

Abstract
Purpose
Methotrexate (MTX) can cause significant clinical neurotoxicity and asymptomatic leukoencephalopathy. We sought to identify clinical, pharmacokinetic, and genetic risk factors for these MTX-related toxicities during childhood acute lymphoblastic leukemia (ALL) therapy and provide data on safety of intrathecal and high-dose MTX rechallenge in patients with neurotoxicity.
Patients and Methods
Prospective brain magnetic resonance imaging was performed at four time points for 369 children with ALL treated in a contemporary study that included five courses of high-dose MTX and 13 to 25 doses of triple intrathecal therapy. Logistic regression modeling was used to evaluate clinical and pharmacokinetic factors, and a genome-wide association study (GWAS) was performed to identify germline polymorphisms for their association with neurotoxicities.
Results
Fourteen patients (3.8%) developed MTX-related clinical neurotoxicity. Of 13 patients rechallenged with intrathecal and/or high-dose MTX, 12 did not experience recurrence of neurotoxicity. Leukoencephalopathy was found in 73 (20.6%) of 355 asymptomatic patients and in all symptomatic patients and persisted in 74% of asymptomatic and 58% of symptomatic patients at the end of therapy. A high 42-hour plasma MTX to leucovorin ratio (measure of MTX exposure) was associated with increased risk of leukoencephalopathy in multivariable analysis (P = .038). GWAS revealed polymorphisms in genes enriched for neurodevelopmental pathways with plausible mechanistic roles in neurotoxicity.
Conclusion
MTX-related clinical neurotoxicity is transient, and most patients can receive subsequent MTX without recurrence of acute or subacute symptoms. All symptomatic patients and one in five asymptomatic patients develop leukoencephalopathy that can persist until the end of therapy. Polymorphisms in genes related to neurogenesis may contribute to susceptibility to MTX-related neurotoxicity.
INTRODUCTION
Methotrexate (MTX) is an essential drug in the treatment of childhood acute lymphoblastic leukemia (ALL). In addition to systemic control of leukemia, it is crucial for prophylaxis and treatment of sanctuary sites, including the CNS. However, MTX can cause acute, subacute, and long-term neurotoxicities.1–6 The mechanism of neurotoxicity is likely through disruption of CNS folate homeostasis and/or direct neuronal damage.7–10 Subacute MTX neurotoxicity typically occurs 2 to 14 days after prolonged low-dose oral, intrathecal, or high-dose MTX and manifests with transient stroke-like symptoms, encephalopathy, seizures, and/or aphasia. Although prior reports have demonstrated that many patients can be safely rechallenged with MTX, some have recurrences of neurotoxicity.2,11 Subsequent MTX is often omitted, potentially increasing relapse risk.
Clinical symptoms of MTX-induced neurotoxicity are often associated with leukoencephalopathy, seen as white matter hyperintensities on T2-weighted and fluid-attenuated inversion recovery magnetic resonance imaging (MRI).1,12 Although leukoencephalopathy is low grade in most patients, fatal diffuse necrotizing leukoencephalopathy has been reported.13,14 Leukoencephalopathy can also develop in asymptomatic children receiving MTX, and its presence has been correlated with increasing MTX exposure.3,15 The clinical significance of these white matter changes is unknown.16 It is also unclear whether patients with asymptomatic leukoencephalopathy are at higher risk of developing symptoms when exposed to additional MTX. Germline polymorphisms may contribute to MTX-induced leukoencephalopathy and neurotoxicities and have included variants in GSTP1,17 MTHFR, and SHMT1.18 A comprehensive genome-wide association study (GWAS) has not been performed.
In this study, we correlate clinical symptoms of MTX-related subacute neurotoxicity with leukoencephalopathy on MRI, provide data on safety of rechallenging patients with additional MTX, and identify clinical, therapy-related, and genetic risk factors for clinical neurotoxicity and leukoencephalopathy.
PATIENTS AND METHODS
Patients and Therapy
From June 2000 to October 2007, 498 children with ALL were enrolled onto the Total Therapy XV study.19 Of these, 408 patients enrolled at St Jude Children's Research Hospital were approached for participation in prospective MRI screening. Data from 369 patients were analyzed in this study (Data Supplement). Treatment details from the protocol specific to MTX have been included here. Two hundred seventy-eight of 369 patients received an upfront window treatment comprising high-dose MTX 1 g/m2 (randomly assigned to 4- v 24-hour infusions).20 During consolidation, patients in the low-risk arm received high-dose MTX 2.5 g/m2 infused over 24 hours for four doses, adjusted to achieve steady-state plasma concentration of 33 μmol/L, and patients in the standard/high-risk arm received 5 g/m2 over 24 hours, adjusted to 65 μmol/L.21 Leucovorin rescue dose for window therapy was 50 mg/m2 at 44 hours, followed by 15 mg/m2 every 6 hours for seven doses. During consolidation, leucovorin was administered at 5 mg/m2 in the low-risk arm and 10 mg/m2 in the standard/high-risk arm for five doses beginning 42 hours after initiation of MTX. Leucovorin doses were increased in patients with a 42-hour MTX level > 1 μmol/L and in those with a history of delayed clearance.21 The first intrathecal therapy was cytarabine alone, and subsequent intrathecal therapies consisted of MTX, hydrocortisone, and cytarabine in age-dependent doses. The total number of triple intrathecal therapies (ITTs) ranged from 13 to 25. No patient received prophylactic cranial irradiation. Low-dose intravenous MTX (40 mg/m2) was administered weekly after reinduction II, with interruption for monthly pulses of dexamethasone/vincristine or cyclophosphamide/cytarabine. Total number of doses of MTX in continuation ranged from 68 to 116.
The study was approved by the institutional review board. Informed consent was obtained from parents/guardians, and assent was obtained from patients when appropriate.
Definition of MTX-Related Clinical Neurotoxicity
A neurotoxic adverse event was attributed to MTX if neurologic symptoms (eg, seizure, stroke, behavioral changes, aphasia) occurred within 2 weeks of receiving MTX (intrathecal or intravenous), and other identifiable causes were reasonably ruled out. Patients with clinical neurotoxicity were evaluated by a pediatric neurologist. Adverse events were graded according to the Common Terminology Criteria for Adverse Events (version 3.0), entered into the database in real time, and retrieved for the purpose of this study.
