

Abstract
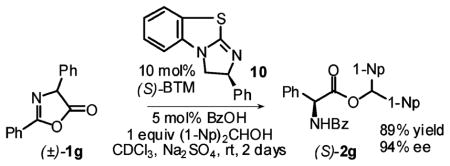
Enantioselective acyl transfer catalyst benzotetramisole (BTM) has been found to promote dynamic kinetic resolution of azlactones providing di(1-naphthyl)methyl esters of α-amino acids with up to 96% ee.
Dynamic kinetic resolution (DKR)1 of azlactones2 by way of their enantioselective alcoholysis (Figure 1) provides an attractive approach to the asymmetric synthesis of α-amino acid derivatives. Enzymatic variant of this transformation,3 while often successful, suffers from one inherent drawback: since enzymes are available in only one enantiomeric form, the reversal of enantioselectivity in a given reaction requires identification of a different enzyme, which is not always a trivial matter. Therefore, the development of nonenzymatic alternatives is of significant practical interest. Several mechanistically different approaches to activating azlactones toward alcoholysis have been employed. In 1997, Seebach et al. reported a Lewis acid-catalyzed version of this reaction using Ti(IV) TADDOLate 3 producing moderate ee’s (up to 68%).4 Promising levels of enantioselectivity (up to 78% ee) were achieved by Fu et al. in 1998 using enantioselective acyl transfer catalyst 4 (Figure 2). However, the reaction was extremely slow (50% conversion/1 week).5 Low enantioselectivities (<39% ee) were reported by Hua et al. in 1999 using a combination of a chiral diketopiperazine, cyclo-[(S)-His-(S)-Phe] 5, with diisopropyl L-tartrate, presumed to activate azlactones via hydrogen bonding.6 More recently (2005–2008), encouraging results have been obtained by Berkessel et al.7 and Connon et al.8 using bifunctional catalysts 6 (72–95% ee) and 7 (78–88% ee), respectively.
Figure 1.

Dynamic kinetic resolution of azlactones.
Figure 2.
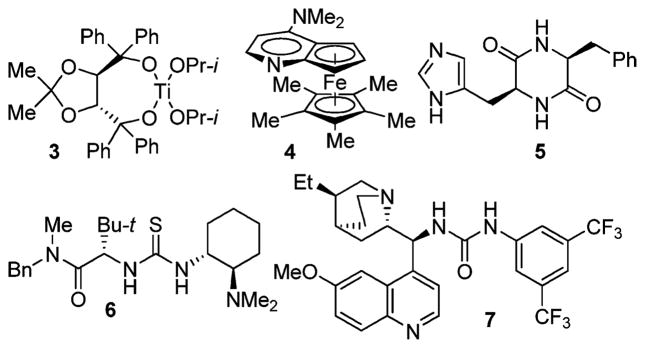
Catalysts previously employed for the DKR of azlactones.
Surprisingly, apart from Fu’s seminal study, there have been no other reported attempts to achieve DKR of azlactones using enantioselective acyl transfer catalysis. Over the past several years, our group has developed amidine-based catalysts 8–11 (Figure 3), which display high enantioselectivity in the acylation of several classes of alcohols and oxazolidinones.9 Recently, Shiina et al.10 have demonstrated the utility of BTM 10 in the kinetic resolution (KR) of α-arylpropionic acids via enantioselective alcoholysis of their mixed anhydrides. We have found that HBTM 11 is also effective in the KR of α-aryloxy- and arylthioalkanoic acids via their symmetrical anhydrides.11 These results have encouraged us to re-examine the possibility of DKR of azlactones via the acyl transfer mechanism.
Figure 3.
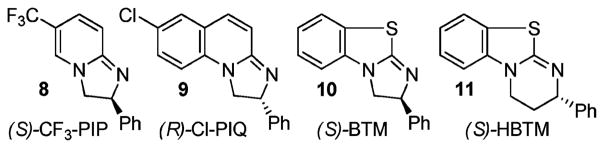
Amidine-based catalysts used in this study.
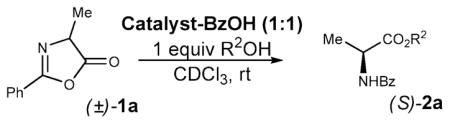
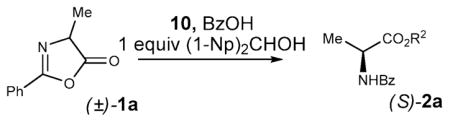
An equimolar combination of HBTM and benzoic acid was found to promote the methanolysis of substrate (±)-1a without any appreciable enantioselectivity (Table 1, entry 1). Switching to benzyl alcohol resulted in a modest ee (entry 2). The reaction with diarylcarbinols was extremely slow (entries 3 and 4). After these disappointing first results, we were pleased to discover that BTM is much more effective in this reaction. Encouraging results were obtained even using methanol (entry 5); however, the bulky di(1-naphthyl)methanol10,11 was required to bring the enantiomeric excess to a respectable 85% (entry 11). A single recrystallization from ethyl acetate produced completely enantiopure material (>99.5% ee). The earlier imidazoline-based catalysts, 8 and 9, proved to be competent but less active and less enantioselective than BTM (entries 12–14).
