and characterization of thiobutyltriphenylphosphonium bromide, a novel thiol reagent targeted to the mitochondrial matrix. Arch Biochem Biophys. 1995;322:60–68. doi: 10.1006/abbi.1995.1436. [DOI] [PubMed] [Google Scholar]
- 140.Murphy MP. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997;15:326–330. doi: 10.1016/S0167-7799(97)01068-8. [DOI] [PubMed] [Google Scholar]
- 141.Smith RA, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 142.Coulter CV, Smith RAJ, Murphy MP. SYNTHESIS, CHARACTERIZATION, AND BIOLOGICAL PROPERTIES OF A FULLERENE TRIPHENYLPHOSPHONIUM SALT. Fullerene Science and Technology. 2001;9:339–350. [Google Scholar]
- 143.Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Murphy MP, Smith RA. Drug delivery to mitochondria: the key to mitochondrial medicine. Adv Drug Deliv Rev. 2000;41:235–250. doi: 10.1016/s0169-409x(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 145.Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 146.Sanjuan-Pla A, Cervera AM, Apostolova N, Garcia-Bou R, Victor VM, Murphy MP, McCreath KJ. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1alpha. FEBS Lett. 2005;579:2669–2674. doi: 10.1016/j.febslet.2005.03.088. [DOI] [PubMed] [Google Scholar]
- 147.Saretzki G, Murphy MP, von Zglinicki T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell. 2003;2:141–143. doi: 10.1046/j.1474-9728.2003.00040.x. [DOI] [PubMed] [Google Scholar]
- 148.Tauskela JS. MitoQ--a mitochondria-targeted antioxidant. IDrugs. 2007;10:399–412. [PubMed] [Google Scholar]
- 149.Villalba JM, Parrado C, Santos-Gonzalez M, Alcain FJ. Therapeutic use of coenzyme Q10 and coenzyme Q10-related compounds and formulations. Expert Opin Investig Drugs. 2010;19:535–554. doi: 10.1517/13543781003727495. [DOI] [PubMed] [Google Scholar]
- 150.Supinski GS, Murphy MP, Callahan LA. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1095–1102. doi: 10.1152/ajpregu.90902.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lowes DA, Wallace C, Murphy MP, Webster NR, Galley HF. The mitochondria targeted antioxidant MitoQ protects against fluoroquinolone-induced oxidative stress and mitochondrial membrane damage in human Achilles tendon cells. Free Radic Res. 2009;43:323–328. doi: 10.1080/10715760902736275. [DOI] [PubMed] [Google Scholar]
- 152.Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- 153.Armstrong JS. Mitochondrial medicine: pharmacological targeting of mitochondria in disease. Br J Pharmacol. 2007;151:1154–1165. doi: 10.1038/sj.bjp.0707288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Boddapati SV, D’Souza GG, Erdogan S, Torchilin VP, Weissig V. Organelle-targeted nanocarriers: specific delivery of liposomal ceramide to mitochondria enhances its cytotoxicity in vitro and in vivo. Nano Lett. 2008;8:2559–2563. doi: 10.1021/nl801908y. [DOI] [PubMed] [Google Scholar]
- 155.Biswas S, Dodwadkar NS, Deshpande PP, Torchilin VP. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J Control Release. 2012;159:393–402. doi: 10.1016/j.jconrel.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Biswas S, Dodwadkar NS, Piroyan A, Torchilin VP. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials. 2012;33:4773–4782. doi: 10.1016/j.biomaterials.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cuchelkar V, Kopeckova P, Kopecek J. Novel HPMA copolymer-bound constructs for combined tumor and mitochondrial targeting. Mol Pharm. 2008;5:776–786. doi: 10.1021/mp800019g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Biswas S, Dodwadkar NS, Sawant RR, Koshkaryev A, Torchilin VP. Surface modification of liposomes with rhodamine-123-conjugated polymer results in enhanced mitochondrial targeting. J Drug Target. 2011;19:552–561. doi: 10.3109/1061186X.2010.536983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Szeto HH. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2006;8:E277–283. doi: 10.1007/BF02854898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8:E521–531. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 162.Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- 163.Yasuzaki Y, Yamada Y, Harashima H. Mitochondrial matrix delivery using MITO-Porter, a liposome-based carrier that specifies fusion with mitochondrial membranes. Biochem Biophys Res Commun. 2010;397:181–186. doi: 10.1016/j.bbrc.2010.05.070. [DOI] [PubMed] [Google Scholar]
