

Abstract
A 6-year-old boy born by a third-degree consanguineous marriage presented with progressive muscle weakness and delayed motor milestones noticed in early infancy with preserved language and social milestones. Examination revealed generalised hypotonia and hyporeflexia. Baseline haematological and biochemical investigations were normal except for mildly elevated creatine kinase. Provisional diagnosis of congenital myopathy was entertained. We performed brain imaging to look for abnormalities associated with congenital muscular dystrophy even though there were only features of myopathy with normal mentation. An MRI of the brain revealed periventricular and subcortical white matter hyperintensities suggestive of leucoencephalopathy. Muscle biopsy findings were consistent with degenerative muscle changes and immunohistochemical staining for merosin was negative, thus confirming the diagnosis of merosin-deficient congenital muscular dystrophy. Supportive care in the form of physiotherapy was initiated. The family was offered genetic counselling in their second pregnancy and immunohistochemistry at 12 weeks confirmed the fetus to be affected, which was then terminated.
Background
This case is being reported for its rarity and to highlight the importance of brain imaging in the diagnosis of subtype of congenital muscular dystrophy even in a child with no clinical features suggestive of central nervous system (CNS) involvement. This variant has its simplicity in diagnosis by muscle biopsy which can be detected prenatally as well.
Case presentation
A 6-year-old boy born out of a third-degree consanguineous marriage presented to us with isolated delayed motor milestones noticed in early infancy. Parents noted a head lag at 3 months of age and subsequent delay in attaining other motor milestones. He was able to sit only with support and was not able to stand or walk. His mentation and speech were normal. Vision and hearing were normal. There was no history of seizures. No respiratory difficulty was noted. His birth and family history were unremarkable.
