

SUMMARY
The cyclic AMP-protein kinase A pathway governs numerous biological features of the fungal pathogen Candida albicans. The catalytic protein kinase A subunits, Tpk1 (orf19.4892) and Tpk2 (orf19.2277), have divergent roles, and most studies indicate a more pronounced role for Tpk2. Here we dissect two Tpk1-responsive properties: adherence and cell wall integrity. Homozygous tpk1/tpk1 mutants are hyperadherent, and a Tpk1 defect enables biofilm formation in the absence of Bcr1, a transcriptional regulator of biofilm adhesins. A quantitative gene expression-based assay reveals that tpk1/tpk1 and bcr1/bcr1 genotypes show mixed epistasis, as expected if Tpk1 and Bcr1 act mainly in distinct pathways. Overexpression of individual Tpk1-repressed genes indicates that cell surface proteins Als1, Als2, Als4, Csh1, and Csp37 contribute to Tpk1-regulated adherence. Tpk1 is also required for cell wall integrity, but has no role in the gene expression response to cell wall inhibition by caspofungin. Interestingly, increased expression of the adhesin gene ALS2 confers a cell wall defect, as manifested in hypersensitivity to the cell wall inhibitor caspofungin and a shallow cell wall structure. Our findings indicate that Tpk1 governs C. albicans cell wall properties through repression of select cell surface protein genes.
Keywords: cyclic AMP, adherence, biofilm, cell wall integrity
INTRODUCTION
The cyclic AMP-protein kinase A pathway has a pivotal role in C. albicans morphogenesis and infection biology (Biswas et al., 2007). The pathway responds to diverse environmental signals, including CO2, bacterial peptidoglycan, and the quorum sensing molecule farnesol (Hogan & Muhlschlegel, 2011). Outputs are equally diverse, and include the yeast-to-hypha morphogenesis program, the white-opaque cell type switch, biofilm formation, binding and damage to epithelial and endothelial cells, and pathogenic potential in several infection models (D’Souza & Heitman, 2001, Leberer et al., 2001, Bahn & Sundstrom, 2001, Cassola et al., 2004, Cloutier et al., 2003). Adenylyl cyclase is thus considered a central integrator of key signals that ultimately governs the way that C. albicans interacts with its host and its competitors (Hogan & Muhlschlegel, 2011).
The intracellular responses to cyclic AMP pathway are mediated by two catalytic subunits of cyclic AMP-dependent protein kinase, Tpk1 and Tpk2 (Cloutier et al., 2003). These protein kinases are activated by the cyclic AMP-induced dissociation of the regulatory subunit, Bcy1 (Cassola et al., 2004), a highly conserved mechanism (D’Souza & Heitman, 2001). Tpk1 and Tpk2 then phosphorylate targets to effect changes in cell biology and gene expression.
Tpk1 and Tpk2 have distinct functions for the most part, as assessed by comparison of tpk1Δ/tpk1Δ and tpk2Δ/tpk2Δ mutant phenotypes. Tpk2 is required for hyphal development in liquid (Bockmuhl et al., 2001) while Tpk1 is required only for hyphal formation on solid media (Bockmuhl et al., 2001). Tpk2 is required for agar invasion; Tpk1 is not (Bockmuhl et al., 2001). Tpk2 governs the qualitative nature of filamentous cells (pseudohyphal to hyphal cell ratio), and thus impacts biofilm formation; Tpk1 does not (Giacometti et al., 2011). The two catalytic subunits also have contrasting roles in glycogen storage as well as resistance to saline, heat, and oxidative stresses (Giacometti et al., 2009). In general, Tpk2 has a more prominent role than Tpk1 in C. albicans biology, most likely because it is the more abundant isoform (Cloutier et al., 2003, Souto et al., 2006, Giacometti et al., 2011).
Our interest is in the C. albicans cell surface and how it contributes to biofilm formation and drug sensitivity. In that context, we find Tpk1 noteworthy, because it is required for normal levels of both resistance to the cell wall inhibitor caspofungin (Blankenship et al., 2010) and, as we show here, adherence to a silicone substrate. Tpk2 does not have measurable impact on either process. These observations prompted us to investigate the mechanistic basis for these roles of Tpk1. Our findings reinforce the idea that Tpk1 has a selective role in C. albicans biology. Most importantly, our results connect defined Tpk1 transcriptional targets to specific biological features, thus strengthening our functional understanding of a central regulatory pathway’s outputs and their relevance to infection biology.
RESULTS
Control of C. albicans silicone adherence by protein kinase genes
In order to define the genetic control of adherence to silicone, we assayed a panel of 70 insertion mutants in protein kinase-related (PK) genes (Blankenship et al., 2010) for altered adherence. A silicone substrate was used to represent the surface of implanted medical devices, such as a venous catheter. Mutants that were hyperfilamentous, aggregated, or grew poorly were not assayed (7 in total). We found that 22 PK mutants had significantly decreased cell-surface adherence, 5 had increased adherence, and 36 showed no significant difference from the wild type under our assay conditions (Figure 1A). Hence, a large fraction of PK mutants have phenotypic impact in this assay. These results are consistent with previous studies in which a high frequency of pronounced phenotypes were found among PK mutants of C. albicans (Blankenship et al., 2010), S. pombe (Bimbo et al., 2005) and N. crassa (Park et al., 2011).
Figure 1. Cell-surface adherence of C. albicans PK mutants.

(A) Adherence to silicone was measured using the Fluxion flow assay. The first bar on the graph represents wild-type level of adherence. Error bars represent standard deviation values. Mutants that filamented, had severe growth defects or aggregated were not assayed; these included homozygous insertion mutants for PBS2, GIN4, Orf19.1874, IME2, KIS1, SWE1, and CBK1. The wild-type strain is DAY286. (B) Adherence to silicone of wild type, tpk1Δ/tpk1Δ and tpk1Δ/tpk1Δ+TPK1Δ strains was measured using the Fluxion flow assay. ** denotes p=0.0011 for wild type versus tpk1Δ/tpk1Δ. The strains used were DAY185, SF1130, and SF1119.
A previous study indicated that a large fraction of PK mutants had defects in cell wall integrity, as evidenced by hypersensitivity to caspofungin (Blankenship et al., 2010). We considered the possibility that cell wall defects and altered adherence may be linked. For example, cell wall perturbation induces ALS1 expression (Blankenship et al., 2010, Bruno et al., 2006, Gregori et al., 2011), and Als1 promotes adherence to silicone (Finkel et al., 2012). In fact, over 29% of the PK mutants with altered adherence are also hypersensitive to cell wall perturbation, including 4 out of 5 of the PK mutants with increased adherence. This finding suggests that altered adherence and cell wall defects may be functionally related.
Elevated adherence has seldom been investigated in C. albicans, and we reasoned that this phenotype may provide unique insight into cell wall regulatory mechanisms. We focused in particular on the tpk1::Tn/tpk1::Tn insertion mutant, because it displayed the most pronounced increase in adherence (Figure 1A). The tpk1::Tn/tpk1::Tn insertion mutant phenotype differed from that of the tpk2::Tn/tpk2::Tn insertion mutant (Figure 1A), thus suggesting that this phenotype is one more reflection of the divergent functions of Tpk1 and Tpk2. We verified the hyperadherent phenotype with assays of a derivative of the original “Ura-blaster” tpk1Δ/tpk1Δ mutant (strain HPY300U), with multiple independent tpk1Δ/tpk1Δ deletion mutants in either the SC5314 or BWP17 strain backgrounds, and through complementation tests (Figure 1B and data not shown). Therefore, Tpk1 is a negative regulator of silicone adherence.
Tpk1 function in biofilm formation
Surface adherence can lead to biofilm formation. Therefore we sought to determine whether increased adherence of a tpk1Δ/tpk1Δ mutant might affect biofilm formation. Wild-type strains form biofilms efficiently under our assay conditions; few planktonic cells are detectable in biofilm supernatants (Nobile et al., 2006b). Thus an increase in adherence over the wild type would not be detectable as an improvement in biofilm formation. In addition, we observed no unusual structural features of tpk1Δ/tpk1Δ mutant biofilms (data not shown). Therefore we turned to a bcr1Δ/bcr1Δ mutant background, which is defective in adherence and biofilm formation. Bcr1 is a transcription factor that is required for expression of numerous cell surface protein genes (Finkel et al., 2012). We reasoned that, if a tpk1Δ/tpk1Δ defect led to increased adherence in a bcr1Δ/bcr1Δ mutant background, then an improvement in biofilm formation might be detectable.
To test this prediction, we compared biofilm formation among a wild-type reference strain, a bcr1Δ/bcr1Δ mutant, a bcr1Δ/bcr1Δ tpk1Δ/tpk1Δ double mutant, and a bcr1Δ/bcr1Δ tpk1Δ/tpk1Δ+TPK1 complemented control strain. The ability to form a biofilm was severely impaired in the bcr1Δ/bcr1Δ strain, as visualized by scanning electron microscopy (SEM), but was restored in bcr1Δ/bcr1Δ tpk1Δ/tpk1Δ double mutant (Figure 2A, B). The complemented bcr1Δ/bcr1Δ tpk1Δ/tpk1Δ+TPK1 strain behaved similarly to the bcr1Δ/bcr1Δ mutant, and thus confirmed that the tpk1Δ/tpk1Δ mutation was responsible for improved biofilm formation (Figure 2A, B). These findings suggest the hypothesis that the increased adherence resulting from a defect in TPK1 can improve biofilm formation.
Figure 2. Relationship between Tpk1 and Bcr1 in biofilm formation.

(A) Apical and cross-sectional views of biofilms grown on silicone squares for 2 days at 37°C in Spider medium, visualized by scanning electron microscopy. (B) Biofilm biomass measurements. Strain genotypes are given beneath each image. The strains used were DAY185, CJN702, SF1151, and SF1147.
