

Abstract
Parkinson’s disease (PD) is a movement disorder caused by neurodegeneration in neocortex, substantia nigra (SN) and brainstem and synucleinopathy. Some inherited PD is caused by mutations in α-synuclein (αSyn), and inherited and idiopathic PD are associated with mitochondrial perturbations. However, the mechanisms of pathogenesis are unresolved. We characterized a human αSyn transgenic mouse model and tested the hypothesis that the mitochondrial permeability transition pore (mPTP) is involved in the disease mechanisms. C57BL/6 mice expressing human A53T-mutant αSyn driven by a Thy1 promoter develop a severe, age-related, fatal movement disorder involving ataxia, rigidity, and postural instability. These mice develop synucleinopathy and neocortical, SN, and cerebello-rubro-thalamic degeneration involving mitochondriopathy and apoptotic and non-apoptotic neurodegeneration. Interneurons undergo apoptotic degeneration in young mice. Mutant αSyn associated with dysmorphic neuronal mitochondria and bound voltage-dependent anion channels. Genetic ablation of cyclophilin D, an mPTP modulator, delayed disease onset and extended lifespans of mutant αSyn mice. Thus, mutant αSyn transgenic mice on a C57BL/6 background develop PD-like phenotypes, and the mPTP is involved in their disease mechanisms.
Keywords: adenine nucleotide translocase, interneuron, porin, ppif, cerebellum, voltage-dependent anion channel
1. Introduction
Parkinson’s disease (PD) is a chronically progressive, age-related, fatally incapacitating movement disorder in humans. Estimates indicate that 4 to 6 million people are diagnosed with PD, and this disease affects about 2% of the population at some time in life (van den Eeden et al., 2003; Dorsey et al., 2007). The greatest prevalence of PD occurs in the USA, with 100–250 cases per 100,000 (van den Eeden et al., 2003), placing PD as the 2nd most common neurodegenerative disease with an adult onset (after Alzheimer’s disease). Progressive resting tremor, rigidity, bradykinesia/akinesia, gait disturbance, and postural instability characterize PD clinically (Olanow and Tatton, 1999; Jankovic, 2008). Cardinal neuropathological features of PD are degeneration and elimination of dopamine neurons in substantia nigra (SN) and in other brainstem regions, dopamine depletion in striatum, and α-synuclein (αSyn) pathology (Lowe et al., 1997; Olanow and Tatton, 1999; Giasson et al., 2000a; Dickson, 2012; Jellinger et al., 2012). The molecular pathogenesis of PD is not understood. At least 2 forms of PD exist: idiopathic (sporadic) and heritable (familial) (Olanow and Tatton, 1999; Schapira, 2006; Klein and Schlossmacher, 2007). The majority of PD cases are sporadic with no known genetic component. Epidemiological studies reveal several risk factors for developing idiopathic PD. Aging and αSyn are the most common risk factors for idiopathic PD (Klein and Schlossmacher, 2007). Pesticides are also linked to the origin of PD (Ascherio et al., 2006; Tanner et al., 2011). Herbicides, well water (contaminated with pesticides), and industrial chemicals are possible environmental agents related to the development of PD (Schapira, 2006).
Mitochondrial mechanisms are believed to be involved in PD pathogenesis (Olanow and Tatton, 1999; Pilsl and Winklhofer, 2012; Coskun et al., 2012). Mitochondria became suspects in PD etiology when heroine abusers presented with PD after exposure to 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP) (Langston et al., 1983) which is converted by monoamine oxidase to a complex I inhibitor and free radical generator (Nicklas et al., 1985; Ramsay et al., 1986; Cleeter et al., 1992). Mitochondrial involvement in idiopathic PD etiology became more compelling by the discovery that complex I activity (NAPH dehydrogenase) is reduced in the SN (Schapira et al., 1989) and skeletal muscle (Bindoff et al., 1989) of PD cases. Complex I inhibitors, notably MPTP and the pesticide rotenone, cause damage to dopaminergic neurons and are the basis of several animal and cell models of PD (Shimohama et al., 2003). Although mitochondrial mechanisms have been implicated in PD pathogenesis for over two decades, direct cause-effect relationships between mitochondrial damage and disease initiation and progression are still unclear. Mutations in nuclear genes encoding definite mitochondrial proteins such as complex I, mitofusin 2, frataxin, and optic atrophy protein 1, as well as DNA polymerase δ, adenine nucleotide translocase 1 and twinkle, have been linked to Leigh’s syndrome, Charcot-Marie-Tooth disease type 2, Friedreich’s ataxia, autosomal dominant optic atrophy, and chronic progressive external opthalmoplegia, respectively (Schapira, 2012), but linkage of these mutations to PD has not been seen. Furthermore, mutations in the mitochondrial DNA encoded complex I subunits cause Leber’s hereditary optic neuropathy (Vilkki et al., 1989) but not PD (Olanow and Tatton, 1999). About 5–10% of people with PD have familial inheritance (Schapira, 2006). Most identified mutations occur in genes encoding proteins that have both non-mitochondrial and putative mitochondrial functions, including αSyn (Polymeropoulos et al., 1997; Singleton et al., 2003), DJ-1 (Bonifati et al., 2003), parkin (Leroy et al., 1998; Kitada et al., 1998), and leucine-rich repeat kinase-2 (Paisan-Ruiz et al., 2004; Zimprich et al., 2004), confounding the interpretation of the intracellular mechanisms leading to PD. However, mutations in phosphatase and tensin homolog-induced putative kinase-1 (PINK1) do directly link mitochondrial dysfunction to the etiology of some early-onset recessive forms of PD (Valente et al., 2004; Hatano et al., 2004; Pilsl and Winklhofer, 2012), but PINK1 null mice do not develop neurodegeneration, despite mitochondrial abnormalities (Gispert et al., 2009). Thus, further interrogation of mitochondrial dysfunction as a general cause of PD is still needed.
