

Abstract
The emergence of multidrug-resistant bacteria has created an urgent need for antibiotics with a novel mechanism of action. The bacterial cell division protein FtsZ is an attractive target for the development of novel antibiotics. The benzo[c]phenanthridinium sanguinarine and the dibenzo[a,g]quinolizin-7-ium berberine are two structurally similar plant alkaloids that alter FtsZ function. The presence of a hydrophobic functionality at either the 1-position of 5-methylbenzo[c]phenanthridinium derivatives or the 2-position of dibenzo[a,g]quinolizin-7-ium derivatives is associated with significantly enhanced antibacterial activity. 3-Phenylisoquinoline represents a subunit within the ring-systems of both of these alkaloids. Several 3-phenylisoquinolines and 3-phenylisoquinolinium derivatives have been synthesized and evaluated for antibacterial activity against Staphylococcus aureus and Enterococcus faecalis, including multidrug-resistant strains of methicillin-resistant S. aureus (MRSA) and vancomycin-resistant E. faecalis (VRE). A number of derivatives were found to have activity against both MRSA and VRE. The binding of select compounds to S. aureus FtsZ (SaFtsZ) was demonstrated and characterized using fluorescence spectroscopy. In addition, the compounds were shown to act as stabilizers of SaFtsZ polymers and concomitant inhibitors of SaFtsZ GTPase activity. Toxicological assessment of select compounds revealed minimal cross-reaction mammalian β-tubulin as well as little or no human cytotoxicity.
Keywords: Antibacterial, Isoquinoline, FtsZ-targeting, Cytotoxicity, Staphylococcus aureus, Enterococcus faecalis
1. Introduction
Infections associated with methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE) represent a serious nosocomial health concern for both patients and healthcare professionals.1,2 Antibacterial agents with novel mechanisms of action represent a critical need in light of the increased incidence of bacterial resistance to current clinical agents. FtsZ is a key bacterial protein involved in microbial cell division (cytokinesis).3,4 It is highly conserved among bacterial pathogens and, in several genetic studies, has been shown to be essential for bacterial viability.5-8 Cell division in bacteria occurs at the site of formation of a cytokinetic Z-ring polymeric structure comprised of FtsZ subunits.9 The vital role that FtsZ plays in bacterial cell division makes this protein a promising therapeutic target. FtsZ-targeting antibacterial agents can exert their disruptive effects on the Z-ring by either stabilizing FtsZ polymers or inhibiting their formation.14-21 Recent advances in the development of small molecules that target FtsZ have been the subject of several recent reviews.10-13,22-24
The benzo[c]phenanthridine sanguinarine (1) and the dibenzo[a,g]quinolizin-7-ium berberine (3) (Fig. 1) are antibacterial plant alkaloids (albeit a weak antibacterial in the case of berberine) that have been identified as small molecules that alter FtsZ Z-ring formation and FtsZ function.15,16,22,25 In addition, the presence of a hydrophobic functionality at either the 1-position of benzo[c]phenanthridines, as in 4, or at the 2-position of dibenzo[a,g]quinolizin-7-ium derivatives, as in 5, significantly enhances antibacterial activity.26-28
Figure 1.

Structures of the alkaloids sanguinarine 1, chelerythrine 2, berberine 3, and their synthetic analogs 4 and 5.
3-Phenylisoquinoline represents a flexible subunit of the scaffold associated with the core structure of each of the compounds illustrated in Figure 1. We report, herein, the synthesis and relative antibacterial activity of several 3-phenylisoquinolines and 3-phenylisoquinolinium derivatives. As the constitutively charged quaternary isoquinolinium derivatives uniformly exhibited enhanced antibacterial activity relative to their non-quaternary isoquinoline precursors, we also explored the effect of varied basic functionalities at the 1-position.
2. Chemistry
3-Bromo-6,7-dimethoxyisoquinolin-1-one was prepared from 5,6-dimethoxy-1-indanone as previously described in the literature.29 Suzuki-coupling of this intermediate with [1,1′]biphenyl-3-ylboronic acid provided 3-[[1,1′]-biphenyl-3-yl]-6,7-dimethoxyisoquinolin-1-one, which was converted to 1a, 8a–11a or 14a as outlined in Scheme 1. Alternatively, 3-bromo-6,7-dimethoxyisoquinolin-1-one was used in a Suzuki-coupling with 3(t-butyl) phenyl boronic acid to provide the intermediate for ultimately preparing 17a. Both 1b and 9b were prepared by reacting either 1a or 9a, respectively, with methyl iodide. The 2-guanidinoethyl derivatives 15a and 20a were prepared from the primary amines, 14a and 17a by reaction with 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine followed by removal of the Boc-protecting groups with trifluoroacetic acid.
Scheme 1.
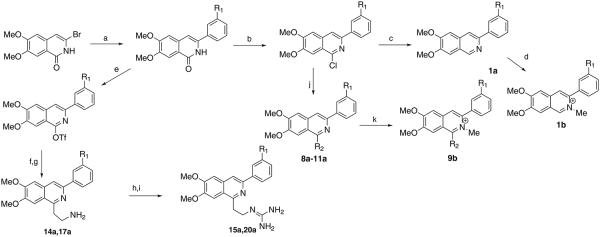
Synthesis of 3-phenylisoquinolines from 6,7-dimethoxy-3-bromoquinolin-1-one. Reagents and conditions: (a) R1-C6H4B(OH)2, Pd(OAc)2, XPhos, K2CO3, ACN/H2O, 100 °C; (b) POCl3, 110 °C; (c) H2, Pd/C (10%), EtOH, rt; (d) MeI, sealed tube 100 °C; (e) Tf2O, Et3N, DCM, −78 °C; (f) potassium t-butyl-N-[2-(trifluoroboranuidyl)ethyl]carbamate, PdCl2(dppf), Cs2CO3, dioxane/H2O, 102 °C; (g) TFA, DCM, 0 °C to rt; (h) 1,3-di-Boc-2-(trifluoromethylsulfonyl) guanidine, Et3N, CH2Cl2, 37 °C; (i) TFA, DCM, 0 °C to rt; (j) R2N, t-BuXPhos precatalyst, LHMDS, rt; for 8a and 9a; R2N, t-BuXPhos precatalyst, NaH/DMSO, rt for 10a; CuCN, DMSO, 140 °C; for 11a; (k) MeI, sealed tube 100 °C.
6,7-Dimethoxy-3-hydroxy-1-methylisoquinoline and 6,7,8-trimethoxy-3-hydroxy-1-methylisoquinoline were used for the preparation of 2a–4a as outlined in Scheme 2. Both of these 3-hydroxy-1-methylisoquinolines were prepared from either 3,4-dimethoxyphenyl acetic acid or 3,4,5-trimethoxyphenyl acetic acid as described in the literature.30 Formation of their triflates and subsequent reaction with either [1,1′]-biphenyl-3-ylboronic acid or 3-(t-butyl) phenyl boronic acid provide the appropriately substituted 3-phenyl-1-methylisoquinoline, which was treated with methyl iodide to provide 2b–4b. Bromination of 2a with NBS provided the bromomethyl derivative used to prepare 12a. Similarly, bromination of 4a provided its bromomethyl derivative, which was subsequently used to form 16a and 18a.
Scheme 2.
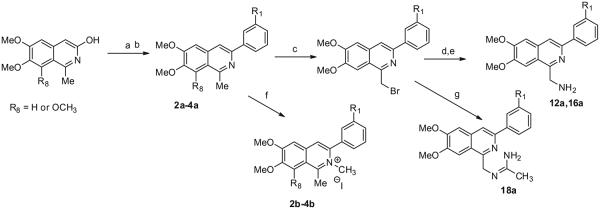
Synthesis of 3-phenylisoquinolines from 6,7-dimethoxy-3-hydroxy-1-methylisoquinoline. Reagents and conditions: (a) Tf2O, Et3N, DCM, −78 °C; (b) R1-C6H4B(OH)2, Pd(OAc)2, XPhos, K2CO3, dioxane/H2O, 102 °C; (c) NBS, AIBN, CCl4, 85 °C; (d) NaN3, DMF, rt; (e) PPh3 polymer bound, THF/H2O, 0 °C to rt; (f) MeI, sealed tube 100 °C; (g) acetamidine HCl, K2CO3, DMF, rt.
The preparation of 6a,b is outlined in Scheme 3. The triflate of 6,7,8-trimethoxy-3-hydroxy-1-methylisoquinoline was coupled with 2-benzyloxy-3,4-dimethoxyphenylboronic acid. Removal of the benzyloxy group, followed by formation of the triflate of the resulting phenol and Suzuki-coupling with [1,1′]-biphenyl-3-ylboronic acid provided 6a, which was treated with methyl iodide to give 6b.
Scheme 3.
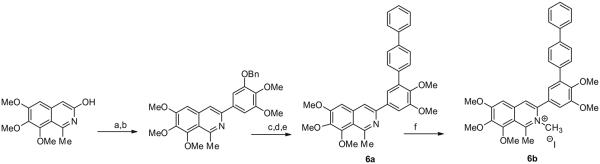
Synthesis of 3-phenylisoquinolines from 6,7-dimethoxy-3-hydroxy-1-methylisoquinoline. Reagents and conditions: (a) Tf2O, Et3N, DCM, −78 °C; (b) 2-benzyloxy- 3,4-dimethoxyphenylboronic acid, Pd(OAc)2, XPhos, K2CO3, ACN/H2O, 90 °C; (c) H2, Pd/C, MeOH; (d) Tf2O, Et3N, DCM; (e) 4-biphenylboronic acid, Pd(OAc)2, XPhos, K2CO3, ACN/H2O, 95 °C; (f) MeI, sealed tube 100 °C.
The preparation of 12a and the 1-guanidinomethyl derivatives 13a and 19a is illustrated in Scheme 4. Compounds 2a and 4a were oxidized to their 1-formyl derivatives with SeO2, which could then be reduced to their respective benzyl alcohols with NaBH4. Treatment with 1,3-bis(t-butoxycarbonyl)guanidine and subsequent removal of the N-Boc-protecting groups with TFA provided the guanidinomethyl derivatives 13a and 19a. Compound 12a was prepared by reaction of 3-([1,1-biphenyl]-3-yl)-6,7-dimethoxy-1-hydroxymethylisoquinoline with diphenylphosphorylazide to form the azide intermediate, which was reduced to the 1-aminomethyl derivative using polymer supported triphenylphosphine.
Scheme 4.
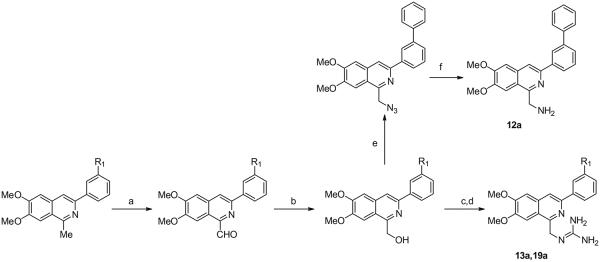
Synthesis of 3-phenylisoquinolines from 6,7-dimethoxy-3-hydroxy-1-methylisoquinoline. Reagents and conditions: (a) SeO2, dioxane, 102 °C; (b) NaBH4, EtOH, 0 °C to rt; (c) 1,3-bis(t-butoxycarbonyl)guanidine, PPh3, DIAD, toluene, 0 °C to rt; (d) TFA, DCM, 0 °C to rt.; (e) diphenylphosphorylazide, THF, DBU 0 °C to rt; (f) Ph3P (polymer supported) in THF:H2O (1:1)
Method used for 5a,b, 7a,b, and 21a are illustrated in Scheme 5. Using the triflate of 6,7-dimethoxy-3-hydroxy-1-methylisoquino-line, an initial Suzuki-coupling with 3-hydroxyphenylboronic acid provided 6,7-dimethoxy-3-(3-hydroxyphenyl)-1-methylisoquinoline, which served as a versatile intermediate for the formation of these compounds. This intermediate was converted to its triflate and then subjected to a second Suzuki-coupling with 4-biphenylboronic acid or 4-t-butylphenylboronic acid provided 5a and 7a, respectively. Both 5a and 7a were converted to the quaternary ammonium derivatives by treatment with methyl iodide at 100 °C in a sealed tube. The 1-methyl substituent of 7a was oxidized to its 1-formyl derivative with SeO2, reduced with sodium borohydride to the 1-hydroxymethyl intermediate, which under Mitsunobu reaction conditions provided the bis-(N-Boc guanidine). Treatment of this compound with trifluoroacetic acid provided 21a.
Scheme 5.

Synthesis of 3-(4′-(t-butyl)-[1,1′-biphenyl]-3-yl)isoquinolines and 3-([1,1′:4′,1″-terphenyl]-3-yl)isoquinolines from 6,7-dimethoxy-3-(3-hydroxyphenyl)-1-methylisoquinoline. Reagents and conditions: (a) Tf2O, Et3N, DCM, −78 °C; (b) 3-hydroxyphenylboronic acid; (c) 4-t-butylphenylboronic acid or 4-biphenylboronic acid, Pd(OAc)2, XPhos, K2CO3, ACN/H2O, 95 °C; (d) MeI, sealed tube 100 °C; (e) SeO2, dioxane, 102 °C; (f) NaBH4, EtOH 0 °C to rt; (g) 1,3-bis(t-butoxycarbonyl)guanidine, PPh3, DIAD, toluene, 0 °C to rt; (h) TFA, DCM, 0 °C to rt.
3. Pharmacology
The relative antistaphylococcal and antienterococcal activities of the 3-phenylisoquinoline and 3-phenylisoquinolinium derivatives synthesized are summarized in Table 1. No significant antibiotic activity was observed for the non-quaternary derivatives 1a–7a against either S. aureus or E. faecalis. Antibacterial activity was observed for N-methyl quaternary ammonium derivatives 1b–7b against S. aureus. Antibacterial potency increased with the increased lipophilicity of the substituent at the 3′-position. With the exception of 1b and 7b, there were relatively minor differences in the MICs observed with methicillin-sensitive S. aureus (MSSA) relative to methicillin-resistant S. aureus (MRSA). The MICs observed with vancomycin-sensitive E. faecalis (VSE) did tend to be greater than those observed with MSSA. Against vancomycin-resistant E. faecalis (VRE), only 5b, 6b, and 7b had MICs within the range of 4–8 μg/mL.
Table 1.
Antistaphylococcal and antienterococcal activities of 3-phenyl-6,7-dimethoxyisoquinoline and 3-phenyl-6,7-dimethoxy-2-methylisoquinolinium derivatives synthesized
| Compound | Y | R8 | R3′ | MICa (μg/mL) |
|||
|---|---|---|---|---|---|---|---|
|
S. aureus
8325-4 (MSSA) |
S. aureus
ATCC 33591 (MRSA) |
E. faecalis
ATCC 19433 (VSE) |
E. faecalis
ATCC 51575 (VRE) |
||||
| 1a | H | H | Phenyl | >64 | >64 | >64 | >64 |
| 1b | H | H | Phenyl | 16 | 64 | >64 | >64 |
| 2a | CH3 | H | Phenyl | >64 | >64 | >64 | >64 |
| 2b | CH3 | H | Phenyl | 16 | 32 | >64 | >64 |
| 3a | CH3 | OCH3 | Phenyl | >64 | >64 | >64 | >64 |
| 3b | CH3 | OCH3 | Phenyl | 8 | 8 | 32 | 32 |
| 4a | CH3 | H | t-Butyl | >64 | >64 | >64 | >64 |
| 4b | CH3 | H | t-Butyl | 16 | 16 | 64 | 64 |
| 5a | CH3 | H | Biphenyl | >64 | >64 | >64 | >64 |
| 5b | CH3 | H | Biphenyl | 1 | 2 | 4 | 8 |
| 6a | CH3 | OCH3 | Biphenylb | >64 | >64 | >64 | >64 |
| 6b | CH3 | OCH3 | Biphenylb | 1 | 1 | 4 | 4 |
| 7a | CH3 | H | 4-(t-Butyl)Ph | >64 | >64 | >64 | >64 |
| 7b | CH3 | H | 4-(t-Butyl)Ph | 1 | 8 | 8 | 8 |
| 8a | NHCH3 | H | Phenyl | >64 | >64 | >64 | >64 |
| 9a | N(CH3)2 | H | Phenyl | >64 | >64 | >64 | >64 |
| 9b | N(CH3)2 | H | Phenyl | 8 | 32 | 64 | 64 |
| 10a | NC(NH2)2 | H | Phenyl | 8 | 4 | 8 | 8 |
| 11a | CN | H | Phenyl | >64 | >64 | >64 | >64 |
| 12a | CH2NH2 | H | Phenyl | 8 | 16 | >64 | >64 |
| 13a | CH2NC(NH2)2 | H | Phenyl | 2 | 2 | 8 | 8 |
| 14a | CH2CH2NH2 | H | Phenyl | 4 | 4 | 16 | >64 |
| 15a | CH2CH2NC(NH2)2 | H | Phenyl | 2 | 2 | 8 | 8 |
| 16a | CH2NH2 | H | t-Butyl | 4 | 8 | 32 | 32 |
| 17a | CH2CH2NH2 | H | t-Butyl | 16 | 16 | 16 | 16 |
| 18a | CH2NC(CH3)NH2 | H | t-Butyl | 8 | 8 | 16 | 16 |
| 19a | CH2NC(NH2)2 | H | t-Butyl | 4 | 8 | 16 | 16 |
| 20a | CH2CH2NC(NH2)2 | H | t-Butyl | 4 | 4 | 16 | 16 |
| 21a | CH2NC(NH2)2 | H | 4-(t-Butyl)Ph | 4 | 4 | 8 | 16 |
| Sanguinarine | 2 | 2 | 8 | 16 | |||
| Chelerythrine | 4 | 4 | 32 | 32 | |||
| Berberine | >64 | >64 | >64 | >64 | |||
| Oxacillin | 0.06 | >64 | 8 | >64 | |||
| Vancomycin | 1 | 2 | 1 | >64 | |||
| Erythromycin | 0.1 | >64 | 1 | >64 | |||
| Tetracycline | 0.06 | 64 | 0.5 | >64 | |||
| Clindamycin | 0.03 | >64 | 2 | >64 | |||
Minimum inhibitory concentration (MIC) assays were conducted in accordance with Clinical and Laboratory Standards Institute (CLSI) guidelines for broth microdilution.31 MIC is defined as the lowest compound concentration at which bacterial growth is ≥90% inhibited.