The analysis of double mutants is in essence an epistasis test, and the results above show that a tpk1Δ/tpk1Δ defect is epistatic to a bcr1Δ/bcr1Δ defect for the biofilm formation phenotype. Epistasis is often interpreted in the context of gene regulation (Roth et al., 2009). Therefore, we also examined epistasis at the level of gene expression. Specifically, we measured expression of a panel of 117 genes through nanoString analysis of RNA levels (Geiss et al., 2008). Our gene set included several Bcr1- and stress-responsive genes as well as the control gene TDH3 for normalization purposes (Table S1). Cells were grown in our biofilm medium (Spider medium) at 37°C; conditions used previously for microarray and nanoString analysis of the bcr1Δ/bcr1Δ strain (Nobile & Mitchell, 2005, Finkel et al., 2012). We compared gene expression levels among four strains: the wild type, a tpk1Δ/tpk1Δ mutant, a bcr1/bcr1Δ mutant, and a bcr11Δ/bcr1Δ tpk1Δ/tpk1Δ double mutant. We used the combined expression levels of all genes in the set as a basis for determining epistasis. Comparison of gene expression in the tpk1Δ/tpk1Δ and bcr1Δ/bcr1Δ strains (Figure 3A) shows many substantial differences in expression levels. Agreement or divergence between these two expression datasets can be represented by the correlation coefficient (R2) value of 0.53. When we compared the tpk1Δ/tpk1Δ bcr1Δ/bcr1Δ double mutant to the tpk1Δ/tpk1Δ mutant, we obtained an R2 value of 0.67. The R2 value of 0.67 is greater than 0.53; a simple interpretation is that the tpk1Δ/tpk1Δ phenotype is partially manifested in the double mutant, and thus that the tpk1Δ/tpk1Δ defect is partially epistatic to the bcr1Δ/bcr1Δ defect. When we compared the tpk1Δ/tpk1Δ bcr1Δ/bcr1Δ double mutant to the bcr1Δ/bcr1Δ mutant, we obtained an R2 value of 0.83 (Figure 3C). Following our previous logic, we infer that the bcr1Δ/bcr1Δ phenotype is partially manifested in the double mutant, and thus that the bcr1Δ/bcr1Δ defect is partially epistatic to the tpk1Δ/tpk1Δ defect. Comparison of the double mutant to the wild-type strain indicates that the two mutations to not precisely counterbalance each other (Figure 3D; R2 of 0.67). Therefore, each mutation is partially epistatic to the other at the level of gene expression. This finding is not consistent with the idea that Tpk1 and Bcr1 act at successive steps of a single pathway. Instead, it is consistent with a model in which Tpk1 and Bcr1 act, for the most part, independently of one another.
Figure 3. Relationship between Tpk1 and Bcr1 in gene expression.

Expression data was generated by nanoString measurement (Table S1) using probes for 117 genes. RNA levels for each gene were normalized to internal control TDH3 RNA levels, and the mean of three biological replicates for each gene was plotted here. (A) Normalized gene expression in a bcr1Δ/bcr1Δ strain (x-axis) versus a tpk1Δ/tpk1 strain (y-axis). (B) Normalized gene expression in a bcr1Δ/bcr1Δ tpk1Δ/tpk1 strain (x-axis) versus a tpk1Δ/tpk1 strain (y-axis). (C) Normalized gene expression in a bcr1Δ/bcr1Δ tpk1Δ/tpk1 strain (x-axis) versus a bcr1Δ/bcr1Δ strain (y-axis). (D) Normalized gene expression in a wild-type strain (x-axis) versus a bcr1Δ/bcr1Δ tpk1Δ/tpk1 strain (y-axis). The strains used were DAY185, CJN702, SF1151, and SF1130.
Identification of TPK- regulated genes
Our findings above suggest that Tpk1 affects a different pathway from Bcr1. Therefore, we sought to define Tpk1 pathway target genes on a genome-wide scale. We conducted a microarray comparison between the tpk1::Tn/tpk1::Tn insertion mutant and the tpk1::Tn/tpk1::Tn+TPK1 complemented strain (NCBI GEO database: GSE38846). Fourteen of the targets identified by microarray were verified by RT-PCR (Figure 4A). Interestingly, 8 out of the 14 genes that were most up-regulated in the tpk1::Tn/tpk1::Tn mutant specify known or predicted cell wall or cell surface localized proteins, including adhesins Als1, Als2, and Als4. We verified that ALS1, ALS2, and ALS4 RNA levels were up-regulated in the Ura-blaster tpk1Δ/tpk1Δ isolate (data not shown). Because our interest is in phenotypes related to the cell wall and cell surface, we focused on the up-regulated genes for functional analysis.
Figure 4. Effect of Tpk1 target genes on adherence and biofilm formation in a bcr1Δ/ bcr1Δ background.

(A) Gene expression measurements for the 14 most highly Tpk1-repressed identified by microarray analysis. The bar graph shows results of QRTPCR assays that were conducted in triplicate samples of the wild type, complemented, and mutant strains. The strains used were DAY286, JJH384, and JJH233. (B and C) Each of the most highly Tpk1-repressed genes was overexpressed from the TDH3 promoter in a bcf1Δ/bcr1Δ background, and strains were assayed for (B) adherence, using a Fluxion flow assay, and (C) biofilm biomass. Error bars represent standard deviation. Statistical significance is relative to bcr1Δ/bcr1Δ. *p≤0.05, **p≤0.005, ***p≤0.001, ***p≤0.0005. The strains used were DAY185, CJN702, SF1255, SF1258, SF1303, SF1294, SF1259, SF1300, SF1284, SF1264, SF1253, SF1275, SF1257, SF1297, and SF1295.
Relationship of Tpk1-repressed genes to adherence
We considered the specific hypothesis that Tpk1 defects cause increased adherence through up-regulation of genes that promote adherence. To test this hypothesis, we created derivatives of the adherence-defective bcr1Δ/bcr1Δ mutant that overexpressed individual Tpk1-repressed genes and assayed each strain for adherence and biofilm formation. We observed that overexpression of ALS1, CSH1, CSP37, ALS2, or ALS4 caused a significant increase in adherence in the bcr1Δ/bcr1Δ background (Figure 4B). In addition, overexpression of each of the five genes in the bcr1Δ/bcr1Δ mutant improved biofilm formation substantially, as determined by SEM visualization (Figure 5) and, in most cases, biofilm biomass measurements (Figure 4C). CSH1 was the one anomalous case: biofilms of the CSH1-overexpressing bcr1Δ/bcr1Δ mutant fragmented under typical handling conditions, so an increase in biomass could not be documented (Figure 4C). Although some of the strains that overexpressed other Tpk1-repressed genes seemed to trend toward increased adherence or biofilm formation, these increases were not statistically significant. We conclude that five Tpk1-repressed genes, ALS1, CSH1, CSP37, ALS2, and ALS4, promote substrate adherence and biofilm formation.
Figure 5. Structure of biofilms of bcr1Δ/bcr1Δ strains that overexpress Tpk1-repressed genes.

Biofilms on silicone squares were visualized in cross sectional views by scanning electron microscopy. Genotypes are given beneath each panel. The strains used were DAY185, CJN702, SF1258, SF1255, SF1271, SF1294, and SF1303.
Relationship of Tpk1-repressed genes to cell wall integrity
We reported previously that a tpk1::Tn/tpk1::Tn insertion mutant is hypersensitive to caspofungin (Blankenship et al., 2010). Our isolate of the original Ura-blaster tpk1Δ/tpk1Δ mutant did not share this phenotype. To determine whether Tpk1 is required for caspofungin tolerance, we examined multiple independent insertion and deletion mutants in both the BWP17 and SC5314 backgrounds. All were caspofungin hypersensitive, and the phenotype was rescued by complementation (Figure 6A and (Blankenship et al., 2010)). The complementation results indicate that Tpk1 defects are the cause of the phenotype. We first considered the possibility that Tpk1 may be required for cell wall integrity gene expression. Microarray analysis indicated that all of the genes that were most highly up- or down-regulated in response to caspofungin in the complemented strain responded similarly in the tpk1::Tn/tpk1::Tn mutant (Figure 7 and NCBI GEO database: GSE38846). In a few cases in which the mutant did not show a significant change in expression of a gene (white bars in Figure 7), the trends and mean expression changes were still similar to that of the complemented strain. Therefore, Tpk1 is not required for cell wall integrity gene expression.
Figure 6. Role of Tpk1 and Tpk1-repressed genes in cell wall integrity.

Serial spot-dilution assays were used to compare growth of strains on control YPD (left panels) and YPD + caspofungin (right panels). (A) Comparison of wild-type (DAY185), tpk1Δ/tpk1Δ mutant, and tpk1Δ/tpk1Δ+TPK1 complemented strains in the BWP17 background. The strains used were DAY185, SF1130, and SF1119. (B) Comparison of wild-type strain SC5314 and its derivatives that overexpress individual Tpk1-repressed genes. The strains used were SC5314, SF1127, SF1190, SF1160, SF1204, SF1129, SF1242, SF1306, SF1280, SF1236, SF1161, SF1279, SF1245, SF1290, and SF1265.
Figure 7. Caspofungin-responsive gene expression in tpk1::Tn/tpk1::Tn mutant and tpk1::Tn/tpk1::Tn+TPK1 complemented strains.

Microarray analysis was used to identify the most highly caspofungin-responsive genes in the tpk1::Tn/tpk1::Tn+TPK1 complemented strain. The fold change, relative to expression in the absence of caspofungin, is presented for both the tpk1::Tn/tpk1::Tn mutant and tpk1::Tn/tpk1::Tn+TPK1 complemented strains. The graph represents significant responses of the complement strain, significant responses of the mutant strain and responses in the mutant that are not statistically significant relative to untreated cells. The strains used were JJH384 and JJH233. Error bars indicate standard deviation values.
We considered a second explanation for the tpk1/tpk1 mutant caspofungin hypersensitivity: altered expression of cell wall and cell surface proteins in tpk1/tpk1 mutants may perturb cell wall structure and cause caspofungin hypersensitivity. To test this hypothesis, we created a panel of strains that overexpressed the top Tpk1-repressed genes in an otherwise wild type background, and assayed their sensitivity to caspofungin. Strikingly, overexpression of ALS2 specifically conferred substantial caspofungin hypersensitivity (Figure 6B). We observed the same phenotype among several ALS2 overexpressing transformants (data not shown). In addition, among the bcr1Δ/bcr1Δ strains that overexpress Tpk1-repressed genes, we observed caspofungin hypersensitivity specifically when ALS2 was overexpressed (data not shown). These findings suggest that one role of Tpk1 in cell wall integrity is to prevent ALS2 expression.
