

Abstract
Knowing the role of MC1R in skin tanning can provide a brand new idea to resolve pigmentary disorders. αMSH has 13 amino acids and is the most essential pigmentary melanocortin responsible for melanin synthesis. One could utilize the compound library to find lead compounds by virtual screening from peptide database and traditional Chinese medicine (TCM) database@Taiwan. Computational simulation provided a convenient technology to survey potential lead. Ligand-based validation set up the reliable model for molecular dynamics simulation. Molecular dynamics simulation approved the binding affinity and stability of the peptides selected by virtual screening. Thus, we concluded that Glu-Glu-Lys-Glu (EEKE), Glu-Gly-Gly-Ser-Val-Glu-Ser (EGGSVES), and Glu-Glu-Asp-Cys-Lys (EEDCK) were potent lead peptides for MC1R to resolve pigmentary disorders.
1. Introduction
Excessive melanin contributes to skin tanning or darkening. Ultraviolet (UV) radiation leads to skin pigmentation by manufacturing melanin in the melanocytes located at the basal layer of epidermis. Expression of the pro-opiomelanocortin (POMC) gene producing α-melanocyte stimulating hormone (αMSH) takes place in keratinocytes. αMSH recognizing melanocortin 1 receptor (MC1R) located on the cell membrane of melanocytes starts a series of signal pathways [1]. The αMSH/MC1R triggers downstream signal transduction which is followed by cyclic adenosine monophosphate (cAMP), protein kinase A (PKA), and cAMP responsive element binding protein/cAMP responsive element (CREB/CRE) pathway [2]. Microphthalmia-associated transcription factor (MITF) begins its function in turn. It is an important protein that controls the activation of following melanotic genes: tyrosinase and tyrosinase related protein 1 and 2 (Trp1 and Trp2) [3, 4].
MSHs belong to the POMC hormone groups and include three types of α, β, and γ-MSH [5]. Generally speaking, αMSH is a pituitary peptide hormone derived from adrenocorticotropic hormone (ACTH) [6]. αMSH which affects skin pigmentation mainly produces locally instead of pituitary origin [7].αMSH has 13 amino acids and is the most essential pigmentary melanocortin responsible for melanin synthesis or melanogenesis [8]. Its amino acid sequences are Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val [9]. Synthetic analogs of αMSH have been developed as useful probes binding to the melanocortin receptor or MC1R which is overexpressed in melanoma lesions [10, 11]. His6-Phe7-Arg8-Trp9 (HFRW) is the most common active motif approved in the literature [12, 13]. Experimental order exchange of αMSH had been demonstrated as high-affinity peptides binding to the MC1R but loss of their agonistic function [14].
Melanocortin receptors belong to class A or rhodopsin of the superfamily of 7-transmembrane G protein-coupled receptors (GPCRs) [15–17]. GPCR receives external signal, for example, hormones and neurotransmitters, vary in molecular size from small peptides to large proteins [18]. There are five known melanocortin receptors, MC1~5R [19]. They have similar structure conformation but participate in unique physiologic functions: pigmentation, adrenal function, cardiovascular regulation, obesity or energy homeostasis, and exocrine gland secretion [20, 21]. MC1R is the irreplaceable target involved in regulating our skin or hair color [22]. The coat color of animals or plumage color of birds is also regulated by MC1R gene [23, 24]. MC1R has 318 amino acids; αMSH is its agonist, and agouti signal protein is its antagonist. They determine the phenotype of our skin and hair by producing black, brown eumelanin or yellow, red pheomelanin [25].
Protein sequence and structure analysis by computational simulation have become popular technology in recent decades [26, 27]. We use computational systems biology or in silico biology to research the protein-molecule or the protein-ligand interaction [28, 29]. Drug discovery integrates systems biology and informatics called computer-aided drug design (CADD) [30, 31]. The advantages of CADD techniques shorten our time to find appropriate drug compound opposite to traditional biochemistry [32, 33]. Quantitative structure activity relationship in silico can tell us the properties between small molecule and target protein [34]. Virtual screening and validations through structure-based or ligand-based analysis constitute to CADD procedures [35, 36]. Virtual screening and data analysis utilize docking and molecular dynamics (MD) simulation [37–39]. How long the compound needs to form stable complex structure with target protein can be predicted by MD [40]. Docking and MD accuracy is relying on a series of statistic or score systems [41]. Ligand-based analysis utilizes mathematical model such as Bayesian algorithm [42, 43]. We can choose best candidates from virtual screening and validations as potential effective drugs [44].
Knowing the role of MC1R in skin tanning can provide a brand new idea to prevent UV darkening [45]. Clinical application of αMSH analog is significant in managing certain dermatologic diseases [46]. CADD has been rapidly applied in small molecular drug design [47–50]. Virtual screening of compound database becomes the first and convenient way for CADD [51–56]. Screening peptides for compounds as a drug is a method to design antimicrobial peptides and potent peptides for peptide receptors, such as GPCRs [57–59]. MC1R is a peptide receptor, and peptide design for its agonist and antagonist can be achieved [60]. Virtual screening from peptide database and traditional Chinese medicine (TCM) database@Taiwan in silico saves our time to filter the functional compounds [61–63]. We attempted to investigate the lead for MC1R to resolve pigmentary disorders.
2. Materials and Methods
2.1. Compound Database
To investigate lead peptides of MC1R from peptide library, we downloaded all the peptides from PepBank (http://pepbank.mgh.harvard.edu/) to conduct MC1R lead peptide screening [64].
2.2. Data Collection
For the purpose of identifying MC1R lead peptides, we obtained the structures and corresponding bioactivities (pIC50) of 18 peptides to construct the data set for ligand-based prediction [65].
2.3. Homology Modeling
The MC1R protein sequence was acquired from the Uniprot Knowledgebase (Q01726, MC1R_Human). The 3D structure of human MC4R was acquired from Protein Data Bank (PDB ID: 2IQP). MC1R sequence and the template structure were aligned by Discovery Studio (DS) 2.5. The rational MC1R model was further examined by Ramachandran plot [66].
2.4. Structure-Based Virtual Screening
The ligands from PepBank and the control ligand, His-Phe-Arg-Trp (HFRW), were prepared for specified modeling methods. We used Chemistry at HARvard Molecular Mechanics (CHARMm) force field to set up the model [67]. Docking and scoring functions were estimated by LigandFit module in DS 2.5. We applied the scoring functions including Dock Score, piecewise linear potentials (-PLP), and potential of mean force (-PMF) [47, 50].