R′4 and R′5 = OCH3.
We evaluated a series of 3-(3′-phenyl)phenylisoquinoline derivatives with varied substituents at the 1-position. Among the 1-amino-3-(3′-phenyl)phenylisoquinolines 8a, 9a and 9b, only the quaternary ammonium derivative 9b exhibited modest activity against S. aureus, but was not significantly active against either strain of E. faecalis. The 1-guanidino derivative 10a was active against both sensitive and resistant strains of both S. aureus and E. faecalis. These data suggest that the presence of a basic substituent at the 1-position that would exist as a protonated species under physiological conditions is associated with antibacterial activity. This structure-activity is further substantiated by the observation that the 1-cyano derivative is inactive, while its reduction product, the aminomethyl derivative 12a is active against S. aureus. The 1-guanidinomethyl- and 1-(2-guanidinoethyl)-derivatives 13a and 15a are active against both strains of S. aureus and E. faecalis. The less basic 1-(2-aminoethyl) derivative, 14a, is active against both strains of S. aureus, but is only modestly active against the vancomycin-sensitive strain of E. faecalis. Compound 21a is analogous to 13a, having a t-butyl substituent at its 4″-position. In general, the addition of this t-butyl group results in 21a exhibiting slightly less antibacterial activity than 13a.
The results observed for a series of 3-(3′-t-butyl)phenylisoquinolines indicate that the 1-aminomethyl analog 16a has greater potency than the 1-(2-aminoethyl) derivative 17a. This differs from the trend observed with the 3-(3-biphenyl)isoquinolines. The more basic amidinomethyl derivative 18a and the guanidinomethyl derivative 19a are modestly more active than 17a in both strains of S. aureus, while the guanidinomethyl derivative 19a had comparable activity to 16a. The guanidinoethyl derivative 20a is the more active of 3-(3′-t-butyl)phenylisoquinolines against MRSA. Compounds 17a–20a have similar activity against VRE and VSE and were only slightly more active than 16a.
While sanguinarine and chelerythrine have significant antibacterial activity against MSSA and MRSA, berberine has much weaker potency against these S. aureus strains. Against both strains of E. faecalis, chelerythrine and berberine do not have remarkable antibacterial activity. Only sanguinarine has comparable activity against both sensitive and resistant strains of both S. aureus and E. faecalis to several of the more potent isoquinoline derivatives evaluated in this study.
4. Biochemistry
4.1. Binding of the isoquinolines to S. aureus FtsZ (SaFtsZ)
We sought to determine whether the antibacterial activity of the 3-phenyl substituted isoquinolines might reflect a corresponding ability of the compounds to target bacterial FtsZ. As a first step toward this end, we explored the potential of the active compounds to bind purified S. aureus FtsZ (SaFtsZ). Specifically, we monitored the intrinsic fluorescence of the compounds as a function of added SaFtsZ. Figure 2 shows representative results for two of the more active compounds (5b and 7b). Note that increasing SaFtsZ concentrations increase the fluorescence emission intensities of both compounds, while also blue-shifting the maximum spectral wavelength by 4–6 nm (Fig. 2A and B). These FtsZ-induced changes in compound fluorescence are indicative of compound binding to the target protein.
Figure 2.

(A and B) Fluorescence emission spectra of 7 μM 5b (A) and 10 μM 7b (B) acquired in the absence and presence of SaFtsZ at concentrations ranging from 2.5 to 30 μM. From bottom to top at 420 nm the spectra correspond to SaFtsZ concentrations of 0, 2.5, 5, 10, 20, and 30 μM. (C and D) Fluorescence profiles of emission intensity at 410 nm for the titration of SaFtsZ into a solution of either 5b (C) or 7b (D). The solid lines represent fits of the experimental data with Eq. (1), which, in turn, yielded the indicated Kd values. The indicated uncertainties in Kd reflect the standard deviations of the experimental data points from the fitted curves. All experiments were conducted at 25 °C.
We determined compound-FtsZ dissociation constants (Kd) for the binding reactions by analyzing the FtsZ-induced changes in compound fluorescence with the following 1:1 binding formalism:
| (1) |
In this relationship, I0 and I are the fluorescence emission intensities of the compound in the absence and presence of protein, respectively; I∞ is the fluorescence emission intensity of the compound in the presence of an infinite protein concentration; and [C]tot and [P]tot are the total concentrations of compound and protein, respectively. The 1:1 binding formalism yielded excellent fits (R2 >0.99) of the 5b and 7b fluorescence intensity profiles (the solid lines in Fig. 2C and D). The Kd values obtained from these fits were similar in magnitude (2.0 ± 0.7 μM for 5b and 5.4 ± 1.5 μM for 7b). Significantly, these Kd values are also similar in magnitude to the corresponding MIC values of the two compounds versus MSSA (MIC = 1 μg/mL = 2.2 μM for 5b and 2.3 μM for 7b).
4.2. Impact of the isoquinolines on SaFtsZ polymerization
We next sought to explore whether the binding of the isoquinoline compounds to FtsZ had an impact on the self-polymerization activity of the protein. In this connection, we utilized a microtiter plate-based light scattering (turbidity) assay in which FtsZ polymerization is detected in solution by a time-dependent increase in light scattering, as reflected by a corresponding increase in solution absorbance at 340 nm (A340). When tested at a concentration of 40 μg/mL, all active isoquinoline compounds with MIC values ≤16 μg/mL (Table 1) were found to stimulate SaFtsZ polymerization, with the time-dependent A340 profiles acquired in the presence of DMSO vehicle and seven of the active compounds (5b, 7b, 15a, 17a, 18a, 19a, and 20a) being shown in Figure 3A as illustrative examples. This behavior is similar to that previously reported for the FtsZ-targeting benzamide analog PC19072314,18 and recapitulated here as a positive control (Fig. 3B). We used the non-FtsZ-targeting drug vancomycin as a negative control in these studies. As expected, vancomycin had no impact on the polymerization of SaFtsZ (Fig. 3B). Similar to vancomycin, and in striking contrast to the active isoquinolines, the inactive isoquinoline compounds (with MIC values >64 μg/mL) did not significantly impact SaFtsZ polymerization, as exemplified by the similar A340 profiles acquired in the presence of vehicle and two of the inactive compounds (4a and 7a) at an equivalent concentration of 40 μg/mL (Fig. 3A). Thus, the SAR of the isoquinolines with regard to antistaphylococcal activity correlates well with the corresponding SAR observed for stimulation of SaFtsZ polymerization. As an additional comparator in these assays, we also included the natural plant alkaloid berberine (3), which is inactive versus S. aureus at concentrations ≤64 μg/mL (Table 1). Like the inactive isoquinolines, berberine did not significantly impact SaFtsZ polymerization at an equivalent concentration of 40 μg/mL (Fig. 3B).
Figure 3.

Impact of isoquinoline compounds on the polymerization SaFtsZ (10 μM) in the presence of 1 mM GTP, as determined by monitoring time-dependent changes in absorbance at 340 nm (A340). (A and B) A340 profiles of SaFtsZ are shown in the presence of DMSO vehicle or the indicated compounds or comparator control agents each at a concentration of 40 μg/mL. (C) A340 profiles of SaFtsZ in the presence of DMSO vehicle or 20a at a concentration of 20, 30, or 40 μg/mL. For comparative purposes, the corresponding profile of 40 μg/mL 20a alone is also included as a no-protein control. (D) A340 profiles of SaFtsZ in the presence of 7b, 17a, or 20a at a concentration of 40 μg/mL. GDP (1 mM) was added at the time indicated by the arrows.
We further investigated whether the stimulatory impact of the active isoquinolines on SaFtsZ polymerization was dependent on compound concentration. Figure 3C illustrates the A340 results obtained in the presence of vehicle and compound 20a at concentrations of 20, 30, and 40 μg/mL. Note that the extent to which 20a enhances SaFtsZ polymerization increases with increasing compound concentration. We observed a similar concentration-dependent behavior with the other active isoquinolines (not shown). As a negative control, Figure 3C also shows the A340 profile of 40 μg/mL 20a in the absence of FtsZ. The lack of A340 change associated with the compound alone confirms that the enhanced light scattering induced by 20a in the presence of FtsZ reflects a corresponding stimulation of FtsZ self-polymerization and not simply non-specific compound aggregation or precipitation.
We next explored the stability of the SaFtsZ polymers induced by the active isoquinolines. In the absence of a polymer-stabilizing agent or compound, addition of GDP has been shown to depolymerize FtsZ polymers formed in the presence of GTP.14 We, therefore, sought to determine the impact, if any, of added GDP (1 mM) on the SaFtsZ polymers formed in the presence of both GTP (1 mM) and the active isoquinolines (40 μg/mL). Figure 3D shows the results for compounds 7b, 17a, and 20a as illustrative examples. Note that addition of 1 mM GDP does not exert a significant impact on the A340 signal, an observation indicating that SaFtsZ polymers induced by the presence of the isoquinoline compounds are stable to the depolymerizing effects of GDP. This behavior is similar to that previously reported for FtsZ-targeting antibacterial compound PC190723 and the FtsZ protein of Bacillus subtilis.14 Viewed as a whole, our polymerization results are consistent with the antibacterial activities of the isoquinolines being related, at least in part, to their stabilizing actions on FtsZ polymerization.
4.3. Impact of the isoquinolines on SaFtsZ GTPase activity
We investigated the impact, if any, of the active isoquinolines on the GTPase activity of SaFtsZ. Table 2 summarizes the results for identical concentrations (40 μg/mL) of the seven active compounds (5b, 7b, 15a, 17a, 18a, 19a, and 20a) used in the SaFtsZ polymerization profiles shown in Figure 3A. Note that the isoquinoline compounds inhibit the GTPase activity of SaFtsZ by as much as 85%, in marked contrast to the non-FtsZ-targeting drug vancomycin, which has no significant impact. This inhibitory activity is consistent with that previously observed for the FtsZ polymer-stabilizing compound PC190723 versus both B. subtilis and S. aureus FtsZ.14,18,32 An analysis of the concentration dependence with which the active isoquinolines 15a and 18a inhibit the GTPase activity of SaFtsZ reveals that both compounds stimulate GTPase activity at lower concentrations, followed by inhibition at higher concentrations, a behavior similar to that previously reported for PC190723 and B. subtilis FtsZ.14 It has been suggested that the increase in FtsZ GTPase activity in the presence of low compound concentrations may simply reflect a polymerization-induced enhancement in GTPase activity relative to that associated with the polymerization of the protein in the absence of added compound (see Fig. 4).14
Table 2.
Impact of select isoquinoline compounds on the GTPase activity of SaFtsZ
| Compound or control agenta | Percent GTPase activityb |
|---|---|
| Vehicle | 100.0 ± 3.3 |
| 5b | 30.2 ± 0.3 |
| 7b | 14.9 ± 1.7 |
| 15a | 56.0 ±1.7 |
| 17a | 59.7 ± 1.0 |
| 18a | 64.8 ± 0.1 |
| 19a | 60.4 ± 1.9 |
| 20a | 66.5 ± 1.7 |
| Vancomycin | 99.7 ± 12.2 |
Vancomycin and all isoquinoline compounds were used at a concentration of 40 μg/mL.
Percent GTPase activity reflects the percentage of the GTPase activity observed in the presence of vehicle (DMSO) alone. Each value represents the mean of two independent assessments, with the indicated uncertainties reflecting the standard deviations from the mean.
Figure 4.

Concentration dependence of the impact of 15a and 18a on the GTPase activity of SaFtsZ (10 μM) in the presence of 1 mM GTP. The indicated GTPase activity reflects the percentage of the control GTPase activity observed in the presence of vehicle (DMSO) alone. Each data point represents the mean of two independent assessments, with the indicated error bars reflecting the standard deviations from the mean.
5. Toxicology
5.1. Impact of the isoquinolines on the polymerization of mammalian tubulin
Tubulin is the closest mammalian functional homolog to bacterial FtsZ. We therefore sought to determine whether the isoquinoline compounds that are potent stimulators of FtsZ polymerization would exert similar effects on mammalian β-tubulin. To this end, we monitored the impact of two such compounds (7b and 19a) on the polymerization of β-tubulin porcine tubulin using an assay similar to that described above for SaFtsZ polymerization. We used the antineoplastic drugs paclitaxel (taxol) and nocodazole as positive controls in these assays. Paclitaxel is a known stimulator of tubulin polymerization and nocodazole is a known inhibitor of tubulin polymerization.33-35 Figure 5 shows the time-dependent A340 profiles of porcine β-tubulin in the absence and presence of 7b (at 40 μg/mL), 19a (at 40 μg/mL), paclitaxel (at 25 μg/mL), or nocodazole (at 10 μg/mL). Both paclitaxel and nocodazole produce their expected impacts on tubulin polymerization dynamics. By contrast, neither 7b nor 19a exert a significant impact. These observations indicate that isoquinoline compounds which profoundly stimulate bacterial FtsZ polymerization (as shown in Fig. 3A) do not appear to cross-react with mammalian tubulin to any significant degree.
Figure 5.

Impact of 7b and 19a on the polymerization of microtubule-associated protein (MAP)-rich porcine β-tubulin (70% tubulin, 30% MAPs), as determined by monitoring time-dependent changes in absorbance at 340 nm (A340). The A340 profiles of tubulin (2 mg/mL) in the presence of DMSO vehicle (●), 40 μg/mL 7b (□), 40 μg/mL 19a (▲), 25 μg/mL paclitaxel (■), or 10 μg/mL nocodazole (○) are depicted.
5.2. Cytotoxicity of the isoquinolines
The non-quaternized isoquinoline derivatives that exhibited significant antibacterial activity did tend to have better solubility properties than the quaternary ammonium derivatives. In addition, it would be expected that these derivatives would be more efficiently absorbed and distributed. Several of these non-quaternized 6,7-dimethoxyisoquinoline derivatives were evaluated for cytotoxicity against mammalian cells. Among the compounds that were evaluated were 10a, 12a, 13a, 15a–17a, and 19a–21a. These data indicate that there is no clear correlation between the observed cytotoxicity to mammalian cells, as reflected in their IC50 values, and the observed antibacterial activity as reflected by their MICs (IC90 values). For 13a, 15a, 16a, 19a–21a, no significant human cell toxicity was observed at the highest concentration tested (10 μM) in HEK293 cells. Compound 12a did exhibit modest human cell toxicity with an IC50 value of 3.0 μM in HEK293 cells, but was less toxic to canine MDCK cells with (IC50 = 7.0 μM). The more toxic derivatives of these 6,7-dimethoxyisoquinoline derivatives were 10a and 17a which had IC50 values that ranged from 2.2 to 3.5 μM in these cells.
6. Conclusions
The data indicate that various 3-phenylisoquinolines and 3-phenylisoquinolinium derivatives can exhibit significant antibacterial activity against methicillin-sensitive S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA). The presence of a basic substituent at the 1-position among the 3-phenylisoquinoline derivatives was associated with increased antibacterial activity. Several 3-phenylisoquinolinium derivatives such as 13a–15a, 20a, and 21a have MIC values that range from 2 to 4 μg/mL against MSSA and MRSA. Few of these compounds exhibited potent antibacterial activity against VRE. However, 10a, 13a, and 15a each has a MIC against VRE of 8 μg/mL, which is significantly lower than that observed for all of the clinical control compounds evaluated (MICs >64 μg/mL).