On examination, his weight was 12.5 kg (below 3rd centile), length was 110 cm (3rd centile) and head circumference was 48 cm (between −2SD and −3SD). His vital signs were normal. He had classical myopathic facies with inverted “v”-shaped upper lip and shallow cheeks. There were no other dysmorphic features and both testes were descended. Neurological examination revealed symmetrical decrease in bulk of muscles of upper and lower limbs with contractures. There was no fasciculation or hypertrophy of any group of muscles. Muscle power was decreased globally (MRC grading 2/5) with hypotonia, diminished deep tendon reflexes and bilateral flexor plantar response with no sensory deficits. Examination of other systems were normal.
Investigations
Investigations revealed normal blood counts and electrolytes with serum creatine phophokinase (CPK) level of 357 IU/L (normal range 5–130 IU/L). Thyroid, renal and liver function tests were normal. EMG (electromyography) was suggestive of myopathic pattern. We performed an MRI of the brain to look for features associated with congenital muscular dystrophy. The MRI showed T2-weighted and fluid-attenuated inversion recovery (FLAIR) hyperintensities involving periventricular and subcortical white matter of both cerebral hemispheres, suggestive of dysmyelination (figures 1 and 2). This gave us a clue to suspect merosin-deficient variety of congenital muscular dystrophy. Muscle biopsy showed evidence of muscular dystrophy with immunohistochemistry (IHC) staining negative for merosin, thus confirming the diagnosis.
Figure 1.
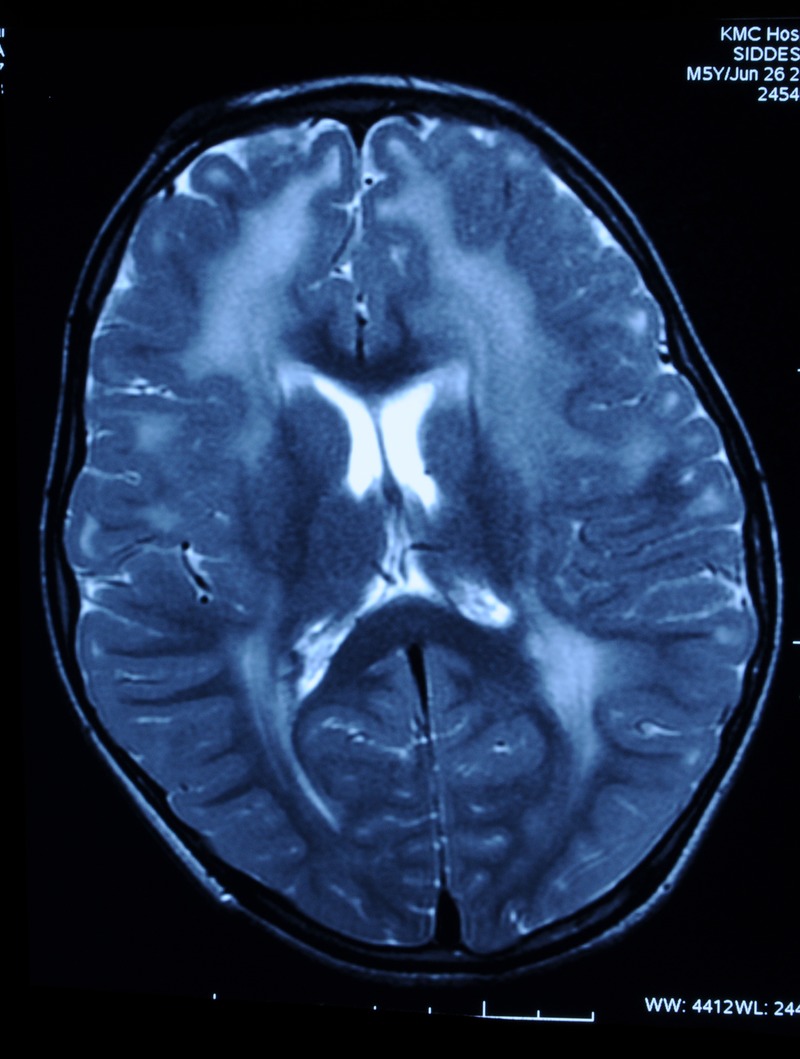
T2-weighted and fluid-attenuated inversion recovery images showing hyperintense signals in the periventricular and subcortical white matter, suggestive of dysmyelination.
Figure 2.
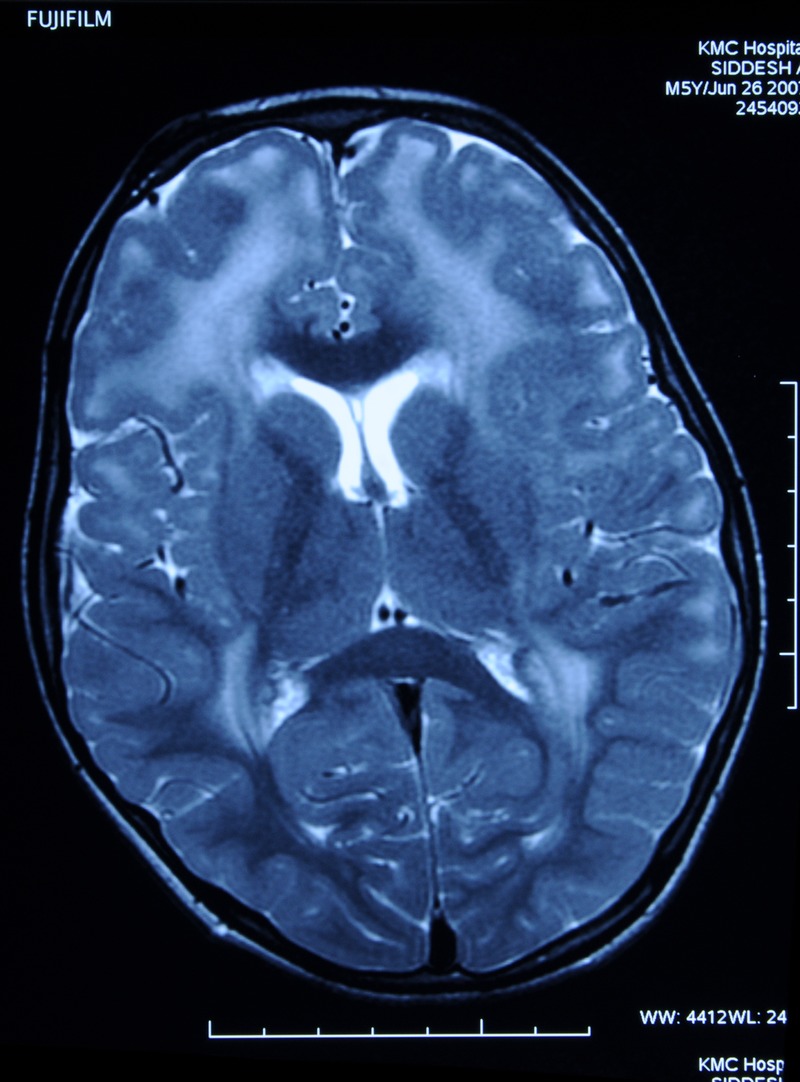
T2-weighted and fluid-attenuated inversion recovery images showing hyperintense signals in the periventricular and subcortical white matter, suggestive of dysmyelination.