2.5. Ligand-Based Validation
Bayesian network constructed the property of descriptors by integrating the data of training set and test set. The data of descriptors and pIC50 were discretized to reduce bias distribution [68]. They were discretized into a maximum of three categories. The training set was defined as linear regression analysis for every pIC50 category after data discretization [69]. We used Banjo package and Bayes Net Toolbox (BNT) package for simulation in our study. The 18 ligands were randomly divided into 13 training sets and 5 test sets for further validation.
2.6. Molecular Dynamics (MD) Simulation
We used Simulation module in DS 2.5 for MD simulation. The cytoplasmic status was simulated with transferable intermolecular potential 3P (TIP3P) water at 0.9% NaCl concentration. Selected protein-ligand complexes from docking were conducted under minimization, heating, equilibration, and production. The minimization phase included 500 steps of deepest descent and 500 steps of conjugated gradient. The heating time from 50 K to 310 K was 50 ps. The equilibration time at 310 K was 150 ps. The production time with constant temperature dynamics method was 10 ns. The temperature decay time was 0.4 ps. The Analyze Trajectory module was used to analyze total energy, root mean square deviations (RMSD), gyrate, mean square deviation (MSD), and solvent accessible surface (SAS) for each ligand and protein-ligand complex. We also illustrated cluster analysis to observe structure features during MD. Illustration of disordered protein was shown to exclude disordered residues [70, 71]. We used LigandPath module to estimate the possible pathway for each ligand. A surface probe was set at 6 Å, and minimum clearance was set at 3 Å.
3. Results
3.1. Homology Modeling
The overall identity of sequence alignment between MC1R and template was 49.8%. The overall similarity was 72.4% (Figure 1). Ramachandran plot of MC1R-modeled structure indicated that 84.7% of residues were in the favored region, 9.9% were in the allowed region, and only 5.3% were in the disallowed region (Figure 2).
Figure 1.
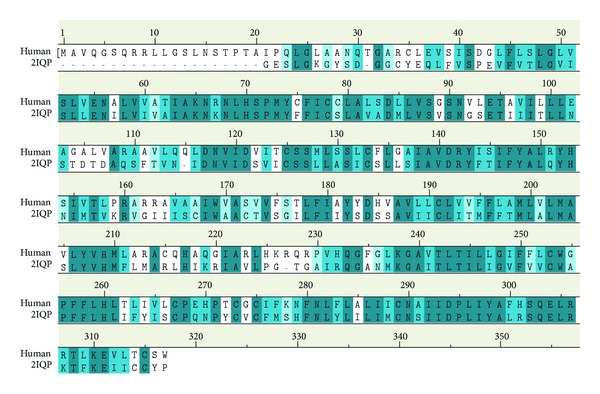
Sequence alignment between MC1R_human and template (2IQP). The identity is 49.8% and the similarity is 72.4%.
Figure 2.
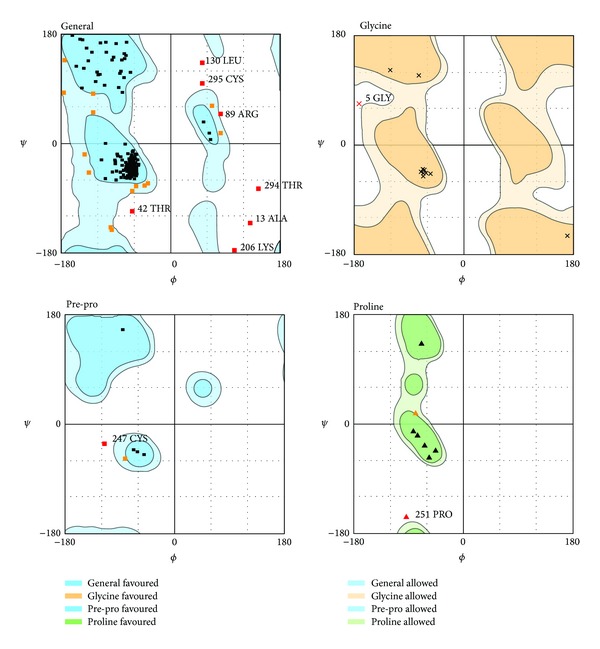
Ramachandran plot of MC1R-modeled structure. Number of residues in favored region (~98.0% expected): 444 (84.7%). Number of residues in allowed region (~2.0% expected): 52 (9.9%). Number of residues in disallowed region: 28 (5.3%).
3.2. Structure-Based Virtual Screening
Dock score, BNT, -PLP1, -PLP2, and -PMF for the top 10 peptides ranked by Dock score were listed in Table 1. Integrating these data, we selected 3 following peptides: Glu-Glu-Lys-Glu (EEKE), and Glu-Gly-Gly-Ser-Val-Glu-Ser (EGGSVES), Glu-Glu-Asp-Cys-Lys (EEDCK) as candidates for further investigation (Figure 3). Docking poses of EEKE, EGGSVES, EEDCK,and the control (HFRW) with MC1R were illustrated in Figure 4. EEKE formed H-bond with Ala88 and Arg89 and formed charge interaction with Arg89 (Figure 4(a)). EGGSVES formed H-bond with Ala88 and Arg89 and formed charge interaction with Arg89 (Figure 4(b)). EEDCK formed charge interaction with Arg89 (Figure 4(c)). The control formed H-bond with Ala88 and Arg89 (Figure 4(d)).
Table 1.
Top 10 candidates of scoring function from PepBank database screening.
| Name | Dock score | BNT | -PLP1 | -PLP2 | -PMF |
|---|---|---|---|---|---|
| EEKE | 345.464 | 7.963191 | 37.91 | 36.24 | 32.59 |
| EGGSVES | 338.21 | 6.979679 | 32.61 | 40.4 | 41.39 |
| EEDCK | 337.005 | 9.512784 | 24.46 | 26.69 | 26.37 |
| GEGEGSGG | 335.016 | 9.346013 | 42.84 | 51.79 | 29.34 |
| SEEEAA | 322.144 | 12.351714 | 58.12 | 67.98 | 24.42 |
| DSGVETS | 316.584 | 8.611215 | 13.43 | 19.48 | 31.47 |
| EGEVGLG | 315.171 | 5.676182 | 28.59 | 30.23 | 22.78 |
| EAGVDAA | 315.157 | 7.491065 | 24.74 | 25.56 | 30.21 |
| DTAGQE | 311.701 | 6.382156 | 33.58 | 29.06 | 34.39 |
| EEKE | 311.503 | 8.708929 | 44.54 | 48.58 | 27.17 |
| His-Phe-Arg-Trp (HFRW)* | 97.949 | 3.431413 | 32.8 | 26.9 | 28.15 |
*Control.
Figure 3.