- 164.Yamada Y, Akita H, Kamiya H, Kogure K, Yamamoto T, Shinohara Y, Yamashita K, Kobayashi H, Kikuchi H, Harashima H. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim Biophys Acta. 2008;1778:423–432. doi: 10.1016/j.bbamem.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 165.Yamada Y, Akita H, Kogure K, Kamiya H, Harashima H. Mitochondrial drug delivery and mitochondrial disease therapy--an approach to liposome-based delivery targeted to mitochondria. Mitochondrion. 2007;7:63–71. doi: 10.1016/j.mito.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 166.Yamada Y, Furukawa R, Yasuzaki Y, Harashima H. Dual function MITO-Porter, a nano carrier integrating both efficient cytoplasmic delivery and mitochondrial macromolecule delivery. Mol Ther. 2011;19:1449–1456. doi: 10.1038/mt.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yamada Y, Nakamura K, Furukawa R, Kawamura E, Moriwaki T, Matsumoto K, Okuda K, Shindo M, Harashima H. Mitochondrial delivery of bongkrekic acid using a MITO-porter prevents the induction of apoptosis in human hela cells. Journal of Pharmaceutical Sciences. 2013:n/a–n/a. doi: 10.1002/jps.23442. [DOI] [PubMed] [Google Scholar]
- 168.Weissig V, Lizano C, Torchilin VP. Selective DNA release from DQAsome/DNA complexes at mitochondria-like membranes. Drug Deliv. 2000;7:1–5. doi: 10.1080/107175400266722. [DOI] [PubMed] [Google Scholar]
- 169.Weissig V, Torchilin VP. Cationic bolasomes with delocalized charge centers as mitochondria-specific DNA delivery systems. Adv Drug Deliv Rev. 2001;49:127–149. doi: 10.1016/s0169-409x(01)00131-4. [DOI] [PubMed] [Google Scholar]
- 170.Weissig V, Torchilin VP. Drug and DNA delivery to mitochondria. Adv Drug Deliv Rev. 2001;49:1–2. [PubMed] [Google Scholar]
- 171.Weissig V, Torchilin VP. Mitochondriotropic cationic vesicles: a strategy towards mitochondrial gene therapy. Curr Pharm Biotechnol. 2000;1:325–346. doi: 10.2174/1389201003378870. [DOI] [PubMed] [Google Scholar]
- 172.D’Souza GG, Rammohan R, Cheng SM, Torchilin VP, Weissig V. DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J Control Release. 2003;92:189–197. doi: 10.1016/s0168-3659(03)00297-9. [DOI] [PubMed] [Google Scholar]
- 173.Weissig V, D’Souza GG, Torchilin VP. DQAsome/DNA complexes release DNA upon contact with isolated mouse liver mitochondria. J Control Release. 2001;75:401–408. doi: 10.1016/s0168-3659(01)00392-3. [DOI] [PubMed] [Google Scholar]
- 174.Weissig V, Lasch J, Erdos G, Meyer HW, Rowe TC, Hughes J. DQAsomes: a novel potential drug and gene delivery system made from Dequalinium. Pharm Res. 1998;15:334–337. doi: 10.1023/a:1011991307631. [DOI] [PubMed] [Google Scholar]
- 175.Brini M, Marsault R, Bastianutto C, Alvarez J, Pozzan T, Rizzuto R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c). A critical evaluation. J Biol Chem. 1995;270:9896–9903. doi: 10.1074/jbc.270.17.9896. [DOI] [PubMed] [Google Scholar]
- 176.Vestweber D, Schatz G. DNA-protein conjugates can enter mitochondria via the protein import pathway. Nature. 1989;338:170–172. doi: 10.1038/338170a0. [DOI] [PubMed] [Google Scholar]
- 177.Seibel P, Trappe J, Villani G, Klopstock T, Papa S, Reichmann H. Transfection of mitochondria: strategy towards a gene therapy of mitochondrial DNA diseases. Nucleic Acids Res. 1995;23:10–17. doi: 10.1093/nar/23.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Flierl A, Jackson C, Cottrell B, Murdock D, Seibel P, Wallace DC. Targeted delivery of DNA to the mitochondrial compartment via import sequence-conjugated peptide nucleic acid. Mol Ther. 2003;7:550–557. doi: 10.1016/s1525-0016(03)00037-6. [DOI] [PubMed] [Google Scholar]