It seemed possible that the cell wall perturbation conferred by a Tpk1 defect might be evident by electron microscopy. Indeed, this analysis revealed that the tpk1Δ/tpk1Δ mutant has a cell wall that is half the depth of the wild-type cell wall (Figure 8A, B). This mutant phenotype was complemented by introduction of a wild-type TPK1 allele (Figure 8C). The shallow cell wall phenotype was also manifested by the strain that overexpressed ALS2 (Figure 8D), but not the related gene ALS4 (Figure 8F). Consistent with the findings of Hoyer and colleagues (Zhao et al., 2005), we were unable to construct a homozygous deletion mutant of ALS2. Deletion of one allele of ALS2 resulted in a heterozygous ALS2 strain with a cell wall depth significantly greater than the wild-type strain (Figure 8E). Overexpression of the other Tpk1-repressed ALS gene, ALS1, caused cell wall depth that was intermediate between that of the tpk1Δ/Δ mutant and the wild type (Figure S1).
Figure 8. Cell wall depth visualization.

Transmission electron microscopy of sectioned cells was used to determine cell wall thickness on 10 unselected cells of each strain, with means and standard deviations indicated. Representative images are shown. (A) The wild-type (SC5314) cell wall measurement was 92nm+/−22nm. (B) The tpk1Δ/tpk1Δ (SF1358) cell wall measurement was 48nm+/−22nm. p=<0.0001 relative to wild type. (C) The tpk1Δ/tpk1Δ +TPK1 (SF1119) cell wall measurement was 74nm+/−31nm. p=0.04 relative to tpk1Δ/tpk1Δ and not statistically significant (p=0.08) from wild type. (D) The wild-type strain overexpressing ALS2 (SF1127) cell wall measurement was 39nm +/− 14nm. p=<0.0001 relative to wild-type. (E) A heterozygous ALS2 deletion strain (SF1332) had a cell wall measurement of 111nm +/− 29nm. p=0.04 relative to wild type. (F) The wild-type strain overexpressing ALS4 (SF1129) cell wall measurement was 75nm +/− 24nm. p= 0.06 relative to wild type.
These results indicate that Tpk1 promotes both cell wall integrity and cell wall depth, and that Tpk1-dependent repression of ALS2 is one mechanism that mediates both functions.
DISCUSSION
Cell-substrate adherence is pivotal for biofilm formation, a major source of C. albicans infection (Finkel & Mitchell, 2011). We have described here a quantitative characterization of protein kinase mutants that identified many new regulators of adherence. Our findings define a function for many protein kinase genes that have not been extensively characterized and, in addition, connect adherence for the first time to several known signalling and cell biological pathways. We have focused in particular on Tpk1, and discern a novel connection between adherence, cell wall integrity, and cell wall structure. Several Tpk1-responsive genes contribute to adherence, and one Tpk1-responsive gene, the adhesin gene ALS2, has significant roles in adherence as well as cell wall integrity and structure (Figure 9).
Figure 9. Model for control of cell wall functions by Tpk1.

Tpk1, one of the cyclic AMP-dependent protein kinase catalytic subunits, governs adherence, cell wall integrity, and cell wall structure. Our findings argue that many of these roles are mediated by Tpk1-dependent repression of select target genes. The target genes ALS1, ALS4, CSH1, and CSP37 act to promote adherence. The target gene ALS2 acts to promote adherence and also governs cell wall integrity and structure.
Previous analysis of tpk1/tpk1 mutants has implicated Tpk1 in diverse metabolic and cellular processes (Souto et al., 2006, Giacometti et al., 2009, Giacometti et al., 2011, Bockmuhl et al., 2001, Cloutier et al., 2003, Cassola et al., 2004, Park et al., 2005). Stress sensitivity is one general theme, and our microarray data provide some functional insight. For example, the tpk1/tpk1 glycogen deficiency (Giacometti et al., 2009) may arise from increased expression of GBD1, which specifies a glycogen debranching enzyme that promotes glycogen degradation (Teste et al., 2000). The tpk1/tpk1 salt sensitivity (Giacometti et al., 2009) may arise from increased expression of GAC1, which specifies a protein phosphatase regulatory subunit whose S. cerevisiae ortholog, when overexpressed, confers salt sensitivity (Wu et al., 2001). We cannot point to a cause of the tpk1/tpk1 peroxide sensitivity (Giacometti et al., 2009), but elevated expression of oxidative stress genes HSP12, GPX2, and orf19.251 may result from elevated endogenous reactive oxygen species levels. Cell-surface interaction is another recurring theme among Tpk1-responsive properties. Tpk1 was known to promote formation of hyphae on solid media (Bockmuhl et al., 2001), not in liquid, and our studies here show that Tpk1 governs surface adherence as well. Interestingly, our microarray data provide convergent evidence for a close Tpk1-cell wall connection, which is related logically to cell-surface interaction. Among 33 significantly up-regulated genes in the tpk1::Tn/tpk1::Tn mutant, there was pronounced enrichment for cell wall-related functions (GO term “fungal-type cell wall,” 8 genes, P-value 4.29 × 10−6). Thus Tpk1 is tied to cell surface properties through both cellular phenotypes and gene expression targets.
Tpk1 is well established as one of the two cyclic AMP-dependent protein kinase catalytic subunits in C. albicans (Cloutier et al., 2003), yet we have found no alteration of adherence or biofilm formation in mutants of the upstream genes CDC25 (orf19.6926) and CDC35/CYR1 (orf19.5148) (unpublished results). We have also used gene expression changes as a measure of phenotype. Among the 46 genes that are up-regulated at least 2-fold in the tpk1::Tn/tpk1::Tn mutant, we have microarray data from a cdc35Δ/cdc35Δ for 23 genes (Harcus et al., 2004). Among those 23 genes, almost all are up-regulated in the cdc35Δ/cdc35Δ mutant compared to the wild type (Figure S2), including CSP37, ECM331, and IHD1. This correlation between Tpk1-repressed genes and Cdc35-repressed genes is consistent with the current understanding of the cyclic AMP pathway. Given that Tpk2 can compensate for the loss of Tpk1 (Bockmuhl et al., 2001), it seems reasonable that some phenotypes of tpk1/tpk1 mutants may be distinct from those of cdc35/cdc35 mutants.
We have used a bcr1Δ/bcr1Δ mutant as an analytical tool in our studies because it has reduced adherence and biofilm formation capability. Nonetheless, because Bcr1 and Tpk1 govern related phenotypes, it may seem possible that they act in the same pathway. Our data provide no support for that idea. Specifically, our nanoString data show little correlation between the genes up- and down-regulated in the two mutants. One of the few prominent exceptions is ALS1, which is down-regulated in the bcr1Δ/bcr1Δ mutant and up-regulated in the tpk1Δ/tpk1Δ mutant (see Table S1). If we examine ALS1 expression specifically, the bcr1Δ/bcr1Δ phenotype is epistatic to the tpk1Δ/tpk1Δ phenotype, in keeping with the idea that Bcr1 functions downstream of Tpk1. That conclusion is consistent with the genome-wide chromatin immunoprecipitation data from Nobile and colleagues (Nobile et al., 2012), which indicates that Bcr1 binds directly to the ALS1 promoter. However, those data also indicate that four additional transcription factors bind to the ALS1 promoter, including the known cyclic AMP pathway target Efg1 (Sonneborn et al., 2000, Bockmuhl & Ernst, 2001, Stoldt et al., 1997). Thus the ALS1 promoter may be a region at which multiple signalling pathways interact, and thus a challenging choice for deductions about pathway relationships. Our overall inference, based on non-correlation of Tpk1- and Bcr1-responsive genes, is that these two gene products largely act independently to govern gene expression.
Analysis of the Tpk1-repressed genes in the chromatin immunoprecipitation dataset from Nobile and colleagues (Nobile et al., 2012) has the potential to define the transcription factors that mediate regulation by Tpk1. Among the 14 Tpk1-repressed genes in Figure 4A, six genes had no detectable binding of any transcription factor studied, two genes were bound only by Ndt80, one gene was bound only by Tec1, one gene was bound by both Ndt80 and Tec1, and four genes were bound by Ndt80, Efg1, Bcr1, and some combination of Brg1 and Tec1. Thus there is no unequivocal candidate for a mediator of Tpk1 responsiveness, but Ndt80 binds to more of these genes than any other transcription factor examined (Nobile et al., 2012). A simple model is that Ndt80 protein levels or activity increases in the absence of Tpk1, and activates a large subset of Tpk1-repressed genes.
Functional analysis indicates that several Tpk1-repressed gene products contribute to adherence. Among them, Als1, Als2, and Als4 are canonical adhesins, in that they are large, GPI-linked cell surface proteins. All three, like other Als family members, have been shown to mediate adherence to several substrates, using both overexpression approaches and underexpression or null mutant approaches (Finkel et al., 2012, Nobile et al., 2006a, Kamai et al., 2002, Zhao et al., 2004, Green et al., 2004, Spellberg et al., 2005, Nobile et al., 2008a, Dwivedi et al., 2011). The other relevant targets, Csh1 and Csp37, do not have structural features of known adhesins, such as a signal sequence or GPI anchor addition signal. However, both have been found associated with the cell wall (Urban et al., 2003, Pitarch et al., 2002, Singleton et al., 2001), so their localization is consistent with a direct role in adherence. In addition, deletion mutants of either gene display reduced adherence (Singleton et al., 2001, Sentandreu et al., 1997). Finally, both genes are down-regulated in several recently characterized biofilm-defective mutants (Nobile et al., 2012). Thus our conclusions regarding Tpk1-repressed genes that govern adherence, based on overexpression assays in a bcr1Δ/bcr1Δ mutant, are consistent with functional assignments from independent studies and null mutant analysis. The mechanistic connection between Tpk1 and adherence fits a well-accepted paradigm: inactivation of Tpk1 causes elevated expression of several surface proteins that may mediate surface interaction through direct binding or, perhaps, through more global effects on cell wall structure.