Scaffold of top 3 candidate peptides: (a) EEKE, (b) EGGSVES, (c) EEDCK, and the control: (d) HFRW.
Figure 4.
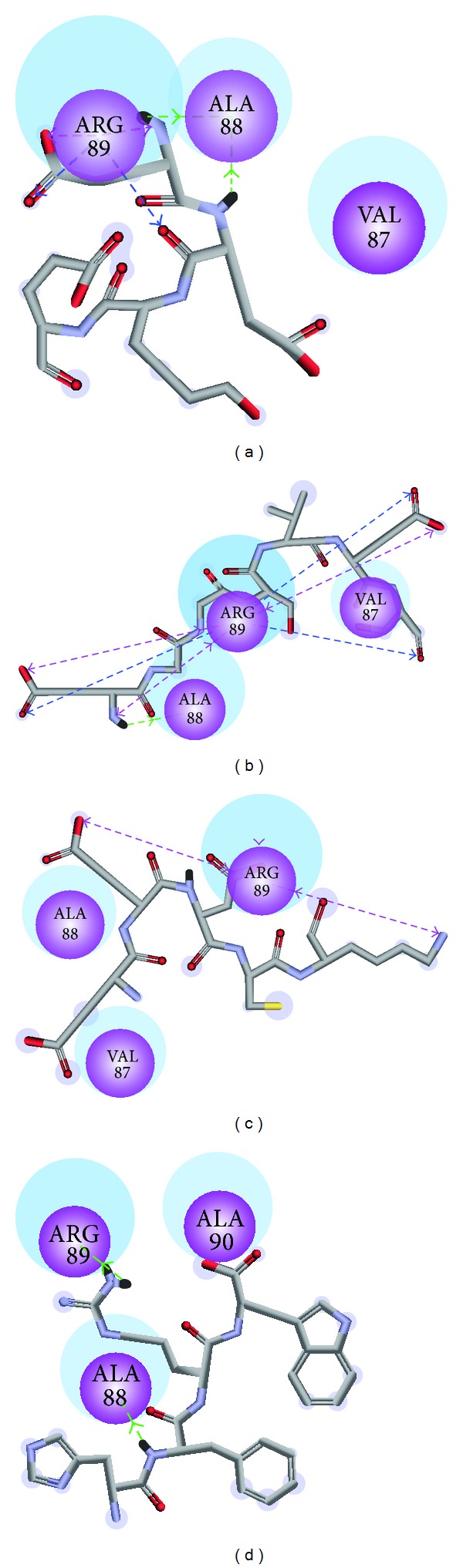
Docking poses of (a) EEKE, (b) EGGSVES, (c) EEDCK, and (d) HFRW. Purple sphere: Residues involved in hydrogen bond (H-bond) or charge interaction. Blue halo around the residue: the solvent accessible surface (SAS) of an interacting residue. The blue halo was proportional to SAS. Green dashed line: H-bond with an amino acid main chain. Blue dashed line: H-bond with an amino acid side chain. Pink dashed line: charge interaction.
3.3. Ligand-Based Validation
We illustrated the correlation of observed and predicted activities using the BNT model. The R 2 value of 0.999 indicated that it is a highly reliable model (Figure 5).
Figure 5.
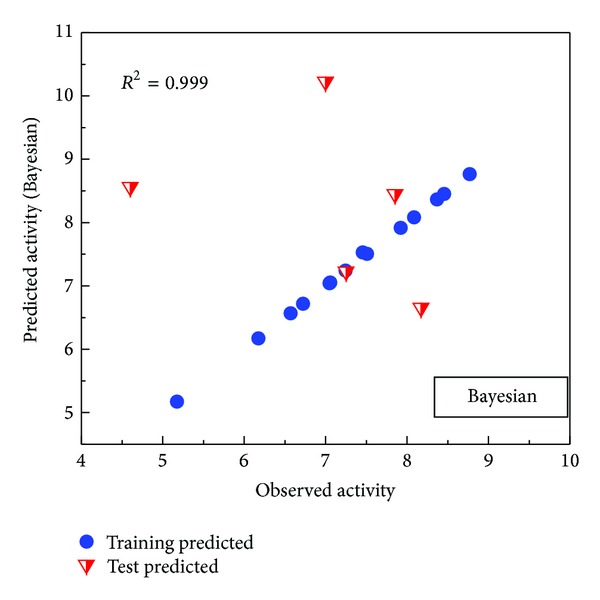
Ligand-based validation: Bayesian network. 18 ligands were randomly divided into 13 training sets and 5 test sets. R 2 = 0.999.
3.4. Molecular Dynamics (MD) Simulation
We analyzed MD trajectories which were generated by Gromacs. Root mean square deviation (RMSD) showed the deviation from the starting structure of each ligand or complex to the end of MD. EGGSVES had larger deviation after 8 ns, but the corresponding ligand-protein complex was relatively stable in the same period. Other peptides were stable during 10 ns MD in contrast (Figure 6). gyrate, or radius of gyration, measured the distance of the atoms relative to the center of each mass. gyrate indicated the compact degree of each ligand or complex. Interestingly, EGGSVES had larger gyrate value after 8 ns, but the corresponding complex was also relatively stable. Other peptides were stable during 10 ns MD in contrast (Figure 7). Mean square deviation (MSD) measured the movement of atoms from their initial positions to the end of MD. MSD indicated the trend of each ligand or complex during MD. All the ligands and complexes had different line graphs, but the long-term trends could be predictable (Figure 8). Solvent accessible surface (SAS) measured the surface area of each ligand or complex in contact with the water. Although the control ligand had larger SAS value compared with other ligands, but the corresponding complex was almost consistent to other complexes (Figure 9). In addition, total energy of each ligand or complex means the total energy of atoms during MD. The total energy would fluctuate, but overall trend was consistent (Figure 10). We performed cluster analysis with RMSD values to identify the representative structure of the complex. The cluster analysis could identify two adjacent structure features for each complex during 5–10 ns MD. EEDCK ligand-protein complex had fluctuated structure features, indicated the complex had undergone many tiny changes (Figure 11). Most residues of MC1R-modeled structure were not in the disordered region (Figure 12). Different ligand pathways for EEKE, EGGSVES, and the control bound with MC1R were illustrated. EEDCK was not shown due to out of the criteria mentioned in materials and methods (Figure 13).
Figure 6.