Table 1.
Catalyst and Alcohol Screening
| |||||
|---|---|---|---|---|---|
| entry | catalyst (mol %) | time, d | R2 | % convna | % ee |
| 1 | 11 (5) | 1 | Me | 54 | <3 |
| 2 | 11 (5) | 1 | PhCH2 | 47 | −25 |
| 3 | 11 (5) | 1 | Ph2CH | 5 | ND |
| 4 | 11 (10) | 1 | 1-Np2CH | <5 | ND |
| 5 | 10 (5) | 2 | Me | 91b | 34b |
| 6 | 10 (5) | 2 | PhCH2 | 94b | 48b |
| 7 | 10 (5) | 2 | 1-NpCH2 | 97b | 51b |
| 8 | 10 (5) | 2 | 2-NpCH2 | 96b | 47b |
| 9 | 10 (5) | 2 | Me2CH | <5 | ND |
| 10 | 10 (5) | 2 | Ph2CH | 91b | 75b |
| 11 | 10 (5) | 2 | 1-Np2CH | 96b | 85b |
| 12 | 10 (10) | 0.4 | 1-Np2CH | 92b | 80b |
| 13 | 8 (10) | 2 | 1-Np2CH | 47 | 59 |
| 14 | 9 (10) | 2 | 1-Np2CH | 47 | −52 |
Conversion was determined by 1H NMR, unless indicated otherwise.
Reported % isolated yields and % ee’s are averages of two runs.
Changing the amount of benzoic acid relative to the catalyst did not have a significant effect on the reaction rate or the enantioselectivity, although in the absence of the acid promoter the reaction did not proceed at all, consistent with Fu’s original report5 (entries 1–4, Table 2). Lower catalyst loadings, down to 2 mol %, were still effective, although required prolonged reaction times (entries 5 and 6). Decreased temperatures proved to be detrimental to the enantioselectivity, while higher temperatures resulted in a higher rates, but the same ee (entries 7–9). Solvents other than chloroform were less effective, in line with our earlier experience with the KR of alcohols9c (entries 10–13).
Table 2.
Variation of Reaction Conditionsa
| ||||||
|---|---|---|---|---|---|---|
| entry | mol % 10/mol % BzOH | time d | temp °C | solvent | % convn | % ee |
| 1 | 10/0 | 1 | 23 | CDCl3 | <5 | ND |
| 2 | 10/5 | 0.4 | 23 | CDCl3 | 90 | 83 |
| 3 | 10/10 | 0.4 | 23 | CDCl3 | 92 | 80 |
| 4 | 10/20 | 0.4 | 23 | CDCl3 | 93 | 82 |
| 5 | 5/5 | 2 | 23 | CDCl3 | 96 | 85 |
| 6 | 2/2 | 4 | 23 | CDCl3 | 94 | 84 |
| 7 | 5/5 | 2 | 0 | CDCl3 | 95 | 72 |
| 8 | 10/5 | 2 | −20 | CDCl3 | 94 | 55 |
| 9 | 5/5 | 1 | 45 | CDCl3 | 91 | 84 |
| 10 | 5/5 | 2 | 23 | C6D6 | 98 | 80 |
| 11 | 5/5 | 2 | 23 | CH2Cl2 | 95 | 74 |
| 12 | 5/5 | 2 | 23 | CH3CN | 85 | 66 |
| 13 | 5/5 | 2 | 23 | THF | <5 | ND |
Conversion in all cases was determined by 1H NMR.
DKR of other azlactones bearing primary alkyl substituents 1b–e produced uniformly good yields and ee’s in the 80–90% range (Table 3, entries 1–5). Isopropyl-substituted substrate 1f proved resistant to alcoholysis under the same conditions (entry 6). On the other hand, excellent enantioselectivity (94% ee) was obtained in the case of 2,4-diphenylazlactone 1g (entry 7). This result was all the more remarkable given the fact that the highest ee previously reported for either enzymatic3a or nonenzymatic7a DKR of this substrate (or any other 4-aryl-substituted azlactones) has been 75%. Variation of electronic properties of the C4-aryl substituent had only a slight effect on the enantioselectivity or reaction rate (entries 8–11). DKR of the 1-naphthyl-substituted substrate 1l, however, proceeded rather slowly and with lower enantioselectivity (entry 12). Substrate 1m was completely unreactive, presumably due to the presence of an ortho-substitutuent (entry 13).
Table 3.