Pharmacokinetic Measurements
Serum MTX and plasma homocysteine concentrations were measured as previously described.20–23 Blood samples for measuring MTX were drawn before high-dose MTX infusion and at 6, 23, and 42 hours from the start of infusion.21 Additional MTX concentrations were measured if the 42-hour level was ≥ 1 μmol/L. Plasma homocysteine concentrations were measured before high-dose MTX infusion and at 23 and 42 hours in courses one and two. The concentration-time course and area under the curve over baseline (μM × hour) for homocysteine were calculated as previously described.22
Detection and Grading of Leukoencephalopathy
Brain MRIs were obtained at four time points during therapy: postinduction between days 33 and 46 (MRI1), postconsolidation (ie, week 1 of reinduction I; MRI2), continuation week 48 (MRI3), and continuation week 120 (MRI4). MRI4 was considered the off-therapy time point for uniformity, although boys received an additional 26 weeks of continuation chemotherapy that did not include ITT or high-dose MTX. Most patients with clinical neurotoxicity underwent additional imaging during or after the event. The protocol noncontrast MRI examinations consisted of sagittal T1, axial T1-weighted inversion recovery, axial T2, axial proton density, and axial fluid-attenuated inversion recovery pulse sequences of the brain obtained on a 1.5 Tesla MRI platform (Data Supplement). Abnormal MRIs were identified by a single neuroradiologist and graded for the extent of leukoencephalopathy by a second neuroradiologist, according to radiographic criteria of Common Terminology Criteria for Adverse Events (version 4.0). In brief, grades 1, 2, and 3 indicate involvement of < one third, one third to two thirds, and > two thirds of the susceptible areas of the cerebrum, respectively. The neuroradiologists were blinded to risk arm of the study.
Statistical Analyses
Logistic regression models were used to determine the association of neurotoxicity and leukoencephalopathy with patient demographics, disease features, and MTX pharmacokinetic parameters. Analyses were performed using SAS (version 9.2; SAS Institute, Cary, NC) and R software for LINUX (version 2.15.0; http://www.r-project.org). The classification and regression tree analysis tool was used as an alternate approach to identify risk factors for leukoencephalopathy.24
Genotyping and GWAS
Genome-wide single-nucleotide polymorphism (SNP) genotyping was performed for 364 patients using Affymetrix 500K/6.0 array sets (Santa Clara, CA). Genotyping of 1,321 candidate SNPs was also performed for 344 patients using the Illumina GoldenGate assay (San Diego, CA) as previously described.23,25 Race was determined by SNP genotype-based ancestry using STRUCTURE.26 GWAS was performed for two phenotypes: leukoencephalopathy (grade 0 v > 0) and clinical neurotoxicity (presence v absence). Multiple logistic regression models were used to test the association between leukoencephalopathy or neurotoxicity and SNP genotype, with genotype treated as an ordinal variable (AA as 0, AB as 1, and BB as 2). Pathway analysis of significant genes was performed using the g:Profiler program.27
RESULTS
Characteristics of Patients With Clinical Neurotoxicity Attributed to MTX
Of 369 patients, 14 (3.8%) developed MTX-related subacute neurotoxic events (Table 1). Four patients were described previously (Nos. 9, 10, 12, and 13).1 Seven patients presented with seizures, six with stroke-like symptoms, and one with ataxia. Most episodes were brief, but ataxia persisted in Patient No. 5 for 4 weeks. All 12 patients with MRIs available at the time of the event had leukoencephalopathy (Fig 1). Overall, leukoencephalopathy was detected in all symptomatic patients at least once during the course of therapy. Screening MRI was available before the event for 10 patients. Pre-existing leukoencephalopathy was evident in seven patients, whereas three patients had normal preceding MRIs. Of 12 patients with end-therapy MRIs, leukoencephalopathy persisted in seven patients and resolved in five.
Table 1.
Patient Characteristics, Details of Clinical Neurotoxic Events, and Rechallenge With MTX
| Patient No. | Age (years) | Sex | CNS Status* | Therapy Arm† | MTX Before Event | Time Point in Therapy | Time From MTX to Event (days) | Neurotoxic Event | Duration of Event | Subsequent No. of High-Dose MTX Doses | Subsequent No. of ITTs | Prophylaxis | Recurrent Neurotoxicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 13 | M | CNS 2 | Standard | High-dose MTX, ITT | Consolidation course one | 4 | Seizure (tonic clonic) | 2 minutes | 3 | 20 | No | |
| 2 | 3 | M | CNS 1 | Low | Low-dose MTX, ITT | Continuation week 40 | 9 | Seizure (complex partial) | 24 hours | 0 | 0 | NA | |
| 3 | 5 | M | CNS 1 | Standard | High-dose MTX, ITT | Consolidation course two | 3 | Seizure (tonic clonic) | 5 minutes | 2 | 13 | Leucovorin after ITT | |
| 4 | 15 | M | CNS 1 | Standard | High-dose MTX, ITT | Consolidation course two | 10 | Stroke-like | 72 hours | 0 (low dose) | 7 | No | |
| 5 | 4 | M | CNS 1 | Standard | ITT | Continuation week 12 | 7 | Ataxia | 4 weeks | 0 | 8 | Leucovorin after ITT | |
| 6 | 10 | F | CNS 1 | Standard | High-dose MTX, ITT | Consolidation course three | 8 | Seizure (complex partial) | 24 hours | 1 | 11 | Leucovorin after ITT | |
| 7 | 11 | M | CNS 2 | Standard | High-dose MTX, ITT | Consolidation course one | 11 | Seizure (tonic clonic) | 20 minutes | 3 | 13 | Leucovorin after ITT | |
| 8 | 2 | F | CNS 2 | Low | Low-dose MTX, ITT | Continuation week 36 | 8 | Seizure (complex partial) | 24 hours | 0 | 3 | No | |
| 9 | 16 | M | CNS 1 | Standard | ITT | Continuation week 13 | 9 | Stroke-like | 24 hours | 0 | 8 | Leucovorin after ITT | |
| 10 | 14 | F | CNS 1 | Standard | High-dose MTX, ITT | Consolidation course one | 7 | Stroke-like | 5 hours | 1 (omit 2)‡ | 9 | Aminophylline | Headache, confusion |
| 11 | 5 | M | CNS 1 | Standard | ITT | Continuation week 29 | 7 | Seizure (complex partial) | 7 days | 0 | 11 | No | |
| 12 | 12 | M | CNS 1 | Standard | High-dose MTX, ITT | Consolidation course one | 10 | Stroke-like | 48 hours | 3‡ | 11 | Aminophylline | |
| 13 | 18 | M | CNS 1 | Standard | High-dose MTX, ITT | Consolidation course one | 10 | Stroke-like (and seizure) | 8 hours | 3 | 20 | No | Stroke (CNS thrombus) |
| 14 | 17 | F | CNS 1 | Standard | Low-dose MTX, ITT | Continuation week 88 | 11 | Stroke-like | 36 hours | 0 | 1 | No |
Abbreviations: CSF, cerebrospinal fluid; ITT, triple intrathecal therapy; MTX, methotrexate; NA: not applicable.
CNS1, < 5 WBC/μL of CSF without blasts; CNS2, < 5 WBC/μL of CSF with any blasts.
Details on risk stratification described by Pui et al.19
Second high-dose MTX and ITT given 1-2 weeks apart.