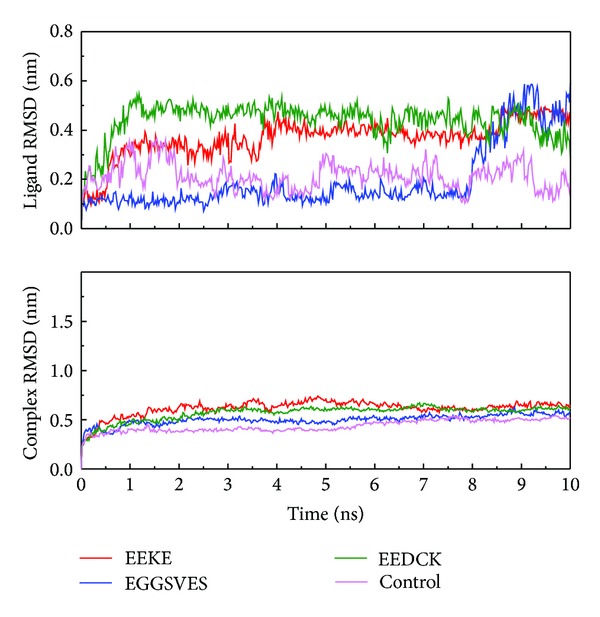
Ligand and complex root mean square deviation (RMSD). Although each ligand alone had larger deviation, the corresponding ligand-protein complex was relatively stable.
Figure 7.
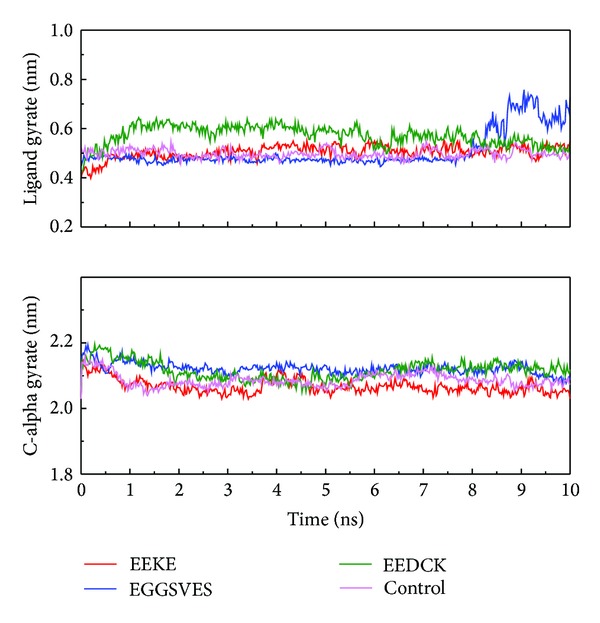
Ligand and C-alpha gyrate. Only EGGSVES had larger gyrate value after 8 ns, other peptides or the complexes were stable in contrast.
Figure 8.
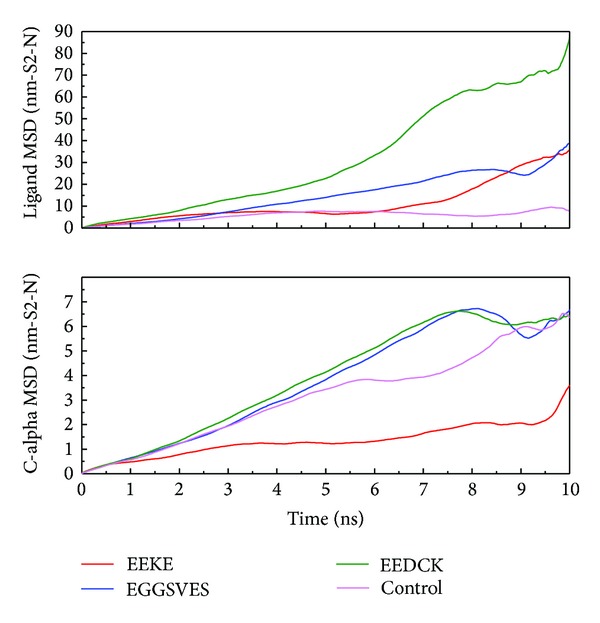
Ligand and C-alpha mean square deviation (MSD). All the ligands and complexes had predictable trends but had larger fluctuation after 8 ns relatively.
Figure 9.
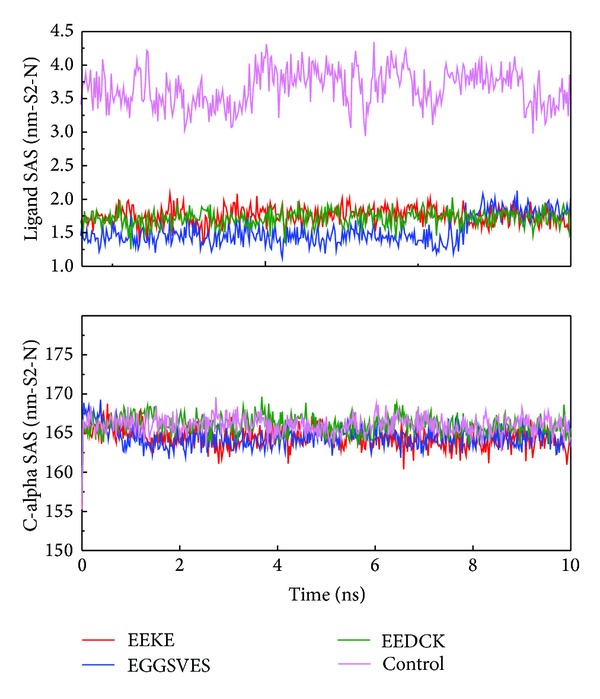
Ligand and C-alpha solvent accessible surface (SAS). The control ligand had larger SAS value compared with other ligands, but its corresponding complex was almost consistent to other complexes.
Figure 10.