Tpk1-mediated cell wall integrity has two surprising features. First, Tpk1 seems to have no role in the cell wall integrity gene expression response: the mutant and complemented strains both induced and repressed the same genes after caspofungin treatment. Second, our studies point toward a single cell surface protein, Als2, whose increased expression mediates caspofungin hypersensitivity and the correlated phenotype, a shallow cell wall. A heterozygous deletion of ALS2 yielded a cell wall depth thicker than the wild type. A correlation between cell wall depth and integrity was first established by Plaine et al. (Plaine et al., 2008) in a survey of cell wall protein gene mutants. The intriguing feature of the cell wall aberration in the tpk1/tpk1 mutant is that it apparently does not trigger a cell wall integrity gene expression response. Many other caspofungin-hypersensitive regulatory mutants have a partially activated response (Blankenship et al., 2010), as expected if their cell wall biosynthetic defect generates the same regulatory signal as addition of an exogenous cell wall inhibitor. Apparently one kind of chronic cell wall defect remains undetected by the C. albicans cell, as assayed by levels of gene expression.
Our observations point toward a novel function for Als2 in cell wall structure. There is extensive similarity between Als2 and Als4 proteins as well as their respective genetic loci (Zhao et al., 2005, Hoyer et al., 2008). However, only one of the two ALS2 alleles could be deleted by Zhao et al. (Zhao et al., 2005), and they proposed that Als2 may be essential for viability. Our findings, based on ALS2 overexpression, suggest specifically that the essential role of Als2 may be in cell wall biogenesis. One might argue that overexpression of any cell surface protein could cause such cell wall aberrations for nonspecific reasons, unrelated to natural protein function. However, Als4 is ~60% identical to Als2, yet its overexpression does not affect either caspofungin sensitivity or cell wall depth. One might suppose that TDH3-ALS2 is overexpressed to a much greater extent than TDH3-ALS1 or TDH3-ALS4, but in fact the opposite is true: TDH3-ALS2 increased ALS2 RNA levels 4-fold, whereas TDH3-ALS1 and TDH3-ALS4 increased ALS1 and ALS4 RNA levels 77- and 97-fold, respectively (unpublished results). Thus our results argue that effects of ALS2 overexpression are specific, and connect Als2 to a process that is essential for viability.
Most studies of Tpk1 and Tpk2 have revealed many prominent roles for Tpk2 yet few for Tpk1 (Souto et al., 2006, Giacometti et al., 2009, Giacometti et al., 2011, Bockmuhl et al., 2001, Cloutier et al., 2003, Cassola et al., 2004, Park et al., 2005). Our findings help to understand this distinction. First, our microarray data reveal that a fairly small set of genes responds to Tpk1 deficiency in growing cells. In fact, a Tpk1 deficiency has little gene expression impact on the perturbations caused by a Bcr1 defect or addition of a cell wall inhibitor. Second, we show that Tpk1 deficiency has profound impact on adherence, but causes a phenotype that is not easily detected as a defect. Rather, the hyperadherence is most readily detected as a restoration of biofilm formation ability. Together, our findings suggest that the cell wall is a major target of Tpk1 activity, and that much of this role is mediated by a few selective Tpk1-responsive gene expression changes.
EXPERIMENTAL PROCEDURES
Media and Strain Construction
C. albicans strains were grown on yeast extract-peptone dextrose (YPD) (2% Bacto Peptone, 2% dextrose,1% yeast extract and supplemented with 80μg/ml uridine for Ura− strains) or on defined synthetic dextrose medium (2% dextrose, 6.7% YNB with ammonium sulfate, and auxotrophic supplements). For transformation procedures cells were selected on SC medium minus the appropriate amino acid(s). caspofungin (Merck) was added to media at 125ng/ml.
All C. albicans strains used in this study are listed in Table 1 and Table S2 and primer sequences are listed in Table 2. Protein kinase insertion mutant strains utilized were generated in (Blankenship et al., 2010) and are listed in Table S2. Unless otherwise indicated, newly constructed strains were derived from BWP17 (Wilson et al., 1999). Strain SF1130, a prototrophic deletion (tpk1Δ/tpk1Δ), was constructed by PCR-directed gene deletion (Wilson et al., 1999) employing long oligonucleotides TPK1-5DR and TPK1-3DR. A double deletion of BCR1 and TPK1, SF1151, was constructed by deleting both TPK1 alleles using PCR directed gene deletion in an auxotrophic bcr1Δ/bcr1Δ (SF803) strain.
Table 1.
Strains used in this study
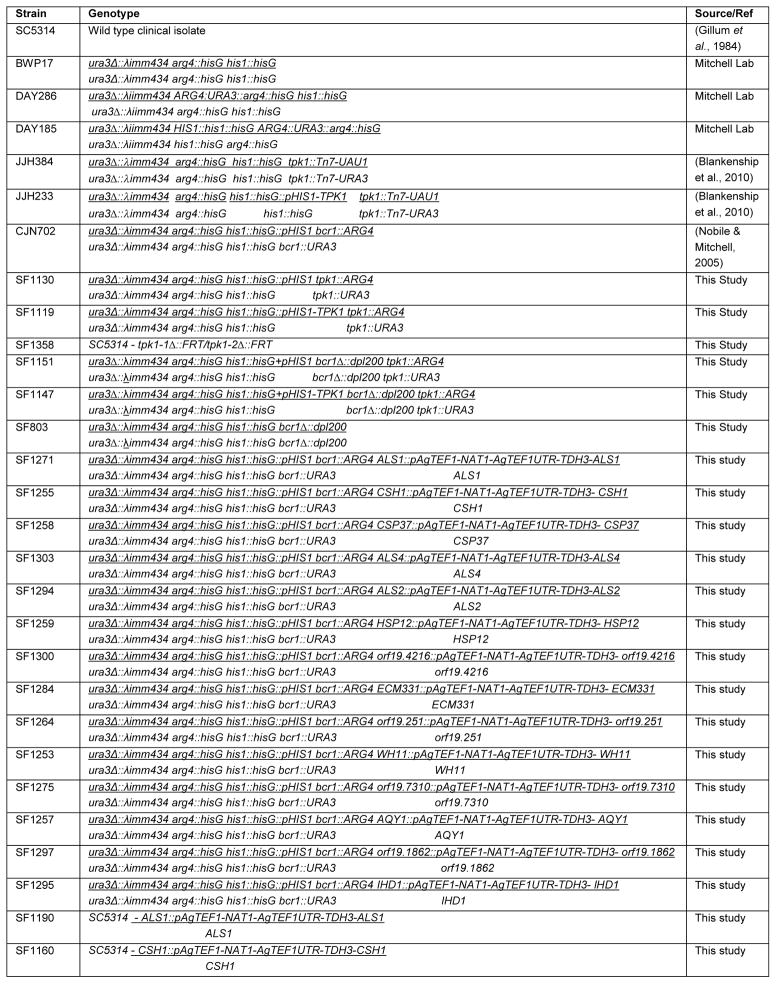
|
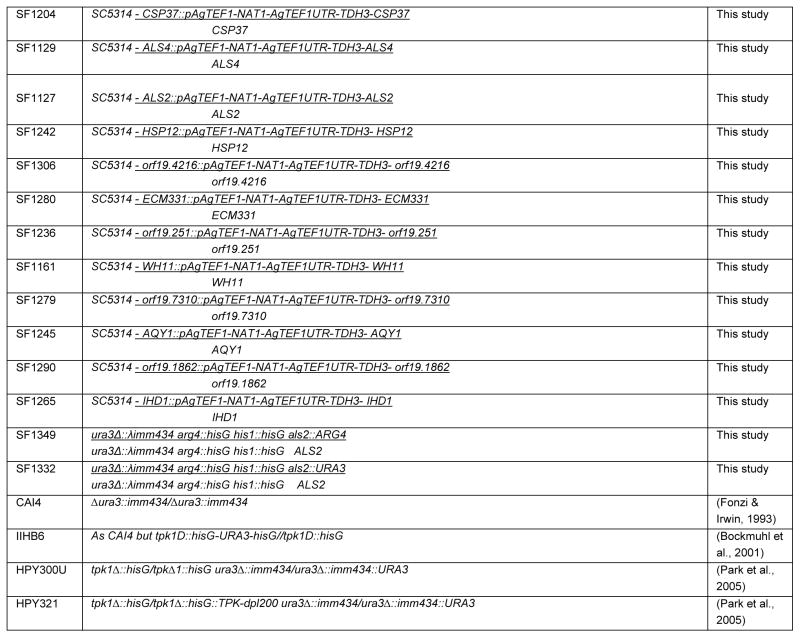 |
Table 2.