Fig 1.

Grades of leukoencephalopathy in symptomatic patients at four screening time points. Boxes outlined in black indicate timing of neurotoxic event and grade of leukoencephalopathy in additional magnetic resonance imaging magnetic resonance imaging (MRI) obtained at time of the event. (*)No leukoencephalopathy on screening MRI before neurotoxic event (n = 3). (†) Leukoencephalopathy present on screening MRI before neurotoxic event (n = 7). (‡) Screening MRI not done (ND) before neurotoxic event (n = 4). DWI, diffusion weighted imaging.
MTX Rechallenge in Patients With Clinical Neurotoxicity
Thirteen patients were rechallenged with high-dose MTX or ITT. One patient (No. 2) was not rechallenged, because he required only one more dose of ITT. High-dose MTX was substituted with low-dose MTX (40 mg/m2) for Patient No. 4. Two patients received aminophylline prophylaxis before subsequent high-dose MTX, and five patients received leucovorin rescue 24 and 36 hours after subsequent ITT. MTX-related neurotoxicity (severe headache and confusion) recurred in one patient (No. 10) when challenged with high-dose MTX. She did not receive additional high-dose MTX but received ITT with leucovorin rescue and experienced occasional headaches for the following 2 to 3 days. The other 12 patients tolerated MTX rechallenge well. Patient No. 13 developed a seizure 5.5 months after his first event but was found to have a CNS thrombus likely related to asparaginase.
Risk Factors for Clinical Neurotoxic Events
To identify risk factors, a logistic regression model with clinical and pharmacokinetic factors as explanatory variables was fitted (Table 2). Univariable analysis revealed that patients age > 10 years were at higher risk for neurotoxic events than those age 1 to 10 years (P = .003). Patients in the standard/high-risk arm were also at higher risk for clinical neurotoxicity than those treated in the low-risk arm (P = .016). No risk factor retained significance in a multivariable model. The ratio of 42-hour MTX level to leucovorin dose was calculated for each consolidation course. This measure of MTX exposure relative to leucovorin did not vary significantly between patients with and without neurotoxicity.
Table 2.
Association of Clinical and MTX Pharmacokinetic Parameters* With MTX-Related Clinical Neurotoxic Events and Leukoencephalopathy
| MTX Toxicity | Summary Statistics |
Univariable Logistic Regression |
Multiple Logistic Regression |
||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Median | Range | OR | 95% CI | P | OR | 95% CI | P | |
| Clinical NT | |||||||||
| Age (1-10 v > 10 years) | 0.19 | 0.06 to 0.57 | .003 | 0.43 | 0.01 to 1.80 | .249 | |||
| No NT: 1-10 years | 283 | 4.2 | 1.0-10.9 | ||||||
| NT: 1-10 years | 6 | 4.3 | 2.1-10.0 | ||||||
| No NT: > 10 years | 72 | 14.3 | 11.0-18.9 | ||||||
| NT: > 10 years | 8 | 14.3 | 11.1-18.2 | ||||||
| Treatment arm (LR v SR/HR) | 0.16 | 0.03 to 0.71 | .016 | 0.42 | 0.06 to 2.91 | .383 | |||
| No NT: LR | 183 | NA | NA | ||||||
| NT: LR | 2 | NA | NA | ||||||
| No NT: SR/HR | 172 | NA | NA | ||||||
| NT: SR/HR | 12 | NA | NA | ||||||
| Total no. of triple IT therapies† | 1.09 | 0.98 to 1.21 | .127 | 1.08 | 0.93 to 1.26 | .313 | |||
| No NT | 355 | 13 | 1.00-24.0 | ||||||
| NT | 14 | 15 | 5.00-23.0 | ||||||
| Course one: homocysteine AUC > baseline (μM · hour) | 1.02 | 0.96 to 1.08 | .489 | 1 | 0.90 to 1.12 | .963 | |||
| No NT | 347 | 10.5 | 0.00-73.2 | ||||||
| NT | 13 | 15.1 | 0.00-22.5 | ||||||
| Course two: homocysteine AUC > baseline (μM · hour) | 1.01 | 0.93 to 1.09 | .886 | 0.95 | 0.82 to 1.09 | .463 | |||
| No NT | 347 | 11.4 | 0.00-61.1 | ||||||
| NT | 13 | 10.9 | 0.00-38.1 | ||||||
| Course one: ratio of 42-hour MTX to LV dose | 1.07 | 0.88 to 1.31 | .482 | 1.18 | 0.91 to 1.53 | .212 | |||
| No NT | 317 | 6.2 | 0.47-16.2 | ||||||
| NT | 13 | 6.7 | 1.84-11.6 | ||||||
| Course two: ratio of 42-hour MTX to LV dose | 0.8 | 0.63 to 1.03 | .082 | 0.82 | 0.58 to 1.15 | .248 | |||
| No NT | 310 | 6.5 | 0.80-22.9 | ||||||
| NT | 13 | 5.2 | 2.37-11.6 | ||||||
| Course three: ratio of 42-hour MTX to LV dose | 0.95 | 0.76 to 1.19 | .664 | 1.02 | 0.78 to 1.34 | .892 | |||
| No NT | 309 | 6.6 | 1.62-18.2 | ||||||
| NT | 11 | 6.7 | 2.38-10.8 | ||||||
| Course four: ratio of 42-hour MTX to LV dose | 1.02 | 0.89 to 1.17 | .766 | 1.05 | 0.91 to 1.20 | .516 | |||
| No NT | 299 | 6.3 | 1.97-48.2 | ||||||
| NT | 11 | 7.8 | 2.77-11.5 | ||||||
| LE | |||||||||
| Age (1-10 v > 10 years) | 1.59 | 0.34 to 1.02 | .059 | 0.81 | 0.10 to 1.80 | .556 | |||
| No LE: 1-10 years | 228 | 4.4 | 1.0-10.9 | ||||||
| LE: 1-10 years | 61 | 3.5 | 1.2-10.8 | ||||||
| No LE: > 10 years | 55 | 14.3 | 11.0-18.8 | ||||||
| LE: > 10 years | 25 | 13.8 | 11.1-18.2 | ||||||
| Treatment arm (LR v SR/HR) | 0.69 | 0.42 to 1.12 | .133 | 0.83 | 0.41 to 1.65 | .588 | |||
| No LE: LR | 149 | NA | NA | ||||||
| LE: LR | 36 | NA | NA | ||||||
| No LE: SR/HR | 134 | NA | NA | ||||||
| LE: SR/HR | 50 | NA | NA | ||||||
| Total no. of triple IT therapies† | 1.06 | 1.01 to 1.11 | .023 | 1.07 | 1.00 to 1.15 | .066 | |||
| No LE | 283 | 12 | 1.00-24.0 | ||||||
| LE | 86 | 15 | 1.00-24.0 | ||||||
| Course one: homocysteine AUC > baseline (μM · hour) | 1.04 | 1.00 to 1.07 | .023 | 1.03 | 0.99 to 1.07 | .145 | |||
| No LE | 276 | 10.1 | 0.00-44.8 | ||||||
| LE | 84 | 11.7 | 0.00-73.2 | ||||||