Because αSyn and mitochondria both seem to have roles in PD, it is possible that interplay between these two entities participates in PD pathogenesis. αSyn is largely a cytoplasmic and nuclear protein that is enriched in nervous tissue axon terminals (Maroteaux et al., 1988; Murphy et al., 2000; Lesuisse and Martin, 2002) and functions in presynaptic vesicle dynamics during activity-dependent neurotransmitter release (Fortin et al., 2005; Chandra et al., 2005); yet, it also has actions at mitochondria. αSyn associates with mitochondrial membranes in neurons (Nakamura et al., 2008) and has been found to be imported into mitochondria to cause complex I impairment (Devi et al., 2008). Human αSyn (hαSyn) with an alanine-53→threonine mutation (A53T) directly interacts with mitochondria in transgenic (tg) mice, and these mice develop mitochondrial abnormalities in association with a severe movement disorder, synucleinopathy and a shortened lifespan (Lee et al., 2002; Martin et al., 2006) but no loss of dopaminergic neurons in substantia nigra (Daher et al., 2012). Unexpectedly, the motor abnormality and neuropathology in mice with prion protein promoter-driven hαSyn-A53T transgene expression is more consistent with a motor neuron disease, rather than a PD phenotype, because of the profound loss of spinal motor neurons (Martin et al., 2006). Tg mice expressing hαSyn-A53T driven by the human neuron-specific thymic antigen-1 (Thy1)-promoter have also been generated, but their neurological and neuropathological characterization is limited (Chandra et al., 2005) and relevance to PD as a disease model is uncertain. The brain distribution of pathology and possible mitochondrial mechanisms of disease in these mice have not been studied. In this study we characterized the neuropathology in Thy1-hαSyn-A53T tg mice and tested the hypothesis that mitochondrial abnormalities are related causally to the disease process in PD-linked mutant αSyn tg mice through the mitochondrial matrix protein cyclophilin D (CyPD) that modulates the mitochondrial permeability transition pore (Crompton, 2004; Baines et al., 2005; Nakagawa et al., 2005; Halestrap, 2009; Bernardi et al., 2006).
2. Materials and Methods
2.1 Transgenic mice
The PD mice studied here were Thy1-hαSyn-A53T tg mice. The original founder mouse of line B6.Cg-Tg[Thy1-SNCA*A53T]M53Sud/J (stock #008135) was purchased from The Jackson Laboratory (Bar Harbor, ME). There is no characterization of the brain neuropathology in these mice. We bred this line, a hybrid of SV129 and C57BL/6 strains, with pure C57BL/6 mice and then progeny were backcrossed at least 7 generations into a pure C57BL/6 strain background with the goal of eliminating the SV129 background. The SV129 genetic background is known to increase susceptibility to excitotoxic and necrotic neurodegeneration (Schauwecker and Steward, 1997; Kofler et al., 2004), and thus we wanted to minimize this prominent strain effect. All mice were genotyped at 1 month of age to identify individuals with the A53T transgene. The primer pair used for PCR was: 5′-GGCACCTAGAGGATCTCGACTAGTGG-3′ (forward) and 5′-GGACCTCGACGCTTAAGGCTTCAGG-3′ (reverse). The agouti fur coat was eventually eliminated and studies were conducted exclusively on Thy1-A53T tg mice on a C57BL/6 background (black fur coats). We evaluated A53T mice at presymptomatic stages of disease (n= 10), early- to mid-stages of disease (n = 10), defined by presence of bradykinesia and ataxia, and at near endstage disease (n =20), defined by postural stiffness and immobility (Supplementary Data Videos 1 and 2). Age-matched non-tg littermates served as controls. The CyPD null mice are described elsewhere (Basso et al., 2005; Martin et al., 2011). The GlyT2-eGFP tg mice, with expression of eGFP only in glycinergic interneurons, are described elsewhere (Zeilhofer et al., 2005; Martin, 2011). The institutional Animal Care and Use Committee approved the animal protocols.
2.2 Brain harvesting and processing for histology
Mice were anesthetized with an overdose of sodium pentobarbital and perfused through the heart with ice-cold phosphate buffer-saline (PBS, 100 mM, pH 7.4) followed by ice-cold 4% paraformaldehyde. After perfusion-fixation, the brain was removed after 2 hours, postfixed overnight in 4% paraformaldehyde, and cryoprotected 24 hours in 20% glycerol-PBS. The brains were frozen and serially sectioned from frontal pole to posterior cerebellum in the coronal plane at 40 μm on a sliding microtome with every section being saved individually in 96-well plates containing antifreeze buffer. The sections were stored at −20°C. For hi stological analyses, sections were selected systematically and stained using cresyl violet (CV) for cell morphology and counting, FD-silver for neurodegeneration, the terminal transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) method (Portera-Cailliau et al., 1997; Martin, 1999; Martin et al., 2006), as an assay for cell death based on the detection of DNA double-strand breaks, and immunohistochemistry.
2.3 Immunohistochemistry
We evaluated the localizations of hαSyn, tyrosine hydroxylase (TH), the interneuron marker parvalbumin (Kita et al., 1990), selected cell death proteins, and putative mitochondrial permeability transition pore (mPTP) proteins in the brains of A53T mice. Immunoperoxidase histochemistry with diaminobenzidine (DAB) as chromogen or immunofluorescence were used as before (Martin et al., 2005, 2006, 2007) to detect hαSyn protein with a hαSyn-specific monoclonal Syn211 antibody (Giasson et al., 2000b; Sigma, St. Louis, MO), TH with a rabbit polyclonal antibody (Novus Biologicals, Littleton, CO), cleaved caspase-3 with a rabbit monoclonal antibody (R&D Systems, Minneapolis, MN), and phospho-p53 (pp53) with a rabbit polyclonal antibody (R&D systems). Autophagy was assessed using an antibody to LC3A (Cell Signaling Technology, Beverly, MA). In some instances the immunoperoxidase sections were counterstained with CV before viewing.
We studied, as done for other mitochondrial proteins (Martin and Liu, 2002; Martin et al., 2007, 2009), VDAC, ANT, and CyPD. Antibodies to these proteins were characterized previously (Martin et al., 2009) by western blotting to determine specificity in brain extracts. Additional negative controls for antibody specificity were incubating sections in the primary antibody diluent without primary antibody with all other steps similar. Immunofluorescence was used for dual labeling to colocalize putative mPTP protein regulators with the mitochondrial marker manganese SOD (or SOD2) using a rabbit polyclonal antibody (Assay Designs-Enzo Life Sciences, Farmingdale, NY). Immunoreactivity was visualized with species-specific secondary antibodies (all raised in goat) conjugated to Alexa Fluor 488 or Alexa Fluor 594 (Invitrogen Corporation, Carlsbad, CA).