Differential diagnosis
A close differential diagnosis for the typical MRI changes of leucoencephalopathy would be leucodystrophies. Clinically, leucodystrophies are characterised by loss of acquired developmental milestones (neuroregression) with the presence of upper motor neuron signs and cognitive impairment often with seizures, in contrast to our case. Although there is significant overlap in symptomatology, certain clinical signs can be clues to particular leucodystrophies. Adrenal insufficiency is seen almost exclusively with X-linked adrenoleucodystrophy (X-ALD). A coexistent peripheral neuropathy often occurs in Krabbe disease and MLD. Megalencephaly is characteristic for Canavan and Alexander disease. MRI changes in leucodystrophies predominantly involve cortical white matter. It was the subcortical white matter involved in our case.
Treatment
As there is no specific treatment for this variant, supportive care in the form of physiotherapy therapy was initiated. Nutritional rehabilitation with ready-to-use therapeutic foods was supplemented as per standard guidelines.
Outcome and follow-up
The family was offered genetic counselling in their second pregnancy, and immunohistochemistry performed on chorionic villous sampling at 12 weeks confirmed the fetus to be affected. Subsequently, termination was performed, but the parents were not screened for carrier status.
Discussion
Congenital muscular dystrophies (CMDs) are a heterogeneous group of degenerative muscle diseases mainly inherited by autosomal recessive pattern. Muntoni and Voit classified CMD phenotypes into four categories, one of which is defect in extracellular matrix protein namely laminin α2, also called merosin, designated as merosin-deficient congenital muscular dystrophy type 1A (MDC1A, MIM#607855).1 It was the first CMD variant for which mutations in Laminin α2 (LAMA2, MIM #156225) was found.
Merosin is a basement membrane structural protein that is expressed in skeletal muscle, Schwann cells, cardiac muscle and placental villi. It binds α-dystroglycan and, in turn, is linked to the subsacrolemmal cytoskeleton via the dystrophin-glycoprotein complex (DGC). The DGC provides a structural link between the cytoskeleton and the proteins of the basement membrane through interaction with extracellular ligands. It is thought to protect the sarcolemma from mechanical stress during muscle contraction. Studies have shown that a deficiency of merosin could disrupt the link between the extracellular matrix and the subsacrolemmal cytoskeleton, thus causing muscle degeneration.2 Some of the researches suggested that myofibre injury may be the result of an inflammatory myopathy, followed by poor regeneration of myofibres caused by a defective basal lamina.3
Children with complete deficiency of merosin, as occurs in majority, present in the neonatal period with profound muscular weakness, severe contractures and respiratory insufficiency with CPK levels typically exceeding 1000 IU/L. A minority can have a partial deficiency, as in our case, presenting in early infancy with delayed motor milestones, hypotonia and variable degree of contractures with mildly elevated CPK. Other manifestations reported in these patients include extra-ocular muscle involvement and myocardial involvement, which were absent in our case. Intelligence is usually normal in MDC1A, as in this case, although some children may have mental retardation and seizures. Diagnosis is usually made by a combination of clinical features, muscle biopsy and mutational analysis. Muscle biopsies show a variable degree of fibre necrosis and regeneration, increased connective tissue and inflammatory infiltrates. Immunohistochemically, laminin α2 chain is deficient with over-expression of α4 and α5 chains in the basal lamina surrounding muscle fibres.4
Majority of merosin-deficient CMD variants often reveal diffuse white matter changes on MRI of the brain, which is referred to as dysmyelinating leucoencephalopathy.5 Studies have shown that these MRI changes are known to progress with age. Though MR findings in early infancy are normal in a child suspected of having CMD, the diagnosis of merosin-deficient CMD should not be discarded. Instead, the study should be repeated at least after the age of 2 years to look for changes in the white matter.6 Thus, MRI in early infancy cannot be used for screening CMD. The exact pathogenesis of these white matter changes in the absence of clinical CNS involvement is unknown. One of the hypotheses states that as merosin is known to promote neurite outgrowth and Schwann cell migration, its deficiency may result in abnormal myelination.7 Another hypothesis postulated by Villanova et al8 states that merosin forms an integral component of basement membrane of cerebral blood vessels, thus its deficiency may result in disruption of blood–brain barrier, leading to vascular hyperpermeability. Thus, increased T2 prolongation time on an MRI may be attributable to increased water content in the white matter owing to an abnormal blood–brain barrier rather than abnormal myelination. The literature supports this hypothesis, and findings report normal brain stem auditory-evoked responses in patients with merosin-deficient CMD. Brain stem auditory-evoked responses are typically abnormal in patients with leucodystrophies. However, further studies are needed to prove this hypothesis.8 MR spectroscopy would also add a value in diagnosing certain leucodystrophies. Some children exhibiting mental retardation often have structural changes visible on MRI including cerebellar hypoplasia, cysts and occasionally neuronal migration abnormalities usually of occipital cortex.9–13
Muscular dystrophy is often viewed with diagnostic apathy as we can offer no curative treatment at present. Supportive care is essential in the form of physiotherapy to allow for maximal functional ability, minimising the development of contractures.14 Ventilatory support and tracheotomy, when necessary, have contributed to a marked increase of the life expectancy for the most severely affected patients. Considerable importance regarding nutritional management is also required. Mutational analysis for LAMA2 gene would add value, but is not easily available. Linkage analysis provides an indirect approach when it is difficult to obtain a molecular diagnosis. Prenatal diagnosis can be performed by either molecular methods or IHC of chorionic villus sampling.