- 179.Samuelson LE, Dukes MJ, Hunt CR, Casey JD, Bornhop DJ. TSPO targeted dendrimer imaging agent: synthesis, characterization, and cellular internalization. Bioconjug Chem. 2009;20:2082–2089. doi: 10.1021/bc9002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene. 2004;23:2746–2756. doi: 10.1038/sj.onc.1207513. [DOI] [PubMed] [Google Scholar]
- 181.Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11:3155–3162. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 182.Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene. 2004;23:2881–2890. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- 183.Balaji KN, Schaschke N, Machleidt W, Catalfamo M, Henkart PA. Surface cathepsin B protects cytotoxic lymphocytes from self-destruction after degranulation. J Exp Med. 2002;196:493–503. doi: 10.1084/jem.20011836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. doi: 10.1083/jcb.201208152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Thekkedath R, Koshkaryev A, Torchilin VP. Lysosome-targeted octadecyl-rhodamine B-liposomes enhance lysosomal accumulation of glucocerebrosidase in Gaucher’s cells in vitro. Nanomedicine. 2012:1–11. doi: 10.2217/nnm.12.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 187.Stoka V, Turk V, Turk B. Lysosomal cysteine cathepsins: signaling pathways in apoptosis. Biol Chem. 2007;388:555–560. doi: 10.1515/BC.2007.064. [DOI] [PubMed] [Google Scholar]
- 188.Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes and oxidative stress in aging and apoptosis. Biochim Biophys Acta. 2008;1780:1291–1303. doi: 10.1016/j.bbagen.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 189.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 190.Turk B, Turk V. Lysosomes as “suicide bags” in cell death: myth or reality? J Biol Chem. 2009;284:21783–21787. doi: 10.1074/jbc.R109.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U, Salvesen GS, Stoka V, Turk V, Turk B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem. 2008;283:19140–19150. doi: 10.1074/jbc.M802513200. [DOI] [PubMed] [Google Scholar]
- 192.Broker LE, Huisman C, Span SW, Rodriguez JA, Kruyt FA, Giaccone G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004;64:27–30. doi: 10.1158/0008-5472.can-03-3060. [DOI] [PubMed] [Google Scholar]
- 193.Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, Senik A. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003;278:31401–31411. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 194.Brunk UT, Svensson I. Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Rep. 1999;4:3–11. doi: 10.1179/135100099101534675. [DOI] [PubMed] [Google Scholar]
- 195.Yuan XM, Li W, Dalen H, Lotem J, Kama R, Sachs L, Brunk UT. Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci U S A. 2002;99:6286–6291. doi: 10.1073/pnas.092135599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Cesen MH, Pegan K, Spes A, Turk B. Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res. 2012;318:1245–1251. doi: 10.1016/j.yexcr.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 197.Johansson AC, Appelqvist H, Nilsson C, Kagedal K, Roberg K, Ollinger K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis. 2010;15:527–540. doi: 10.1007/s10495-009-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, Weber E, Multhoff G, Rohde M, Jaattela M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–435. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nylandsted J, Rohde M, Brand K, Bastholm L, Elling F, Jaattela M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc Natl Acad Sci U S A. 2000;97:7871–7876. doi: 10.1073/pnas.97.14.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mahat RI, Monera OD, Smith LC, Rolland A. Peptide-based gene delivery. Curr Opin Mol Ther. 1999;1:226–243. [PubMed] [Google Scholar]
- 201.Nigg EA. Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature. 1997;386:779–787. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- 202.Bremner KH, Seymour LW, Pouton CW. Harnessing nuclear localization pathways for transgene delivery. Curr Opin Mol Ther. 2001;3:170–177. [PubMed] [Google Scholar]