Formation of the N-methylisoquinolinium derivatives of those compounds that did not have a functional group at the 1-position that could be protonated at physiological pH increased antibacterial activity in each instance. In general the more lipophilic compounds, such as 3-(3′-biphenyl)isoquinoline or 3-3′-terphenyl)isoquinoline, exhibited the greater antibacterial activity. Several quaternary ammonium derivatives, 5b, 6b, and 7b had MICs of 1 μg/mL against MSSA and 1–8 μg/mL against MRSA. Compounds 5b, 6b, and 7b have MIC values that range from 4 to 8 μg/mL, which is lower than the MICs observed for the clinical compounds evaluated in this study.
In vitro studies with purified SaFtsZ suggest that the antibacterial activity of the compounds may be related to their stabilizing impact on FtsZ polymerization. Importantly, however, the compounds do not impact the polymerization dynamics of mammalian tubulin to any significant degree. Several of the non-quaternized isoquinoline derivatives in this study were also shown not to be highly toxic to mammalian cells. This degree of target specificity bodes well for desirable toxicological profiles on the part of the more active compounds that may have beneficial physicochemical and pharmacokinetic properties.
7. Experimental
7.1. Chemistry: general methods
All reactions, unless otherwise stated, were done under nitrogen atmosphere. Reaction monitoring and follow-up were done using aluminum backed Silica G TLC plates with UV254 (Sorbent Technologies), visualizing with ultraviolet light. Flash column chromatography was done on a Combi Flash Rf Teledyne ISCO using hexane, ethyl acetate, dichloromethane, and methanol. The 1H (400 MHz) and 13C (100 MHz) NMR spectra were done in CDCl3, methanol-d4, and DMSO-d6 and recorded on a Bruker Avance III (400 MHz) Multinuclear NMR Spectrometer. Data is expressed in parts per million relative to the residual nondeuterated solvent signals, spin multiplicities are given as s (singlet), δ (doublet), dd (doublet of doublets), t (triplet), dt (doublet of triplets), q (quartet), m (multiplet), and bs (broad singlet), and coupling constants (J) are reported in Hertz. Melting points were determined using Mel-temp II apparatus and are uncorrected. IR data was recorded on a Thermo Nicolet Avatar Model 360 FTIR. HRMS experiments were conducted by Washington University Resource for Biomedical and Bioorganic Mass Spectrometry Department of Chemistry.
7.2. General procedure for the synthesis of compound (1)
7.2.1. 3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1(2H)one
3-Bromo-6,7-dimethoxyisoquinolin-1(2H)-one (550 mg, 1.94 mmol) was combined with 3-biphenyl boronic acid (768 mg, 3.88 mmol), Pd(OAc)2 (43.5 mg, 0.194 mmol), XPhos (185 mg, 0.388 mmol), and K2CO3 (1.07 g, 7.76 mmol) in a flask and degassed. ACN (15 mL) and H2O (7.5 mL) were then added and solution was heated at 100 °C for 1.5 h. Reaction mixture was cooled to RT then diluted with EtOAc and washed with NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 70% EtOAc/hexane yielding product as a white solid (540 mg, 78% yield); mp 229–231 °C; 1H NMR (400 MHz) (CDCl3) δ 9.98 (bs, 1H), 7.97 (m, 1H), 7.78 (s, 1H), 7.75–7.67 (m, 4H), 7.60 (t, J = 16.0 Hz, 1H), 7.50–7.46 (m, 2H), 7.43–7.39 (m, 1H), 7.01 (s, 1H), 6.81 (s, 1H), 4.05 (s, 3H), 3.96 (s, 3H); 13C NMR (100 MHz) (CDCl3). 163.5, 153.9, 149.3, 142.0, 140.4, 138.5, 135.1, 133.9, 129.5, 128.9, 127.8, 127.8, 127.2, 125.1, 125.0, 119.0, 107.4, 106.6, 104.2, 56.1, 56.1; HRMS (ESI) Calcd for C23H20NO3 (M+H)+ 358.1438. Found 358.1432.
7.2.2. 3-([1,1′-Biphenyl]-3-yl)-1-chloro-6,7-dimethoxyisoquino line
3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1(2H)-one (130 mg, 0.36 mmol) was refluxed at 110 °C in POCl3 (3 mL) for 3 h. POCl3 was then removed under vacuum. Chromatography achieved using ISCO max gradient 70% EtOAc/hexane yielding product as a beige solid (117 mg, 85% yield); mp 143–144 °C; 1H NMR (400 MHz) (CDCl3) δ 8.32 (t, J = 4.0 Hz, 1H), 8.10–8.07 (m, 1H), 7.96 (s, 1H), 7.73–7.71 (m, 2H), 7.66–7.64 (m, 1H), 7.57 (t, J = 12.0 Hz, 2H), 7.52–7.48 (m, 2H), 7.42–7.38 (m, 1H), 7.18 (s, 1H), 4.11 (s, 3H), 4.08 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 153.7, 151.1, 149.2, 149.1, 141.8, 141.2, 138.9, 135.3, 129.2, 128.8, 127.5, 127.4, 127.3, 125.6, 125.6, 121.8, 115.4, 105.4, 104.6, 56.2; HRMS (ESI) Calcd for C23H19ClNO2 (M+H)+ 376.1099. Found 376.1087.
7.2.3. 3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinoline (1a)
3-([1,1′-Biphenyl]-3-yl)-1-chloro-6,7-dimethoxyisoquinoline (60 mg, 0.16 mmol) was dissolved in EtOH (5 mL) and Pd/C (10%, 20 mg) was added. Flask was then degassed to remove air and reaction was then stirred under an H2(g) atmosphere overnight at RT. Catalyst was then filtered out and solvent evaporated. Chromatography achieved using ISCO max gradient 70% EtOAc/hexane yielding product as a beige oil (18 mg, 33% yield); 1H NMR (400 MHz) (CDCl3) δ 9.09 (s, 1H), 8.25 (t, J = 8.0 Hz, 1H), 8.00–7.98 (m, 1H), 7.93 (s, 1H), 7.66–7.64 (m, 2H), 7.57–7.55 (m, 1H), 7.49 (t, J = 12.0 Hz, 1H), 7.42–7.38 (m, 2H), 7.32–7.28 (m, 1H), 7.17 (s, 1H), 7.07 (s, 1H), 3.98 (s, 6H); 13C NMR (100 MHz) (CDCl3) δ 53.3, 150.4, 150.2, 149.9, 141.8, 141.3, 140.4, 133.4, 129.2, 128.8, 128.7, 127.3, 127.0, 125.8, 125.8, 123.9, 115.7, 105.3, 105.0, 56.1, 56.1; HRMS (ESI) Calcd for C23H20NO2 (M+H)+ 342.1489. Found 342.1485.
7.2.4. 3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxy-2-methylisoquinolin-2-ium iodide (1b)
3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinoline (9 mg, 0.026 mmol) and MeI (1 mL) were heated in a sealed tube overnight at 100 °C. Solvent was then evaporated and residue was taken back up in DCM. Ether was then used to crash out solid which was filtered and dried to yield product as a tan solid (13 mg, quantitative); mp 205–208 °C; 1H NMR (400 MHz) (CDCl3) δ 10.90 (s, 1H), 8.21 (s, 1H), 7.92 (s, 1H), 7.86–7.84 (m, 1H), 7.69–7.62 (m, 4H), 7.51–7.41 (m, 4H), 7.25 (s, 1H), 4.40 (s, 3H), 4.16 (s, 3H), 4.13 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 153.6, 150.5, 150.2, 141.4, 140.2, 139.2, 129.3, 129.2, 129.0, 128.4, 128.0, 127.8, 127.2, 126.9, 107.8, 56.7, 53.1, 46.0; HRMS (ESI) Calcd for C24H22INO2 (M–I)+ 356.1651. Found 356.1647.
7.3. General procedure for the synthesis of compound (2)
7.3.1. 6,7-Dimethoxy-1-methylisoquinolin-3-yl trifluorometha nesulfonate
6,7-Dimethoxy-1-methylisoquinolin-3-ol (540 mg, 2.47 mmol) and Et3N (0.7 mL, 4.94 mmol) in DCM were cooled to −78 °C. Tf2O (0.5 mL, 2.96 mmol) was slowly added to the mixture and was stirred for 30 min at −78 °C. Reaction was then quickly diluted with additional DCM and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a white solid (737 mg, 85% yield); mp 142–142 °C; 1H NMR (400 MHz) (CDCl3) δ 7.28 (m, 2H), 7.10 (s, 1H), 4.06 (m, 6H), 2.88 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 156.5, 153.8, 150.7, 150.6, 135.5, 123.3, 107.6, 105.4, 103.7, 56.2, 56.1, 22.0; HRMS (ESI) Calcd for C13H13F3NO5S (M+H)+ 352.0461. Found 352.0459.
7.3.2. 3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxy-1-methylisoquino line (2a)
6,7-Dimethoxy-1-methylisoquinolin-3-yl trifluoromethanesulfonate (575 mg, 1.64 mmol), 3-biphenylboronic acid (390 mg, 1.968 mmol), Pd(OAc)2 (37 mg, 0.16 mmol), XPhos (156 mg, 0.33), and K2CO3 (792 mg, 5.74 mmol) were combined in a flask with ACN (9 mL) and H2O (3 mL) and degassed. Reaction mixture was then refluxed at 100 °C for 5 h. Solution was cooled to RT then diluted with EtOAc and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as white solid (473 mg, 81% yield); mp 106–108 °C; 1H NMR (400 MHz) (CDCl3) δ 8.36 (m, 1H), 8.11–8.09 (m, 1H), 7.89 (s, 1H), 7.75–7.73 (m, 2H), 7.64–7.62 (m, 1H), 7.58 (t, J = 12.0 Hz, 2H), 7.50 (t, J = 16.0 Hz, 1H), 7.40 (t, J = 12.0 Hz, 1H), 7.33 (s, 1H), 7.16 (s, 1H), 4.09 (s, 3H), 4.08 (s, 3H), 3.01 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 156.0, 152.7, 149.9, 149.2, 141.6, 141.5, 140.7, 133.5, 129.1, 128.7, 127.3, 127.3, 126.8, 125.8, 122.4, 114.5, 105.7, 104.0, 56.0, 30.9, 22.8; HRMS (ESI) Calcd for C24H22NO2 (M+H)+ 356.1645. Found 356.1638.
7.3.3. 3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxy-1,2-dimethyliso quinolin-2-ium iodide (2b)
3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxy-1-methylisoquinoline (35 mg, 0.099 mmol) and MeI (1.5 mL) were heated in a sealed tube for 3 h. Solvent was then evaporated. Chromatography achieved using silica column max gradient 10% MeOH/DCM yielding product as a pale yellow solid (5 mg, 10% yield); mp 224–225 °C; 1H NMR (400 MHz) (CDCl3) δ 7.88 (s, 1H), 7.60 (s, 1H), 7.44–7.30 (m, 4H), 7.28 (m, 1H), 7.24 (s, 1H), 7.15–7.02 (m, 4H), 4.50 (s, 3H), 4.05 (s, 3H), 4.02 (s, 3H), 3.35 (s, 3H); 13C NMR (CDCl3) δ 157.4, 155.5, 152.7, 145.4, 141.4, 138.9, 135.4, 133.9, 129.5, 129.0, 128.7, 128.3, 128.1, 128.1, 127.2, 123.5, 123.4, 106.4, 104.9, 58.0, 56.6, 44.5, 20.0; HRMS (ESI) Calcd for C25H24INO2 (M–I)+ 370.1807. Found 370.1793.
7.4. General procedure for synthesis of compound (3)
7.4.1. 6,7,8-Trimethoxy-1-methylisoquinolin-3-yl trifluoromethanesulfonate
6,7,8-Trimethoxy-1-methylisoquinolin-3-ol (200 mg, 0.80 mmol) and Et3N (0.22 mL, 1.60 mmol) in anhydrous DCM (15 mL) were cooled to −70 °C and Tf2O (0.15 mL, 0.88 mmol) was slowly added. The reaction mixture was stirred at −70 to −40 °C for 30 min then diluted with DCM and washed with saturated NaHCO3 followed by brine. Organic layer was collected, dried over MgSO4, and concentrated. Chromatography achieved using ISCO max gradient 100% DCM yielding product as a white solid (210 mg, 69% yield); mp 46–47 °C; 1H NMR (400 MHz) (CDCl3) δ 7.13 (s, 1H), 6.83 (s, 1H), 3.96 (s, 3H), 3.94 (s, 3H), 3.88 (s, 3H), 2.96 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 158.4, 157.3, 151.1, 151.0, 143.1, 137.7, 120.4, 119.3–117.2 (m), 107.2, 102.0, 61.3, 61.1, 56.1, 26.5; HRMS (ESI) Calcd for C14H15F3NO6S (M+H)+ 382.0567. Found 382.0560.
7.4.2. 3-([1,1′-Biphenyl]-3-yl)-6,7,8-trimethoxy-1-methyliso quinoline (3a)
6,7,8-Trimethoxy-1-methylisoquinolin-3-yl trifluoromethanesulfonate (100 mg, 0.26 mmol), [1,1′-biphenyl]-3-ylboronic acid (78 mg, 0.39 mmol), Pd(OAc)2 (4 mg, 0.02 mmol), XPhos (12 mg, 0.03 mmol), and K2CO3 (90 mg, 0.65 mmol) were combined in a flask with ACN (6 mL) and H2O (3 mL) and degassed. Reaction mixture was then refluxed at 100 °C for 5 h. The reaction mixture was cooled to room temperature then diluted with EtOAc and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 50% EtOAc/hexane yielding product as white solid (65 mg, 64% yield); mp 45–46 °C; 1H NMR (400 MHz) (CDCl3) δ 8.36–8.34 (m, 1H), 8.12–8.10 (m, 1H), 7.83 (s, 1H), 7.75–7.72 (m, 2H), 7.66–7.63 (m, 1H), 7.60–7.55 (m, 1H), 7.52–7.48 (m, 2H), 7.42–7.37 (m, 1H), 6.99 (s, 1H), 4.07 (s, 3H), 4.06 (s, 3H), 4.00 (s, 3H), 3.20 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 156.9, 156.4, 151.1, 149.1, 142.6, 141.7, 141.4, 139.7, 136.1, 129.1, 128.7, 127.3, 127.3, 127.2, 125.9, 125.9, 118.1, 114.7, 102.3, 61.3, 61.1, 56.0, 26.9; HRMS (ESI) Calcd for C25H24NO3 (M+H)+ 386.1751. Found 386.1746.
7.4.3. 3-([1,1′-Biphenyl]-3-yl)-6,7,8-trimethoxy-1,2-dimethyliso quinolin-2-ium iodide (3b)
3-([1,1′-Biphenyl]-3-yl)-6,7,8-trimethoxy-1-methylisoquino-line (30 mg, 0.08 mmol) in MeI (1.5 mL) was stirred in a sealed vial at 70 °C overnight. After cooling to RT, acetone (5 mL) was added and solids were collected by filtration yielding product as an off-white solid (10 mg, 25% yield); mp 179–180 °C; 1H NMR (400 MHz) (CDCl3) δ 7.91 (s, 2H), 7.82–7.78 (m, 2H), 7.72–7.69 (m, 2H), 7.65–7.60 (m, 1H), 7.52–7.48 (m, 2H), 7.43–7.39 (m, 1H), 7.18 (s, 1H), 4.28 (s, 3H), 4.15 (s, 3H), 4.10 (s, 3H), 4.05 (s, 3H), 3.62 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 161.1, 158.7, 146.1, 145.6, 142.1, 139.3, 136.2, 133.9, 129.6, 129.0, 128.8, 128.8, 128.1, 128.1, 127.3, 123.9, 119.4, 103.4, 62.4, 61.5, 57.5, 44.5, 21.8; HRMS (ESI) Calcd for C26H26INO3 (M–I)+ 400.1913. Found 400.1899.