Learning points.
Congenital muscular dystrophy includes a vast spectrum of subtypes.
Muscle biopsy with immunohistochemistry and appropriate genetic study are required for the definitive diagnosis of the subtype but are not easily available.
MRI of the brain in congenital muscular dystrophy provides some valuable clue for diagnosis of merosin-deficient subtype even in a child with normal mentation.
Brain imaging cannot be used for screening congenital muscular dystrophy in early infancy as white matter changes are known to progress with age typically after infancy.
This variant of congenital muscular dystrophy can be easily detected prenatally as well.
Acknowledgments
The authors are thankful to Dr Girish KM, associate professor, department of medical genetics for his prenatal diagnosis and his valuable guidance.
Footnotes
Contributors: SK was involved in the care of the patient presented and wrote the first draft of the manuscript which was subsequently edited and finalised by SA. SM and MK were involved in data collection and review of literature.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004;14:635–49 [DOI] [PubMed] [Google Scholar]
- 2.Pilar AC, Mena S, Eric H, et al. MR imaging findings in children with merosin-deficient congenital muscular dystrophy. AJNR Am J Neuroradiol 1999;20:324–6 [PMC free article] [PubMed] [Google Scholar]
- 3.Pegoraro E, Mancias P, Swerdlow SH, et al. Congenital muscular dystrophy with primary laminin alpha2 (merosin) deficiency presenting as inflammatory myopathy. Ann Neurol 1996;40:782–91 [DOI] [PubMed] [Google Scholar]
- 4.Allamand V, Guicheney P. Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2gene coding for α2 chain of laminin). Eur J Hum Genet 2002;10:91–4 [DOI] [PubMed] [Google Scholar]
- 5.Kaye EM. Update on genetic disorders affecting white matter. Pediatr Neurol 2001;24:11–24 [DOI] [PubMed] [Google Scholar]
- 6.Trevisan CP, Martinello F, Ferruzza E, et al. Brain alterations in the classical form of congenital muscular dystrophy: clinical and neuroimaging follow-up of 12 cases and correlation with the expression of merosin in muscle. Childs Nerv Syst 1996;12:604–10 [DOI] [PubMed] [Google Scholar]
- 7.Engvall E, Earwicker D, Day A, et al. Merosin promotes cell attachment and neurite outgrowth and is a component of the neurite promoting factor of RN22 schwannoma cells. Exp Cell Res 1992;198:115–23 [DOI] [PubMed] [Google Scholar]
- 8.Villanova M, Malandrini A, Toti PE, et al. Localization of merosin in the normal human brain: implications for congenital muscular dystrophy with merosin deficiency. J Submicrosc Cytol Pathol 1996;28:1–4 [PubMed] [Google Scholar]
- 9.Jones K, Morgan G, Johnson H, et al. The expanding phenotype of laminin α2 chain (merosin) abnormalities: case series and review. J Med Genet 2001;38:649–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan E, Topaloglu H, Sewry C, et al. Late onset muscular dystrophy with cerebral white matter changes due to partial merosin deficiency. Neuromuscul Disord 1997;7:85–9 [DOI] [PubMed] [Google Scholar]
- 11.Leyten QH, Gabreels FJM, Renier WO, et al. White matter abnormalities in congenital muscular dystrophy. J Neurol Sci 1995;129:162–9 [DOI] [PubMed] [Google Scholar]
- 12.Klein A, Clement E, Mercuri E, et al. Differential diagnosis of congenital muscular dystrophies. Eur J Paediatr Neurol 2008;12:371–7 [DOI] [PubMed] [Google Scholar]
- 13.Lamer S, Carlier RY, Pinard JM, et al. Congenital muscular dystrophy: use of brain MR imaging findings to predict merosin deficiency. Radiology 1998;206:811–16 [DOI] [PubMed] [Google Scholar]
- 14.Emery AE. The muscular dystrophies. Lancet 2002;359:687–95 [DOI] [PubMed] [Google Scholar]