| Course two: homocysteine AUC > baseline (μM · hour) | 1.03 | 0.99 to 1.06 | .132 | 0.98 | 0.93 to 1.04 | .592 | |||
| No LE | 276 | 11.3 | 0.00-50.7 | ||||||
| LE | 84 | 11.5 | 0.00-61.1 | ||||||
| Course one: ratio of 42-hour MTX to LV dose | 1.1 | 1.00 to 1.21 | .04 | 1.12‡ | 1.10 to 1.25 | .038 | |||
| No LE | 256 | 6.1 | 0.47-16.2 | ||||||
| LE | 74 | 6.7 | 1.18-15.5 | ||||||
| Course two: ratio of 42-hour MTX to LV dose | 0.98 | 0.90 to 1.07 | .635 | 0.97 | 0.87 to 1.08 | .567 | |||
| No LE | 251 | 6.5 | 0.80-22.9 | ||||||
| LE | 72 | 6.2 | 2.37-15.9 | ||||||
| Course three: ratio of 42-hour MTX to LV dose | 1.01 | 0.92 to 1.11 | .822 | 1.01 | 0.90 to 1.13 | .889 | |||
| No LE | 249 | 6.6 | 1.62-17.0 | ||||||
| LE | 71 | 6.7 | 1.86-18.2 | ||||||
| Course four: ratio of 42-hour MTX to LV dose | 1.06 | 0.99 to 1.13 | .118 | 1.05 | 0.98 to 1.13 | .192 | |||
| No LE | 240 | 6.3 | 1.97-19.3 | ||||||
| LE | 70 | 6.4 | 2.52-48.2 | ||||||
NOTE. Additional clinical variables that were tested and were not significantly associated with clinical NT or LE in univariable analyses were sex, race, immunophenotype, CNS status, and leukocyte count at diagnosis.
Abbreviations: AUC, area under the curve; HR, high risk; IT, intrathecal; LE, leukoencephalopathy; LR, low risk; LV, leucovorin; MTX, methotrexate; NA, not applicable; NT, neurotoxicity (clinical); OR, odds ratio; SR, standard risk.
Obtained during consolidation phase.
Total No. of triple IT therapies before last screening magnetic resonance imaging.
OR > 1 indicates 1.12-fold higher risk of LE in those with higher ratio of plasma MTX (in μM) to LV dose (in mg/m2 per total course).
Incidence of Leukoencephalopathy
Of 369 patients, 86 (23.3%) had evidence of leukoencephalopathy on at least one screening MRI. These included 73 (20.6%) of 355 asymptomatic patients and 13 (92.9%) of 14 patients with clinical neurotoxicity. Figure 2 shows the grades of leukoencephalopathy at various time points. No patient had radiographic grade 3 or 4 leukoencephalopathy. In asymptomatic patients, leukoencephalopathy was detected in 12.3% at MRI1, in 20.1% at MRI2, in 19.1% at MRI3, and in 15.9% at MRI4. Of 62 asymptomatic patients who developed leukoencephalopathy at any time during therapy and for whom MRI4 was available, leukoencephalopathy persisted in 46 (74.2%). In the 13 symptomatic patients with positive screening MRIs, leukoencephalopathy was detected in 50%, 100%, 72.7%, and 58.3% at the four time points, respectively. Thus, leukoencephalopathy was more prevalent in symptomatic versus asymptomatic patients at all four time points (P < .001). The presence of leukoencephalopathy on screening MRI1 and MRI2 indicated neurotoxic events with 50% and 100% sensitivity, respectively, but the positive predictive values were only 15.1% and 13.2% at the two time points, respectively (Data Supplement).
Fig 2.

Number and percentage of patients with leukoencephalopathy at four screening time-points for (A) asymptomatic and (B) symptomatic patients. Incidence of leukoencephalopathy was highest after four doses of high-dose methotrexate (at time of magnetic resonance imaging [MRI] postconsolidation [MRI2]) and progressively decreased over time. At all four screening time points, incidence of leukoencephalopathy was higher in symptomatic than asymptomatic patients (P < .001). MRI1, MRI postinduction; MRI2, MRI postconsolidation; MRI3, MRI at continuation week 48; MRI4, MRI at continuation week 120.
Natural History and Risk Factors for Leukoencephalopathy
To assess the natural history of leukoencephalopathy, we studied 74 patients with leukoencephalopathy for whom screening MRIs were available for at least three time points (Fig 3). In 30 patients (40.5%), the grade of leukoencephalopathy improved over time, including in 17 patients (23%) in whom leukoencephalopathy resolved completely. Leukoencephalopathy remained stable in 33 patients (46.6%) and worsened from grade 1 to 2 in 11 patients (14.9%). Thus, in the majority (77%) who developed leukoencephalopathy in this subset of 74 patients, MRI abnormalities were still evident at week 120.
Fig 3.
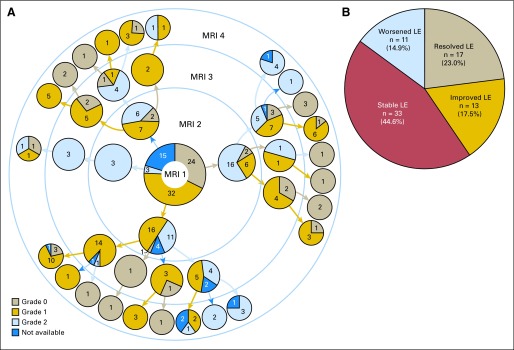
Natural history of leukoencephalopathy (LE) until completion of therapy. Magnetic resonance imaging (MRI) available for 74 patients with LE at minimum of three time points. (A) Detailed grading of MRIs for individual patients at four screening time points. Numbers of patients and grades of LE are indicated in pie charts. (B) Over time, MRI changes of LE resolved, improved, worsened, or were stable in 23%, 17.5%, 14.9%, and 44.6% of patients, respectively. MRI1, MRI postinduction; MRI2, MRI postconsolidation; MRI3, MRI at continuation week 48; MRI4, MRI at continuation week 120.
Table 2 and the Data Supplement show risk factors for developing leukoencephalopathy. In the univariable model, higher cumulative number of ITTs was associated with increased risk of leukoencephalopathy (P = .023). Clinical features were also analyzed at individual screening time points (Data Supplement). Higher MTX level at 42 hours (relative to leucovorin rescue) and higher homocysteine concentration (area under the curve over baseline) in course one were associated with increased risk of leukoencephalopathy. Of all risk factors, only the ratio of 42-hour plasma MTX concentration to leucovorin dose at the first course of high-dose MTX retained significance in a multivariable model (P = 0·038).