7.5. General procedure for synthesis of compound (4)
7.5.1. 3-(3-(t-Butyl)phenyl)-6,7-dimethoxy-1-methylisoquino line (4a)
6,7-Dimethoxy-1-methylisoquinolin-3-yl trifluoromethanesulfonate (7.3.1) (300 mg, 0.85 mmol), 3-t-butylphenylboronic acid (183 mg, 1.02 mmol), Pd(OAc)2 (19 mg, 0.09 mmol), XPhos (81 mg, 0.17 mmol), and K2CO3 (354 mg, 2.55 mmol) were combined in a flask with dioxane (9 mL) and H2O (3 mL) and degassed. Reaction mixture was then refluxed at 100 °C for 2 h. Solution was cooled to RT then diluted with EtOAc and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 40% EtOAc/hexane yielding product as a clear oil (245 mg, 83% yield); 1H NMR (400 MHz) (CDCl3) δ 8.14 (s, 1H), 7.92–7.89 (m, 1H), 7.81 (s, 1H), 7.45–7.44 (m, 2H), 7.31 (s, 1H), 7.15 (s, 1H), 4.08 (s, 3H), 4.07 (s, 3H), 3.00 (s, 3H), 1.44 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 155.9, 152.6, 151.4, 149.9, 149.7, 134.0, 133.5, 128.4, 125.1, 124.2, 123.9, 122.2, 114.5, 105.7, 103.9, 56.0, 56.0, 34.9, 31.5, 22.8; HRMS (ESI) Calcd for C22H26NO2 (M+H)+ 336.1958. Found 336.1954.
7.5.2. 3-(3-(t-Butyl)phenyl)-6,7-dimethoxy-1,2-dimethylisoqui nolin-2-ium iodide (4b)
3-(3-(t-Butyl)phenyl)-6,7-dimethoxy-1-methylisoquinoline (23.5 mg, 0.07 mmol) was heated in MeI (0.5 mL) in a sealed tube overnight at 100 °C. Solvent was then evaporated and residue was taken back up in DCM. Ether was then used to crash out solid which was filtered and dried to yield product as an off-white solid (23 mg, 70% yield); mp 209–211 °C; 1H NMR (400 MHz) (CD3OD) δ 8.04 (s, 1H), 7.81 (s, 1H), 7.74–7.71 (m, 1H), 7.69 (t, J = 4.0 Hz, 1H), 7.61–7.57 (m, 2H), 7.45–7.43 (m, 1H), 4.15 (s, 3H), 4.12 (s, 6H), 3.28 (s, 3H), 1.43 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 159.4, 157.6, 154.6, 153.9, 147.6, 136.9, 135.3, 130.2, 128.6, 127.8, 124.7, 124.6, 107.1, 106.6, 57.5, 57.2, 43.9, 35.9, 31.7, 18.2; HRMS (ESI) Calcd for C23H28INO2 (M–I)+ 350.2115. Found 350.2107.
7.6. General procedure for synthesis of compound (5)
7.6.1. 3-(6,7-Dimethoxy-1-methylisoquinolin-3-yl)phenol
6,7-Dimethoxy-1-methylisoquinolin-3-yl trifluoromethanesulfonate (200 mg, 0.57 mmol), 3-hydroxyphenylboronic acid (157 mg, 1.14 mmol), Pd(OAc)2 (13 mg, 0.057 mmol), XPhos (54 mg, 0.114 mmol), and Cs2CO3 (650 mg, 1.995 mmol) were combined in a flask with ACN (9 mL) and H2O (3 mL) and degassed. Reaction mixture was then refluxed at 100 °C for 5 h. Solution was cooled to RT then diluted with EtOAc and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 50% EtOAc/hexane yielding product as white solid (78 mg, 46% yield); mp 111–113 °C; 1H NMR (400 MHz) (CDCl3) δ 7.66–7.63 (m, 2H), 7.38 (d, J = 4.0 Hz, 1H), 7.20 (t, J = 8.0 Hz, 1H), 7.17 (s, 1H), 7.00 (s, 1H), 6.73–6.71 (m, 1H), 3.95 (s, 3H), 3.94 (s, 3H), 2.87 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 156.7, 156.1, 152.9, 149.9, 149.2, 141.5, 133.6, 129.9, 122.4, 118.7, 115.5, 115.3, 114.6, 105.7, 103.9, 56.1, 56.0, 22.2; HRMS (ESI) Calcd for C18H18NO3 (M+H)+ 296.1281. Found 296.1274.
7.6.2. 3-(6,7-Dimethoxy-1-methylisoquinolin-3-yl)phenyl trifluoromethanesulfonate
3-(6,7-Dimethoxy-1-methylisoquinolin-3-yl)phenol (175 mg, 0.59 mmol) and Et3N (0.16 mL, 1.18 mmol) in DCM were cooled to −78 °C. Tf2O (0.12 mL, 0.708 mmol) was slowly added to the mixture and was stirred for 30 min at −78 °C. Reaction was then quickly diluted with additional DCM and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a white solid (230 mg, 91% yield); mp 81–82 °C; 1H NMR (400 MHz) (CDCl3) δ 8.14 (d, J = 8.0 Hz, 1H), 8.09 (s, 1H), 7.82 (s, 1H), 7.56 (t, J = 16.0 Hz, 1H), 7.31 (s, 1H), 7.29 (dd, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.15 (s, 1H), 4.08 (s, 3H), 4.07 (s, 3H), 2.98 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 156.3, 152.9, 150.3, 150.2, 146.6, 143.0, 133.2, 130.3, 126.3, 122.8, 120.3, 119.6–117.2 (m), 114.8, 105.8, 103.9, 56.1, 56.0, 22.7; HRMS (ESI) Calcd for C19H17F3NO5S (M+H)+ 428.0774. Found 428.0762.
7.6.3. 3-([1,1′:4′,1″-Terphenyl]-3-yl)-6,7-dimethoxy-1-methyliso quinoline (5a)
3-(6,7-Dimethoxy-1-methylisoquinolin-3-yl)phenyl trifluoromethanesulfonate (80 mg, 0.19 mmol), [1,1′-biphenyl]-4-ylboronic acid (56 mg, 0.28 mmol), Pd(OAc)2 (2 mg, 0.01 mmol), XPhos (9 mg, 0.02 mmol), and K2CO3 (65 mg, 0.47 mmol) were combined in a flask with ACN (6 mL) and H2O (3 mL) and degassed. Reaction mixture was then refluxed at 100 °C for 5 h. Solution was cooled to room temperature then diluted with EtOAc and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as white solid (64 mg, 79% yield); mp 150–153 °C; 1H NMR (400 MHz) (CDCl3) δ 8.40 (m, 1H), 8.12–8.10 (m, 1H), 7.90 (s, 1H), 7.84–7.82 (m, 2H), 7.75–7.64 (m, 5H), 7.59 (t, J = 7.7 Hz, 1H), 7.52–7.47 (m, 2H), 7.41–7.38 (m, 1H), 7.34 (s, 1H), 7.18 (s, 1H), 4.09 (s, 3H), 4.08 (s, 3H), 3.01 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 156.0, 152.7, 149.9, 149.1, 141.1, 140.8, 140.8, 140.4, 140.1, 133.5, 129.2, 128.8, 127.7, 127.5, 127.3, 127.1, 126.7, 125.9, 125.7, 122.4, 114.6, 105.7, 104.0, 56.0, 56.0, 22.8; HRMS (ESI) Calcd for C30H26NO2 (M+H)+ 432.1958. Found 432.1950.
7.6.4. 3-([1,1′:4′,1″-Terphenyl]-3-yl)-6,7-dimethoxy-1,2-dimethy lisoquinolin-2-ium iodide (5b)
3-([1,1′:4′,1″-Terphenyl]-3-yl)-6,7-dimethoxy-1-methylisoquinoline (60 mg, 0.14 mmol) in MeI (1.5 mL) was stirred in a sealed vial at 70 °C overnight. After cooling to RT, acetone (5 mL) was added and solids were collected by filtration to yield product as an off-white solid (40 mg, 50% yield); mp 222–224 °C; 1H NMR (400 MHz) (CDCl3) δ 8.21(s, 1H), 7.97 (m, 1H), 7.86–7.82 (m, 3H), 7.76–7.68 (m, 2H), 7.70–7.60 (m, 4H), 7.56 (t, J = 7.7 Hz, 1H), 7.51–7.47 (m, 2H), 7.41–7.30 (m, 2H), 4.29 (s, 3H), 4.11 (s, 3H), 4.01 (s, 3H), 3.38 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 157.4, 155.4, 152.6, 145.3, 140.9, 140.3, 137.7, 135.4, 134.0, 129.5, 128.9, 128.7, 128.0, 127.6, 127.6, 127.0, 123.5, 123.3, 106.6, 104.8, 58.1, 56.6, 44.6, 19.9; HRMS (ESI) Calcd for C31H29INO2 (M–I)+ 446.2120. Found 446.2104.
7.7. General procedure for synthesis of compound (6)
7.7.1. 6,7,8-Trimethoxy-1-methylisoquinolin-3-yl trifluoromethanesulfonate
6,7,8-Trimethoxy-1-methylisoquinolin-3-ol (200 mg, 0.80 mmol) and Et3N (0.22 mL, 1.60 mmol) in anhydrous DCM (15 mL) were cooled to −70 °C and Tf2O (0.15 mL, 0.88 mmol) was slowly added. The reaction mixture was stirred at −70 to −40 °C for 30 min then diluted with DCM and washed with saturated NaHCO3 followed by brine. Organic layer was collected, dried over MgSO4, and concentrated. Chromatography achieved using ISCO max gradient 100% DCM yielding product as a white solid (210 mg, 69% yield); mp 46–47 °C; 1H NMR (400 MHz) (CDCl3) δ 7.13 (s, 1H), 6.83 (s, 1H), 3.96 (s, 3H), 3.94 (s, 3H), 3.88 (s, 3H), 2.96 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 158.4, 157.3, 151.1, 150.9, 143.1, 137.7, 120.4, 119.3–117.2 (m), 107.2, 102.0, 61.3, 61.1, 56.1, 26.5; HRMS (ESI) Calcd for C14H15F3NO6S (M+H)+ 382.0567. Found 382.0560.
7.7.2. 3-(3-(Benzyloxy)-4,5-dimethoxyphenyl)-6,7,8-trimethoxy-1-methylisoquinoline
A flask containing 6,7,8-trimethoxy-1-methylisoquinolin-3-yl trifluoromethanesulfonate (1.7 g, 4.46 mmol), (3-(benzyloxy)-4,5-dimethoxyphenyl)boronic acid (1.54 g, 5.35 mmol), K2CO3 (1.54 mg, 11.2 mmol), and XPhos (212 mg, 0.45 mmol) in ACN (20 mL) and H2O (10 mL) was degassed and then Pd(OAc)2 (50 mg, 0.22 mmol) was added. The resulting solution was carefully degassed again. Reaction was then heated at 90 °C for 4 h. After cooling to RT, the reaction mixture was diluted with EtOAc and washed with saturated NaHCO3 followed by brine. Organic layer was collected, dried over sodium sulfate, and concentrated. Chromatography achieved using ISCO max gradient 20% EtOAc/hexane yielding product as a light yellow oil (2.03 g, 96% yield); 1H NMR (400 MHz) (CDCl3) δ 7.66 (s, 1H), 7.73–7.55 (m, 2H), 7.44–7.39 (m, 4H), 7.37–7.33 (m, 1H), 6.95 (s, 1H), 5.28 (s, 2H), 4.05 (s, 3H), 4.04 (s, 3H), 4.02 (s, 3H), 3.99 (s, 3H), 3.94 (s, 3H), 3.15 (s, 3H); 13C NMR (CDCl3) δ 156.6, 156.1, 153.7, 152.7, 150.9, 149.1, 142.4, 139.5, 137.4, 135.9, 135.3, 128.5, 127.9, 127.5, 117.9, 113.8, 106.6, 104.6, 102.2, 71.4, 61.2, 61.0, 60.9, 56.3, 55.9, 27.3; HRMS (ESI) Calcd for C28H30NO6 (M+H)+ 476.2073. Found 476.2078.
7.7.3. 2,3-Dimethoxy-5-(6,7,8-trimethoxy-1-methylisoquinolin-3-yl)phenol
3-(3-(Benzyloxy)-4,5-dimethoxyphenyl)-6,7,8-trimethoxy-1-methylisoquinoline (2.3 g, 4.84 mmol) was suspended in MeOH (250 mL) followed by addition of Pd/C (10% wt.) (200 mg). The reaction flask was sealed with septum and purged with N2 (3x) followed by H2 (3x). Reaction mixture was then stirred at RT under H2 balloon for 3 h. Reaction was monitored by TLC and stopped once the starting material was consumed. Reaction mixture was then passed through a pad of Celite and washed with MeOH. The filtrate was concentrated yielding the crude product as a grey foam which was taken forward without further purification (1.67 g, 90% yield); 1H NMR (400 MHz) (CDCl3) δ 7.69 (s, 1H), 7.39 (d, J = 1.9 Hz, 1H), 7.31 (d, J = 2.0 Hz, 1H), 6.94 (s, 1H), 5.83 (s, 1H), 4.05 (s, 3H), 4.04 (s, 3H), 4.03 (s, 3H), 3.99 (s, 3H), 3.97 (s, 3H), 3.15 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 156.6, 156.1, 152.6, 150.9, 149.4, 149.0, 142.4, 136.0, 135.9, 135.8, 118.0, 113.9, 106.5, 103.2, 102.2, 61.2, 61.0, 56.0, 55.9, 27.2; HRMS (ESI) Calcd for C21H24NO6 (M+H)+ 386.1604. Found 386.1606.
7.7.4. 2,3-Dimethoxy-5-(6,7,8-trimethoxy-1-methylisoquinolin-3-yl)phenyl trifluoromethanesulfonate
2,3-Dimethoxy-5-(6,7,8-trimethoxy-1-methylisoquinolin-3-yl)phenol (1.66 g, 4.31 mmol) in DCM (100 mL) and triethylamine (1.20 mL, 8.62 mmol) was cooled to –70 °C and triflic anhydride (0.80 mL, 4.74 mmol) was added slowly. The resulting reaction mixture was stirred at –70 -to –30 °C for 30 min. Reaction was then diluted with DCM and washed with saturated NaHCO3 followed by brine. Organic layer was collected, dried over sodium sulfate, and concentrated. Chromatography achieved using ISCO max gradient 20% EtOAc/hexane yielding product as a clear golden oil (2.21 g, 99% yield); 1H NMR (400 MHz) (CDCl3) δ 7.72 (d, J = 1.9 Hz, 1H), 7.59 (s, 1H), 7.45 (d, J = 1.8 Hz, 1H), 6.89 (s, 1H), 3.96 (s, 6H), 3.95 (s, 3H), 3.92 (s, 3H), 3.90 (s, 3H), 3.06 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 157.1, 156.3, 153.9, 151.0, 147.2, 142.9, 142.8, 141.5, 135.8, 135.7, 118.3, 114.0, 112.3, 110.8, 102.3, 61.4, 61.2, 61.1, 56.4, 56.0, 27.2; HRMS (ESI) Calcd for C22H23F3NO8S (M+H)+ 518.1096. Found 518.1091.
7.7.5. 3-(5,6-Dimethoxy-[1,1′:4′,1″-terphenyl]-3-yl)-6,7,8-trimethoxy-1-methylisoquinoline (6a)
2,3-Dimethoxy-5-(6,7,8-trimethoxy-1-methylisoquinolin-3-yl)phenyl trifluoromethanesulfonate (100 mg, 0.19 mmol), [1,1′-biphenyl]-4-ylboronic acid (58 mg, 0.29 mmol), K2CO3 (66 mg, 0.48 mmol), and XPhos (10 mg, 0.02 mmol) in ACN (4 mL) and H2O (2 mL) were degassed then Pd(OAc)2 (3.0 mg, 0.065 mmol) was added and solution was carefully degassed again. The reaction mixture was warmed to 100 °C and stirred for 1 h. After cooling to RT, the reaction mixture was diluted with EtOAc and washed with saturated NaHCO3 followed by brine. Organic layer was collected, dried over sodium sulfate, and concentrated. Chromatography achieved with ISCO max gradient 30% EtOAc/hexane yielding product as a clear oil (56 mg, 57% yield); 1H NMR (400 MHz) (CDCl3) δ 7.82 (d, J = 2.0 Hz, 1H), 7.78–7.69 (m, 8H), 7.52–7.47 (m, 2H), 7.41–7.37 (m, 1H), 6.96 (s, 1H), 4.09 (s, 3H), 4.06 (s, 3H), 4.03 (s, 3H), 3.99 (s, 3H), 3.71 (s, 3H), 3.17 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 156.7, 156.1, 153.4, 151.0, 149.1, 147.1, 142.4, 141.0, 139.9, 137.6, 136.0, 135.8, 135.4, 129.8, 128.8, 127.3, 127.1, 126.8, 121.1, 118.0, 113.9, 110.5, 102.2, 61.2, 61.1, 60.8, 56.1, 55.9, 27.3; HRMS (ESI) Calcd for C33H32NO5 (M+H)+ 522.2280. Found 522.2288.