Primers used in this study
| Primer Name | Primer Sequence | |
|---|---|---|
| TPK1-5DR | AGATCAATATATCCTATCGTTATCCTCCTCCTTCTCCTTTCAACTTTTGAAAAAGGTGATATTATTCAACACTGTTTCTGTTTTATCAACCAAAACCAGGTTTCCCAGTCACGACGTT | This study |
| TPK1-3DR | TCTTTTTTCTTGTTCTTTCTTGCAAATTAATTATACATCTATAAAACTAGTTATCATAATTAACATTGTTGTGCCAATAAATACAATTTTATTTTTACATGTGGAATTGTGAGCGGATA | This study |
| TDH3F | ATCCCACAAGGACTGGAGA | Blankenship et al., 2010 |
| TDH3R | GCAGAAGCTTTAGCAACGTG | Blankenship et al., 2010 |
| ALS1F | GACTAGTGAACCAACAAATACCAGA | Green et al., 2004 |
| ALS1R | CCAGAAGAAACAGCAGGTGA | Green et al., 2004 |
| ALS2F | CCAAGTATTAACAAAGTTTCAATCACTTAT | Green et al., 2004 |
| ALS2R | TCTCAATCTTAAATTGAACGGCTTAC | Green et al., 2004 |
| ALS3F | CCACTTCACAATCCCCATC | Green et al., 2004 |
| ALS3R | CAGCAGTAGTAGTAACAGTAGTAGTTTCATC | Green et al., 2004 |
| ALS4F | CCCAGTCTTTCACAAGCAGTAAAT | Green et al., 2004 |
| ALS4R | GTAAATGAGTCATCAACAGAAGCC | Green et al., 2004 |
| ALS5F | TGACTACTTCCAGATTTATGCCGAG | Green et al., 2004 |
| ALS5R | ATTGATACTGGTTATTATCTGAGGGAGAAA | Green et al., 2004 |
| ALS6F | GACTCCACAATCATCTAGTAGCTTGGTTT | Green et al., 2004 |
| ALS6R | CAATTGTCACATCATCTTTTGTTGC | Green et al., 2004 |
| ALS7F | GAAGAGAACTAGCGTTTGGTCTAGTTGT | Green et al., 2004 |
| ALS7R | TGGCATACTCCAATCATTTATTTCA | Green et al., 2004 |
| ALS9F | CCATATTCAGAAACAAAGGGTTC | Green et al., 2004 |
| ALS9R | AACTGAAACTGCTGGATTTGG | Green et al., 2004 |
| ORF19.251F | GTCATGGTCCTGCCATTTTT | This study |
| ORF19.251R | AGCACCTTCCTTTTCAGCAA | This study |
| ORF19.4477F | TGGTGTTTTGTGTCGTCCAT | This study |
| ORF19.4477R | CACCATGCCAATGAAACTTG | This study |
| ORF19.7310F | TCAGCAACAAGGTTGACGAG | This study |
| ORF19.7310R | GAGTAGCCTGAGACCCAACG | This study |
| ORF19.1862F | ACAACGTGGTGCTGAAGGTA | This study |
| ORF19.1862R | GTGGGATCCACCAATTTCAC | This study |
| ORF19.5760F | TGATGCCTCAGGTACTGCTG | This study |
| ORF19.5760R | CCTGAAGCAGCAGTTGTCAT | This study |
| ORF19.3160F | AAAGGGCAAGGAACAAGTCA | This study |
| ORF19.3160R | TTCAGCAGCCTTTCCAATTT | This study |
| ORF19.3548.1F | TTGGTGATAAAATCGAATCCAA | This study |
| ORF19.3548.1R | GCATCAGAAGCTTTTTGGACA | This study |
| ORF19.2531F | TGAAAAATGACGCCGATAAA | This study |
| ORF19.2531R | TCGTAATGTTTGTGCCATTCA | This study |
| ORF19.4216F | AAAGGGCAAGGAACAAGTCA | This study |
| ORF19.4216R | TTCAGCAGCCTTTCCAATTT | This study |
| ORF19.2849F | TTGCTAACCAAGACCCAACC | This study |
| ORF19.2849R | AATTGGTGGGACAGCTTGAG | This study |
| ECM331F | CCAAACTTGAAATCCGTTGG | This study |
| ECM331R | TGGACCAGAAATGGTGACAG | This study |
| AQY1F | TTGCTAACCAAGACCCAACC | This study |
| AQY1R | AATTGGTGGGACAGCTTGAG | This study |
| HSP12-O/EXP-F | GTGATAATAAGTGTGAAGAGTAGTAATTGAAGTTTGGTGTGTCTAGGTGGTGTTTAAGACTGTTTAGTATTTTCGCAAGAATTTTGTTAACTTTGTGTGTATCAAGCTTGCCTCGTCCCC | This study |
| HSP12-O/EXP-R | TGGTGTTTGTCGAAAAAAAACAAGATTCAAAGAATTTACCTCTACTAGAGAAATTGACTCTATTTATATTGTTTTTCGAGAGGGGGGCAAAGGTTGCCATATTTGAATTCAATTGTGATG | This study |
| ORF19.7310-O/EXP-F | CCTTGGTTTTCGGACATGAAAATTGATACTAATAAGTGTGTTTTTATTACCAAACCCAGTAAAAGTTGCAAATACAATTCTCATCAAAGTTTCTTTGAGAATCAAGCTTGCCTCGTCCCC | This study |
| ORF19.7310-O/EXP-R | AAAATTGTTTTAAATCATCATTTGTCCATTGATCAAAACCATAATTGGCAACCACAAAAGTGATAGATACATATAACCATACAATGATTGGGAAATACATATTTGAATTCAATTGTGATG | This study |
| IHD1-O/EXP-F | CTATTAAATTGACCCCATTACTACATAGATTTTTAGTTCATAGTCTTTATGGTTTGCTTTGTTTCTTTAATCGCATTGATAATTCAATATTTGTGTTACAATCAAGCTTGCCTCGTCCCC | This study |
| IHD1-O/EXP-R | TTTGGTATGCGTTAGAACAGTGGTCGTTACAAATTTGGTCAGCTCTTTTTGCGGCAAGAGCAATTTGTAAAAGTGCAAAGAATAATGGAGTTGGTCTCATATTTGAATTCAATTGTGATG | This study |
| ORF19.251-O/EXP-F | GCGTTGTTATATTGTTGATGAATCGAAGAAAACTAGATTACTAATTGAAATGGTACAAAATAGTTCAACCGTTTCAAGTTTTCAATTATCAGAATTTTCGATCAAGCTTGCCTCGTCCCC | This study |
| ORF19.251-O/EXP-R | CAAAAGGATGTAAAGCTTCAACAACGAAAACACCAGTCTTTTTACCGTCACTGTAAAAAGTTTCGTTATAAGAAGTAAGAGCGAGTAAAACTTTGACCATATTTGAATTCAATTGTGATG | This study |
| AQY1-O/EXP-F | TATACTTTCTAGAAATAAGAAAAGGGTTACCCTATCACTATTCTTCTCTTTGGTTAGTTACCAAAACCAAATTCTACTTCTTGGACAACTTCTTTTCCCT ATCAAGCTTGCCTCGTCCCC | This study |
| AQY1-O/EXP-R | GCGACACATTGACAGAATCGTCATATTTTGGTTCATAGACTGGTCTTTGAGCCTCAACGTCGTTAGGAGTATTATCAATGCTAGAACTTTCGGCAACCAT ATTTGAATTCAATTGTGATG | This study |
| ORF19.1862-O/EXP-F | TTGTGGATCCAATACAGCTATTGTAGTGGTACTTACTGAAATGTAATCACTTCGCATTATTCAAGATCTTTTTCTATTGTGGCTGGTTTTTGGTGATTGC ATCAAGCTTGCCTCGTCCCC | This study |
| ORF19.1862-O/EXP-R | TTATATTTGGCTAACAAATCGGCATCATCGATGAAAATGACAAAATCGTTTTCAGCACCTTTGTAGAATAATTTATGTGGAGAAGTTTTAGCCATATTTGAATTCAATTGTGATG | This study |
| ORF19.4216-O/EXP-F | AAACAGCCTAGTGATAATAAGTGTGAAGAGTAGTAATTGAAGTTTGGTGTGTCTAGGTGGTGTTTAAGACTGTTTAGTATTTTCGCAAGAATTTTGTTAACT ATCAAGCTTGCCTCGTCCCC | This study |
| ORF19.4216-O/EXP-R | TTATGGTGTATGTCGAAAAAAAACAAGATTCAAAGAATTTACCTCTACTAGAGAAATTGACTCTATTTATATTGTTTTTCGAGAGGGGGGCAAAGGTTGCCAT ATTTGAATTCAATTGTGATG | This study |
| CSP37-O/EXP-F | GAATAGTCACAGAGAGAGAGAGAAGGATTGCTATTTTTATTTTTTTGGGGGCATAATCACTTATTATACGTCACCACCACCACTAATGGATATATGATTC ATCAAGCTTGCCTCGTCCCC | This study |
| CSP37-O/EXP-R | GGTGTTGTTGTCTTGGTAAGATTGGTTGAACATATTGATCATAATAATAAACCCCACCGACAAGGGCAGCAGTACCTAATAAATATTTTCCAGCAGACATATTTGAATTCAATTGTGATG | This study |
| NAT-OE-R-DET | GAA ACA ACA ACG AAA CCA GC | Nobile et al., 2006 |
| ALS2-F-O/EXP | ACTTCTTCAAATACTAAAACGTTTTCACATAAACTGCAAGCCTCAAATTGATAGTATTCTACAGTATAAATGCTGAACTAACTTATTTTTACAATTACAA ATCAAGCTTGCCTCGTCCCC | This study |
| ALS2-R-O/EXP | TTGTCCATGTCAACGAATCAAAACTATTGAAAACACCCGTAATAACTTTTGCAGTAGCAACTGATACACAGAGGCTTAGCAACAAAAATTGTAAAAGCAT ATTTGAATTCAATTGTGATG | This study |
| ALS4-F-O/EXP | TTGATAGTTCTCTCCAGTATAAATGCTGAATTAACTCATTTTTACAATTACAAATTAGTAATTATGCATTTGAAAATGGTGCTTACGTTTGATTTCAATG ATCAAGCTTGCCTCGTCCCC | This study |
| ALS4-R-O/EXP | TGGTCCAAGTTAACGAATTAAAACTATCGAAAATGCCCGTAATAACCTTTGCCGTAGCAACTGATACACAGAGGCTTAGCAACAAAAATTGTAAAAGCAT ATTTGAATTCAATTGTGATG | This study |
| ALS1-O/EXP-F | GAATCTGCAATGAAAACGTTAAGACATTGGAATTTTTCATCAAATTTACGATGAATTGCTAATCATCTTTGGAGATATTCGTAGTAAGATCTTCAACCCA ATCAAGCTTGCCTCGTCCCC | This study |
| ALS1-O/EXP-R | TGGACCAAGTTAATGAATTAAAACTATCAAAAACACCAGTGATTGTCTTTGCACTTGCAATTGACAAATATAGGAATAACAATGTAAATTGTTGAAGCAT ATTTGAATTCAATTGTGATG | This study |
| WH11-O/EXP-F | CTAAATTTCTTCTTATTTCTACATTTAAGGGAACACCCTATTCAACCTATGGAATTGTGATTTCACCTTGAAATCCTCGGGATCTGCATAAACTAAGTTG ATCAAGCTTGCCTCGTCCCC | This study |
| WH11-O/EXP-R | TAACCTTATCCTTGAATTGTTCTGGAGTGGATTTTTGAGAATCTGGAGTTAATTTGGATTCGATTTTATCACCAATATCTTTTCTACCTAAGTCGGACAT ATTTGAATTCAATTGTGATG | This study |
| CSH1-F-O/EXP | ACGGAATGCCATCAACTGTTGTGAGAAACTTGGGACTATAGTATAGAGAACATTCAAGCTTGGTGTTGTTATAGATGATTTGATTAATAATGTAAAAAAT ATCAAGCTTGCCTCGTCCCC | This study |
| CSH1-R-O/EXP | CGATGTCACCATTAAAACCTCTCCAACTGGATCCCAATCTCATAGTACCGACAGCAACAGTGTTGACCTTCAAACCAGATTTACCTAATCTGGTGACCAT ATTTGAATTCAATTGTGATG | This study |
| ECM331-F-O/EXP | GGTGGTAGTAATGACAATTTGATAGTATTGTTACTTTTCGTGTTTTCTAACTCATTTATACAACATTCCTTTTGTTGCATTACTTTACTATAGCAACAAT ATCAAGCTTGCCTCGTCCCC | This study |
| ECM331-R-O/EXP | TGGAAGTTTTAGAGAATGAACATTTGTTTGATGAGTCTGCTGCTGAAACTGATGTCAATAAGGCAGCGACTATTGGTAAAAGAAATGACTTAATTTGCAT ATTTGAATTCAATTGTGATG | This study |
| TPK1-SAT1-F1 | ACTGTCACGCAA GGTACCAAGTTTTAACTAACTA | This study |
| TPK1-SAT1-R1 | AACTTCTCGAGCAGAAACAGTGTTGAATAATATCACCTT | This study |
| TPK1-SAT1-F2 | TAATTAATTTGCCCGCGGAAGAAAGAACAAGAAA | This study |
| TPK1-SAT1-R2 | AGACTTGACAAACGAGCTCTTTAGTATCACCGTA | This study |
To complement deletions, PCR fragments of the gene to be complemented, including 1000bp upstream of the start codon and approximately 700bp downstream of the stop codon, were amplified from BWP17 genomic DNA. Complementation primers were 60–70bp in length and comprised a 20–30bp gene specific sequence and a 40mer sequence (upstream primer-TTCACACAGGAAACAGCTATGACCATGATTACGCCAAGCT, downstream primer-TCGACCATATGGGAGAGCTCCCAACGCGTTGGATGCATAG) to direct in vivo recombination into plasmid pDDB78 (Ma et al., 1987). The complementation PCR product was co-transformed into the S. cerevisiae BY4741Δtrp strain with an EcoRI/NotI restricted pDDB78 plasmid. The resulting complementing clone was amplified in E. coli, digested with NruI to direct insertion of the complement to the HIS1 locus and transformed into the deletion strains. Presence of wild-type was detected by colony PCR. NruI-digested pDDB78 was also transformed into the deletion strains to generate prototrophic marker-matched strains for comparison with complemented strains.