2.4 Regional cell counting and neocortical measurements
Profile counting of Nissl-stained sections was done to estimate the numbers of neurons in the substantia nigra pars compacta (SNc), red nucleus magnocellular division (RN), thalamic ventrolateral nucleus (VL), and cerebellar deep interposed nucleus (DCN) in Thy1-A53T tg and littermate non-tg mice at 1 and 12 months of age (n=4–6 mice/age/genotype). Neurons in these regions were counted in anatomical level-matched sections (2–3 sections/region) at 1000x magnification. Corresponding to the mouse brain stereotaxic atlas (Franklin and Paxinos, 1997), SNc neurons were counted at bregma −292, −3.16, and −3.52; RN neurons were counted at bregma −3.52 and −3.80; VL neurons were counted at bregma −0.94 and −1.22; and DCN neurons were counted at bregma −5,88 and −6.12. Strict morphological criteria were applied when classifying normal appearing neurons, including a round, open, euchromatic nucleus (not condensed and darkly stained), globular Nissl staining of the cytoplasm, clear vacuole-free cytoplasm, and a cell body diameter of ~10–20 μm. With these criteria, degenerating neurons with necrotic, apoptotic, and necrotic-apoptotic hybrids as well as astrocytes, oligodendrocytes, and microglia were excluded from the counts.
Dying cells in TUNEL preparations were counted in SNc and cerebral cortex of A53T and control mice in six non-overlapping microscopic fields at 1000x magnification. Four to five sections through the brain regions of interest were assessed. Only cells with clearly discernible TUNEL+ nuclei were counted throughout the depth of the section.
Immunopositive cell bodies in immunoperoxidase- and immunofluorescence- stained sections were also counted. Cleaved caspase-3+ cells were counted in six non-overlapping, microscopic fields of striatum and cerebellar cortex at 400x magnification. Phospho-p53+ cells were counted in six non-overlapping, microscopic fields of striatum at 1000x magnification. Parvalbumin+ cells were counted in four non-overlapping, microscopic fields of striatum at 200x magnification.
Neocortical gray mantle and subcortical white matter thicknesses were measured by ocular filar micrometry in brain sections at a level of bregma −0.58. Somatosensory (S1) cortex and the subjacent external capsule were analyzed in five different sections in each mouse.
2.5 Silver staining
Silver staining was used to visualize degenerating neuronal elements in brain sections of Thy1-A53T α-Syn mice and non-tg age-matched control mice. Sections were processed using the FD NeuroSilver kit (FD Neurotechnologies Inc, Baltimore, MD).
2.6 Western blotting
The expression of hαSyn and the mitochondrial proteins CyPD, ANT, and VDAC in mouse brain regions were evaluated by immunoblotting. HαSyn A53T tg mice at different stages of disease, including pre-symptomatic, early- to mid-symptomatic, and end-stage (n=8/disease stage), were deeply anesthetized, decapitated, and the brain and spinal cord were removed quickly and rinsed in ice-cold Hanks buffer before snap freezing. Before freezing, the brain was microdissected ice-cold Hanks buffer into olfactory bulb, cerebral cortex, hippocampus, striatum, diencephalon, brainstem, and cerebellum. Crude tissue extracts were prepared and protein fractions were subjected to SDS-PAGE and immunoblotting using enhanced chemilumesence detection as described (Martin et al., 2003). The reliability of sample loading and electroblotting in each experiment was evaluated by staining nitrocellulose membranes with Ponceau S before immunoblotting. If transfer was not uniform, blots were discarded and gels were run again. Monoclonal antibody Syn211 (Giasson et al., 2000b) was used to detect hαSyn. For CyPD, a mouse monoclonal antibody (clone E11AE12BD4, MitoSciences, Eugene, OR) was used. For ANT, a mouse monoclonal antibody (MitoSciences, clone 5F51BB5AG7) and a rabbit polyclonal antibody (Santa Cruz, H-188) were used. For VDAC, two mouse monoclonal antibodies (MitoSciences, clone 20B12AF2 and Calbiochem, clone 89-173/016) were used. ANT and VDAC antibodies were not isoform specific. The antibodies were used at concentrations for visualizing protein immunoreactivity within the linear range. To quantify protein immunoreactivity, films were scanned and densitometry was performed as described (Martin et al., 2003). Protein levels were expressed as relative optical density measurements. Immunodensities were normalized to Ponceau S stained proteins.
2.7 Immunoprecipitation
Immunoprecipitation and western blot analysis of CNS region homogenates from A53T mice and age-matched non-tg mice were used to identify interactions between hαSyn protein and proteins putatively involved in mPTP operation. Protein (100 μg) from mitochondria-enriched membrane fractions from non-tg and A53T mouse brain was immunoprecipitated using 5 μg of monoclonal Syn211 hαSyn antibody. After immunocapture, the samples were subjected to SDS-PAGE and electroelution for western blot detection of CyPD and VDAC using enhanced chemiluminescence detection.
2.8 Mouse crossing experiments
Mice homozygous for the CyPD targeted mutation on a C57/BL6 background are viable, with normal growth and appearance, and are fertile (Basso et al., 2005; Martin et al., 2011). Mitochondria isolated from heart and CNS of CyPD−/− (ppif−/−) mice are devoid of CyPD, resistant to mitochondria swelling and mitochondrial permeability transition, and are protected from mitochondrial Ca2+ overload and oxidative stress (Baines et al., 2005; Martin et al., 2009; Martin et al., 2011). Adult mouse neurons without CyPD are protected from necrosis and apoptosis (Martin et al., 2009; Martin et al., 2011; Martin, 2011). We crossed A53T tg mice to ppif−/− mice (Basso et al., 2005) to test the hypothesis that CyPD, and possibly the mPTP, has a role in neurodegeneration cell death caused by hαSyn-A53T. Mice with targeted deletions of both CyPD alleles were crossed to A53T mice and F1 offspring positive for the hαSyn-A53T transgene were crossed with F1 siblings lacking the transgene. F2 generations yielded progeny carrying all PCR-confirmed CyPD genotypes (ppif+/+, ppif+/−, and ppif−/−) with or without the hαSyn-A53T transgene. These mice were maintained for 3 generations and followed longitudinally.
2.9 Statistical analyses
For histological and western blot measurements group means and variances were evaluated statistically by one-way ANOVA followed by a Newman-Keuls post-hoc test.
2.10 Photography and figure construction
Marker comparisons between tg and non-tg mice were made from sections that were imaged under identical conditions and analyzed using identical parameters. Original images used for figure construction were generated using digital photography. Digital images were captured as TiF files using a SPOT digital camera and SPOT Advanced software (Diagnostic Instruments) or a Nikon digital camera (DXM1200) and ACT-1 software. Images were altered slightly for brightness and contrast using ArcSoft PhotoStudio 2000 or Adobe Photoshop software without changing the content and actual result. Figure composition was done using CorelDraw software with final figures being converted to TiF files. Files of composite figures were adjusted for brightness and contrast in Adobe Photoshop.