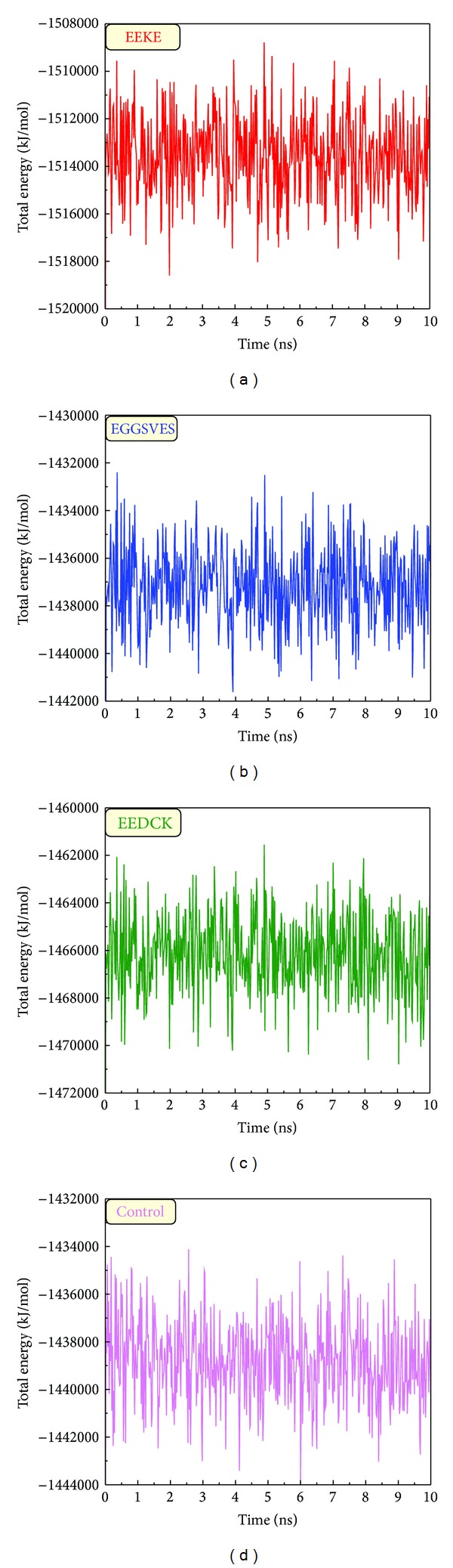
Total energy of (a) EEKE, (b) EGGSVES, (c) EEDCK, and (d) the control (HFRW).
Figure 11.

RMSD values (left upper portion) and graphical depiction of clusters (right lower portion). (a) EEKE, (b) EGGSVES, (c) EEDCK, and (d) the control (HFRW).
Figure 12.

Disorder disposition of MC1R-modeled structure. Most residues are in the non-disordered region (below the red line).
Figure 13.
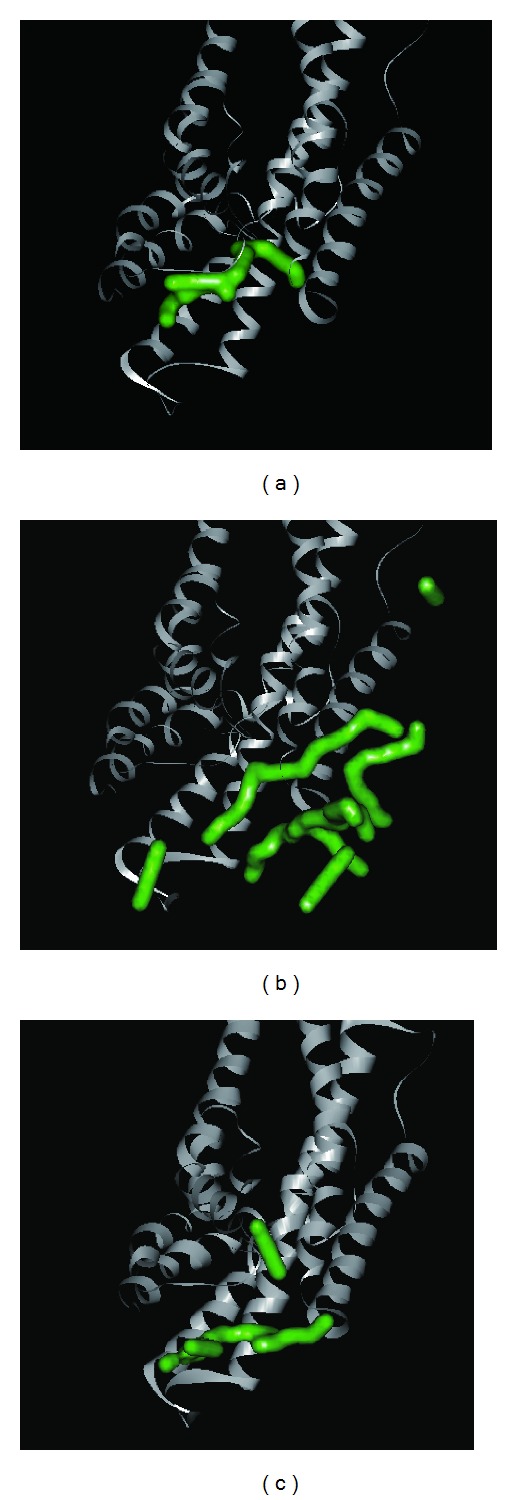
3D simulation of ligand pathway for (a) EEKE, (b) EGGSVES, and (c) the control (HFRW) bound with MC1R. EEDCK is not shown here.
4. Discussion
4.1. Compound Database
MC1R is a peptide receptor, so we utilized the PepBank which was established by Massachusetts General Hospital, Harvard University, to conduct MC1R lead peptide screening. The PepBank database has contained nearly 20000 bioactive peptides. It is a useful and convenient peptide database.
4.2. Homology Modeling
The high percentage of identity and similarity of sequence alignment between MC1R (Q01726) and template (2IQP) indicated that the sequence alignment was reasonable. The high percentage of residues in the favored and allowed region indicated that the MC1R-modeled structure was reliable.
4.3. Structure-Based Virtual Screening
His-Phe-Arg-Trp (HFRW) is the most common key motif for MC1R. We originally expected the outcome peptide sequence from virtual screening should be similar to the control (HFRW). However, Dock score, BNT, -PLP1, -PLP2, and-PMF of the top 10 candidates were almost better than the control. The top 3 candidates which we selected were EEKE, EGGSVES, and EEDCK. Their sequences were quite different from the control. We could speculate that these candidates at least had similar function as the control and might have better affinity for MC1R. Comparing the docking poses of the top 3 candidates and the control with MC1R had the common finding. All of EEKE, EGGSVES, and the control formed H-bond with Ala88 and Arg89. All of EEKE, EGGSVES, and EEDCK formed charge interaction with Arg89. We could conclude that Ala88 and Arg89 were key residues for the top 3 candidates and the control.
4.4. Molecular Dynamics (MD) Simulation
RMSD, gyrate, MSD and, SAS were utilized to analyze the stability of each ligand or complex. Although some ligands were not stable during MD, the ligand-protein complexes were stable relatively. So either of EEKE, EGGSVES, EEDCK, or HFRW could form stable complex with MC1R.
Further analyzing the figure of ligand RMSD, EEDCK had the largest average deviation than other candidates. Interestingly, the deviation of EEKE and EGGSVES exceeded EEDCK after 9 ns. However, analyzing the figure of complex RMSD, EEKE had the largest average deviation than other candidates. Although EGGSVES alone had larger deviation at 8-9 ns, the EGGSVES-MC1R complex did not have substantial change at the same time. It showed that deviation of individual ligand did not affect the stability of its corresponding complex.