7.7.6. 3-(5,6-Dimethoxy-[1,1′:4′,1″-terphenyl]-3-yl)-6,7,8-trimethoxy-1,2-dimethylisoquinolin-2-ium iodide (6b)
A solution of 3-(5,6-dimethoxy-[1,1′:4′,1″-terphenyl]-3-yl)-6,7,8-trimethoxy-1-methylisoquinoline (50 mg, 0.096 mmol) in MeI (1.5 mL) was stirred in a sealed vial at 70 °C overnight. After cooling to RT, acetone (5 mL) was added and solids were collected by filtration to yield product as a white solid (32 mg, 50% yield); mp 222–224 °C; 1H NMR (400 MHz) (CDCl3) δ 7.88 (s, 1H), 7.84 (d, J = 2.0 Hz, 1H), 7.78–7.64 (m, 6H), 7.51–7.46 (m, 2H), 7.41–7.38 (m, 1H), 7.12 (s, 1H), 7.09 (d, J = 2.0 Hz, 1H), 4.35 (s, 3H), 4.14 (s, 3H), 4.11 (s, 3H), 4.09 (s, 3H), 4.05 (s, 3H), 3.78 (s, 3H), 3.60 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 161.7, 157.9, 153.8, 148.4, 145.6, 140.6, 140.5, 136.0, 135.9, 129.6, 128.8, 127.5, 127.1, 127.0, 125.1, 122.9, 119.5, 114.1, 102.5, 63.2, 61.5, 60.9, 59.7, 57.4, 57.2, 44.5; HRMS (ESI) Calcd for C34H34INO5 (M–I)+ 536.2437. Found 536.2418.
7.8. General procedure for synthesis of compound 7
7.8.1. 3-(4′-(t-Butyl)-[1,1′-biphenyl]-3-yl)-6,7-dimethoxy-1-methylisoquinoline (7a)
3-(6,7-Dimethoxy-1-methylisoquinolin-3-yl)phenyl trifluoromethanesulfonate (7.6.2) (130 mg, 0.3 mmol), 4-t-butylphenylboronic acid (108 mg, 0.6 mmol), Pd(OAc)2 (7 mg, 0.03 mmol), XPhos (29 mg, 0.06 mmol), and K2CO3 (147 mg, 1.05 mmol) in ACN (3 mL) and H2O (1.5 mL) were combined in a flask and degassed. Reaction mixture was heated to 95 °C for 2 h. Solution was then cooled to RT, diluted with EtOAc, and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 40% EtOAc/hexane yielding product as a white solid (121 mg, 97% yield); mp 185–187 °C; 1H NMR (400 MHz) (CDCl3) δ 8.35–8.34 (m, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.88 (s, 1H), 7.68 (d, J = 8.0 Hz, 2H), 7.63 (d, J = 8.0 Hz, 1H), 7.56 (t, J = 16.0 Hz, 1H), 7.53 (d, J = 8.0 Hz, 2H), 7.33 (s, 1H), 7.16 (s, 1H), 4.09 (s, 3H), 4.08 (s, 3H), 3.00 (s, 3H), 1.41 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 156.0, 152.7, 150.3, 149.8, 149.3, 141.5, 140.6, 138.6, 133.5, 129.0, 127.0, 126.7, 125.7, 125.7, 125.5, 122.3, 114.5, 105.7, 104.0, 56.0, 34.6, 31.4, 22.7; HRMS (ESI) Calcd for C28H30NO2 (M+H)+ 412.2271. Found 412.2259.
7.8.2. 3-(4′-(t-Butyl)-[1,1′-biphenyl]-3-yl)-6,7-dimethoxy-1,2-dimethylisoquinolin-2-ium iodide (7b)
3-(4′-(t-Butyl)-[1,1′-biphenyl]-3-yl)-6,7-dimethoxy-1-methylisoquinoline (15 mg, 0.036 mmol) was heated in MeI (0.5 mL) in a sealed tube overnight at 100 °C. Solvent was then evaporated and residue was taken back up in DCM. Ether was then used to crash out solid which was filtered and dried to yield product as a tan solid (15 mg, 75% yield); mp 202–204 °C; 1H NMR (400 MHz) (CDCl3) δ 8.21 (s, 1H), 7.86 (s, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.70–7.68 (m, 3H), 7.57 (d, J = 8.0 Hz, 1H), 7.53–7.48 (m, 3H), 7.38 (s, 1H), 4.24 (s, 3H), 4.09 (s, 3H), 3.99 (s, 3H), 3.36 (s, 3H), 1.38 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 157.5, 152.7, 151.4, 145.6, 136.0, 135.4, 133.9, 129.5, 128.4, 128.2, 127.9, 126.8, 126.0, 123.5, 106.3, 105.1, 57.8, 56.6, 44.5, 34.6, 31.3, 20.1; HRMS (ESI) Calcd for C28H32INO2 (M–I)+ 426.2433. Found 426.2431.
7.9. General procedure for synthesis of 3-([1,1′-biphenyl]-3-yl)-6,7-dimethoxy-N-methylisoquinolin-1-amine (8a)
3-([1,1′-Biphenyl]-3-yl)-1-chloro-6,7-dimethoxyisoquinoline (7.2.2) (20 mg, 0.053 mmol) and chloro(2-di-t-butylphosphino-2′,4′,6′-tri-i-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl]PdII) (t-buXPhos precatalyst) (4 mg, 0.0053 mmol) were combined in a flask and air was evacuated and replaced with N2. Methylamine (2 M in THF) (2 mL) followed by LHMDS (1 M in THF) (0.02 mL, 0.08 mmol) was then added, and reaction was allowed to stir overnight at RT. Reaction mixture was then diluted with EtOAc and washed with NH4Cl. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 50% EtOAc/hexane yielding product as a clear oil (20 mg, quantitative); 1H NMR (400 MHz) (CDCl3) δ 8.31 (t, J = 4.0 Hz, 1H), 8.06 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.64–7.62 (m, 2H), 7.51–7.49 (m, 1H), 7.46–7.43 (m, 1H), 7.41–7.37 (m, 2H), 7.34 (s, 1H), 7.31–7.27 (m, 1H), 6.99 (s, 1H), 6.91 (s, 1H), 4.92 (bs, 1H), 3.93 (s, 6H), 3.21 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 154.5, 152.2, 149.1, 141.7, 141.4, 141.0, 134.1, 128.9, 128.7, 127.2, 126.7, 125.5, 125.5, 112.1, 106.6, 106.6, 101.2, 56.1, 55.9, 29.0; HRMS (ESI) Calcd for C24H23N2O2 (M+H)+ 371.1754. Found 371.1746.
7.10. General procedure for synthesis of compound (9)
7.10.1. 3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxy-N,N-dimethylisoquinolin-1-amine (9a)
3-([1,1′-Biphenyl]-3-yl)-1-chloro-6,7-dimethoxyisoquinoline (7.2.2) (20 mg, 0.053 mmol) and chloro(2-di-t-butylphosphino-2′,4′,6′-tri-i-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl]PdII) (t-buXPhos precatalyst) (4 mg, 0.0053 mmol) were combined in a flask and air was evacuated and replaced with N2. Dimethylamine (2 M in THF) (2 mL) followed by LHMDS (1 M in THF) (0.02 mL, 0.08 mmol) was then added, and reaction was allowed to stir overnight at RT. Reaction mixture was then diluted with EtOAc and washed with NH4Cl. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 40% EtOAc/hexane yielding product as a tan oil (20 mg, quantitative); 1H NMR (400 MHz) (CDCl3) δ 8.30 (t, J = 4.0 Hz, 1H), 8.05 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.64–7.62 (m, 2H), 7.58 (s, 1H), 7.52–7.49 (m, 1H), 7.47–7.44 (m, 1H), 7.42–7.38 (m, 2H), 7.36 (s, 1H), 7.32–7.28 (m, 1H), 7.03 (s, 1H), 3.96 (s, 3H), 3.95 (s, 3H), 3.06 (s, 6H); 13C NMR (100 MHz) (CDCl3) δ 152.2, 148.9, 147.2, 145.9, 141.7, 141.5, 140.7, 135.6, 128.9, 128.7, 127.3, 127.2, 126.8, 125.5, 125.43, 115.9, 110.4, 106.1, 105.0, 56.0, 55.9, 43.0; HRMS (ESI) Calcd for C25H25N2O2 (M+H)+ 385.1911. Found 385.1903.
7.10.2. 3-([1,1′-Biphenyl]-3-yl)-1-(dimethylamino)-6,7-dimethoxy-2-methylisoquinolin-2-ium iodide (9b)
3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxy-N,N-dimethylisoquinolin-1-amine (28 mg, 0.073 mmol) was heated in MeI (0.5 mL) in a sealed tube overnight at 100 °C. Solvent was then evaporated and residue was taken back up in DCM. Ether was then used to crash out solid which was filtered and dried to yield product as a tan solid (20 mg, 71% yield); mp 158–161 °C; 1H NMR (400 MHz) (CDCl3) δ 7.87 (t, J = 4.0 Hz, 1H), 7.79–7.77 (m, 1H), 7.75 (s, 1H), 7.73–7.69 (m, 3H), 7.67–7.63 (m, 1H), 7.56 (s, 1H), 7.52–7.48 (m, 2H), 7.44–7.42 (m, 1H), 7.39 (s, 1H), 4.15 (s, 3H), 4.12 (s, 3H), 3.97 (s, 3H), 3.63 (s, 6H); 13C NMR (100 MHz) (CDCl3) δ 158.7, 156.9, 151.9, 144.9, 142.5, 139.6, 136.9, 134.2, 130.0, 129.1, 129.0, 128.2, 127.6, 127.3, 120.1, 119.4, 106.9, 106.6, 57.1, 45.6, 45.2, 34.5; HRMS (ESI) Calcd for C26H27IN2O2 (M–I)+ 399.2073. Found 399.2071.
7.11. General procedure for synthesis of 2-(3-([1,1′-biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)guanidine (10a)
Guanidine HCl (38 mg, 0.4 mmol) was added to a suspension of NaH 60% dispersion in mineral oil (10 mg, 0.4 mmol) in anhydrous DMSO (5 mL). Reaction was heated at 60 °C for 30 min then 3-([1,1′-biphenyl]-3-yl)-1-chloro-6,7-dimethoxyisoquinoline (7.2.2) (50 mg, 0.13 mmol) and chloro(2-di-t-butylphosphino-2′,4′,6′-tri-i-propyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl]PdII) (t-buXPhos precatalyst) (9 mg, 0.013 mmol) were then quickly added, and the reaction was heated at 100 °C overnight. Reaction mixture was cooled to RT, diluted with EtOAc, and washed with H2O. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 5% MeOH/DCM yielding product as a tan solid (20 mg, 38%); mp 255–257 °C; 1H NMR (400 MHz) (DMSO-d6) δ 11.36 (bs, 1H), 8.26–8.19 (m, 3H), 7.96 (d, J = 8.0 Hz, 1H), 7.81–7.74 (m, 3H), 7.64 (t, J = 16.0 Hz, 1H), 7.55–7.50 (m, 3H), 7.43 (t, J = 12.0 Hz, 1H), 4.05 (s, 3H), 3.98 (s, 3H); 13C NMR (100 MHz) (DMSO-d6) δ 156.3, 153.4, 150.6, 145.0, 141.0, 140.0, 139.0, 135.1, 129.7, 129.0, 127.7, 126.9, 125.2, 124.5, 113.4, 113.2, 106.6, 102.9, 56.9, 55.8; HRMS (ESI) Calcd for C24H23N4O2 (M+H)+ 399.1816. Found 399.1823.
7.12. General procedure for synthesis of 3-([1,1′-biphenyl]-3-yl)-6,7-dimethoxyisoquinoline-1-carbonitrile (11a)
3-([1,1′-Biphenyl]-3-yl)-1-chloro-6,7-dimethoxyisoquinoline (7.2.2) (50 mg, 0.13 mmol) and CuCN (24 mg, 0.27 mmol) in DMSO (2 mL) were heated at 140 °C for 3 h. Reaction mixture was then cooled to RT, diluted with EtOAc, and washed with H2O. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a beige solid (13 mg, 27% yield); mp 171–174 °C; 1H NMR (400 MHz) (CDCl3) δ 8.25 (t, J = 4.0 Hz, 1H), 8.10 (s, 1H), 8.00–7.98 (m, 1 H), 7.64–7.62 (m, 2H), 7.60–7.57 (m, 1H), 7.50 (t, J = 16.0 Hz, 1H), 7.43–7.39 (m, 3H), 7.34–7.30 (m, 1H), 7.11 (s, 1H), 4.03 (s, 3H), 4.00 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 154.3, 152.6, 150.7, 142.0, 141.0, 138.5, 134.1, 131.6, 129.3, 128.8, 127.9, 127.5, 127.3, 125.7, 125.6, 125.5, 118.9, 116.6, 105.2, 102.7, 56.5, 56.3; HRMS (ESI) Calcd for C24H19N2O2 (M+H)+ 367.1441. Found 367.1435.
7.13. General procedure for synthesis of compound (12)
7.13.1. 3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinoline-1-carbaldehyde
3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxy-1-methylisoquinoline (2a) (200 mg, 0.56 mmol) and SeO2 (75 mg, 0.68 mmol) in anhydrous dioxane (10.5 mL) were refluxed at 102 °C for 3 h. Solution was then cooled to RT and filtered to remove precipitate. Resulting filtrate was concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a yellow solid (175 mg, 84% yield); mp 153–154 °C; 1H NMR (400 MHz) (CDCl3) δ 10.50 (s, 1H), 8.77 (s, 1H), 8.44 (s, 1H), 8.23–8.18 (m, 2H), 7.75–7.66 (m, 3H), 7.63 (t, J = 16.0 Hz, 1H), 7.52 (t, J = 16.0 Hz, 2H), 7.44–7.42 (m, 1H), 7.22 (s, 1H), 4.14 (s, 3H), 4.09 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 196.8, 153.3, 152.8, 149.6, 147.1, 142.0, 141.2, 139.3, 135.4, 129.3, 128.8, 127.6, 127.5, 127.3, 125.7, 125.6, 122.2, 120.0, 105.0, 103.6, 56.3, 56.1; HRMS (ESI) Calcd for C24H20NO3 (M+H)+ 370.1438. Found 370.1431.
7.13.2. (3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)methanol
3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinoline-1-carbaldehyde (160 mg, 0.43 mmol) in ethanol (7 mL) was treated slowly with NaBH4 (50 mg, 1.302 mmol)) at RT. Reaction was stirred for 1 h then acetone (2 mL) was added and solution was filtered through filter paper. Filtrate was concentrated then re-dissolved in DCM and washed with H2O. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 50% EtOAc/hexane yielding product as a yellow solid (98 mg, 61% yield); mp 164–165 °C; 1H NMR (400 MHz) (CDCl3) δ 8.36–8.35 (m, 1H), 8.13–8.11 (m, 1H), 7.98 (s, 1H), 7.73 (d, J = 8.0 Hz, 2H), 7.68–7.66 (m, 1H), 7.60 (t, J = 16.0 Hz, 1H), 7.51 (t, J = 16.0 Hz, 2H), 7.42 (t, J = 12.0 Hz, 1H), 7.22 (s, 1H), 7.09 (s, 1H), 5.35 (bs, 1H), 5.32 (s, 2H), 4.08 (s, 3H), 4.07 (s, 3H); 13C NMR (100 MHz) (CDCl3) δ 154.7, 141.8, 141.2, 129.2, 128.8, 127.4, 127.3, 127.3, 125.6, 125.5, 119.8, 115.4, 105.9, 101.3, 61.4, 56.2, 56.1; HRMS (ESI) Calcd for C24H22NO3 (M+H)+ 372.1594. Found 372.1587.
7.13.3. 3-([1,1′-Biphenyl]-3-yl)-1-(azidomethyl)-6,7-dimethoxyi soquinoline
(3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)methanol (90 mg, 0.24 mmol) in anhydrous THF (5 mL) was cooled to 0 °C. Diphenylphosphorylazide (0.21 mL, 0.97 mmol) was then added followed by drop wise addition of DBU (0.15 mL, 0.97 mmol). Reaction was kept stirring at 0 °C for 4 h then allowed to warm to RT overnight. Reaction mixture was then poured into 0.5 N HCl and extracted with EtOAc. Organic layer was washed with brine, dried over sodium sulfate, and concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a clear oil (75 mg, 78% yield); 1H NMR (400 MHz) (CDCl3) δ 8.43 (s, 1H), 8.14 (d, J = 8.0 Hz, 1H), 8.02 (s, 1H), 7.75 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 1H), 7.59 (t, J = 16.0 Hz, 1H), 7.51 (t, J = 16.0 Hz, 2H), 7.43–7.39 (m, 1H), 7.32 (s, 1H), 7.20 (s, 1H), 4.92 (s, 2H), 4.08 (s, 6H); 13C NMR (100 MHz) (CDCl3) δ 153.1, 152.2, 150.5, 149.0, 141.7, 141.3, 139.9, 134.4, 129.2, 127.4, 127.3, 127.1, 125.7, 125.7, 125.6, 121.5, 116.1, 105.9, 102.8, 56.2, 56.1, 53.9; IR (thin film NaCl) 2936, 2099, 1620, 1593, 1574, 1507, 1467, 1427, 1408, 1364, 1301, 1247, 1225, 1162, 1078, 1056, 997, 880, 836, 802, 759, 736, 700; HRMS (ESI) Calcd for C24H21N4O2 (M+H)+ 397.1659. Found 397.1650.