3. Results
3.1 Thy1-A53T tg mice develop profound fatal age-related disease mimicking neurological aspects of PD
Thy1-A53T tg C57BL/6 mice were born in normal-sized litters and had no overt phenotype at birth. Juvenile mice also appeared overtly normal as did young adult mice. Thy1-A53T tg mice at 2 and 4 months of age had normal spontaneous motor activity (Fig. 1A). However tg mice at 6 months of age showed deficits in motor activity compared to non-tg littermate controls and Thy1-A53T tg mice at 2 and 4 months of age (Fig. 1A). Motor activity progressively deteriorated to complete immobility by 12 months of age (Fig. 1A,B, Supplementary Videos 1 and 2). This period of progressive neurological deterioration was characterized by prominent motor phenotypes. A53T mice began to show spasticity and tremors at about 6 months of age. Rigidity emerged at about 8 months of age coinciding with the development of bradykinesia and gait abnormalities such as jerking movements (Supplementary Video 1). Mice that were about 1 year old showed 100% penetrance in the development of rigidity, postural instability, and immobility (Fig. 1B, Supplementary Video 2). There was no indication of flaccid paralysis at any stage in the disease process. Thy1-A53T tg mice had a much shortened lifespan. Most mice died between 10 and 12 months of age (Fig. 1C).
Figure 1.
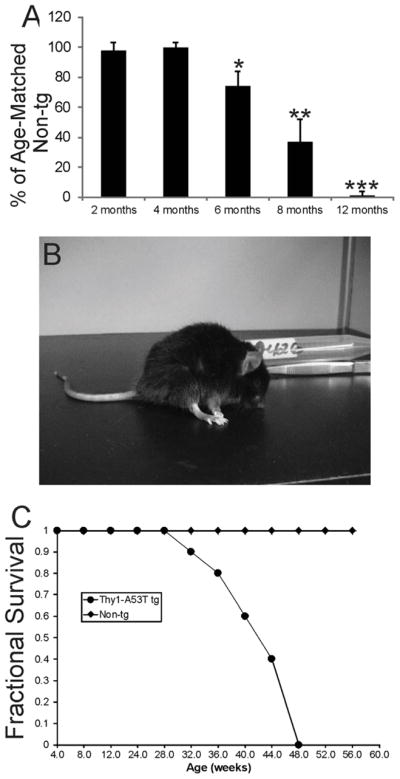
Thy1-A53T hαSyn C57BL/6 tg mice develop a PD-like neurological phenotype. A. Thy1-A53T mice develop over the first year of life an age-dependent loss of motor activity in the running wheel. Values are mean ± SD (n = 6 mice/group). Significant differences from age-matched control are indicated: *p < 0.05, **p < 0.01, and ***p < 0.001 (n= 6). B. Photograph of an endstage Thy1-A53T mouse in an akinetic, rigid, and stooped posture (see Video 2). C. Thy1-A53T mice develop fatal disease and have a shortened lifespan compared to non-tg littermate controls.
3.2 Expression and localization of hαSyn protein in Thy1-A53T tg mice
Monoclonal Syn211 antibody with putative specificity for hαSyn (with no detection of endogenous mouse αSyn) (Giasson et al., 2000b) was used to map by western blotting the regional expression of hαSyn-A53T transgene in the CNS of tg mice. Syn211 antibody had confirmed specificity for hαSyn because it did not detect endogenous α-Syn in non-tg mouse brain (Fig. 2A). In contrast, in A53T mouse brain extracts, Syn211 antibody detected monomeric hαSyn at ~16 kDa and higher molecular weight forms at ~60 kDa and larger (Fig. 2A). Microdissected brain regions and spinal cord samples showed robust expression of A53T-hαSyn throughout the CNS in A53T mice at 6 months of age with early disease (Fig. 2B) and at 12 months of age with endstage disease (Fig. 2C). hαSyn was detected in cerebral cortex, olfactory bulb, striatum, diencephalon, brainstem, cerebellum, and spinal cord. The individual CNS regions showed different propensities for the development of higher molecular weight species of hαSyn early in the disease (Fig. 2B). High molecular weight (aggregated) forms of A53T-hαSyn were seen most prominently in spinal cord, brainstem, cerebellum, and hippocampus early in disease. At endstage disease high molecular weight forms of A53T-hαSyn were detected in all CNS regions examined (Fig. 2C).
Figure 2.

Expression, aggregation, and localization of hαSyn in Thy1-A53T hαSyn C57BL/6 tg mouse CNS. A. Western blot demonstrating the specificity of monoclonal Syn211 antibody for hαSyn. In young tg mouse whole brain extracts subjected to SDS-PAGE, hαSyn was detected as a monomer at ~14–16 kDa (arrow) and, at lower amounts, as stable high molecular forms greater than 112 kDa, consistent with the size and in vivo aggregation propensity of hαSyn (Giasson et al., 2002). These immunoreactive proteins were not detected in non-tg mouse brain (right lane). Ponceau S stained membrane shows protein loading. B. Thy1-A53T hαSyn tg mice at early symptomatic stages of disease expressed monomeric hαSyn (arrow) in all CNS regions analyzed as well as apparent stable dimeric and oligomeric forms in many regions, except olfactory bulb and cerebral cortex. Stable higher molecular weight aggregates were detected robustly in hindbrain and spinal cord (and also in hippocampus). Ponceau S stained membrane shows protein loading. C. In Thy1-A53T hαSyn tg mice at endstage disease, detection of hαSyn aggregates was shifted to the much higher molecular weight forms in most brain regions, while monomer levels (arrow) tended to be lower than in mice at earlier disease. Ponceau S stained membrane shows protein loading. D–F. Localization of hαSyn in Thy1-A53T tg mouse somatosensory cerebral cortex by immunohistochemistry (with DAB as chromogen, brown staining) and cresyl violet counterstaining (blue) at low (D) and higher magnifications (E,F). hαSyn immunoreactivity was seen throughout the neuropil in all six layers of cerebral cortex in tg mice (D), but no immunoreactivity was detected in non-tg mice (D, inset). Scale bars = 120 μm (D), 62 μm (D inset). At higher magnification hαSyn immunoreactivity was seen in the cytoplasm and nucleus of subsets of neurons in superficial layers (E, arrows) and deeper layers (F, arrows) as well as in the neuropil. Cerebrocortical neurons developed cytoplasmic and intranuclear inclusions positive for hαSyn (F, left inset, arrow). Axons in subcortical white matter were also hαSyn+ (F, right inset). Scale bars = 25 μm (E and F), 14 μm (F left inset), 62 μm (F right inset). G–I. Localization of hαSyn in Thy1-A53T tg mouse basal ganglia circuit regions. hαSyn immunoreactivity was enriched in subsets of nigral neurons (G, arrows), while midbrain sections in non-tg mice were completely blank after probing with Syn211 antibody (H). hαSyn immunoreactivity was present in the neuropil of striatum (I) and globus pallidus (I upper inset). Some neuronal cell bodies in striatum were hαSyn+ (I, arrows), but most neuronal cell bodes were not positive. Neuronal cell bodies in ventrolateral nucleus (VL) of motor thalamus were enriched in hαSyn immunoreactivity. Scale bars = 33 μm (G), 21 μm (H), 24 μm (I), 125 μm (I upper inset), 44 μm (I lower inset). J. Localization of hαSyn in Thy1-A53T tg mouse cerebellum. hαSyn immunoreactivity was found in the neuropil of the molecular layer (ML) and granule cell layer (GCL) of cerebellar cortex. Purkinje cell bodies in the Purkinje cell layer (PCL) were not immunoreactive for hαSyn. Cerebellar sections in non-tg mice were completely blank after probing with hαSyn antibody (inset). Scale bars = 23 μm, 47 μm (inset) . K. In the granule cell layer of Thy1-A53T tg mouse cerebellum, numerous clusters of hαSyn immunoreactivity, likely corresponding to mossy fiber rosettes, were seen in the neuropil, and granule cell bodies, although not hαSyn+, had their surface contours decorated with hαSyn immunoreactivity (inset). Cell bodies in the granule cell layer that were much larger than granule cells and less numerous were hαSyn+ and were likely Golgi cells (inset arrow). Scale bar = 25 μm, 20 μm (inset). L. Numerous large neuronal cell bodies in the brainstem reticular formation (arrows) were positive for hαSyn. Scale bar = 30 μm.
Immunohistochemistry was used to localize hαSyn protein in different brain regions in Thy1-A53T tg mice. Monoclonal Syn211 antibody showed immunohistochemical specificity for hαSyn because it did not detect endogenous αSyn in perfusion-fixed non-tg mouse brain sections, including the cerebral cortex (Fig. 2D inset), SN (Fig. 2H), and cerebellar cortex (Fig. 2J inset). In contrast, in A53T tg mouse cerebral cortex, hαSyn immunoreactivity was localized to neurons and the neuropil (Fig. 2D–F). Glial cells did not appear immunoreactive for A53T-hαSyn during the course of disease (data not shown). hαSyn+ neuronal cell bodies were observed in cerebral cortical superficial (Fig. 2E) and deep (Fig. 2F) layers where it was localized to the cytoplasm and nucleus in a subset (about 35%) of cortical neurons. Cortical neurons showed prominent cytoplasmic and intranuclear aggregates of hαSyn (Fig. 2F left inset). Axons in subcortical white matter were also positive for hαSyn (Fig. 2F right inset). Basal ganglia and related compartments of the brain were enriched in hαSyn. Neurons in the SNc were strongly immunoreactive for hαSyn (Fig. 2G). The striatum had robust hαSyn immuoreactivity in the neuropil (Fig. 2I), consistent with expression of hαSyn in nigrostriatal SNc projection neurons (Fig. 2G), but the majority of striatal neuron cell bodies were negative (Fig. 2I). However, a minor subset of neuronal cell bodies was positive for hαSyn (Fig. 2I) which were subsequently identified as interneurons (see below). The globus pallidus was enriched in punctate immunoreactivity for hαSyn that was found in the neuropil and decorating the surfaces of pallidal neurons (Fig. 2I upper inset). The ventral anterior-ventral lateral nuclei of thalamus contained numerous hαSyn+ neurons (Fig. 2I lower inset). The cerebellum and brainstem also had prominent hαSyn immunoreactivity. The cerebellar cortex showed fine hαSyn punctate labeling in the molecular layer (Fig. 2J), but Purkinje cells were generally negative for hαSyn (Fig. 2J). The granule cell layer had prominent large clusters of immunoreactivity for hαSyn (Fig. 2K). These clusters were consistent with the appearance of mossy fiber terminals (Weissman et al., 2011). Granule cell bodies were uniformly negative for hαSyn (Fig. 2K), but their surfaces were decorated with hαSyn immunoreactivity (Fig. 2K inset). In contrast, larger cells near the granule cell layer-Purkinje cell layer border were hαSyn+ (Fig. 2K inset). These cells were subsequently identified as Golgi interneurons, consistent with the known size and locations of these neurons (Crook et al., 2006; Simat et al., 2007; Sillitoe et al., 2008). In brainstem, numerous neurons in the pontine and medullary reticular formation, including the pontine nuclei and gigantocellular reticular nucleus, were hαSyn+ (Fig. 2L). These neuronal cell bodies are known to be the origin of mossy fiber terminals in the cerebellar granule cell layer (Voogd et al., 1985) and would be congruent with the presence of hαSyn+ mossy fiber ending in this layer (Fig. 2K)
3.3 Nigral neuron degeneration in Thy1-A53T tg mice
Thy1-A53T tg C57BL/6 mice were assessed for degeneration of neurons in the SNc using Nissl staining and immunohistochemistry for TH (Fig. 3). CV-stained sections of midbrain suggested loss of neurons in the SNc in A53T tg mice at endstage disease compared to age-match non-tg littermate controls (Fig. 3A). Closer inspection supported this impression noted at low magnification as there was an apparent dropout of larger neurons in the SNc in of A53T tg mice when compared to controls (Fig. 3C,D). Counts of SNc neurons in CV-stained sections of midbrain revealed a major (~60%) loss of neurons (Fig. 3E). The loss of SNc neurons was age-related because 1 month-old A53T tg mice had a normal complement of SNc neurons (Fig. 3E). Immunohistochemical staining for TH in midbrain and forebrain sections corroborated the loss of SNc neurons in A53T tg mice. Mice at endstage showed diminished TH immunoreactivity in the SNc compared to age-matched non-tg littermate controls (Fig. 3F–I). This finding was reflected by an attenuation of TH immunostaining of neuronal cell bodies and the neuropil (Fig. 3H,I). The loss of TH immunoreactivity in the SNc was corroborated by dissipation of TH immunoreactivity in the striatum of A53T tg mice (Fig. 3J,K). TH immunoreactivity in the striatal neuropil of tg mice (Fig. 3K) was weak compared to the rich TH staining of controls (Fig. 3J). Moreover, dystrophic TH+ axons were seen in the striatum of tg mice (Fig. 3K inset). The dissipation of TH in the forebrain of A53T mice was more dramatic in the dorsal striatum compared to the olfactory tubercle of ventral striatum (Fig. 3J,K) that receives dopaminergic innervation from the ventral tegmental area instead of the SNc (Fallon and Loughlin, 1985).