GWAS
Genotypic analyses were adjusted for genetically determined ancestry, age, treatment arm, and CNS status. To identify SNPs related to leukoencephalopathy, genotype frequencies for patients with leukoencephalopathy (n = 85) versus those without leukoencephalopathy (n = 279) were analyzed on Affymetrix 500K/6.0 arrays. Of 347 SNPs associated with the presence of leukoencephalopathy (P < .001), 148 were annotated to genes. We also compared the SNP genotypes of 14 patients with clinical neurotoxicity with the genotypes of 350 asymptomatic patients. Of 206 SNPs associated with clinical neurotoxicity (P < .001), 103 were annotated to genes. Table 3 lists SNPs with P values < .0001 in both analyses. Pathway analyses of these genes per Gene Ontology biologic processes revealed over-representation of the neuron projection development pathway (GO:0031175; P = .036) and axon guidance pathway (GO:007411; P = .047). The Data Supplement provides results of association analyses of SNPs from the Illumina array (including candidate SNPs previously related to MTX disposition and toxicity).
Table 3.
SNPs on Affymetrix Arrays Associated With Leukoencephalopathy and Clinical Neurotoxicity*
| SNP ID | Chromosome | Position | Gene | MAF | Risk Allele | OR | 95% CI | P |
|---|---|---|---|---|---|---|---|---|
| Leukoencephalopathy | ||||||||
| rs4145201 | 6 | 17007422 | — | 0.33 | C | 0.42 | 0.29 to 0.61 | 5.14E−06 |
| rs556269 | 1 | 164839754 | FMO9P† | 0.41 | T | 2.31 | 1.60 to 3.34 | 7.79E−06 |
| rs32571 | 5 | 14227106 | TRIO | 0.48 | C | 0.43 | 0.29 to 0.63 | 1.73E−05 |
| rs7590550 | 2 | 202266660 | MPP4 | 0.16 | G | 2.65 | 1.69 to 4.15 | 2.28E−05 |
| rs245311 | 5 | 127194498 | LOC728586 | 0.29 | T | 2.72 | 1.70 to 4.35 | 2.88E−05 |
| rs10842702 | 12 | 26331326 | SSPN | 0.35 | C | 0.44 | 0.30 to 0.65 | 3.03E−05 |
| rs33005 | 5 | 14259537 | TRIO | 0.49 | G | 0.45 | 0.30 to 0.65 | 3.20E−05 |
| rs9545873 | 13 | 81288492 | — | 0.42 | T | 2.09 | 1.47 to 2.97 | 3.75E−05 |
| rs1904006 | 10 | 53479033 | PRKG1 | 0.17 | C | 0.37 | 0.23 to 0.60 | 4.03E−05 |
| rs6632675 | X | 13245592 | — | 0.18 | C | 0.48 | 0.34 to 0.68 | 4.08E−05 |
| rs2065920 | 1 | 164869647 | FMO9P† | 0.36 | C | 0.48 | 0.34 to 0.69 | 4.46E−05 |
| rs5762295 | 22 | 26373959 | — | 0.03 | G | 8.11 | 2.97 to 22.2 | 4.52E−05 |
| rs1465614 | 2 | 16516774 | — | 0.24 | C | 0.42 | 0.28 to 0.64 | 4.90E−05 |
| rs16985255 | 22 | 26371883 | — | 0.03 | G | 7.00 | 2.73 to 17.9 | 5.03E−05 |
| rs1448686 | 8 | 137104691 | — | 0.41 | A | 0.47 | 0.32 to 0.68 | 5.66E−05 |
| rs17584752 | 4 | 108120536 | DKK2 | 0.19 | C | 0.42 | 0.28 to 0.64 | 5.71E−05 |
| rs11986485 | 8 | 41659544 | ANK1 | 0.29 | C | 0.38 | 0.24 to 0.61 | 6.55E−05 |
| rs13267761 | 8 | 138930469 | FLJ45872 | 0.27 | C | 0.46 | 0.31 to 0.67 | 6.83E−05 |
| rs9466410 | 6 | 22722709 | — | 0.25 | C | 0.35 | 0.20 to 0.58 | 7.17E−05 |
| rs6840582 | 4 | 122669925 | LOC729112 | 0.28 | G | 2.59 | 1.62 to 4.15 | 7.27E−05 |
| rs7320755 | 13 | 109829093 | COL4A2 | 0.32 | C | 0.45 | 0.31 to 0.67 | 7.49E−05 |
| rs6841032 | 4 | 122670192 | LOC729112 | 0.29 | G | 2.59 | 1.61 to 4.16 | 7.97E−05 |
| rs9936750 | 16 | 53729375 | — | 0.18 | G | 2.48 | 1.58 to 3.90 | 8.13E−05 |
| rs11185944 | 10 | 91731927 | LOC119358 | 0.35 | G | 2.06 | 1.44 to 2.96 | 8.51E−05 |
| rs1034893 | 17 | 9013280 | NTN1 | 0.17 | A | 0.27 | 0.14 to 0.52 | 8.92E−05 |
| rs17133261 | 5 | 100579903 | — | 0.07 | T | 3.71 | 1.92 to 7.18 | 9.80E−05 |
| Clinical Neurotoxicity | ||||||||
| rs12379211 | 9 | 119127189 | ASTN2 | 0.08 | C | 0.11 | 0.04 to 0.32 | 3.65E−05 |
| rs226945 | 6 | 3662178 | — | 0.09 | T | 11.7 | 3.57 to 38.2 | 4.79E−05 |
| rs17626001 | 14 | 42139233 | — | 0.07 | C | 0.09 | 0.03 to 0.28 | 5.17E−05 |
| rs226962 | 6 | 3669960 | PXDC1 | 0.09 | A | 0.07 | 0.02 to 0.25 | 5.32E−05 |
| rs682518 | 6 | 150770134 | IYD | 0.08 | G | 7.66 | 2.81 to 20.9 | 6.84E−05 |
| rs665670 | 6 | 150775949 | — | 0.14 | T | 9.17 | 3.03 to 27.7 | 8.70E−05 |
| rs7887242 | X | 20931587 | — | 0.18 | T | 4.33 | 2.08 to 9.03 | 9.13E−05 |
| rs10846690 | 12 | 123652370 | — | 0.13 | T | 9.84 | 3.13 to 31.0 | 9.28E−05 |
| rs10886214 | 10 | 85117719 | — | 0.21 | T | 8.43 | 2.89 to 24.6 | 9.64E−05 |
Abbreviations: MAF, minor allele frequency; OR, odds ratio; SNP, single-nucleotide polymorphism.
P < .0001.
Pseudogene.