7.13.4. (3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)methanamine (12a)
3-([1,1′-Biphenyl]-3-yl)-1-(azidomethyl)-6,7-dimethoxyisoquinoline (70 mg, 0.18 mmol) was combined in a flask with polymer supported (3 mmol/g loading) PPh3 (88.5 mg, 0.26 mmol), THF (3 mL), and H2O (0.3 mL). Reaction was stirred overnight at RT. Resin was then filtered off and filtrate concentrated. Chromatography achieved using silica column max gradient 10% MeOH/DCM/0.1% NH4OH yielding product as a tan oil (60 mg, 92% yield); 1H NMR (400 MHz) (DMSO-d6) δ 8.58 (s, 1H), 8.43 (s, 1H), 8.34 (d, J = 4.0 Hz, 1H), 7.88 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 4.0 Hz, 1H), 7.67 (t, J = 16.0 Hz, 1H), 7.59 (t, J = 12.0 Hz, 2H), 7.54 (s, 1H), 7.54–7.46 (m, 2H), 4.66 (s, 2H), 4.04 (s, 3H), 4.02 (s, 3H); 13C NMR (100 MHz) (DMSO-d6) δ 152.8, 150.1, 146.6, 140.6, 140.2, 139.6, 129.3, 128.9, 127.6, 126.9, 126.6, 125.4, 124.5, 120.3, 115.1, 106.1, 102.9, 55.9, 55.7, 48.6; HRMS (ESI) Calcd for C24H23N2O2 (M+H)+ 371.1754. Found 371.1748.
7.14. General procedure for synthesis of 2-((3-([1,1′-biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)methyl)guanidine trifluo roacetate (13a)
(3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)methanol (7.13.2) (30 mg, 0.081 mmol), PPh3 (32 mg, 0.12 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (42 mg, 0.162 mmol) in anhydrous toluene (3 mL) at 0 °C was added diisopropylazodicarboxylate (0.024 mL, 0.12 mmol) drop wise over 15 min. Reaction was stirred for 3 h at RT then 2 drops H2O were added, and the solution was concentrated. Solid was then dissolved in DCM and passed through silica column and resulting crude product was then re-dissolved in anhydrous DCM (1.5 mL) and cooled to 0 °C. TFA (1.5 mL) was then added. Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Solid was then taken back up in DCM and precipitate was filtered off yielding product as a white solid (40 mg, 93% yield over 2 steps); mp 210–213 °C; 1H NMR (400 MHz) (CD3OD) δ 8.40 (m, 1H), 8.22 (s, 1H), 8.20 (m, 1H), 7.76–7.74 (m, 2H), 7.68 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.61 (t, J = 16.0 Hz, 1H), 7.53–7.49 (m, 2H), 7.46 (s, 1H), 7.42–7.38 (m, 2H), 5.07 (s, 2H), 4.07 (s, 3H), 4.05 (s, 3H); 13C NMR (100 MHz) (CD3OD) δ 159.6, 155.0, 152.4, 151.8, 149.4, 143.1, 142.5, 141.3, 135.9, 130.3, 130.0, 128.5, 128.2, 128.1, 126.8, 122.1, 117.2, 107.4, 103.2, 56.7, 56.6, 45.1; HRMS (ESI) Calcd for C25H25N4O2 (M+H)+ 413.1972. Found 413.1973.
7.15. General procedure for synthesis of compound (14)
7.15.1. 3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl trifluoromethanesulfonate
3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1(2H)-one (7.2.1) (234 mg, 0.66 mmol) and Et3N (0.182 mL, 1.31 mmol) in anhydrous DCM (20 mL) were cooled to −78 °C. Tf2O (0.132 mL, 0.79 mmol) was slowly added to the mixture and was stirred for 30 min at −78 °C. Reaction was then quickly diluted with additional DCM and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 50% EtOAc/hexane yielding product as a white solid (255 mg, 79% yield); mp 127–129 °C; 1H NMR (400 MHz) (CDCl3) δ 8.15 (t, J = 4.0 Hz, 1H), 7.87 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.84 (s, 1H), 7.58–7.52 (m, 2H), 7.51–7.49 (m, 1H), 7.42–7.36 (m, 3H), 7.31–7.26 (m, 1H), 7.12 (s, 1H), 7.02 (s, 1H), 3.91 (s, 6H); 13C NMR (100 MHz) (CDCl3) δ 154.4, 151.6, 151.4, 147.0, 141.7, 140.9, 137.8, 137.4, 129.3, 128.9, 127.7, 127.5, 127.1, 125.3, 125.2, 117.3, 115.9, 114.4, 105.4, 100.6, 56.3, 56.2; HRMS (ESI) Calcd for C24H19F3NO5S (M+H)+ 490.0931. Found 490.0910.
7.15.2. t-Butyl (2-(3-([1,1′-biphenyl]-3-yl)-6,7-dimethoxyisoqui nolin-1-yl)ethyl)carbamate
3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl trifluoromethanesulfonate (250 mg, 0.51 mmol), potassium t-butyl N-[2-(trifluoroboranuidyl)ethyl]carbamate (256 mg, 1.02 mmol), Pd(PPh3)4 (59 mg, 0.051 mmol), and K2CO3 (246 mg, 1.785 mmol) were combined in a flask with dioxane (8 mL) and H2O (2 mL) and degassed. Reaction mixture was then refluxed at 102 °C overnight. Solution was cooled to RT then diluted with EtOAc and washed with saturated NH4Cl. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a white fluffy solid (166 mg, 67% yield); mp 66–69 °C; 1H NMR (400 MHz) (CDCl3) δ 8.37–8.36 (m, 1H), 8.14 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.94 (s, 1H), 7.75–7.72 (m, 2H), 7.65 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.60 (t, J = 12.0 Hz, 1H), 7.53–7.49 (m, 2H), 7.44–7.39 (m, 2H), 7.17 (s, 1H), 5.63 (bs, 1H), 4.10 (s, 3H), 4.08 (s, 3H), 3.90 (q, J = 16.0 Hz, 2H), 3.49 (t, J = 12.0 Hz, 2H), 1.44 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 152.8, 150.2, 141.7, 141.4, 140.5, 129.2, 128.8, 127.4, 127.3, 127.0, 125.6, 125.5, 122.4, 114.6, 105.8, 103.3, 56.2, 56.1, 38.5, 34.7, 28.5; HRMS (ESI) Calcd for C30H33N2O4 (M+H)+ 485.2435. Found 485.2428.
7.15.3. 2-(3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)ethanamine (14a)
To a cooled solution of t-butyl (2-(3-([1,1′-biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)ethyl)carbamate (166 mg, 0.34 mmol) in anhydrous DCM (1.5 mL) was added trifluoroacetic acid (1.5 mL). Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Chromatography achieved using ISCO max gradient 10% MeOH/DCM yielding product as a tan fluffy solid (131 mg, quantitative); mp 187–189 °C; 1H NMR (400 MHz) (CD3OD) δ 8.19 (t, J = 4.0 Hz, 1H), 8.08 (s, 1H), 7.93 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.64–7.59 (m, 3H), 7.50 (t, J = 16.0 Hz, 1H), 7.40–7.35 (m, 4H), 7.31–7.27 (m, 1H), 3.95 (s, 3H), 3.93 (s, 3H), 3.62–3.55 (m, 4H); 13C NMR (100 MHz) (CD3OD) δ 155.8, 154.9, 152.8, 143.3, 142.3, 136.4, 130.5, 130.0, 128.7, 128.5, 128.2, 126.9, 126.7, 123.3, 117.8, 107.4, 104.0, 56.7, 39.0, 31.1; HRMS (ESI) Calcd for C25H25N2O2 (M+H)+ 385.1911. Found 385.1912.
7.16. General procedure for synthesis of compound (15)
7.16.1. 1,3-di-Boc-2-(2-(3-([1,1′-biphenyl]-3-yl)-6,7-dimethoxyi soquinolin-1-yl)ethyl)guanidine
2-(3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)ethanamine (50 mg, 0.13 mmol), 1,3-di-Boc-2-(trifluoromethylsulfonyl)-guanidine (61 mg, 0.156 mmol), and Et3N (0.02 mL, 0.156 mol) in anhydrous DCM (3 mL) were stirred for 1 h at 37 °C. Reaction mixture was then diluted with DCM and washed with NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 40% EtOAc/hexane yielding product as a white fluffy solid (80 mg, 99% yield); mp 69–72 °C; 1H NMR (400 MHz) (CDCl3) δ 9.03 (bs, 1H), 8.78 (bs, 1H), 8.25–8.24 (m, 1H), 8.07 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.82 (s, 1H), 7.64–7.61 (m, 2H), 7.55–7.52 (m, 1H), 7.57 (t, J = 16.0 Hz, 1H), 7.41–7.37 (m, 2H), 7.31–7.26 (m, 2H), 7.06 (s, 1H), 4.04–4.01 (m, 2H), 3.98 (s, 3H), 3.96 (s, 3H), 3.50 (t, J = 12.0 Hz, 2H), 1.42 (s, 9H), 1.23 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 163.5, 156.3, 155.9, 152.8, 152.8, 151.9, 150.1, 149.1, 141.5, 140.4, 133.8, 129.0, 128.8, 127.3, 126.9, 126.1, 125.9, 122.2, 121.0, 115.1, 105.8, 103.2, 86.0, 82.9, 56.2, 56.0, 53.4, 39.2, 34.0, 28.3, 27.9; HRMS (ESI) Calcd for C36H43N4O6 (M+H)+ 627.3177. Found 627.3164.
7.16.2. 2-(2-(3-([1,1′-Biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)ethyl)guanidine (15a)
To a cooled solution of 1,3-di-Boc-2-(2-(3-([1,1′-biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)ethyl)guanidine (80 mg, 0.12 mmol) in anhydrous DCM (2 mL) was added trifluoroacetic acid (2 mL). Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Chromatography achieved using ISCO max gradient 10% MeOH/DCM yielding product as a white solid (36 mg, 67% yield); mp 239–241 °C; 1H NMR (400 MHz) (CD3OD) δ 8.18 (s, 1H), 8.15 (t, J = 4.0 Hz, 1H), 7.88–7.86 (m, 1H), 7.71–7.69 (m, 1H), 7.66–7.64 (m, 2H), 7.57 (t, J = 16.0 Hz, 1H), 7.46 (s, 2H), 7.42–7.38 (m, 2H), 7.32–7.28 (m, 1H), 3.99 (s, 3H), 3.98 (s, 3H), 3.79 (t, J = 12.0 Hz, 2H), 3.64 (t, J = 12.0 Hz, 2H); 13C NMR (100 MHz) (CD3OD) δ 158.8, 155.4, 153.5, 143.5, 141.9, 137.6, 130.7, 130.1, 129.2, 127.5, 127.2, 123.6, 119.3, 107.5, 104.7, 57.0, 56.8, 41.3, 33.0; HRMS (ESI) Calcd for C26H27N4O2 (M+H)+ 427.2129. Found 427.2132.
7.17. General procedure for synthesis of compound (16)
7.17.1. 1-(Bromomethyl)-3-(3-(t-butyl)phenyl)-6,7-dimethoxyi soquinoline
3-(3-(t-Butyl)phenyl)-6,7-dimethoxy-1-methylisoquinoline (7.5.1) (400 mg, 1.19 mmol), NBS (223 mg, 1.25 mmol), and AIBN (20 mg, 0.119 mmol) in CCl4 (7 mL) were heated at 85 °C for 2 h. Reaction mixture was then cooled to RT and diluted with hexane. Solid precipitate was filtered off and filtrate was concentrated. Chromatography achieved using ISCO max gradient 15% EtOAc/hexane yielding product as a white solid (350 mg, 71% yield); mp 167–170 °C; 1H NMR (400 MHz) (CDCl3) δ 8.15–8.14 (m, 1H), 7.94–7.91 (m, 2 H), 7.48–7.45 (m, 3H), 7.18 (s, 1H), 5.10 (s, 2H), 4.11 (s, 3H), 4.07 (s, 3H), 1.45 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 153.0, 152.9, 151.5, 150.1, 150.0, 139.2, 134.6, 128.5, 125.5, 124.2, 123.9, 121.5, 116.6, 105.8, 103.6, 56.2, 56.1, 34.9, 33.0, 31.5; HRMS (ESI) Calcd for C22H25BrNO2 (M+H)+ 414.1063. Found 414.1057.
7.17.2. 1-(Azidomethyl)-3-(3-(t-butyl)phenyl)-6,7-dimethoxyi soquinoline
1-(Bromomethyl)-3-(3-(t-butyl)phenyl)-6,7-dimethoxyisoquinoline (130 mg, 0.31 mmol) and sodium azide (25 mg, 0.38 mmol) were combined in anhydrous DMF (3 mL) and stirred at RT overnight. Reaction was then diluted with EtOAc and washed with saturated NaHCO3 followed by 10% LiCl solution. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 25% EtOAc/hexane to yield product as a clear oil (114 mg, 97% yield); 1H NMR (400 MHz) (CDCl3) δ 8.24 (s, 1H), 7.96 (s, 2H), 7.48–7.46 (m, 2H), 7.30 (s, 1H), 7.20 (s, 1H), 4.91 (s, 2H), 4.08 (s, 6H), 1.45 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 153.0, 152.1, 151.6, 150.4, 149.7, 139.1, 134.5, 128.5, 125.5, 124.0, 121.3, 116.0, 105.8, 102.7, 56.1, 53.8, 34.9, 31.5; IR (thin film NaCl) 3065, 3005, 2961, 2867, 2835, 2254, 2099, 1621, 1574, 1505, 1468, 1426, 1408, 1363, 1302, 1247, 1207, 1162, 1086, 1056, 1033, 997, 911, 877, 835, 798, 769, 732, 701, 647, 597; HRMS (ESI) Calcd for C22H25N4O2 (M+H)+ 377.1972. Found 377.1966.
7.17.3. (3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl)methanamine (16a)
1-(Azidomethyl)-3-(3-(t-butyl)phenyl)-6,7-dimethoxyisoquinoline (110 mg, 0.29 mmol) was combined in a flask with polymer supported (3 mmol/g loading) PPh3 (145 mg, 0.44 mmol), THF (5 mL), and H2O (0.5 mL). Reaction was stirred overnight at RT. Resin was then filtered off and filtrate concentrated. Chromatography achieved using silica column max gradient 10% MeOH/DCM/0.1% NH4OH yielding product as a white solid (68 mg, 67% yield); mp 173–174 °C; 1H NMR (400 MHz) (CDCl3) δ 8.08 (s, 1H), 7.87–7.84 (m, 1H), 7.74 (s, 1H), 7.36–7.35 (m, 2H), 7.12 (s, 1H), 7.06 (s, 1H), 4.10 (s, 2H), 3.97 (s, 3H), 3.93 (s, 3H), 1.34 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 156.1, 152.7, 151.5, 150.0, 149.0, 139.5, 133.8, 128.5, 125.4, 124.0, 123.6, 120.6, 114.8, 105.8, 102.2, 56.1, 56.0, 44.0, 34.9, 31.5; HRMS (ESI) Calcd for C22H27N2O2 (M+H)+ 351.2067. Found 351.2069.
7.18. General procedure for synthesis of compound (17)
7.18.1. 3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1(2H)one
3-Bromo-6,7-dimethoxyisoquinolin-1(2H)-one (300 mg, 1.06 mmol) was combined with 3-t-butylphenyl boronic acid (226 mg, 1.27 mmol), Pd(OAc)2 (24 mg, 0.11 mmol), XPhos (100 mg, 0.21 mmol), and K2CO3 (437 mg, 3.17 mmol) in a flask and degassed. ACN (9 mL) and H2O (3 mL) were then added and solution was heated at 100 °C for 2 h. Reaction mixture was cooled to RT then diluted with EtOAc and washed with NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 70% EtOAc/hexane yielding product as a white solid (276 mg, 78% yield); mp 211–214 °C; 1H NMR (400 MHz) (CDCl3) δ 9.14 (bs, 1H), 7.81 (s, 1H), 7.67–7.66 (m, 1H), 7.53–7.50 (m, 1H), 7.47–7.45 (m, 2H), 7.00 (s, 1H), 6.73 (s, 1H), 4.05 (s, 6H), 1.42 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 163.1, 153.9, 152.3, 149.2, 138.8, 134.3, 134.0, 129.0, 126.5, 123.3, 123.0, 118.8, 107.3, 106.5, 104.0, 56.2, 56.1, 35.0, 31.4; HRMS (ESI) Calcd for C21H24NO3 (M+H)+ 338.1751. Found 338.1744.