Figure 3.

The substantia nigra compacta (SNc) undergoes age-related degeneration in Thy1-A53T hαSyn tg mice. A, B. Low magnification images of cresyl violet (Nissl) stained midbrain hemisections of age-matched non-tg (A) and Thy1-A53T (B) mice suggesting neuronal dropout in the SNc of tg mice. The nearby red nucleus (RN) also appears affected in tg mice. Scale bar = 400 μm. C,D. Nissl staining of age-matched mouse midbrain sections shows the SNc of non-tg mice (C) populated with large darkly staining neurons and the SNc of A53T tg mice (D) depopulated of neurons and having reactive changes. Remaining SNc neurons in A53T tg mice contained round cytoplasmic inclusions (D left inset) or displayed chromatolytic changes in the cell body (D right inset). Scale bars: C (same for D) = 20 μm, D left inset (same for right inset) = 12 μm. E. Counts of SNc neurons in Nissl-stained sections revealed a significant loss (asterisk, p < 0.01) in Thy1-A53T tg mice at 12 months of age but not in tg mice at 1 month of age. Values are mean ± SD (n = 6 mice/group). F,G. Low magnification images of tyrosine hydroxylase (TH) immunohistochemically-stained midbrain hemisections of age-matched non-tg (F) and Thy1-A53T (G) mice showing attenuation of TH immunoreactivity (brown labeling) in the SN of A53Tmice. Scale bar in F (same for G) = 400 μm. H,I. TH immunoreactivity was enriched in SNc neuronal cell bodies (H arrows), processes, and neuropil in non-tg mice. TH immunoreactivity was attenuated in SNc neuronal cell bodies, processes, and neuropil in Thy1-A53T tg mice (I) compared to the staining in non-tg mice (H). Scale bar in H (same for I) = 25 μm. J,K. Low magnification images of TH immunohistochemically-stained forebrain hemisections of age-matched non-tg (J) and Thy1-A53T (K) mice showing attenuation of TH immunoreactivity (brown labeling) in the striatum of tg mice. TH immunoreactivity was highly enriched in the striatal neuropil of non-tg mice (J inset) compared to the striatum of tg mice (K inset). The loss of striatal neuropil TH immunoreactivity in A53T mice was accompanied by accumulation of numerous dystrophic axons (K inset). The olfactory tubercle (OT) in tg mice was mostly spared compared to the dorsal striatum, consistent with its dopaminergic innervation derived from the ventral tegmental area instead of the SNc. Scale bars: J (same for K) = 800 μm, J inset (same for K inset) = 50 μm.
3.4 Nigral neurons in Thy1-A53T tg mice undergo apoptotic cell death
Additional evidence for the degeneration of the ventral midbrain in Thy1-A53T tg C57BL/6 mice was the presence of conspicuous argyrophilia within the SNc and surrounding gray matter and white matter regions in silver-stained sections (Fig. 4A). Nearby regions that showed marked argyrophilic degeneration were the red nucleus (RN), superior cerebellar peduncle (scp), medial lemniscus, and cerebral peduncle, but the adjacent substantia nigra reticulata appeared mostly free of degeneration (Fig. 4A). This degeneration was evidenced by silver-positive axons, cell bodies, and puncta (Fig. 4B), while age-matched non-tg control sections were blank for argyrophilia (Fig. 4C). Some silver+ degenerating neurons (Fig. 4B inset) had cell body and dendrite morphologies typical of dopaminergic SNc neurons (Fallon and Loughlin, 1985). The presence of robust degeneration in the SNc is consistent with the high expression of mutant hαSyn in neurons in this region (Figs. 2G,4D). Some degenerating SNc neurons in Thy1-A53T tg showed cytoplasmic Lewy-body-like inclusions that were hαSyn+ (Fig. 4D inset). These degenerating SNc neurons also appeared to receive axosomatic innervation from hαSyn+ neurons (Fig. 4D inset).
Figure 4.

Substantia nigra compacta (SNc) neurons undergo apoptotic cell death in Thy1-A53T hαSyn tg mice. A. Low magnification image of a silver-stained midbrain section of a Thy1-A53T mouse demonstrating degeneration (dark gray/black staining) of the SNc and nearby structures including the red nucleus (RN) and major white matter axonal pathways such as the superior cerebellar peduncle (scp), medical lemniscus (ml), and cerebral peduncle (cp). The substantia nigra reticulata (SNr) appeared unaffected as evidenced by the negligible argyrophilia. Scale bar = 312 μm. B,C. At higher magnification, silver staining revealed ubiquitous degeneration (black staining) of axons and neuronal cell bodies in the SNc, RN, and scp. Silver impregnated profiles of degenerating SNc neuronal cell bodies and processes could be seen detail in tg mice (B inset). Age-matched non-tg mouse midbrain sections were essentially blank for argyrophilia (C). Scale bars = 25 μm (B), 14 μm (B inset), 17 μm (C). D. Immunohistochemistry for hαSyn (brown) and Nissl counterstaining (blue) showed that subsets of SNc neurons had strong sustained expression of hαSyn (hatched arrows), but nearby neurons were not hαSyn+ (open arrows) in symptomatic Thy1-A53T hαSyn tg mice. Inset shows a degenerating nigral neuron with a condensed nucleus, a cytoplasmic hαSyn+ inclusion (hatched arrow), and apparent innervation from hαSyn+ axon terminals (solid arrow) in a tg mouse at endstage disease. Scale bar = 7 μm, 10 μm (inset). E. Immunohistochemistry for cleaved caspase-3 (brown) showed that subsets of SNc cells were positive for cleaved caspase-3 (hatched arrows) in symptomatic Thy1-A53T hαSyn tg mice. Some cleaved caspase-3+ cells appeared shrunken and rounded-up and were separating from the extracellular matrix (as seen by the white pericellular halo, open arrows), features typical of apoptosis (Martin et al., 1998; Martin and Liu, 2002). Nissl staining revealed shrunken cells with apoptotically condensed chromatin (E inset arrow) in the SNc of A53T mice. Scale bar = 10 μm, 7 μm (inset). F. Cell death detection by DNA fragmentation demonstrated TUNEL+ cells (open arrows) and apoptotic fragments (hatched arrows) in the SNc of symptomatic Thy1-A53T hαSyn tg mice. Scale bar = 7 μm. G. Counting of TUNEL+ cells in SNc revealed significant elevations above age-matched non-tg control levels in Thy1-A53T tg mice at 8 and 12 months of age but not in tg mice at 1 month of age (*, p < 0.05; **, p < 0.01). Values are mean ± SD (n = 6 mice/group). H,I. Immunohistochemistry for activated phospho-p53 (brown) showed that subsets of SNc neurons accumulated phospho-p53-immunoreactivity in their cell bodies and nuclei (H, arrow). Midbrain sections of age-matched non-tg mice were generally blank for activated p53 immunoreactivity, except for occasional phospho-p53+ intravascular cells (I, arrow). Scale bar (in I same for H) = 10 μm.