Structural Variation of Azlactone Substratesa
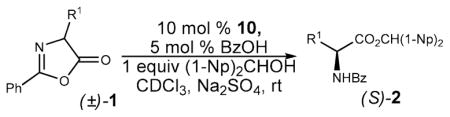
| |||||
|---|---|---|---|---|---|
| entry | substrate | time d | R1 | % yieldb | % eeb |
| 1 | 1a | 0.4 | Me | 90 | 83 |
| 2 | 1b | 7 | Me2CHCH2 | 97 | 80 |
| 3 | 1c | 3 | MeSCH2CH2 | 92 | 90 |
| 4 | 1d | 4 | PhCH2 | 94 | 83 |
| 5 | 1e | 4 | CH2=CHCH2 | 88 | 83 |
| 6 | 1f | 4 | Me2CH | <5 | ND |
| 7 | 1g | 2 | Ph | 89 | 94 |
| 8 | 1h | 2 | p-MeOC6H4 | 87 | 91 |
| 9 | 1i | 2 | p-ClC6H4 | 88 | 96 |
| 10 | 1j | 2 | p-BrC6H4 | 90 | 95 |
| 11 | 1k | 2 | 2-naphthyl | 88 | 91 |
| 12 | 1l | 7 | 1-naphthyl | 46 | 76 |
| 13 | 1m | 2 | 2,4-(MeO)2C6H3 | 0 | ND |
| 14c | 1h | 2 | p-MeOC6H4 | 86 | 92 |
| 15c | 1i | 2 | p-ClC6H4 | 86 | 97 |
| 16c | 1n | 2 | p-FC6H4 | 90 | 95 |
Initially, azlactones 1a–m used in this study were synthesized in a separate step and purified by recrystallization before subjecting them to the DKR conditions. Later, however, we found that in situ cyclization of N-benzoyl-α-amino acids with DCC followed by addition of di-(1-naphthyl)methanol and the catalyst works just as well as the earlier, more time-consuming protocol (cf. entries 14–16 vs 8 and 9, Table 3). Coupled with the one-step synthesis of starting materials via the Ben-Ishai amidoalkylation12 of simple arenes, this procedure provides an attractive route to enantioenriched arylglycine derivatives13 illustrated in Scheme 1. Hydrogenolysis of dinaphthylmethyl ester 2i proceeded without appreciable erosion of enantiomeric purity.
Scheme 1.
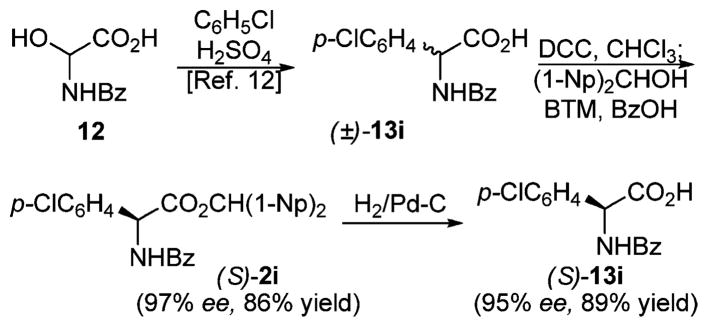
Asymmetric Synthesis of Arylglycines
It is of interest to analyze the mechanism of enantiodifferentiation in the DKR of azlactones. Berkessel et al.7a proposed that the thiourea-catalyzed version of this process occurs via irreversible, hydrogen bonding-assisted attack of the alcohol on the less hindered face of the substrate, which results in the highest enantioselectivity being observed in the case of the bulkiest C4-substituents (cf. general structure 1, R1 = isobutyl (1b), isopropyl (1f), or tert-butyl). The situation is clearly different in our case. The strong dependence of the enantioselectivity on the alcohol nucleophile indicates that enantiodifferentiation occurs in the second step of the catalytic cycle (Figure 4). The absolute sense of asymmetric induction and structure-selectivity trends can be explained by transition state model 15, which invokes hydrogen bonding between the benzamide group and the reacting carbonyl. The involvement of π–π interactions with the C2-phenyl group on the catalyst and/or lower steric repulsion may explain the higher enantioselectivity generally observed in the DKR of the aryl-substituted azlactones 1g–k compared to the alkyl-substituted ones 1a–e. The low reactivity of substrate bearing bulky C4 substituents (cf. 1f, 1l, and 1m) is also consistent with this proposal.
Figure 4.
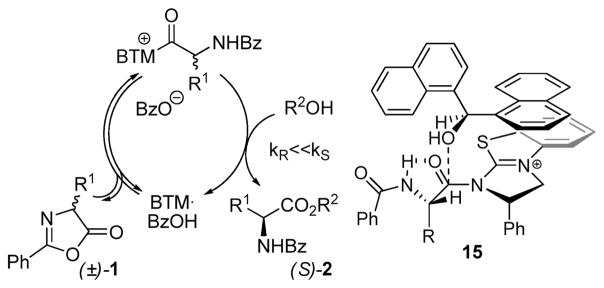
Proposed catalytic cycle and transition state (benzoate anion omitted for clarity).