Further analyzing the figure of ligand gyrate, EEDCK still had the largest average value than other candidates. Although EGGSVES alone had larger fluctuation at 8-9 ns, the EGGSVES-MC1R complex (c-alpha) did not have substantial change at the same time. It showed that gyrate of individual ligand did not affect the stability of its corresponding complex, either.
From the additional analysis of the figure of ligand MSD, EEDCK still had the largest deviation than other candidates. However, from results of the complex (c-alpha) MSD, EEKE had the lowest average deviation than other candidates. We speculated that EEKE-MC1R complex vibrated back and forth contributing to distinct presentation in complex RMSD and c-alpha MSD. EGGSVES vibrated in opposite direction might explain the upward slope in ligand RMSD and the downward slope in ligand MSD at 8-9 ns.
Further analyzing ligand SAS, the control had the largest average value than the 3 candidates. The result might be related to the hydrophobic side chain of the control.
Further analyzing the figure of total energy, EEKE had the lowest total energy (−1514000 kJ/mol), and EGGSVES had the highest total energy (−1437000 kJ/mol). We speculated that EEKE-MC1R complex only vibrated back and forth contributing to the lowest total energy.
When individual ligand bound to MC1R, MD was convenient to analyze the change of the ligand or the ligand-protein complex. MD could evaluate whether the ligand or the complex was stable or not under dynamic condition. MD might help us understand what happened during conjugation of the ligand and protein. Comparing the figures of RMSD and gyrate, EGGSVES had larger change after 8 ns. The change implied that the structure of EGGSVES underwent some kind of deviation or even translocation. This change did not affect the conjugation of EGGSVES with MC1R because the ligand-protein complex was stable relatively. Comprehensive assessment of the methods of MD, such as RMSD, gyrate, MSD, SAS, and total energy, indicated that all the 3 candidates and the control could form stable complexes with MC1R [72–81].
5. Conclusion
MC1R is important for skin tanning. αMSH is a melanotropin which can bind to MC1R. His-Phe-Arg-Trp (HFRW) is a key motif for conjugating with MC1R. We tried to find potential peptides that can also bind to MC1R by virtual screening of peptide database. Glu-Glu-Lys-Glu (EEKE), Glu-Gly-Gly-Ser-Val-Glu-Ser (EGGSVES), and Glu-Glu-Asp-Cys-Lys (EEDCK) had better affinity for MC1R. The binding affinity was further validated by molecular dynamics. Thus, we concluded that EEKE, EGGSVES, and EEDCK were more potent lead peptides for MC1R to resolve pigmentary disorders.
Acknowledgments
This study was supported by Grants from the National Science Council of Taiwan (NSC102-2325-B039-001 and NSC102-2221-E-468-027-), Asia University (ASIA101-CMU-2, 102-Asia-07), and China Medical University Hospital (DMR-103-001, DMR-103-058, and DMR-103-096). This study was also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH102-TD-B-111-004) and Taiwan Department of Health Cancer Research Center of Excellence MOHW103-TD-B-111-03.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Johnson BE, Mandell G, Daniels F., Jr. Melanin and cellular reactions to ultraviolet radiation. Nature. 1972;235(57):147–149. doi: 10.1038/newbio235147a0. [DOI] [PubMed] [Google Scholar]
- 2.Cui R, Widlund HR, Feige E, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007;128(5):853–864. doi: 10.1016/j.cell.2006.12.045. [DOI] [PubMed] [Google Scholar]
- 3.Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature. 2007;445(7130):843–850. doi: 10.1038/nature05660. [DOI] [PubMed] [Google Scholar]
- 4.Fisher DE, James WD. Indoor tanning—science, behavior, and policy. The New England Journal of Medicine. 2010;363(10):901–903. doi: 10.1056/NEJMp1005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schally AV, Arimura A, Kastin AJ. Hypothalamic regulatory hormones. Science. 1973;179(4071):341–350. doi: 10.1126/science.179.4071.341. [DOI] [PubMed] [Google Scholar]
- 6.Proulx-Ferland L, Labrie F, Dumont D. Corticotropin-releasing factor stimulates secretion of melanocyte-stimulating hormone from the rat pituitary. Science. 1982;217(4554):62–63. doi: 10.1126/science.6283632. [DOI] [PubMed] [Google Scholar]
- 7.Lugcr TA, Scholzen T, Grabbe S. The role of a-melanocyte-stimulating hormone in cutaneous biology. Journal of Investigative Dermatology Symposium Proceedings. 1997;2(1):87–93. doi: 10.1038/jidsymp.1997.17. [DOI] [PubMed] [Google Scholar]
- 8.Lerner AB, McGuire JS. Melanocyte-stimulating hormone and adrenocorticotrophic hormone. Their relation to pigmentation. The The New England Journal of Medicine. 1964;270:539–546. doi: 10.1056/NEJM196403122701101. [DOI] [PubMed] [Google Scholar]
- 9.Harris JI, Lerner AB. Amino-acid sequence of the α-melanocyte-stimulating hormone. Nature. 1957;179(4574):1346–1347. doi: 10.1038/1791346a0. [DOI] [PubMed] [Google Scholar]
- 10.Singh A, Wilczynski A, Holder JR, et al. Incorporation of a bioactive reverse-turn heterocycle into a peptide template using solid-phase synthesis to probe melanocortin receptor selectivity and ligand conformations by 2D1H NMR. Journal of Medicinal Chemistry. 2011;54(5):1379–1390. doi: 10.1021/jm101425m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barkey NM, Tafreshi NK, Josan JS, et al. Development of melanoma-targeted polymer micelles by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. Journal of Medicinal Chemistry. 2011;54(23):8078–8084. doi: 10.1021/jm201226w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jayawickreme CK, Quillan JM, Graminski GF, Lerner MR. Discovery and structure-function analysis of α-melanocyte-stimulating hormone antagonists. Journal of Biological Chemistry. 1994;269(47):29846–29854. [PubMed] [Google Scholar]
- 13.Lim S, Li S, Lee C, Yoon C, Baik J, Lee W. Minimization of MC1R selectivity by modification of the core structure of α-MSH-ND. Chemistry and Biology. 2001;8(9):857–870. doi: 10.1016/s1074-5521(01)00057-6. [DOI] [PubMed] [Google Scholar]
- 14.Brabez N, Lynch RM, Xu L, et al. Design, synthesis, and biological studies of efficient multivalent melanotropin ligands: tools toward melanoma diagnosis and treatment. Journal of Medicinal Chemistry. 2011;54(20):7375–7384. doi: 10.1021/jm2009937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourne HR, Meng EC. Rhodopsin sees the light. Science. 2000;289(5480):733–734. doi: 10.1126/science.289.5480.733. [DOI] [PubMed] [Google Scholar]