Thy1-A53T tg mice showed evidence for apoptotic cell death in the SNc. hαSyn+ neurons had nuclei condensed into masses consistent with apoptosis (Fig. 4D inset). Immunohistochemistry revealed subsets of attritional SNc cells positive for cleaved caspase-3. Some cleaved caspase-3+ SNc cells were round and had a distinct halo of separation from the extracellular matrix (Fig. 4E), indicative of apoptosis (Martin et al., 1998). Nissl staining confirmed the presence of apoptotic cells within the SNc of Thy1-A53T tg mice (Fig. 4E inset). TUNEL was used to demonstrate the presence of cell death in the SNc (Fig. 4F). TUNEL+ cell numbers were increased significantly in the SNc of Thy1-A53T tg mice at 8 and 12 months of age compared to age-matched non-tg littermate controls (Fig. 4G). No difference between tg and non-tg mouse SNc were seen with TUNEL at 1 month of age (Fig. 4G). A robust activation of p53, identified by immunohistochemistry for phospho-p53, was observed in subsets of SNc neurons in Thy1-A53T tg mice (Fig. 4H), but not in littermate non-tg controls (Fig. 4I).
3.5 The cerebral cortex degenerates in Thy1-A53T tg mice
Nissl-stained sections through the forebrain of Thy1-A53T tg C57BL/6 mice suggested pathology in cerebral cortex (Fig. 5A,B). The ventricular system appeared dilated in mice at endstage disease (Fig. 5B) compared to age matched non-tg controls (Fig. 5A). These sections also suggested thinning of the cortical mantle in tg mice (Fig. 5A,B). Ocular filar measurements revealed significant reductions in the thicknesses of the sensorimotor cortical gray matter (Fig. 5C) and subcortical white matter (Fig. 5D) in tg mice. Silver staining demonstrated axonal degeneration in the cortical gray matter and subcortical white matter (Fig. 5F) and in the cerebral peduncle (Fig. 4A) which contains corticobulbar and corticospinal axons. TUNEL was used to assay for cell death in the cerebral cortex (Fig. 5F). TUNEL+ cell numbers were increased significantly in layer V of Thy1-A53T tg mice at 8 and 12 months of age compared to non-tg littermate controls (Fig. 5G). Cleaved caspase-3 immunostaining was present in the cerebral cortex of symptomatic tg mice (Fig. 5H), while being undetectable in age-matched non-tg control cerebral cortex (Fig. 5H upper right inset). In tg mice, cleaved caspase-3 immunoreactivity was found throughout the cortical neuropil and in pyramidal neurons in layer V (Fig. 5H lower right inset). Some cortical neurons that were cleaved caspase-3+ were found to have intranuclear hαSyn aggregates (Fig. 5H left inset). Nissl-staining revealed morphological evidence for apoptosis in cortical neurons of Thy1-A53T tg mice (Fig. 5J), and LC3A immunohistochemistry indicated enhanced autophagy in cortical pyramidal neurons of tg mice (Fig. 5J) compared to age-matched non-tg mice (Fig. 5K).
Figure 5.

The cerebral cortex degenerates in Thy1-A53T hαSyn tg mice. A, B. Low magnification images of cresyl violet (Nissl) stained forebrain sections of age-matched non-tg (A) and Thy1-A53T (B) mice suggesting atrophy of the neocortex in tg mice. The lateral ventricle (lv) was dilated and the cortical mantle appeared thinned in A53T mice at endstage disease (B) compared to age-matched non-tg mice (A). Scale bar (in A, same for B) = 273 μm C. Ocular filar micrometer measurements revealed a significant reduction (asterisk, p < 0.05) in gray matter cortical thickness in endstage Thy1-A53T hαSyn tg mice compared to age-matched non-tg littermate mice (n=6/group). Values are mean ± SD. D. Ocular filar micrometer measurements revealed a significant reduction (asterisk, p < 0.01) in subcortical white matter thickness in endstage Thy1-A53T hαSyn tg mice compared to age-matched non-tg littermate mice (n=6/group). Values are mean ± SD. E. Silver staining revealed degeneration axons (arrows) in cortical gray matter and subcortical white matter in A53T tg mice. Scale bar = 35 μm. F. Cells with TUNEL+ nuclei (arrows) were observed in the neocortex of symptomatic Thy1-A53T hαSyn tg mice. Scale bar = 10 μm. G. Counting of TUNEL+ cells in motor-sensory cortex revealed significant elevations above age-matched non-tg control levels in Thy1-A53T tg mice at 8 and 12 months of age but not in tg mice at 1 month of age (*, p < 0.01; **, p < 0.001). Values are mean ± SD (n = 6 mice/group). H. Immunohistochemistry for cleaved caspase-3 (brown) showed positive staining throughout the cortical neuropil and in some neuronal cell bodies in symptomatic Thy1-A53T hαSyn tg mice, while the cerebral cortex of age-matched non-tg littermates was blank (H upper right inset). Layer V pyramidal neurons were positive for cleaved caspase-3 (arrows) in A53T mice (lower right inset). Some cleaved caspase-3+ cortical neurons contained prominent intranuclear hαSyn+ inclusions (left inset) as seen by immunofluorescence. Scale bars = 75 μm, 100 μm (upper right inset), 25 μm (lower right inset), 8 μm (lower right inset). I. Nissl staining identified layer V pyramidal neuron cell bodies undergoing shrinkage, chromatin clumping, and separation for the extracellular matrix (arrow). Scale bar = 4 μm. J,K. Immunohistochemistry for autophagy marker LC3A (brown) showed enhanced LC3A immunostaining in layer V cortical neurons in A53T tg mice compared to age-matched non-tg littermates. Scale bar (in J, same for K) = 35 μm.