In conclusion, we have developed a new, highly enantioselective method for the DKR of azlactones. It is especially suited for the C4-aryl-substituted substrates, thus complementing the previously available enzymatic and nonenzymatic protocols. Further exploration of its substrate scope is under active investigation in our laboratory.
Supplementary Material
Acknowledgments
We thank Washington University for the financial support of this study. Mass spectrometry was provided by the Wash. U. Mass Spectrometry Resource, an NIH Research Resource (Grant No. P41RR0954).
Footnotes
Supporting Information Available: Experimental procedures and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a recent review of dynamic kinetic resolution, see: Pellissier H. Tetrahedron. 2008;64:1563.
- 2.Fisk JS, Mosey RA, Tepe JJ. Chem Soc Rev. 2007;36:1432. doi: 10.1039/b511113g. [DOI] [PubMed] [Google Scholar]
- 3.See, e.g., Crich JZ, Brieva R, Marquart P, Gu RL, Flemming S, Sih CJ. J Org Chem. 1993;58:3252.Brown SA, Parker MC, Turner NJ. Tetrahedron: Asymmetry. 2000;11:1687. and references cited therein.
- 4.(a) Seebach D, Jaeschke G, Gottwald K, Mastsuda K, Formisano R, Chaplin DA, Breuning M, Bringmann G. Tetrahedron. 1997;53:7539. [Google Scholar]; (b) Gottwald K, Seebach D. Tetrahedron. 1999;55:723. [Google Scholar]
- 5.Liang J, Ruble JC, Fu GC. J Org Chem. 1998;63:3154. [Google Scholar]
- 6.Xie L, Hua W, Chan ASC, Leung YC. Tetrahedron: Asymmetry. 1999;10:4715. [Google Scholar]
- 7.(a) Berkessel A, Cleeman F, Mukherjee S, Müller TN, Lex J. Angew Chem. 2005;44:807. doi: 10.1002/anie.200461442. [DOI] [PubMed] [Google Scholar]; (b) Berkessel A, Mukherjee S, Cleemann F, Müller TN, Lex J. Chem Commun. 2005:1898. doi: 10.1039/b418666d. [DOI] [PubMed] [Google Scholar]; (c) Berkessel A, Mukherjee S, Müller TN, Cleeman F, Roland K, Brandenburg M, Neudörfl JM, Lex J. Org Biomol Chem. 2006;4:4319. doi: 10.1039/b607574f. [DOI] [PubMed] [Google Scholar]
- 8.Peschiulli A, Quigley C, Tallon S, Gun’ko YuK, Connon SJ. J Org Chem. 2008;73:6409. doi: 10.1021/jo801158g. [DOI] [PubMed] [Google Scholar]
- 9.(a) Birman VB, Uffman EW, Jiang H, Li X, Kilbane CJ. J Am Chem Soc. 2004;126:12226. doi: 10.1021/ja0491477. [DOI] [PubMed] [Google Scholar]; (b) Birman VB, Jiang H. Org Lett. 2005;7:3445. doi: 10.1021/ol051063v. [DOI] [PubMed] [Google Scholar]; (c) Birman VB, Li X. Org Lett. 2006;8:1351. doi: 10.1021/ol060065s. [DOI] [PubMed] [Google Scholar]; (d) Birman VB, Jiang H, Li X, Guo L, Uffman EW. J Am Chem Soc. 2006;128:6536. doi: 10.1021/ja061560m. [DOI] [PubMed] [Google Scholar]; (e) Birman VB, Guo L. Org Lett. 2006;8:4859. doi: 10.1021/ol061906y. [DOI] [PubMed] [Google Scholar]; (f) Birman VB, Li X. Org Lett. 2008;10:1115. doi: 10.1021/ol703119n. [DOI] [PubMed] [Google Scholar]; Zhang Y, Birman VB. Adv Synth Catal. 2009;351:2525. doi: 10.1002/adsc.200900383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiina I, Nakata K, Onda Y. Eur J Org Chem. 2008:5887. [Google Scholar]
- 11.Yang X, Birman VB. Adv Synth Catal. 2009;351:2301. doi: 10.1002/adsc.200900451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Ishai D, Satati I, Bernstein Z. Tetrahedron. 1976;32:1571. [Google Scholar]
- 13.For a review of asymmetric routes to α-arylglycines, see: Williams RM, Hendrix JA. Chem Rev. 1992;92:889.For more recent examples, see: Beenen MA, Weix DJ, Ellman JA. J Am Chem Soc. 2006;128:6304. doi: 10.1021/ja060529h. and references cited therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.