DISCUSSION
To our knowledge, this study includes the largest cohort of patients with ALL in a contemporary therapeutic protocol who underwent serial radiologic screening for leukoencephalopathy to correlate radiographic findings with symptomatic neurotoxicity and to identify risk factors for leukoencephalopathy. Consistent with previous studies, MTX-related subacute neurotoxicity occurred in 3.8% of patients.2,4 The incidence was as high as 19% in children receiving a regimen with suboptimal leucovorin rescue and correlated with the MTX dose to leucovorin ratio.3,5 In our study, the ratio of 42-hour MTX level to leucovorin dose was not associated with increased risk of neurotoxic events, indicating that adequate leucovorin rescue may attenuate neurotoxic effects of MTX. Most episodes of clinical neurotoxicity were brief, and all but one patient were successfully rechallenged with high-dose MTX and/or ITT after resolution of symptoms. Aminophylline, via competitive inhibition of adenosine, is a candidate for secondary prophylaxis for MTX-related neurotoxicity, but the benefit of this modality is unclear.1,28 The only patient with recurrent MTX-related symptoms in our cohort had received prophylaxis with aminophylline. In general, the most common modification made by physicians after the first MTX-related neurotoxic event is removal of MTX from ITT and administration of only hydrocortisone and cytarabine.5 In our opinion, this is unnecessary, because our patients received one to 20 additional doses of ITT (some with leucovorin rescue) without recurrence of neurotoxicity.
At the time of the neurotoxic event, patients underwent diagnostic MRI, which almost always revealed at least grade 1 or 2 leukoencephalopathy, and most patients tolerated subsequent high-dose or intrathecal MTX. In addition, MRIs of 73 asymptomatic patients (20.6%) showed leukoencephalopathy. The sensitivity of MRI1 to predict clinical neurotoxicity was only 50%. Although screening MRI2 detected leukoencephalopathy in all patients who developed symptoms, it was not a good predictive tool, because eight of 14 patients developed symptoms before MRI2. Thus, leukoencephalopathy at the time of a neurotoxic event supports the diagnosis of MTX toxicity, but MRI screening is not useful to predict clinical neurotoxicity.
In prior studies, the incidence of asymptomatic leukoencephalopathy varied with type and timing of imaging, and cumulative dose of MTX, and ranged from 9% to 86% during active therapy.15,29,30 For example, quantitative MRI segmentation techniques detected leukoencephalopathy in 86% of patients after six to seven courses of high-dose MTX (5 g/m2).31 However, these advanced imaging modalities are not widely available and are used primarily in research settings. Segmentation was not used in our study. The absence of a true baseline MRI is a limitation of our study. The first MRI was performed after window MTX and up to three doses of ITT. It was not feasible to schedule baseline neuroimaging before initiation of therapy. However, three of 10 patients who developed neurotoxic symptoms had negative MRIs after their first dose of high-dose MTX but before their neurotoxic event, so a negative MRI cannot be assumed to presage a lack of neurotoxicity.
Leukoencephalopathy developed in 23.3% of all patients and 20.6% of asymptomatic patients. The incidence (especially grade 2) was highest after four doses of high-dose MTX and gradually decreased over time. Of those who developed leukoencephalopathy, 69% had persistent abnormal findings on MRI at the end of therapy. A longer follow-up period is required to monitor for resolution and study impact of leukoencephalopathy on long-term adverse effects, especially neurocognitive functioning. Although there was a higher risk (odds ratio, 1.12) for leukoencephalopathy associated with a higher 42-hour MTX to leucovorin ratio at consolidation course one, the clinical importance of leukoencephalopathy is unclear. Although this metric was not significantly associated with symptomatic neurotoxicity, the number of neurotoxic events was low, and the direction of association and magnitude of the odds ratio (1.18) of the MTX to leucovorin ratio was similar. Interestingly, the association of MTX/leucovorin was most evident with the first course of high-dose MTX of consolidation, suggesting that after that time, perhaps leucovorin dosing was adequate to decrease neurotoxicity or leukoencephalopathy associated with high-dose MTX. None of the patients in this study developed severe leukoencephalopathy (≥ grade 3), which may be partly explained by the omission of prophylactic cranial irradiation. In addition, all patients received a minimum of five doses of leucovorin, and most doses of MTX in consolidation were targeted to achieve desired concentrations, thereby avoiding high plasma MTX concentrations.21 Some other groups have included only three doses of leucovorin after fixed doses of high-dose MTX.32
We identified several SNPs that strongly influence the risk of leukoencephalopathy and/or symptomatic neurotoxicity. None of the SNPs reached genome-wide significance (P < 5.0 × 10−8), likely because of limited sample size and lack of patients with severe leukoencephalopathy. Of significant SNPs (P < .0001) that were annotated to known genes, 73% (eight of 11) were in genes important for neurogenesis. TRIO, PRKG1, ANK1, COL4A2, NTN1, and ASTN2 are involved in neuronal development and migration and/or axon guidance.33–37 SSPN is implicated in glial cell death38 and DKK2 in Wnt/β-catenin signaling, which influences neural development.39 In the absence of a validation cohort and functional studies, findings of the GWAS remain speculative. Although consequences of these polymorphisms and the molecular mechanism of neurotoxicity are unclear, over-representation of genes involved in neurogenesis points to plausible mechanisms linking these variants with neurotoxicity. For example, viral-induced neuronal cell death can be mediated through TRIO signaling40 and hypoxia-induced glial cell death by downregulation of SSPN.38 SNPs in ASTN2 are associated with autism,41 migraine,42 and attention-deficit hyperactivity disorder (ADHD)43 and SNPs in PRKG1 with ADHD44 and Alzheimer's disease.45 Interestingly, seven of 14 patients with clinical neurotoxicity in our cohort were also diagnosed with ADHD (four before ALL diagnosis, three post-therapy). Of these seven patients, six had inherited the risk allele (C) in rs12379211 in ASTN2. Because of the small number of patients and incomplete data on ADHD diagnoses in all patients, the significance of this association requires validation. Childhood ALL survivors are at risk of neurocognitive impairments, particularly attention disorders.46 A recent investigation using a candidate SNP approach reported association of attention problems in survivors with polymorphisms in genes involved in oxidative stress and CNS integrity.47 Additional studies will investigate whether patients who develop leukoencephalopathy during therapy are at higher risk than others for developing neurocognitive impairments.
Supplementary Material
Acknowledgment
We thank Julie Groff (Department of Biomedical Communications, St Jude Children's Research Hospital) for assistance with illustrations and Vani Shanker (Department of Scientific Editing, St Jude Children's Research Hospital) for assistance with editing the manuscript. Neither of these individuals received compensation apart from salary for their contributions.
Glossary Terms
- Genome-wide association study:
Hypothesis-free study that evaluates the association of genetic variations throughout the entire genome with traits, using high-throughput genotyping technologies to assay single-nucleotide polymorphisms.