- 16.Palczewski K, Kumasaka T, Hori T, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 17.Vinós J, Jalink K, Hardy RW, Britt SG, Zuker CS. A G protein-coupled receptor phosphatase required for rhodopsin function. Science. 1997;277(5326):687–690. doi: 10.1126/science.277.5326.687. [DOI] [PubMed] [Google Scholar]
- 18.Venkatakrishnan AJ, Deupi X, Lebon G, et al. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494(7436):185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 19.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257(5074):1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 20.Lembo PMC, Grazzini E, Cao J, et al. The receptor for the orexigenic peptide melanin-concentrating hormone is a G-protein-coupled receptor. Nature Cell Biology. 1999;1(5):267–271. doi: 10.1038/12978. [DOI] [PubMed] [Google Scholar]
- 21.Borowsky B, Durkin MM, Ogozalek K, et al. Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nature Medicine. 2002;8(8):825–830. doi: 10.1038/nm741. [DOI] [PubMed] [Google Scholar]
- 22.Lalueza-Fox C, Römpler H, Caramelli D, et al. A melanocortin 1 receptor allele suggests varying pigmentation among Neanderthals. Science. 2007;318(5855):1453–1455. doi: 10.1126/science.1147417. [DOI] [PubMed] [Google Scholar]
- 23.Mundy NI, Badcock NS, Hart T, Scribner K, Janssen K, Nadeau NJ. Conserved genetic basis of a quantitative plumage trait involved in mate choice. Science. 2004;303(5665):1870–1873. doi: 10.1126/science.1093834. [DOI] [PubMed] [Google Scholar]
- 24.Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP. A single amino acid mutation contributes to adaptive beach mouse color pattern. Science. 2006;313(5783):101–104. doi: 10.1126/science.1126121. [DOI] [PubMed] [Google Scholar]
- 25.Saito Y, Nothacker H-P, Wang Z, Lin SHS, Leslie F, Civelli O. Molecular characterization of the melanin-concentrating-hormone receptor. Nature. 1999;400(6741):265–269. doi: 10.1038/22321. [DOI] [PubMed] [Google Scholar]
- 26.Sotomayor M, Schulten K. Single-molecule experiments in vitro and in silico. Science. 2007;316(5828):1144–1148. doi: 10.1126/science.1137591. [DOI] [PubMed] [Google Scholar]
- 27.Levitt M, Warshel A. Computer simulation of protein folding. Nature. 1975;253(5494):694–698. doi: 10.1038/253694a0. [DOI] [PubMed] [Google Scholar]
- 28.Palsson B. The challenges of in silico biology. Nature Biotechnology. 2000;18(11):1147–1150. doi: 10.1038/81125. [DOI] [PubMed] [Google Scholar]
- 29.von Mering C, Krause R, Snel B, et al. Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 2002;417(6887):399–403. doi: 10.1038/nature750. [DOI] [PubMed] [Google Scholar]
- 30.Palsson B. In silico biology through ‘omics’. Nature Biotechnology. 2002;20(7):649–650. doi: 10.1038/nbt0702-649. [DOI] [PubMed] [Google Scholar]
- 31.Kitano H. Computational systems biology. Nature. 2002;420(6912):206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 32.di Ventura B, Lemerle C, Michalodimitrakis K, Serrano L. From in vivo to in silico biology and back. Nature. 2006;443(7111):527–533. doi: 10.1038/nature05127. [DOI] [PubMed] [Google Scholar]
- 33.van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nature Reviews Drug Discovery. 2003;2(3):192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- 34.Puzyn T, Rasulev B, Gajewicz A, et al. Using nano-QSAR to predict the cytotoxicity of metal oxide nanoparticles. Nature Nanotechnology. 2011;6(3):175–178. doi: 10.1038/nnano.2011.10. [DOI] [PubMed] [Google Scholar]
- 35.Liao G, Wang J, Guo J, et al. In silico genetics: identification of a functional elements regulating HZ-Eα gene expression. Science. 2004;306(5696):690–695. doi: 10.1126/science.1100636. [DOI] [PubMed] [Google Scholar]
- 36.Pellecchia M. Fragment-based drug discovery takes a virtual turn. Nature Chemical Biology. 2009;5(5):274–275. doi: 10.1038/nchembio0509-274. [DOI] [PubMed] [Google Scholar]
- 37.Bajorath J. Integration of virtual and high-throughput screening. Nature Reviews Drug Discovery. 2002;1(11):882–894. doi: 10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- 38.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432(7019):862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naylor E, Arredouani A, Vasudevan SR, et al. Identification of a chemical probe for NAADP by virtual screening. Nature Chemical Biology. 2009;5(4):220–226. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan Y, Kollman PA. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282(5389):740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 41.Karplus M, Petsko GA. Molecular dynamics simulations in biology. Nature. 1990;347(6294):631–639. doi: 10.1038/347631a0. [DOI] [PubMed] [Google Scholar]
- 42.Bash PA, Singh UC, Langridge R, Kollman PA. Free energy calculations by computer simulation. Science. 1987;236(4801):564–568. doi: 10.1126/science.3576184. [DOI] [PubMed] [Google Scholar]
- 43.Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294(5550):2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- 44.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nature Structural Biology. 2002;9(9):646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 45.D’Orazio JA, Nobuhisa T, Cui R, et al. Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature. 2006;443(7109):340–344. doi: 10.1038/nature05098. [DOI] [PubMed] [Google Scholar]
- 46.Harms J, Lautenschlager S, Minder CE, Minder EI. An α-melanocyte-stimulating hormone analogue in erythropoietic protoporphyria. The New England Journal of Medicine. 2009;360(3):306–307. doi: 10.1056/NEJMc0805682. [DOI] [PubMed] [Google Scholar]
- 47.Chen CY. Weighted equation and rules—a novel concept for evaluating protein-ligand interaction. Journal of Biomolecular Structure and Dynamics. 2009;27(3):271–282. doi: 10.1080/07391102.2009.10507315. [DOI] [PubMed] [Google Scholar]
- 48.Chang KW, Tsai TY, Chen KC, et al. iSMART: an integrated cloud computing web server for traditional Chinese medicine for online virtual screening, de novo evolution and drug design. Journal of Biomolecular Structure and Dynamics. 2011;29(1):243–250. doi: 10.1080/073911011010524988. [DOI] [PubMed] [Google Scholar]
- 49.Tsai TY, Chang KW, Chen CY. IScreen: world’s first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan. Journal of Computer-Aided Molecular Design. 2011;25(6):525–531. doi: 10.1007/s10822-011-9438-9. [DOI] [PubMed] [Google Scholar]