- 203.Escriou V, Carriere M, Scherman D, Wils P. NLS bioconjugates for targeting therapeutic genes to the nucleus. Adv Drug Deliv Rev. 2003;55:295–306. doi: 10.1016/s0169-409x(02)00184-9. [DOI] [PubMed] [Google Scholar]
- 204.Lanford RE, Kanda P, Kennedy RC. Induction of nuclear transport with a synthetic peptide homologous to the SV40 T antigen transport signal. Cell. 1986;46:575–582. doi: 10.1016/0092-8674(86)90883-4. [DOI] [PubMed] [Google Scholar]
- 205.Collas P, Alestrom P. Nuclear localization signal of SV40 T antigen directs import of plasmid DNA into sea urchin male pronuclei in vitro. Mol Reprod Dev. 1996;45:431–438. doi: 10.1002/(SICI)1098-2795(199612)45:4<431::AID-MRD4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 206.Akita H, Kudo A, Minoura A, Yamaguti M, Khalil IA, Moriguchi R, Masuda T, Danev R, Nagayama K, Kogure K, Harashima H. Multi-layered nanoparticles for penetrating the endosome and nuclear membrane via a step-wise membrane fusion process. Biomaterials. 2009;30:2940–2949. doi: 10.1016/j.biomaterials.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 207.Sessler RJ, Noy N. A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Mol Cell. 2005;18:343–353. doi: 10.1016/j.molcel.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 208.Ullman E, Pan JA, Zong WX. Squamous cell carcinoma antigen 1 promotes caspase-8-mediated apoptosis in response to endoplasmic reticulum stress while inhibiting necrosis induced by lysosomal injury. Mol Cell Biol. 2011;31:2902–2919. doi: 10.1128/MCB.05452-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sneh-Edri H, Likhtenshtein D, Stepensky D. Intracellular targeting of PLGA nanoparticles encapsulating antigenic peptide to the endoplasmic reticulum of dendritic cells and its effect on antigen cross-presentation in vitro. Mol Pharm. 2011;8:1266–1275. doi: 10.1021/mp200198c. [DOI] [PubMed] [Google Scholar]
- 210.Mukai Y, Yoshinaga T, Yoshikawa M, Matsuo K, Yoshikawa T, Niki K, Yoshioka Y, Okada N, Nakagawa S. Induction of endoplasmic reticulum-endosome fusion for antigen cross-presentation induced by poly (gamma-glutamic acid) nanoparticles. J Immunol. 2011;187:6249–6255. doi: 10.4049/jimmunol.1001093. [DOI] [PubMed] [Google Scholar]
- 211.Shahzidi S, Cunderlikova B, Wiedlocha A, Zhen Y, Vasovic V, Nesland JM, Peng Q. Simultaneously targeting mitochondria and endoplasmic reticulum by photodynamic therapy induces apoptosis in human lymphoma cells. Photochem Photobiol Sci. 2011;10:1773–1782. doi: 10.1039/c1pp05169e. [DOI] [PubMed] [Google Scholar]
- 212.Sahay G, Gautam V, Luxenhofer R, Kabanov AV. The utilization of pathogen-like cellular trafficking by single chain block copolymer. Biomaterials. 2010;31:1757–1764. doi: 10.1016/j.biomaterials.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Shen P, Ohta S, Inasawa S, Yamaguchi Y. Selective labeling of the endoplasmic reticulum in live cells with silicon quantum dots. Chem Commun (Camb) 2011;47:8409–8411. doi: 10.1039/c1cc12713f. [DOI] [PubMed] [Google Scholar]
- 214.Pollock S, Antrobus R, Newton L, Kampa B, Rossa J, Latham S, Nichita NB, Dwek RA, Zitzmann N. Uptake and trafficking of liposomes to the endoplasmic reticulum. FASEB J. 2010;24:1866–1878. doi: 10.1096/fj.09-145755. [DOI] [PubMed] [Google Scholar]
- 215.Bathori G, Cervenak L, Karadi I. Caveolae--an alternative endocytotic pathway for targeted drug delivery. Crit Rev Ther Drug Carrier Syst. 2004;21:67–95. doi: 10.1615/critrevtherdrugcarriersyst.v21.i2.10. [DOI] [PubMed] [Google Scholar]
- 216.Mudhakir D, Akita H, Tan E, Harashima H. A novel IRQ ligand-modified nano-carrier targeted to a unique pathway of caveolar endocytic pathway. J Control Release. 2008;125:164–173. doi: 10.1016/j.jconrel.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 217.Helms JB, Zurzolo C. Lipids as targeting signals: lipid rafts and intracellular trafficking. Traffic. 2004;5:247–254. doi: 10.1111/j.1600-0854.2004.0181.x. [DOI] [PubMed] [Google Scholar]