- Magnetic resonance imaging:
A procedure in which radio waves and a powerful magnet linked to a computer are used to create detailed pictures of areas inside the body. These pictures can show the difference between normal and diseased tissue.
- Single-nucleotide polymorphism:
Natural variations in the genomic DNA sequence present in greater than 1% of the population, with single-nucleotide polymorphisms representing DNA variations in a single nucleotide. Single-nucleotide polymorphisms are being widely used to better understand disease processes, thereby paving the way for genetic-based diagnostics and therapeutics.
Footnotes
Supported by National Institutes of Health Grants No. P30-CA021765, R01-CA90246 (W.E.R.), CA36401 (W.E.E.), and GM92666 (M.V.R.) and by the American Lebanese Syrian Associated Charities.
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The author(s) indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: Deepa Bhojwani, Wilburn E. Reddick, Sima Jeha, John T. Sandlund, William E. Evans, Ching-Hon Pui, Mary V. Relling
Provision of study materials or patients: Raja B. Khan, Hiroto Inaba, Jeffrey E. Rubnitz, Monika L. Metzger, Scott C. Howard, Raul C. Ribeiro, Sima Jeha, John T. Sandlund, Ching-Hon Pui
Collection and assembly of data: Deepa Bhojwani, Noah D. Sabin, Deqing Pei, Raja B. Khan, John C. Panetta, Hiroto Inaba, Jeffrey E. Rubnitz, Monika L. Metzger, Scott C. Howard, Raul C. Ribeiro, Wilburn E. Reddick, Sima Jeha, John T. Sandlund, William E. Evans, Ching-Hon Pui, Mary V. Relling
Data analysis and interpretation: Deepa Bhojwani, Noah D. Sabin, Deqing Pei, Jun J. Yang, John C. Panetta, Kevin R. Krull, Cheng Cheng, Ching-Hon Pui, Mary V. Relling
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Inaba H, Khan RB, Laningham FH, et al. Clinical and radiological characteristics of methotrexate-induced acute encephalopathy in pediatric patients with cancer. Ann Oncol. 2008;19:178–184. doi: 10.1093/annonc/mdm466. [DOI] [PubMed] [Google Scholar]
- 2.Rubnitz JE, Relling MV, Harrison PL, et al. Transient encephalopathy following high-dose methotrexate treatment in childhood acute lymphoblastic leukemia. Leukemia. 1998;12:1176–1181. doi: 10.1038/sj.leu.2401098. [DOI] [PubMed] [Google Scholar]
- 3.Winick NJ, Bowman WP, Kamen BA, et al. Unexpected acute neurologic toxicity in the treatment of children with acute lymphoblastic leukemia. J Natl Cancer Inst. 1992;84:252–256. doi: 10.1093/jnci/84.4.252. [DOI] [PubMed] [Google Scholar]
- 4.Dufourg MN, Landman-Parker J, Auclerc MF, et al. Age and high-dose methotrexate are associated to clinical acute encephalopathy in FRALLE 93 trial for acute lymphoblastic leukemia in children. Leukemia. 2007;21:238–247. doi: 10.1038/sj.leu.2404495. [DOI] [PubMed] [Google Scholar]
- 5.Mahoney DH, Jr, Shuster JJ, Nitschke R, et al. Acute neurotoxicity in children with B-precursor acute lymphoid leukemia: An association with intermediate-dose intravenous methotrexate and intrathecal triple therapy—A Pediatric Oncology Group study. J Clin Oncol. 1998;16:1712–1722. doi: 10.1200/JCO.1998.16.5.1712. [DOI] [PubMed] [Google Scholar]
- 6.Buizer AI, de Sonneville LM, van den Heuvel-Eibrink MM, et al. Behavioral and educational limitations after chemotherapy for childhood acute lymphoblastic leukemia or Wilms tumor. Cancer. 2006;106:2067–2075. doi: 10.1002/cncr.21820. [DOI] [PubMed] [Google Scholar]
- 7.Cole PD, Beckwith KA, Vijayanathan V, et al. Folate homeostasis in cerebrospinal fluid during therapy for acute lymphoblastic leukemia. Pediatr Neurol. 2009;40:34–41. doi: 10.1016/j.pediatrneurol.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Vezmar S, Schüsseler P, Becker A, et al. Methotrexate-associated alterations of the folate and methyl-transfer pathway in the CSF of ALL patients with and without symptoms of neurotoxicity. Pediatr Blood Cancer. 2009;52:26–32. doi: 10.1002/pbc.21827. [DOI] [PubMed] [Google Scholar]
- 9.Vezmar S, Becker A, Bode U, et al. Biochemical and clinical aspects of methotrexate neurotoxicity. Chemotherapy. 2003;49:92–104. doi: 10.1159/000069773. [DOI] [PubMed] [Google Scholar]
- 10.Kishi S, Griener J, Cheng C, et al. Homocysteine, pharmacogenetics, and neurotoxicity in children with leukemia. J Clin Oncol. 2003;21:3084–3091. doi: 10.1200/JCO.2003.07.056. [DOI] [PubMed] [Google Scholar]
- 11.Rollins N, Winick N, Bash R, et al. Acute methotrexate neurotoxicity: Findings on diffusion-weighted imaging and correlation with clinical outcome. AJNR Am J Neuroradiol. 2004;25:1688–1695. [PMC free article] [PubMed] [Google Scholar]
- 12.Asato R, Akiyama Y, Ito M, et al. Nuclear magnetic resonance abnormalities of the cerebral white matter in children with acute lymphoblastic leukemia and malignant lymphoma during and after central nervous system prophylactic treatment with intrathecal methotrexate. Cancer. 1992;70:1997–2004. doi: 10.1002/1097-0142(19921001)70:7<1997::aid-cncr2820700732>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 13.Oka M, Terae S, Kobayashi R, et al. MRI in methotrexate-related leukoencephalopathy: Disseminated necrotising leukoencephalopathy in comparison with mild leukoencephalopathy. Neuroradiology. 2003;45:493–497. doi: 10.1007/s00234-003-0983-3. [DOI] [PubMed] [Google Scholar]
- 14.Robain O, Dulac O, Dommergues JP, et al. Necrotising leukoencephalopathy complicating treatment of childhood leukaemia. J Neurol Neurosurg Psychiatry. 1984;47:65–72. doi: 10.1136/jnnp.47.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddick WE, Glass JO, Helton KJ, et al. Prevalence of leukoencephalopathy in children treated for acute lymphoblastic leukemia with high-dose methotrexate. AJNR Am J Neuroradiol. 2005;26:1263–1269. [PMC free article] [PubMed] [Google Scholar]
- 16.Reddick WE, Conklin HM. Impact of acute lymphoblastic leukemia therapy on attention and working memory in children. Expert Rev Hematol. 2010;3:655–659. doi: 10.1586/ehm.10.65. [DOI] [PubMed] [Google Scholar]
- 17.Kishi S, Cheng C, French D, et al. Ancestry and pharmacogenetics of antileukemic drug toxicity. Blood. 2007;109:4151–4157. doi: 10.1182/blood-2006-10-054528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vagace JM, Caceres-Marzal C, Jimenez M, et al. Methotrexate-induced subacute neurotoxicity in a child with acute lymphoblastic leukemia carrying genetic polymorphisms related to folate homeostasis. Am J Hematol. 2011;86:98–101. doi: 10.1002/ajh.21897. [DOI] [PubMed] [Google Scholar]