7.18.2. 3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl trifluoromethanesulfonate
3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1(2H)-one (125 mg, 0.37 mmol) and Et3N (0.1 mL, 0.45 mmol) in anhydrous DCM (15 mL) were cooled to −78 °C. Tf2O (0.07 mL, 0.74 mmol) was slowly added to the mixture and was stirred for 30 min at −78 °C. Reaction was then quickly diluted with additional DCM and washed with saturated NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a white solid (155 mg, 89% yield); mp 131–134 °C; 1H NMR (400 MHz) (CDCl3) δ 8.20 (s, 1H), 7.98 (s, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.49–7.41 (m, 2H), 7.30 (s, 1H), 7.20 (s, 1H), 4.07 (s, 6H), 1.44 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 154.3, 151.8, 154.5, 154.5, 147.8, 137.5, 137.1, 128.5, 126.1, 123.8, 123.5, 117.3, 115.8, 114.3, 105.4, 100.6, 56.3, 56.2, 34.9, 31.3; HRMS (ESI) Calcd for C22H23F3NO5S (M+H)+ 470.1244. Found 470.1234.
7.18.3. t-Butyl (2-(3-(3-(t-butyl)phenyl)-6,7-dimethoxyisoqui nolin-1-yl)ethyl)carbamate
3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl trifluoromethanesulfonate (155 mg, 0.33 mmol), potassium t-butyl N-[2-(trifluoroboranuidyl)ethyl]carbamate (166 mg, 0.66 mmol), Pd(PPh3)4 (38 mg, 0.033 mmol), and K2CO3 (159 mg, 1.16 mmol) were combined in a flask with dioxane (5 mL) and H2O (2.5 mL) and degassed. Reaction mixture was then refluxed at 102 °C overnight. Solution was cooled to RT then diluted with EtOAc and washed with saturated NH4Cl. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a clear oil (99 mg, 64% yield); 1H NMR (400 MHz) (CDCl3) δ 8.22 (s, 1H), 7.94–7.92 (m, 1H), 7.87 (s, 1H), 7.47–7.45 (m, 2H), 7.39 (s, 1H), 7.16 (s, 1H), 5.75 (bs, 1H), 4.08 (s, 3H), 4.07 (s, 3H), 3.92 (q, J = 16.0 Hz, 2H), 3.48 (t, J = 12.0 Hz, 2H), 1.45 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 156.3, 152.7, 151.5, 150.0, 139.6, 133.7, 128.5, 125.2, 123.8, 123.7, 122.2, 114.3, 105.8, 103.2, 79.0, 56.1, 56.0, 38.2, 34.9, 34.5, 31.5, 28.5; HRMS (ESI) Calcd for C28H37N2O4 (M+H)+ 465.2748. Found 465.2738.
7.18.4. 2-(3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl)ethanamine (17a)
To a cooled solution of t-butyl (2-(3-(3-(t-butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl)ethyl)carbamate (99 mg, 0.21 mmol) in anhydrous DCM (2 mL) was added trifluoroacetic acid (2 mL). Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Chromatography achieved using ISCO max gradient 5% MeOH/DCM yielding product as a tan fluffy solid (78 mg, quantitative); mp 97–100 °C; 1H NMR (400 MHz) (CD3OD) δ 8.13 (s, 1H), 8.07 (t, J = 4.0 Hz, 1H), 7.84 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.57–7.55 (m, 1H), 7.51–7.47 (m, 3H), 4.08 (s, 3H), 4.06 (s, 3H), 3.76 (t, J = 12.0 Hz, 2H), 3.69–3.65 (m, 2H), 1.45 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 156.2, 154.6, 153.2, 153.0, 148.8, 136.9, 129.8, 127.2, 125.5, 125.2, 123.1, 118.2, 107.4, 104.1, 56.9, 56.8, 39.2, 35.8, 31.8, 30.8; HRMS (ESI) Calcd for C23H29N2O2 (M+H)+ 365.2224. Found 365.2226.
7.19. General procedure for synthesis of N′-((3-(3-(t-butyl) phenyl)-6,7-dimethoxyisoquinolin-1-yl)methyl)acetimidamide (18a)
1-(Bromomethyl)-3-(3-(t-butyl)phenyl)-6,7-dimethoxyisoquinoline (7.17.1) (25 mg, 0.06 mmol), acetamidine HCl (7 mg, 0.072 mmol), and K2CO3 (17 mg, 0.12 mmol) in anhydrous DMF (2 mL) were heated at 50 °C for 2 h. Reaction mixture was then cooled to RT, diluted with EtOAc, and washed with 10% LiCl solution. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 10% MeOH/DCM yielding product as an off-white solid (18 mg, 78% yield); mp 137–140 °C; 1H NMR (400 MHz) (CD3OD) δ 8.22 (t, J = 4.0 Hz, 1H), 8.13 (s, 1H), 7.94–7.72 (m, 1H), 7.50 (dt, J = 8.0 Hz, J = 4.0 Hz, 1H), 7.46–7.43 (m, 2H), 7.40 (s, 1H), 5.16 (s, 2H), 4.08 (s, 3H), 4.04 (s, 3H), 2.44 (s, 3H), 1.45 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 167.3, 155.0, 152.8, 152.4, 150.7, 150.1, 140.3, 136.1, 129.6, 126.5, 124.8, 124.6, 122.2, 117.1, 107.3, 103.2, 56.7, 56.6, 46.3, 35.8, 31.9, 19.3; HRMS (ESI) Calcd for C24H30N3O2 (M+H)+ 392.2333. Found 392.2328.
7.20. General procedure for synthesis of compound (19)
7.20.1. 3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinoline-1-carbaldehyde
3-(3-(t-Butyl)phenyl)-6,7-dimethoxy-1-methylisoquinoline (7.5.1) (153 mg, 0.46 mmol) and SeO2 (61 mg, 0.55 mmol) in anhydrous dioxane (5 mL) were refluxed at 102 °C for 3 h. Solution was then cooled to RT and filtered to remove precipitate. Resulting filtrate was concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a yellow solid (97 mg, 62% yield); mp 178–179 °C; 1H NMR (400 MHz) (CDCl3) δ 10.40 (s, 1H), 8.67 (s, 1H), 8.14 (s, 1H), 8.08 (s, 1H), 7.90 (dt, J = 4.0 Hz, J = 4.0 Hz, 1H), 7.42–7.40 (m, 2H), 7.13 (s, 1H), 4.04 (s, 3H), 4.00 (s, 3H), 1.36 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 197.0, 153.2, 152.7, 151.8, 150.4, 147.0, 138.5, 135.4, 128.7, 125.9, 124.1, 123.8, 122.1, 120.1, 105.0, 103.5, 56.3, 56.1, 35.0, 31.5; HRMS (ESI) Calcd for C22H24NO3 (M+H)+ 350.1751. Found 350.1746.
7.20.2. (3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl)methanol
3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinoline-1-carbalde-hyde (97 mg, 0.23 mmol) in ethanol (5 mL) was treated slowly with NaBH4 (26 mg, 0.68 mmol) at RT. Reaction was stirred for 1 h then acetone (2 mL) was added and solution was filtered through filter paper. Filtrate was concentrated then re-dissolved in DCM and washed with H2O. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 50% EtOAc/hexane yielding product as a pale yellow solid (50 mg, 63% yield); mp 181–182 °C; 1H NMR (400 MHz) (CDCl3) δ 8.07 (m, 1H), 7.86–7.83 (m, 1H), 7.82 (s, 1H), 7.39–7.37 (m, 2H), 7.13 (s, 1H), 6.98 (s, 1H), 5.12 (s, 2H), 3.99 (s, 3H), 3.98 (s, 3H), 1.35 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 154.5, 153.1, 151.6, 150.3, 148.2, 138.9, 133.9, 128.5, 125.6, 124.0, 123.7, 119.6, 115.3, 105.8, 101.2, 61.3, 56.1, 56.1, 34.9, 31.5; HRMS (ESI) Calcd for C22H26NO3 (M+H)+ 352.1907. Found 352.1905.
7.20.3. 2-((3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl)methyl)guanidine (19a)
(3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl)methanol (50 mg, 0.14 mmol), PPh3 (56 mg, 0.21 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (74 mg, 0.28 mmol) in anhydrous toluene (4 mL) at 0 °C was added diisopropylazodicarboxylate (0.04 mL, 0.21 mmol) drop wise over 15 min. Reaction was stirred for 3 h at RT then 2 drops H2O were added, and the solution was concentrated. Solid was then dissolved in DCM and passed through silica column and resulting crude product was then re-dissolved in anhydrous DCM (1.5 mL) and cooled to 0 °C. TFA (1.5 mL) was then added. Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Chromatography achieved using ISCO max gradient 10% MeOH/DCM yielding product as an off-white solid (52 mg, 93% yield over two steps); mp 194–196 °C; 1H NMR (400 MHz) (CDCl3) δ 9.69 (bs, 1H), 7.96 (s, 1H), 7.93 (s, 1H), 7.76–7.75 (m, 1H), 7.47–7.42 (m, 3H), 7.18 (s, 1H), 4.87 (d, J = 4.0 Hz, 2H), 4.10 (s, 3H), 4.07 (s, 3H), 1.39 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 159.1, 154.1, 151.9, 151.8, 151.4, 148.2, 137.7, 135.3, 128.8, 126.0, 123.9, 123.5, 121.5, 117.1, 105.6, 102.6, 56.3, 56.2, 44.7, 34.8, 31.3; HRMS (ESI) Calcd for C23H29N4O2 (M+H)+ 393.2285. Found 393.2287.
7.21. General procedure for synthesis of compound (20)
7.21.1. 1,3-di-Boc-2-(2-(3-(3-(t-Butyl)phenyl)-6,7-dimethoxyi soquinolin-1-yl)ethyl)guanidine
2-(3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl)ethan-amine (7.18.4) (67 mg, 0.18 mmol), 1,3-di-Boc-2-(trifluoromethylsulfonyl)-guanidine (86 mg, 0.22 mmol), and Et3N (0.03 mL, 0.22 mmol) in anhydrous DCM (5 mL) were stirred for 1 h at 37 °C. Reaction mixture was then diluted with DCM and washed with NaHCO3. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 40% EtOAc/hexane yielding product as a clear oil (75 mg, 67% yield); 1H NMR (400 MHz) (CDCl3) δ 11.48 (bs, 1H), 9.04 (t, J = 12.0 Hz, 1H), 8.15 (s, 1H), 8.04–8.02 (m, 1H), 7.85 (s, 1H), 7.44–7.43 (m, 2H), 7.35 (s, 1H), 7.16 (s, 1H), 4.20–4.15 (m, 2H), 4.08 (s, 3H), 4.07 (s, 3H), 3.57 (t, J = 16.0 Hz, 2H), 1.54 (s, 9H), 1.43 (s, 9H), 1.39 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 163.8, 156.3, 155.7, 152.7, 152.6, 151.2, 149.9, 149.7, 139.7, 133.7, 128.3, 125.1, 124.4, 123.8, 122.0, 114.6, 105.8, 103.1, 82.7, 79.1, 56.1, 56.0, 38.9, 34.9, 34.2, 31.5, 28.4, 28.0; HRMS (ESI) Calcd for C34H47N4O6 (M+H)+ 607.3490. Found 607.3485.
7.21.2. 2-(2-(3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl)ethyl)guanidine (20a)
To a cooled solution of 1,3-di-Boc-2-(2-(3-(3-(t-Butyl)phenyl)-6,7-dimethoxyisoquinolin-1-yl)ethyl)guanidine (53 mg, 0.087 mmol) in anhydrous DCM (1 mL) was added trifluoroacetic acid (1 mL). Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Chromatography achieved using ISCO max gradient 10% MeOH/DCM yielding product as a tan fluffy solid (36 mg, quantitative); mp 91–94 °C; 1H NMR (400 MHz) (CD3OD) δ 8.30 (s, 1H), 7.94 (t, J = 4.0 Hz, 1H), 7.74–7.69 (m, 2H), 7.67 (s, 1H), 7.63 (s, 1H), 7.60–7.56 (m, 1H), 4.13 (s, 6H), 3.85 (s, 4H), 1.45 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 159.1, 158.8, 154.5, 154.5, 153.8, 145.3, 139.1, 130.2, 128.8, 126.5, 126.2, 123.5, 121.3, 107.5, 105.0, 57.3, 57.0, 41.7, 35.9, 31.9, 31.7; HRMS (ESI) Calcd for C24H31N4O2 (M+H)+ 407.2442. Found 407.2443.
7.22. General procedure for synthesis of compound (21)
7.22.1. 3-(4′-(t-Butyl)-[1,1′-biphenyl]-3-yl)-6,7-dimethoxyiso quinoline-1-carbaldehyde
3-(4′-(t-Butyl)-[1,1′-biphenyl]-3-yl)-6,7-dimethoxy-1-methylisoquinoline (7.8.1) (100 mg, 0.24 mmol) and SeO2 (32 mg, 0.29 mmol) in anhydrous dioxane (5 mL) were refluxed at 102 °C for 3 h. Solution was then cooled to RT and filtered to remove precipitate. Resulting filtrate was concentrated. Chromatography achieved using ISCO max gradient 30% EtOAc/hexane yielding product as a yellow solid (80 mg, 77% yield); mp 165–168 °C; 1H NMR (400 MHz) (CDCl3) δ 10.40 (s, 1H), 8.68 (s, 1H), 8.34 (m, 1H), 8.14 (s, 1H), 8.07 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 8.0 Hz, 3H), 7.52 (t, J = 16.0 Hz, 1H), 7.45 (d, J = 8.0 Hz, 2H), 7.13 (s, 1H), 4.05 (s, 3H), 4.00 (s, 3H), 1.32 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 196.8, 153.3, 152.8, 149.8, 141.8, 139.2, 138.3, 135.4, 129.3, 127.4, 127.0, 125.8, 125.6, 125.4, 122.2, 120.0, 105.0, 103.6, 56.3, 56.0, 34.6, 31.4; HRMS (ESI) Calcd for C28H28NO3 (M+H)+ 426.2064. Found 426.2041.
7.22.2. (3-(4′-(t-Butyl)-[1,1′-biphenyl]-3-yl)-6,7-dimethoxyiso quinolin-1-yl)methanol
3-(4′-(t-Butyl)-[1,1′-biphenyl]-3-yl)-6,7-dimethoxyisoquino-line-1-carbaldehyde (76 mg, 0.18 mmol) in methanol (5 mL) was treated slowly with NaBH4 (20 mg, 0.53 mmol) at RT. Reaction was stirred for 1 h then acetone(2 mL) was added and solution was filtered through filter paper. Filtrate was concentrated then re-dissolved in DCM and washed with H2O. Organic layer was dried over sodium sulfate and concentrated. Chromatography achieved using ISCO max gradient 50% EtOAc/hexane yielding product as a pearly, gold solid (59 mg, 78% yield); mp 104–106 °C; 1H NMR (400 MHz) (CDCl3) δ 8.25–8.24 (m, 1H), 7.99–7.97 (m, 1H), 7.85 (s, 1H), 7.57–7.54 (m, 3H), 7.47 (t, J = 12.0 Hz, 1H), 7.43 (d, J = 8.0 Hz, 2H), 7.08 (s, 1H), 6.96 (s, 1H), 5.22 (t, J = 12.0 Hz, 1H), 5.10 (d, J = 4.0 Hz, 1H), 3.96 (s, 3H), 3.95 (s, 3H), 1.31 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 154.6, 153.2, 150.5, 150.4, 147.7, 141.7, 139.6, 138.3, 133.8, 129.1, 127.1, 126.9, 125.8, 125.4, 125.3, 119.8, 115.3, 105.9, 101.3, 61.4, 56.1, 56.1, 34.6, 31.4; HRMS (ESI) Calcd for C28H30NO3 (M+H)+ 428.2220. Found 428.2213.