The SAT1 flipper (Reuss et al., 2004) was used to generate a TPK1 gene deletion in the clinical isolate SC5314, SF1358. The TPK1 deletion cassette was constructed by insertion of a KpnI-XhoI fragment (approx 500bp in length of the region upstream of TPK1) and a SacII-SacI fragment (approximately 500bp in length of the region downstream of TPK1) into pSFS-2A. The resulting plasmid was linearized with KpnI and SacI and transformed into C. albicans SC5314 and plated on YPD containing 200μg/ml clonNAT and screened by PCR. Positive transformants were grown overnight in YPD containing maltose as the sole carbon source to induce excision of the cassette and plated on YPD. Colonies that could no longer grow on 100μg/ml NAT were identified as having excised the cassette and were transformed again with linearized plasmid to obtain homozygous deletions.
To construct over-expressing strains, the NAT1-TDH3 promoter plasmid pCJN542 (Nobile et al., 2008b) was employed. Primers were designed to have 100bp of homology to 500bp upstream into the promoter region and 100bp of homology from the start codon of the gene to be overexpressed. In cases where there was a gene upstream (within the 500bp upstream) a smaller fragment was generated to ensure other orfs were not compromised. PCR products were generated using pCJN542 as the template. Homology allowed for homologous recombination of the cassette directly upstream of the gene to be overexpressed with the TDH3 promoter driving expression in place of the genes natural promoter. Transformants were plated on YPD+400μg/ml clonNAT plates and transformants were PCR checked using a gene-specific forward detection primer and a reverse primer specific to the NAT gene. Genotypes of all strains constructed were verified by PCR and many were also verified by RT-PCR. In all cases, marker-matched strains were generated and most appropriate wild-type strains were used as reference strains (e.g. DAY185 for prototrophic mutant strains) for phenotype assays.
RNA Isolation
Overnight cultures of cells were diluted to an OD600nm of 0.2 in 100 ml fresh YPD medium (or other appropriate medium for the experiment). Cultures were grown at 30°C with shaking to an OD600nm of 1 or at 37°C for 8 hours. For cell wall integrity studies the 100ml cultures were the split into two cultures. 125ng/ml of caspofungin was added. To the other an equal volume of water was added and the cultures were incubated for 30 minutes. Cells were harvested by vacuum filtration and flash frozen in a dry ice/EtOH bath. Cells were frozen on filters at −80°C.
RNA was extracted using the RNeasy Kit (Qiagen Cat.No. 74104) following the manufacturer’s protocol for Purification of Total RNA from Yeast with the following modifications. Cells were resuspended from filters with 1.5 ml ice-cold dH2O followed by 10–15 seconds of vigorous vortexing. Resuspended cells were transferred to a 1.5 ml tube and spun down according to the manufacturer’s protocol. For cell disruption, the manufacturer’s protocol for mechanical disruption was used and cells were beaten with a Next Advance Bullet Blender for 3 min at 4°C to maximize cell lysis.
Microarray
Microarray expression analysis was performed as previously described (Nantel et al., 2006). Individual hybridization experiments were performed from multiple independent RNA samples of JJH384 and JJH233 (tpk1::Tn/tpk1::Tn and tpk1::Tn/tpk1::Tn +TPK1) with and without caspofungin treatment. Microarray data was verified by RT-PCR of a number of the top targets found by microarray. The microarray data has been deposited in the NCBI GEO database under accession number GSE38846.
Quantitative RT PCR
10 μg total RNA was treated with the DNA-free kit (Ambion) followed by first-strand cDNA synthesis from half of the DNA-free RNA using the AffinityScript multiple temperature cDNA synthesis kit (Stratagene). Absence of DNA contamination was confirmed using control sets for which reverse transcriptase was omitted from the cDNA reaction.
Primer3 software (http://frodo.wi.mit.edu/) was used to design primers for all genes including TDH3 which was used as a reference gene for normalisation. Primers to distinguish between the ALS genes were as per (Green et al., 2004) (Table 2). 2X iQ SYBR Green Supermix (Bio-Rad), 1 μl of first-strand cDNA reaction mixture, and 0.1 μM of primers were mixed in a total volume of 50μl per reaction. Real-time PCR was performed in triplicate using an iCycler iQ real-time PCR detection system (Bio-Rad). The program for amplification had an initial denaturation step at 95°C for 5 min, followed by 40 cycles of 95°C for 45 s and 58°C for 30 s. Product amplification was detected using SYBR Green fluorescence during the 58°C step, and specificity of the reaction was monitored by melt-curve analysis following the real-time program. Gene expression was determined using Bio-Rad iQ5 software (ΔΔCT method).
Cell-Surface Adherence
The method outlined by (Finkel et al., 2012) was followed to assess cell-surface adherence for a library of protein kinase insertion mutants constructed in (Blankenship et al., 2010). Briefly, the Fluxion Bioflux (TM) 200 (Fluxion Biosciences, San Francisco, CA), a flow device consisting of microfluidic channels permitting steady flow through a flow chamber comprised of a glass coverslip fused to a fluidic channel constructed from polydimethylsiloxane, was employed to analyse cell to surface adherence of the strains. Overnight cultures of wild-type and mutant strains were diluted to an OD600nm of 0.2. Cell culture was passed through the channels at a shear force of 3Pa. Experiments were performed at 30°C. After 30 minutes of flow time the number of adhering cells was recorded. Adherent cells (stationary cells attached to the surface) were recorded by photographing four set locations along the flow channel. Channels were visualized using a Nikon Eclipse TS100 microscope and photographs were taken using a QICAM (QImaging) camera (Fast1394). Experiments were performed at n=4.
In vitro Biofilm Assays
Strains were grown in YPD overnight at 30°C. Overnight cultures were diluted to an OD600nm = 0.5 in 2 ml of Spider medium in a sterile 12-well plate containing a silicone square (Bentec Medical Inc) that had been pre-treated with fetal bovine serum overnight and washed with PBS. The plate was incubated for 90 mins at 60 rpm at 37°C to allow initial cell adherence. Squares were washed in 1 ml PBS to remove un-adhered cells and then placed in a new 12 well plate containing 2 ml of Spider medium. The plate was incubated at 37°C at 60 rpm for 24 or 48hours.
Biofilm Biomass Assays
Silicone squares were weighed prior to in vitro biofilm assay setup. Biofilms were grown for 48 hrs (as per In vitro biofilm assay method above). The silicone squares were removed from wells, dried overnight at 37°C and re-weighed. Total biomass was calculated by subtracting the weight of the silicone prior to biofilm growth from the weight of the silicone after biofilm growth. The average biomass for each strain was calculated using independent samples and normalized to that of an un-inoculated silicone square.
Caspofungin Sensitivity Assays
Strains were grown overnight in YPD medium at 30°C with shaking. Cells were diluted to an OD600nm of 3 in H2O. Five-fold dilutions were made and plated on YPD control plates and on YPD containing 125 ng/ml caspofungin. Plates were incubated at 30°C for 24–48 hours and photographed.
Scanning Electron Microscopy
Biofilms were setup in 48 well plates ensuring channels between wells had carefully been constructed. The biofilm protocol was as per the in vitro biofilm assays methodology. Growth medium was removed from the well taking care not to remove the medium below the level of the biofilm. A fixative containing 2% glutaraldehyde and 0.1% ruthenium red, buffered with PBS was added. Cells were fixed for at least 1 hour. After primary fixation, the fixative was to the level of the biofilm, and the biofilm washed with at least 3 changes of PBS. As a secondary fixative, a 1% solution of OsO4 buffered with PBS was carefully added. The biofilm was fixed for 1 hour, and washed with 3 changes of dH2O. A continuous gradient from 100% dH2O to 100% EtOH was produced and suspended in a syringe on a rack above a stir plate. A hot metal probe was used to cut a V-shaped drain into the side of the wells containing the biofilms. The 48 multiwell plate was placed at an angle into a lid of a 150 mm Petri dish. A needle, connected to a tube, attached to the syringe containing the gradient, was placed into the adjacent connected well. A small stir rod was placed in the adjacent connected well, and the stir plate was turned on. The valve of the gradient was opened, and the content of the gradient was slowly introduced into the adjacent connected well. The gradient diffused into the well with the biofilm, and exited through the v-shaped drain into the 150 mm Petri dish lid. After the gradient was delivered, several mls of 100% EtOH was added to the syringe and delivered to the adjacent connected well. After complete dehydration, the biofilm and substrate were dried in a Pelco CPD2 critical point dryer using CO2, at 1200 psi, and 42°C. Dried biofilms were attached to SEM stubs. For cross section views, a sharp razor blade was used to cut the biofilm, and scrape away the biofilm from the foreground. The biofilms were coated with gold using a Pelco SC-6 sputter coater, and were examined using a Hitachi 2460N Scanning Electron Microscope. Digital images were obtained using Quartz PCI Image management system software.