- 19.Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730–2741. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mikkelsen TS, Sparreboom A, Cheng C, et al. Shortening infusion time for high-dose methotrexate alters antileukemic effects: A randomized prospective clinical trial. J Clin Oncol. 2011;29:1771–1778. doi: 10.1200/JCO.2010.32.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pauley JL, Panetta JC, Crews KR, et al. Between-course targeting of methotrexate exposure using pharmacokinetically guided dosage adjustments. Cancer Chemother Pharmacol. 2013;72:369–378. doi: 10.1007/s00280-013-2206-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruhs H, Becker A, Drescher A, et al. Population PK/PD model of homocysteine concentrations after high-dose methotrexate treatment in patients with acute lymphoblastic leukemia. PLoS One. 2012;7:e46015. doi: 10.1371/journal.pone.0046015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trevino LR, Shimasaki N, Yang W, et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol. 2009;27:5972–5978. doi: 10.1200/JCO.2008.20.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Breiman L, Friedman JH, Olshen RA, et al. Classification and Regression Trees. Belmont, CA: Wadsworth International Group; 1984. [Google Scholar]
- 25.Kawedia JD, Kaste SC, Pei D, et al. Pharmacokinetic, pharmacodynamic, and pharmacogenetic determinants of osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2011;117:2340–2347. doi: 10.1182/blood-2010-10-311969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reimand J, Arak T, Vilo J. G:Profiler: A Web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39:W307–W315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernini JC, Fort DW, Griener JC, et al. Aminophylline for methotrexate-induced neurotoxicity. Lancet. 1995;345:544–547. doi: 10.1016/s0140-6736(95)90464-6. [DOI] [PubMed] [Google Scholar]
- 29.Chu WC, Chik KW, Chan YL, et al. White matter and cerebral metabolite changes in children undergoing treatment for acute lymphoblastic leukemia: Longitudinal study with MR imaging and 1H MR spectroscopy. Radiology. 2003;229:659–669. doi: 10.1148/radiol.2293021550. [DOI] [PubMed] [Google Scholar]
- 30.Pääkkö E, Harila-Saari A, Vanionpää L, et al. White matter changes on MRI during treatment in children with acute lymphoblastic leukemia: Correlation with neuropsychological findings. Med Pediatr Oncol. 2000;35:456–461. doi: 10.1002/1096-911x(20001101)35:5<456::aid-mpo3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 31.Reddick WE, Glass JO, Helton KJ, et al. A quantitative MR imaging assessment of leukoencephalopathy in children treated for acute lymphoblastic leukemia without irradiation. AJNR Am J Neuroradiol. 2005;26:2371–2377. [PMC free article] [PubMed] [Google Scholar]
- 32.Möricke A, Reiter A, Zimmermann M, et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: Treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood. 2008;111:4477–4489. doi: 10.1182/blood-2007-09-112920. [DOI] [PubMed] [Google Scholar]
- 33.Bateman J, Van Vactor D. The Trio family of guanine-nucleotide-exchange factors: Regulators of axon guidance. J Cell Sci. 2001;114:1973–1980. doi: 10.1242/jcs.114.11.1973. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Z, Wang Z, Gu Y, et al. Regulate axon branching by the cyclic GMP pathway via inhibition of glycogen synthase kinase 3 in dorsal root ganglion sensory neurons. J Neurosci. 2009;29:1350–1360. doi: 10.1523/JNEUROSCI.3770-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hakanen J, Duprat S, Salminen M. Netrin1 is required for neural and glial precursor migrations into the olfactory bulb. Dev Biol. 2011;355:101–114. doi: 10.1016/j.ydbio.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 36.Wilson PM, Fryer RH, Fang Y, et al. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J Neurosci. 2010;30:8529–8540. doi: 10.1523/JNEUROSCI.0032-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Susuki K, Rasband MN. Spectrin and ankyrin-based cytoskeletons at polarized domains in myelinated axons. Exp Biol Med (Maywood) 2008;233:394–400. doi: 10.3181/0709-MR-243. [DOI] [PubMed] [Google Scholar]
- 38.Zhou D, Wang J, Zapala MA, et al. Gene expression in mouse brain following chronic hypoxia: Role of sarcospan in glial cell death. Physiol Genomics. 2008;32:370–379. doi: 10.1152/physiolgenomics.00147.2007. [DOI] [PubMed] [Google Scholar]
- 39.Diep DB, Hoen N, Backman M, et al. Characterisation of the Wnt antagonists and their response to conditionally activated Wnt signalling in the developing mouse forebrain. Brain Res Dev Brain Res. 2004;153:261–270. doi: 10.1016/j.devbrainres.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Lee JW, Yeo SG, Kang BH, et al. Echovirus 30 induced neuronal cell death through TRIO-RhoA signaling activation. PLoS One. 2012;7:e36656. doi: 10.1371/journal.pone.0036656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glessner JT, Wang K, Cai G, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freilinger T, Anttila V, de Vries B, et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet. 2012;44:777–782. doi: 10.1038/ng.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lionel AC, Crosbie J, Barbosa N, et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med. 2011;3:95ra75. doi: 10.1126/scitranslmed.3002464. [DOI] [PubMed] [Google Scholar]
- 44.Neale BM, Medland S, Ripke S, et al. Case-control genome-wide association study of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49:906–920. doi: 10.1016/j.jaac.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fallin MD, Szymanski M, Wang R, et al. Fine mapping of the chromosome 10q11-q21 linkage region in Alzheimer's disease cases and controls. Neurogenetics. 2010;11:335–348. doi: 10.1007/s10048-010-0234-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conklin HM, Krull KR, Reddick WE, et al. Cognitive outcomes following contemporary treatment without cranial irradiation for childhood acute lymphoblastic leukemia. J Natl Cancer Inst. 2012;104:1386–1395. doi: 10.1093/jnci/djs344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krull KR, Bhojwani D, Conklin HM, et al. Genetic mediators of neurocognitive outcomes in survivors of childhood acute lymphoblastic leukemia. J Clin Oncol. 2013;31:2182–2188. doi: 10.1200/JCO.2012.46.7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.