7.22.3. 2-((3-(4′-(t-Butyl)-[1,1′-biphenyl]-3-yl)-6,7-dimethoxyi soquinolin-1-yl)methyl)guanidine trifluoroacetate (21a)
To a solution of (3-(4′-(t-Butyl)-[1,1′-biphenyl]-3-yl)-6,7-dimethoxyisoquinolin-1-yl)methanol (39 mg, 0.091 mmol), PPh3 (35 mg, 0.14 mmol), and 1,3-bis(t-butoxycarbonyl)guanidine (47 mg, 0.18 mmol) in anhydrous toluene (3 mL) at 0 °C was added diisopropylazodicarboxylate (0.03 mL, 0.14 mmol) drop wise over 15 min. Reaction was stirred for 3 h at RT then 2 drops H2O were added, and the solution was concentrated. Solid was then dissolved in DCM and passed through silica column and resulting crude product was then re-dissolved in anhydrous DCM (1.5 mL) and cooled to 0 °C. TFA (1.5 mL) was then added. Reaction was taken off ice bath and stirred at RT for 2 h then solvents were evaporated. Solid was then taken back up in DCM and precipitate was filtered off yielding product as a grayish white solid (22 mg, 42% yield over 2 steps); mp 119–122 °C; 1H NMR (400 MHz) (CD3OD) δ 8.25 (m, 1H), 8.08 (s, 1H), 8.04 (d, J = 4.0 Hz, 1H), 7.58–7.56 (m, 3H), 7.47 (t, J = 16.0 Hz, 1H), 7.43 (d, J = 8.0 Hz, 2H), 7.33 (s, 1H), 7.29 (s, 1H), 4.95 (s, 2H), 3.95 (s, 3H), 3.93 (s, 3H), 1.28 (s, 9H); 13C NMR (100 MHz) (CD3OD) δ 160.1, 159.6, 155.1, 152.5, 151.9, 151.7, 149.5, 142.9, 141.1, 139.5, 130.3, 128.0, 127.8, 126.8, 126.6, 126.1, 122.1, 117.3, 107.4, 103.2, 56.7, 56.6, 45.1, 35.4, 31.8; HRMS (ESI) Calcd for C29H33N4O2 (M+H)+ 469.2598. Found 469.2599.
7.23. Minimum inhibitory concentration (MIC) assays
MIC assays were conducted in accordance with Clinical and Laboratory Standards Institute (CLSI) guidelines for broth microdilution.31 The following bacterial strains were included in these assays: S. aureus 8325-4 (MSSA), S. aureus ATCC 33591 (MRSA), E. faecalis ATCC 19433 (VSE), and E. faecalis ATCC 51575 (VRE). Log-phase bacteria were added to 96-well microtiter plates (at 105 CFU/mL) containing two-fold serial dilutions of compound or comparator drug (at concentrations ranging from 64 to 0.031 μg/mL) in cation-adjusted Mueller-Hinton (CAMH) broth (for the S. aureus assays) or brain–heart infusion (BHI) broth (for the E. faecalis assays). In the MRSA assays, the CAMH broth was supplemented with 2% NaCl. The final volume in each well was 0.1 mL, and the microtiter plates were incubated aerobically for 24 h at 37 °C. Bacterial growth was then monitored by measuring OD600 using a VersaMax® plate reader (Molecular Devices, Inc.), with the MIC being defined as the lowest compound concentration at which growth was ≥90% inhibited.
7.24. FtsZ binding assays
The binding of 5b and 7b to SaFtsZ was assayed by monitoring protein-induced changes in the intrinsic fluorescence of the compounds. SaFtsZ was expressed in Escherichia coli and purified as described elsewhere.36 In these experiments, aliquots (1.5–3 μL) of a 250 μM SaFtsZ stock solution were sequentially added to a buffered solution (150 μL) containing 5b (7 μM) or 7b (10 μM). After each protein addition, the reaction was allowed to equilibrate for 3 min, and the emission spectrum was then acquired from 510 to 370 nm in 1-nm increments. Each spectrum acquired in this manner was corrected by subtraction of the corresponding background spectrum resulting from the titration of protein into buffer alone. The excitation wavelength was set at 265 nm, the bandwidth was set at 5 nm in both the excitation and emission directions, and the time constant was set at 1 s. All measurements were acquired at 25 °C using an AVIV model ATF 105 spectrofluorimeter (AVIV Biomedical, Inc.) equipped with a thermoelectrically controlled cell holder. A quartz ultra-micro cuvette was used in each experiment, with the pathlength being 10 mm in the excitation direction and 2 mm in the emission direction. Buffer conditions were 50 mM Tris HCl (pH 7.4), 50 mM KCl, and 2 mM magnesium acetate.
7.25. FtsZ polymerization assays
Polymerization of SaFtsZ was monitored using a microtiter plate-based light scattering (turbidity) assay. Test compound or comparator drug (at concentrations ranging from 0 to 40 μg/mL) was combined with either 1 mM GTP or 10 μM SaFtsZ in 100 μL of reaction solution and pre-equilibrated for 10 min at room temperature. Reaction solutions contained 50 mM Tris HCl (pH 7.4), 50 mM KCl, 2 mM magnesium acetate, and 5 mM CaCl2. Reaction solutions were assembled in half-volume, flat-bottom, 96-well microtiter plates, and the polymerization reactions were initiated by addition of either the GTP or the FtsZ (with the polymerization profiles obtained either way being similar). Polymerization was continuously monitored at 25 °C by measuring the absorbance at 340 nm (A340) in a VersaMax® plate reader over a time period of 60 min. In the stability studies of the SaFtsZ polymers, acquisition was interrupted long enough for the addition of 1 mM GDP to the reaction mix and then resumed.
7.26. FtsZ GTPase assays
The impact of the synthesized compounds on the GTPase activity of SaFtsZ was assayed by measuring the inorganic phosphate (Pi) released upon GTP hydrolysis by FtsZ in the absence or presence of compound via an end-point malachite green colorimetric assay. This assay is based on the spectrophotometric detection of the green complex formed between malachite green molybdate and Pi under acidic conditions. Duplicate reactions of 20 μL were assembled in 96-well plates containing 10 μM FtsZ and either DMSO vehicle or compound (at concentrations ranging from 0.1 to 200 μg/mL) in buffer containing 50 mM Tris HCl (pH 7.4), 50 mM KCl, and 2 mM magnesium acetate, and 5 mM CaCl2. The reactions were pre-equilibrated for 10 min at room temperature, whereupon the GTPase activity was then initiated by the addition of 250 μM GTP (Roche Diagnostics GmbH, Mannheim, Germany) and shifting the plates to 37 °C. The GTPase reactions were allowed to proceed for 2 h, and terminated by the addition of 80 μL of a malachite green (Sigma, St. Louis, MO) reagent, which had been previously prepared by mixing a solution of 0.045% (w/v) malachite green (made in water) with a solution of 4.2% (w/v) ammonium molybdate (made in 4 M HCl) at a ratio of 3:1, and filtering through a 0.22-μm filter. After addition of the malachite green reagent to the 96-well plates, the plates were incubated at room temperature for one minute, and the absorbance at 620 nm was recorded using a VersaMax® plate reader. The concentration of Pi released in each reaction was determined by using a phosphate standard curve, which was obtained by diluting a 200 μM KH2PO4 stock solution to achieve final phosphate concentrations ranging from 0 to 60 μM. The Pi released in the presence of each compound is reported as a percentage of Pi released in the presence of vehicle (DMSO) alone.
7.27. Tubulin polymerization assays
Polymerization of microtubule-associated protein (MAP)-rich porcine β-tubulin containing 70% β-tubulin and 30% MAPs (Cyto-skeleton, Inc.) was monitored using a microtiter plate-based light scattering (turbidity) assay similar to that described above for FtsZ polymerization. Test compound or comparator drug was combined with 1 mM GTP and 2 mg/mL porcine β-tubulin in 100 μL of reaction solution containing 80 mM PIPES NaOH (pH 7.0), 2 mM MgCl2, and 1 mM EGTA. Reactions were assembled in half-volume, flat-bottom, 96-well microtiter plates, and polymerization was continuously monitored at 37 °C by measuring A340 in a VersaMax® plate reader over a time period of 60 min.
7.28. Cytotoxicity assays
Cytotoxicity was determined using the MTT-microtiter plate tetrazolinium cytotoxicity assay. The human embryonic kidney 293 (HEK293) cell line was provided by Dr. Zue-Hung Hsu (formerly at Columbia University, presently at Beijing National academy). The Madin-Darby Canine Kidney (MDCK) epithelial cells were obtained from Professor Patrick Sinko (Rutgers University). The cytotoxicity assay was performed using 96-well microtiter plates. Cells were grown in suspension at 37 °C in 5% CO2 and maintained by regular passage in DMEM media. For determination of IC50, cells were exposed continuously for four days to varying concentrations of drug in triplicate wells, each seeded with 1500 cells. Each assay was performed with a control that did not contain any drug. The MTT assays were performed at the end of the fourth day.
Acknowledgments
This study was supported by research agreements between TAXIS Pharmaceuticals, Inc. and both Rutgers, The State University of New Jersey (E.J.L.) and the University of Medicine and Dentistry of New Jersey (D.S.P). We would like to thank Drs. Steve Tuske and Eddy Arnold (Center for Advanced Biotechnology and Medicine, Rutgers University) for their assistance with the expression and purification of S. aureus FtsZ. S. aureus 8325-4 was the generous gift of Dr. Glenn W. Kaatz (John D. Dingell VA Medical Center, Detroit, MI). We also thank Angela Liu from the Department of Pharmacology for performing the cytotoxicity studies. The Bruker Avance III 400 MHz NMR spectrometer used in this study was purchased with funds from NCRR Grant No. 1S10RR23698-1A1. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource with support from the NIH National Center for Research Resources Grant No. P41RR0954.
References and notes
- 1.Leavis HL, Willems RJL, Top J, Spalburg E, Mascini EM, Fluit AC, Hoepelman A, de Neeling AJ, Bonten MJM. Emerg. Infect. Dis. 2003:9–1108. doi: 10.3201/eid0909.020383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. J. Am. Med. Assoc. 2007:298–1763. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 3.Addinall SG, Holland B. J. Mol. Biol. 2002:318–219. doi: 10.1016/S0022-2836(02)00024-4. [DOI] [PubMed] [Google Scholar]
- 4.Margolin W. Nat. Rev. Mol. Cell Biol. 2005:6–862. doi: 10.1038/nrm1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Addinall SG, Bi E, Lutkenhaus J. J. Bacteriol. 1996:178–3877. doi: 10.1128/jb.178.13.3877-3884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinho MG, Errington J. Mol. Microbiol. 2003:50–871. doi: 10.1046/j.1365-2958.2003.03719.x. [DOI] [PubMed] [Google Scholar]
- 7.Lutkenhaus J, Addinall SG. Annu. Rev. Biochem. 1997:66–93. doi: 10.1146/annurev.biochem.66.1.93. [DOI] [PubMed] [Google Scholar]
- 8.Lowe J, van den Ent F, Amos LA. Annu. Rev. Biophys. Biomol. Struct. 2004:33–177. doi: 10.1146/annurev.biophys.33.110502.132647. [DOI] [PubMed] [Google Scholar]
- 9.Lock RL, Harry EJ. Nat. Rev. 2008:7–324. doi: 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- 10.Kapoor S, Panda D. Expert Opin. Ther. Targets. 2009;13:1037. doi: 10.1517/14728220903173257. [DOI] [PubMed] [Google Scholar]
- 11.Foss MH, Eun Y-J, Weibel DB. Biochemistry. 2011:50–7719. doi: 10.1021/bi200940d. [DOI] [PubMed] [Google Scholar]
- 12.Schaffner-Barbero C, Martin-Fontecha M, Chacon P, Andreu JM. ACS Chem. Biol. 2012:7–269. doi: 10.1021/cb2003626. [DOI] [PubMed] [Google Scholar]
- 13.Margalit DN, Romberg L, Mets RB, Hebert AM, Mitchision TJ, Kirschner MW, RayChaudhuri D. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11821. doi: 10.1073/pnas.0404439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreu JM, Schaffner-Barbero C, Huecas S, Alonso D, Lopez-Rodriguez ML, Ruiz-Avila LB, Núñez-Ramírez R, Llorca O, Martín-Galiano AJ. J. Biol. Chem. 2010:285–14239. doi: 10.1074/jbc.M109.094722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beuria TK, Santra MK, Panda D. Biochemistry. 2005:44–16584. doi: 10.1021/bi050767+. [DOI] [PubMed] [Google Scholar]
- 16.Domadia PN, Bhunia A, Sivaraman J, Swarup S, Dasgupta D. Biochemistry. 2008:47–3225. doi: 10.1021/bi7018546. [DOI] [PubMed] [Google Scholar]
- 17.Haydon DJ, Bennett JM, Brown D, Collins I, Galbraith G, Lancett P, Macdonald R, Stokes NR, Chauhan PK, Sutariya JK, Nayal N, Srivastava A, Beanland J, Hall R, Henstock V, Noula C, Rockley C, Czaplewski L. J. Med. Chem. 2010:53–3927. doi: 10.1021/jm9016366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG. Science. 2008:321–1673. doi: 10.1126/science.1159961. [DOI] [PubMed] [Google Scholar]
- 19.Kumar K, Awasthi D, Lee S-Y, Zanardi I, Ruzsicska B, Knudson S, Tonge PJ, Slayden RA, Ojima I. J. Med. Chem. 2011:54–374. doi: 10.1021/jm1012006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Läppchen T, Hartog AF, Pinas VA, Koomen G-J, den Blaauwen T. Biochemistry. 2005:44–7879. doi: 10.1021/bi047297o. [DOI] [PubMed] [Google Scholar]
- 21.Urgaonkar S, La Pierre HS, Meir I, Lund H, RayChaudhuri D, Shaw JT. Org. Lett. 2005:7–5609. doi: 10.1021/ol052269z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Awasthi D, Kumar K, Ojima I. Expert Opin. Ther. Pat. 2011:21–657. doi: 10.1517/13543776.2011.568483. [DOI] [PubMed] [Google Scholar]
- 23.Huang Q, Tonge PJ, Slayden RA, Kirikae T, Ojima I. Curr. Top. Med. Chem. 2007:7–527. doi: 10.2174/156802607780059790. [DOI] [PubMed] [Google Scholar]
- 24.Kumar K, Awasthi D, Berger WT, Tonge PJ, Slayden RA, Ojima I. Future Med. Chem. 2010:2–1305. doi: 10.4155/fmc.10.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boberek JM, Stach J, Good L. PLoS One. 2010;5:e13745. doi: 10.1371/journal.pone.0013745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parhi A, Kelley C, Kaul M, Pilch DS, LaVoie EJ. Bioorg. Med. Chem. Lett. 2012;22 doi: 10.1016/j.bmcl.2012.09.097. in press. http://dx.doi.org/10.1016Zj.bmcl.2012.09.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parhi A, Lu S, Kelley C, Kaul M, Pilch DS, LaVoie EJ. Bioorg. Med. Chem. Lett. 2012:22–6962. doi: 10.1016/j.bmcl.2012.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) LaVoie EJ, Parhi A, Pilch DS, Kaul M. U.S. Patent 20,120,022,061.; (b) LaVoie EJ, Parhi A, Pilch DS, Kaul M. U.S. Patent 20,120,059,026.
- 29.Simchen G, Kramer W. Chem. Ber. 1969:102–3656. [Google Scholar]
- 30.Kanojia RM, Press JB, Lever OW, Jr., Williams L, McNally JJ, Tobia AJ, Falotico R, Moore JB., Jr. J. Med. Chem. 1988:31–1363. doi: 10.1021/jm00402a020. [DOI] [PubMed] [Google Scholar]
- 31.Clinical and Laboratory Standards Institute; Wayne, PA: 2009. CLSI methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. approved standard-eighth edition. [Google Scholar]
- 32.Stokes NR, Sievers J, Barker S, Bennett JM, Brown DR, Collins I, Errington VM, Foulger D, Hall M, Halsey R, Johnson H, Rose V, Thomaides HB, Haydon DJ, Czaplewski LG, Errington J. J. Biol. Chem. 2005:280–39709. doi: 10.1074/jbc.M506741200. [DOI] [PubMed] [Google Scholar]
- 33.Carlier MF, Pantaloni D. Biochemistry. 1983:22–4814. doi: 10.1021/bi00289a031. [DOI] [PubMed] [Google Scholar]
- 34.Kumar N. J. Biol. Chem. 1981:256–10435. [PubMed] [Google Scholar]
- 35.Lin CM, Hamel E. J. Biol. Chem. 1981:256–9242. [PubMed] [Google Scholar]
- 36.Kaul M, Parhi AK, Zhang Y, LaVoie EJ, Tuske S, Arnold E, Kerrigan JE, Pilch DS. J. Med. Chem. 2012;55 doi: 10.1021/jm3012728. in press. http://dx.doi.org/10.1021/jm30127728. [DOI] [PMC free article] [PubMed] [Google Scholar]