NanoString analysis of gene expression
bcr1Δ/bcr1Δ and bcr1Δ/bcr1Δtpk1Δ/tpk1Δ strains were grown in Spider medium for 8 hours at 37°C with shaking. Samples containing 100 ng of C. albicans total RNA were mixed with custom designed probe code set and incubated at 65°C overnight (12–18 hours). The hybridized samples were processed on a nanoString prep station using the manufacturer’s default program. The resultant cartridges were then transferred to the nanoString digital analyzer and scanned for 600 fields per sample. The raw counts were first adjusted for technical variability using the positive and negative controls of irrelevant RNA sequences included in the code set. The technically adjusted counts were then normalized for total input C. albicans RNA using TDH3. The normalized counts were used to compare gene expression levels among different samples.
Transmission Electron Microscopy
Cells were fixed for at least 1 hour in 2% glutaraldehyde, and 0.1% ruthenium red solution buffered with PBS. After washing in 3 changes of PBS, samples were placed in a 1% osmium tetroxide solution buffered with PBS for 1 hour. The cells were dehydrated using ethanol. The dehydration was in 5-minute steps at concentrations of 50%, 70%, 95%, and 3 10-minute steps of 100% ethanol. Propylene oxide was used as a transitional solvent. The propylene oxide was changed twice, at 10 minutes for each change. The sample was placed in a 1:2 mixture of Spurr’s resin and propylene oxide, and held at room temperature overnight. The next day the sample was transferred to a 1:1 mixture of Spurr’s resin and propylene oxide and held at room temperature. After 8 hours, the mixture was changed to a 1:2 mixture of Spurr’s resin and propylene oxide, and held with the tubes open, at room temperature, overnight in a desiccator. The Spurr’s resin and propylene oxide mixture was removed and replaced with 100% Spurr’s resin. The sample was infiltrated with the resin for an additional 8 hours, placed in embedding molds, and polymerized for 24 hours at 60°C. Thin sections were cut using a Reichert-Jung Ultracut E ultramicrotome and a DDK Diamond knife. Thin sections on copper grids were stained with 1% uranyl acetate and Reynold’s lead citrate. Sections were viewed on a Hitachi H-7100 TEM transmission electron microscope. Digital images were obtained using AMT Advantage 10 CCD Camera and NIH Image software.
Supplementary Material
Figure S1. Cell Wall Depth Determinations. Transmission electron microscopy of sectioned cells was used to determine cell wall thickness on 10 unselected cells of each strain, with means and standard deviations indicated. Representative images are shown. The strains used were SC5314, SF1358, SF1127, SF1129, and SF1190.
Figure S2. Gene Expression Comparison between tpk1/tpk1 and cdc35/cdc35 Strains. All 46 genes that were up-regulated at least 2-fold in the tpk1::Tn/tpk1::Tn mutant were compared for expression levels to published data for a cdc35Δ/cdc35Δ mutant (Harcus et al., 2004). For 23 genes, data were available (left portion of graph). For 23 genes, reliable determinations were not available from the cdc35Δ/cdc35Δ dataset (right portion of graph).
Complete NanoString Gene Expression Data. Tab “Raw and Normalized Data”: The raw data, in a form of digital counts for each of the 117 genes in every sample, were first adjusted for binding efficiency and background subtraction using the manufacturer included positive and negative controls, following nCounter data analysis guidelines. Data sets were normalized to the TDH3 control. Tab “Normalized to TDH3”: TDH3-Normalized data set. The average of three determinations for a gene in a mutant was calculated. Tab “Calculations”: The normalized data sets were used to determine if the expression level of a gene in a mutant was up (red shading) or down (green shading) regulated by more than 2 fold.
Strain Table for Insertion Mutant Protein Kinase Library.
Acknowledgments
We are grateful to members of the 2008 Molecular Mycology Class at the Marine Biological Laboratory for preparation of RNA for initial microarray experiments. We are grateful to Tatyana Aleynikova for preparation and management of laboratory stocks and supplies, and to Jonathan Finkel for his work on biofilm SEM imaging. This work was funded by NIH research grant R01 AI067703 (APM), a National University of Ireland Travelling Studentship (SF), and by support from the Canadian Institutes of Health Research (AN).
Footnotes
NCBI GEO database: GSE38846
Complete microarray data can also be downloaded from http://www.cmu.edu/bio/faculty/mitchell.html.
References
- Bahn YS, Sundstrom P. CAP1, an adenylate cyclase-associated protein gene, regulates bud-hypha transitions, filamentous growth, and cyclic AMP levels and is required for virulence of Candida albicans. J Bacteriol. 2001;183:3211–3223. doi: 10.1128/JB.183.10.3211-3223.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bimbo A, Jia Y, Poh SL, Karuturi RK, den Elzen N, Peng X, Zheng L, O’Connell M, Liu ET, Balasubramanian MK, Liu J. Systematic deletion analysis of fission yeast protein kinases. Eukaryot Cell. 2005;4:799–813. doi: 10.1128/EC.4.4.799-813.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S, Van Dijck P, Datta A. Environmental sensing and signal transduction pathways regulating morphopathogenic determinants of Candida albicans. Microbiol Mol Biol Rev. 2007;71:348–376. doi: 10.1128/MMBR.00009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship JR, Fanning S, Hamaker JJ, Mitchell AP. An extensive circuitry for cell wall regulation in Candida albicans. PLoS Pathog. 2010;6:e1000752. doi: 10.1371/journal.ppat.1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockmuhl DP, Ernst JF. A potential phosphorylation site for an A-type kinase in the Efg1 regulator protein contributes to hyphal morphogenesis of Candida albicans. Genetics. 2001;157:1523–1530. doi: 10.1093/genetics/157.4.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockmuhl DP, Krishnamurthy S, Gerads M, Sonneborn A, Ernst JF. Distinct and redundant roles of the two protein kinase A isoforms Tpk1p and Tpk2p in morphogenesis and growth of Candida albicans. Mol Microbiol. 2001;42:1243–1257. doi: 10.1046/j.1365-2958.2001.02688.x. [DOI] [PubMed] [Google Scholar]
- Bruno VM, Kalachikov S, Subaran R, Nobile CJ, Kyratsous C, Mitchell AP. Control of the C. albicans cell wall damage response by transcriptional regulator Cas5. PLoS Pathog. 2006;2:e21. doi: 10.1371/journal.ppat.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassola A, Parrot M, Silberstein S, Magee BB, Passeron S, Giasson L, Cantore ML. Candida albicans lacking the gene encoding the regulatory subunit of protein kinase A displays a defect in hyphal formation and an altered localization of the catalytic subunit. Eukaryot Cell. 2004;3:190–199. doi: 10.1128/EC.3.1.190-199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier M, Castilla R, Bolduc N, Zelada A, Martineau P, Bouillon M, Magee BB, Passeron S, Giasson L, Cantore ML. The two isoforms of the cAMP-dependent protein kinase catalytic subunit are involved in the control of dimorphism in the human fungal pathogen Candida albicans. Fungal Genet Biol. 2003;38:133–141. doi: 10.1016/s1087-1845(02)00520-0. [DOI] [PubMed] [Google Scholar]
- D’Souza CA, Heitman J. Conserved cAMP signaling cascades regulate fungal development and virulence. FEMS Microbiol Rev. 2001;25:349–364. doi: 10.1111/j.1574-6976.2001.tb00582.x. [DOI] [PubMed] [Google Scholar]
- Dwivedi P, Thompson A, Xie Z, Kashleva H, Ganguly S, Mitchell AP, Dongari-Bagtzoglou A. Role of Bcr1-activated genes Hwp1 and Hyr1 in Candida albicans oral mucosal biofilms and neutrophil evasion. PLoS One. 2011;6:e16218. doi: 10.1371/journal.pone.0016218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel JS, Mitchell AP. Genetic control of Candida albicans biofilm development. Nat Rev Microbiol. 2011;9:109–118. doi: 10.1038/nrmicro2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel JS, Xu W, Huang D, Hill EM, Desai JV, Woolford CA, Nett JE, Taff H, Norice CT, Andes DR, Lanni F, Mitchell AP. Portrait of Candida albicans Adherence Regulators. PLoS Pathog. 2012;8:e1002525. doi: 10.1371/journal.ppat.1002525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- Giacometti R, Kronberg F, Biondi RM, Passeron S. Catalytic isoforms Tpk1 and Tpk2 of Candida albicans PKA have non-redundant roles in stress response and glycogen storage. Yeast. 2009;26:273–285. doi: 10.1002/yea.1665. [DOI] [PubMed] [Google Scholar]
- Giacometti R, Kronberg F, Biondi RM, Passeron S. Candida albicans Tpk1p and Tpk2p isoforms differentially regulate pseudohyphal development, biofilm structure, cell aggregation and adhesins expression. Yeast. 2011;28:293–308. doi: 10.1002/yea.1839. [DOI] [PubMed] [Google Scholar]
- Green CB, Cheng G, Chandra J, Mukherjee P, Ghannoum MA, Hoyer LL. RT-PCR detection of Candida albicans ALS gene expression in the reconstituted human epithelium (RHE) model of oral candidiasis and in model biofilms. Microbiology. 2004;150:267–275. doi: 10.1099/mic.0.26699-0. [DOI] [PubMed] [Google Scholar]