- 50.Chen CY. A novel integrated framework and improved methodology of computer-aided drug design. Current Topics in Medicinal Chemistry. 2013;13(9):965–988. doi: 10.2174/1568026611313090002. [DOI] [PubMed] [Google Scholar]
- 51.Tou WI, Chang SS, Lee CC, Chen CY. Drug design for neuropathic pain regulation from traditional Chinese medicine. Scientific Reports. 2013;3:p. 844. doi: 10.1038/srep00844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen KC, Chang SS, Huang HJ, et al. Three-in-one agonists for PPAR-alpha, PPAR-gamma, and PPAR-delta from traditional Chinese medicine. Journal of Biomolecular Structure and Dynamics. 2012;30(6):662–683. doi: 10.1080/07391102.2012.689699. [DOI] [PubMed] [Google Scholar]
- 53.Chen KC, Sun MF, Yang SC, et al. Investigation into potent inflammation inhibitors from traditional Chinese medicine. Chemical Biology and Drug Design. 2011;78(4):679–688. doi: 10.1111/j.1747-0285.2011.01202.x. [DOI] [PubMed] [Google Scholar]
- 54.Chang SS, Huang HJ, Chen CY. Two birds with one stone? Possible dual-targeting H1N1 inhibitors from traditional Chinese medicine. PLoS Computational Biology. 2011;7(12) doi: 10.1371/journal.pcbi.1002315.e1002315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang TT, Sun MF, Chen HY, et al. Screening from the world’s largest TCM database against H1N1 virus. Journal of Biomolecular Structure and Dynamics. 2011;28(5):773–786. doi: 10.1080/07391102.2011.10508605. [DOI] [PubMed] [Google Scholar]
- 56.Yang SC, Chang SS, Chen HY, Chen CYC. Identification of potent EGFR inhibitors from TCM database@Taiwan. PLoS Computational Biology. 2011;7(10) doi: 10.1371/journal.pcbi.1002189.e1002189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fjell CD, Hiss JA, Hancock REW, Schneider G. Designing antimicrobial peptides: form follows function. Nature Reviews. 2012;11(1):37–51. doi: 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- 58.Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends in Pharmacological Sciences. 2012;33(5):268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mooney C, Haslam NJ, Holton TA, et al. PeptideLocator: prediction of bioactive peptides in protein sequences. Bioinformatics. 2013;29(9):1120–1126. doi: 10.1093/bioinformatics/btt103. [DOI] [PubMed] [Google Scholar]
- 60.Hruby VJ. Designing peptide receptor agonists and antagonists. Nature Reviews. 2002;1(11):847–858. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 61.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;353(6348):82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 62.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley T, Cuervo JH. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature. 1991;353(6348):84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 63.Chen CYC. TCM database@Taiwan: the world’s largest traditional Chinese medicine database for drug screening in Silico. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0015939.e15939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duchrow T, Shtatland T, Guettler D, Pivovarov M, Kramer S, Weissleder R. Enhancing navigation in biomedical databases by community voting and database-driven text classification. BMC Bioinformatics. 2009;10, article 1471:p. 317. doi: 10.1186/1471-2105-10-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Doedens L, Opperer F, Cai M, et al. Multiple N -methylation of MT-II backbone amide bonds leads to melanocortin receptor subtype hMC1R selectivity: pharmacological and conformational studies. Journal of the American Chemical Society. 2010;132(23):8115–8128. doi: 10.1021/ja101428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294(5540):93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 67.Brooks BR, Brooks CL, III, Mackerell AD, Jr., et al. CHARMM: the biomolecular simulation program. Journal of Computational Chemistry. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Friedman N, Linial M, Nachman I, Pe’er D. Using Bayesian networks to analyze expression data. Journal of Computational Biology. 2000;7(3-4):601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- 69.Yu J, Smith VA, Wang PP, Hartemink AJ, Jarvis ED. Advances to Bayesian network inference for generating causal networks from observational biological data. Bioinformatics. 2004;20(18):3594–3603. doi: 10.1093/bioinformatics/bth448. [DOI] [PubMed] [Google Scholar]
- 70.Chen CY, Tou WI. How to design a drug for the disordered proteins? Drug Discovery Today. 2013;18(19-20):910–915. doi: 10.1016/j.drudis.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 71.Tou WI, Chen CY. May disordered protein cause serious drug side effect? Drug Discovery Today. 2013 doi: 10.1016/j.drudis.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 72.Liao WL, Tsai Fj. Personalized medicine: a paradigm shift in healthcare. BioMedicine. 2013;3(2):66–72. [Google Scholar]
- 73.Tsai FJ. Biomedicine brings the future nearer. BioMedicine. 2011;1(1):p. 1. [Google Scholar]
- 74.Tsai FJ. Rare diseases: a mysterious puzzle. BioMedicine. 2013;3(2):p. 65. [Google Scholar]
- 75.Chou IC, Lin WD, Wang CH, et al. Möbius syndrome in a male with XX/XY mosaicism. BioMedicine. 2013;3(2):102–104. [Google Scholar]
- 76.Lee CC, Tsai CH, Wan L, et al. Increased incidence of Parkinsonism among Chinese with β-glucosidase mutation in central Taiwan. BioMedicine. 2013;3(2):92–94. [Google Scholar]
- 77.Lin DY, Tsai FJ, Tsai CH, Huang CY. Mechanisms governing the protective effect of 17β-estradiol and estrogen receptors against cardiomyocyte injury. BioMedicine. 2011;1(1):21–28. [Google Scholar]
- 78.Wang CH, Lin WD, Bau DT, et al. Appearance of acanthosis nigricans may precede obesity: an involvement of the insulin/IGF receptor signaling pathway. BioMedicine. 2013;3(2):82–87. [Google Scholar]
- 79.Chang YM, Velmurugan BK, Kuo WW, et al. Inhibitory effect of alpinate Oxyphyllae fructus extracts on Ang II-induced cardiac pathological remodeling-related pathways in H9c2 cardiomyoblast cells. BioMedicine. 2013;3(4):148–152. [Google Scholar]
- 80.Chou IC, Lin WD, Wang CH, et al. Association analysis between Tourette's syndrome and two dopamine genes (DAT1, DBH) in Taiwanese children. BioMedicine. 2013;3(2):88–91. [Google Scholar]
- 81.Lin WY, Liu HP, Chang JS, et al. Genetic variations within the PSORS1 region affect Kawasaki disease development and coronary artery aneurysm formation. BioMedicine. 2013;3(2):73–81. [Google Scholar]