- Gregori C, Glaser W, Frohner IE, Reinoso-Martin C, Rupp S, Schuller C, Kuchler K. Efg1 Controls caspofungin-induced cell aggregation of Candida albicans through the adhesin Als1. Eukaryot Cell. 2011;10:1694–1704. doi: 10.1128/EC.05187-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcus D, Nantel A, Marcil A, Rigby T, Whiteway M. Transcription profiling of cyclic AMP signaling in Candida albicans. Mol Biol Cell. 2004;15:4490–4499. doi: 10.1091/mbc.E04-02-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan DA, Muhlschlegel FA. Candida albicans developmental regulation: adenylyl cyclase as a coincidence detector of parallel signals. Curr Opin Microbiol. 2011;14:682–686. doi: 10.1016/j.mib.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Hoyer LL, Green CB, Oh SH, Zhao X. Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family--a sticky pursuit. Med Mycol. 2008;46:1–15. doi: 10.1080/13693780701435317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamai Y, Kubota M, Hosokawa T, Fukuoka T, Filler SG. Contribution of Candida albicans ALS1 to the pathogenesis of experimental oropharyngeal candidiasis. Infect Immun. 2002;70:5256–5258. doi: 10.1128/IAI.70.9.5256-5258.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberer E, Harcus D, Dignard D, Johnson L, Ushinsky S, Thomas DY, Schroppel K. Ras links cellular morphogenesis to virulence by regulation of the MAP kinase and cAMP signalling pathways in the pathogenic fungus Candida albicans. Mol Microbiol. 2001;42:673–687. doi: 10.1046/j.1365-2958.2001.02672.x. [DOI] [PubMed] [Google Scholar]
- Ma H, Kunes S, Schatz PJ, Botstein D. Plasmid construction by homologous recombination in yeast. Gene. 1987;58:201–216. doi: 10.1016/0378-1119(87)90376-3. [DOI] [PubMed] [Google Scholar]
- Nantel A, Rigby T, Hogues H, Whiteway M. Microarrays for studying pathogenicity in Candida albicans. In: Kavanaugh K, editor. Medical mycology: cellular and molecular techniques. Hoboken (New Jersey): Wiley Press; 2006. pp. 181–210. [Google Scholar]
- Nobile CJ, Andes DR, Nett JE, Smith FJ, Yue F, Phan QT, Edwards JE, Filler SG, Mitchell AP. Critical role of Bcr1-dependent adhesins in C. albicans biofilm formation in vitro and in vivo. PLoS Pathog. 2006a;2:e63. doi: 10.1371/journal.ppat.0020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile CJ, Fox EP, Nett JE, Sorrells TR, Mitrovich QM, Hernday AD, Tuch BB, Andes DR, Johnson AD. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell. 2012;148:126–138. doi: 10.1016/j.cell.2011.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile CJ, Mitchell AP. Regulation of cell-surface genes and biofilm formation by the C. albicans transcription factor Bcr1p. Curr Biol. 2005;15:1150–1155. doi: 10.1016/j.cub.2005.05.047. [DOI] [PubMed] [Google Scholar]
- Nobile CJ, Nett JE, Andes DR, Mitchell AP. Function of Candida albicans adhesin Hwp1 in biofilm formation. Eukaryot Cell. 2006b;5:1604–1610. doi: 10.1128/EC.00194-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile CJ, Schneider HA, Nett JE, Sheppard DC, Filler SG, Andes DR, Mitchell AP. Complementary adhesin function in C. albicans biofilm formation. Curr Biol. 2008a;18:1017–1024. doi: 10.1016/j.cub.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile CJ, Solis N, Myers CL, Fay AJ, Deneault JS, Nantel A, Mitchell AP, Filler SG. Candida albicans transcription factor Rim101 mediates pathogenic interactions through cell wall functions. Cell Microbiol. 2008b;10:2180–2196. doi: 10.1111/j.1462-5822.2008.01198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park G, Servin JA, Turner GE, Altamirano L, Colot HV, Collopy P, Litvinkova L, Li L, Jones CA, Diala FG, Dunlap JC, Borkovich KA. Global analysis of serine-threonine protein kinase genes in Neurospora crassa. Eukaryot Cell. 2011;10:1553–1564. doi: 10.1128/EC.05140-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Myers CL, Sheppard DC, Phan QT, Sanchez AA, EEJ, Filler SG. Role of the fungal Ras-protein kinase A pathway in governing epithelial cell interactions during oropharyngeal candidiasis. Cell Microbiol. 2005;7:499–510. doi: 10.1111/j.1462-5822.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- Pitarch A, Sanchez M, Nombela C, Gil C. Sequential fractionation and two-dimensional gel analysis unravels the complexity of the dimorphic fungus Candida albicans cell wall proteome. Mol Cell Proteomics. 2002;1:967–982. doi: 10.1074/mcp.m200062-mcp200. [DOI] [PubMed] [Google Scholar]
- Plaine A, Walker L, Da Costa G, Mora-Montes HM, McKinnon A, Gow NA, Gaillardin C, Munro CA, Richard ML. Functional analysis of Candida albicans GPI-anchored proteins: roles in cell wall integrity and caspofungin sensitivity. Fungal Genet Biol. 2008;45:1404–1414. doi: 10.1016/j.fgb.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss O, Vik A, Kolter R, Morschhauser J. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene. 2004;341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Roth FP, Lipshitz HD, Andrews BJ. Q&A: epistasis. J Biol. 2009;8:35. doi: 10.1186/jbiol144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentandreu M, Nieto A, Iborra A, Elorza MV, Ponton J, Fonzi WA, Sentandreu R. Cloning and characterization of CSP37, a novel gene encoding a putative membrane protein of Candida albicans. J Bacteriol. 1997;179:4654–4663. doi: 10.1128/jb.179.15.4654-4663.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton DR, Masuoka J, Hazen KC. Cloning and analysis of a Candida albicans gene that affects cell surface hydrophobicity. J Bacteriol. 2001;183:3582–3588. doi: 10.1128/JB.183.12.3582-3588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneborn A, Bockmuhl DP, Gerads M, Kurpanek K, Sanglard D, Ernst JF. Protein kinase A encoded by TPK2 regulates dimorphism of Candida albicans. Mol Microbiol. 2000;35:386–396. doi: 10.1046/j.1365-2958.2000.01705.x. [DOI] [PubMed] [Google Scholar]
- Souto G, Giacometti R, Silberstein S, Giasson L, Cantore ML, Passeron S. Expression of TPK1 and TPK2 genes encoding PKA catalytic subunits during growth and morphogenesis in Candida albicans. Yeast. 2006;23:591–603. doi: 10.1002/yea.1377. [DOI] [PubMed] [Google Scholar]
- Spellberg BJ, Ibrahim AS, Avenissian V, Filler SG, Myers CL, Fu Y, Edwards JE., Jr The anti-Candida albicans vaccine composed of the recombinant N terminus of Als1p reduces fungal burden and improves survival in both immunocompetent and immunocompromised mice. Infect Immun. 2005;73:6191–6193. doi: 10.1128/IAI.73.9.6191-6193.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoldt VR, Sonneborn A, Leuker CE, Ernst JF. Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J. 1997;16:1982–1991. doi: 10.1093/emboj/16.8.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teste MA, Enjalbert B, Parrou JL, Francois JM. The Saccharomyces cerevisiae YPR184w gene encodes the glycogen debranching enzyme. FEMS Microbiol Lett. 2000;193:105–110. doi: 10.1111/j.1574-6968.2000.tb09410.x. [DOI] [PubMed] [Google Scholar]
- Urban C, Sohn K, Lottspeich F, Brunner H, Rupp S. Identification of cell surface determinants in Candida albicans reveals Tsa1p, a protein differentially localized in the cell. FEBS Lett. 2003;544:228–235. doi: 10.1016/s0014-5793(03)00455-1. [DOI] [PubMed] [Google Scholar]
- Wilson RB, Davis D, Mitchell AP. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J Bacteriol. 1999;181:1868–1874. doi: 10.1128/jb.181.6.1868-1874.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Hart H, Cheng C, Roach PJ, Tatchell K. Characterization of Gac1p, a regulatory subunit of protein phosphatase type I involved in glycogen accumulation in Saccharomyces cerevisiae. Mol Genet Genomics. 2001;265:622–635. doi: 10.1007/s004380100455. [DOI] [PubMed] [Google Scholar]
- Zhao X, Oh SH, Cheng G, Green CB, Nuessen JA, Yeater K, Leng RP, Brown AJ, Hoyer LL. ALS3 and ALS8 represent a single locus that encodes a Candida albicans adhesin; functional comparisons between Als3p and Als1p. Microbiology. 2004;150:2415–2428. doi: 10.1099/mic.0.26943-0. [DOI] [PubMed] [Google Scholar]
- Zhao X, Oh SH, Yeater KM, Hoyer LL. Analysis of the Candida albicans Als2p and Als4p adhesins suggests the potential for compensatory function within the Als family. Microbiology. 2005;151:1619–1630. doi: 10.1099/mic.0.27763-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cell Wall Depth Determinations. Transmission electron microscopy of sectioned cells was used to determine cell wall thickness on 10 unselected cells of each strain, with means and standard deviations indicated. Representative images are shown. The strains used were SC5314, SF1358, SF1127, SF1129, and SF1190.
Figure S2. Gene Expression Comparison between tpk1/tpk1 and cdc35/cdc35 Strains. All 46 genes that were up-regulated at least 2-fold in the tpk1::Tn/tpk1::Tn mutant were compared for expression levels to published data for a cdc35Δ/cdc35Δ mutant (Harcus et al., 2004). For 23 genes, data were available (left portion of graph). For 23 genes, reliable determinations were not available from the cdc35Δ/cdc35Δ dataset (right portion of graph).
Complete NanoString Gene Expression Data. Tab “Raw and Normalized Data”: The raw data, in a form of digital counts for each of the 117 genes in every sample, were first adjusted for binding efficiency and background subtraction using the manufacturer included positive and negative controls, following nCounter data analysis guidelines. Data sets were normalized to the TDH3 control. Tab “Normalized to TDH3”: TDH3-Normalized data set. The average of three determinations for a gene in a mutant was calculated. Tab “Calculations”: The normalized data sets were used to determine if the expression level of a gene in a mutant was up (red shading) or down (green shading) regulated by more than 2 fold.
Strain Table for Insertion Mutant Protein Kinase Library.