In comparison to neocortex the hippocampus displayed less remarkable neuropathology in Thy1-A53T tg mice. Nissl staining and cleaved caspase-3 immunohistochemistry yielded scant indication of disease, but silver staining showed modest neuropil punctate degeneration in the stratum radiatum of CA3 and stratum lacunosum-moleculare of CA1 (data not shown).
3.6 Striatal interneurons degenerate in Thy1-A53T tg mice
In the course of surveying tg mouse forebrain sections stained immunohistochemicaly for hαSyn we found occasional cell bodies in striatum that were positive for hαSyn (Fig. 2I). These cells were identified as striatal interneurons because they were positive for the interneuron marker parvalbumin (Fig. 6A). The majority of medium-sized striatal neurons were not positive for hαSyn, although they received extensive innervation from apparent presynaptic boutons that were positive for hαSyn (Fig. 6A). In the course of the disease in Thy1-A53T tg mice a small subset of striatal neurons was positive for phospho-p53 (Fig. 6B,F) and cleaved caspase-3 (Fig. 6C,D,F) as visualized by immunoperoxidase histochemistry and immunofluorescence. Some of the cleaved caspase-3+ neurons were large compared to the majority of medium sized neurons (Fig. 6C). Immunoreactivity for cleaved caspese-3 was undetectable in parallel-processed sections from non-tg littermates (Fig. 6E). Cell counting revealed an age-related increase in the number of striatal neurons that were positive for cleaved caspase-3 or pp53 (Fig. 6F), along with a corresponding decrease in the number of parvalbumin+ interneurons in striatum of tg mice (Fig. 6G).
Figure 6.

Striatal interneurons degenerate in Thy1-A53T tg mice. A. Immunofluorescence showing that parvalbumin+ striatal interneurons express hαSyn in Thy1-A53T tg mice (arrow). hαSyn was present in the cytoplasm and nucleus of parvalbumin+ neurons and was enriched in the neuropil, but the majority of striatal neurons were hαSyn− (asterisks). Scale bar = 6 μm. B. Immunoperoxidase staining shows that a minority of striatal neurons in Thy1-A53T tg mice accumulates phospho-p53 (pp53) immunoreactivity (arrow). Scale bar = 8 μm. C. Immunoperoxidase staining (brown) identifies some large striatal neurons that are cleaved caspase-3+ in A53T tg mice. Scale bar = 5 μm. D,E. Immunofluorescence confirms the cleaved caspase-3 staining in subsets of striatal neurons (D, arrows) in tg mice, while striatal neurons in littermate non-tg mice were negative for cleaved caspase-3 (E). Scale bar (in A, same for E) = 12 μm F. Graph showing the numbers of cleaved caspase-3+ and phospho-p53 (pp53)+ cells in the striatum of Thy1-A53T tg mice at 1 and 12 months of age. Values are mean ± SD. Non-tg littermates were negative. G. Graph showing the numbers of parvalbumin+ cells in the striatum of Thy1-A53T tg mice and non-tg littermates at 1 and 12 months of age. Values are mean ± SD. Asterisk denotes significantly different from control (p < 0.01).
3.7 Cerebellar interneurons degenerate early in the course of the disease in Thy1-A53T tg mice
Silver-stained sections of the cerebellum of Thy1-A53T tg mice at endstage disease displayed remarkable argyrophilic degeneration (Fig. 7A), while age-matched non-tg mouse sections had scant argyrophilic degeneration (Fig. 7B). Prominent axonal and terminal degeneration was observed in cerebellar cortex of tg mice (Fig. 7A). Degenerating axons and putative terminals were concentrated in the white matter and the granule cell layer, respectively (Fig. 7A). The molecular layer, although overall less argyrophilic than deeper layers, also contained discernable degenerating axons and cell bodies (Fig. 7A). Immunohistochemical staining for cleaved caspase-3 disclosed several salient findings in cerebellar cortex (Fig. 7C), while non-tg control sections were blank for cleaved caspase-3 immunoreactivity (Fig. 7C inset). The molecular layer neuropil was enriched in cleaved caspase-3 immunoreactivity, but the Purkinje cell bodies were not obviously labeled (Fig. 7C). In the granule cell layer, subsets of cells larger than granule cells were cleaved caspase-3+ (Fig. 7C,D). These latter cells were often found at the granule cell layer-Purkinje cell layer border (Fig. 7C,D) and were likely to be Golgi cells (Zeilhofer et al., 2005; Simat et al., 2007; Sillitoe et al., 2008). The dendritic arbors of Golgi cells immunoreactive for cleaved caspase-3 would contribute to the observed immunostaining (Fig. 7C) in the molecular layer neuropil (Sillitoe et al., 2008). Counts of cleaved caspase-3+ putative Golgi cells revealed significant increases above non-tg age-matched controls as early as 1 month of age, and progressive increases in the number of positive cells were seen thereafter (Fig. 7E). Silver-stained sections of cerebellum from 1-month-old Thy1-A53T tg mice showed isolated degenerating cell bodies in the granule cell layer, apoptotic cells in the molecular layers, and terminal degeneration at the granule cell layer-Purkinje cell layer border (Fig. 7F). Neurons thought to be Golgi cells in cerebellar cortex were hαSyn+ (Fig. 7F inset). To identify that early-degenerating cells in the cerebellum were Golgi cells, Thy1-A53T tg mice were crossed to GlyT2-eGFP tg which, among other cells throughout the CNS (Zeilhofer et al., 2005), express eGFP in cerebellar Golgi cells (Simat et al., 2007; Sillitoe et al., 2008). Subsets of eGFP-positive Golgi cells that expressed hαSyn aggregates in their cytoplasm were found to be apoptotic in Thy1-A53T tg mice (Fig. 7G).
Figure 7.

Cerebellar interneurons and deep nuclei degenerate in Thy1-A53T tg mice. A,B. Silver-stained sections of cerebellum from endstage Thy1-A53T tg mice displayed robust degeneration (black staining) of axons in white matter (wm, arrows) and ubiquitous puncta in the granule cell layer (GL). Occasional degeneration of cell bodies was seen in the molecular layer (ML, open arrows). Age-matched non-tg mouse cerebellar sections were blank for argyrophilia (B). Scale bars = 35 μm (A), 25 μm (B). C,D. Immunohistochemistry for cleaved caspase-3 (brown) showed positive staining throughout the molecular layer (ML) neuropil and in some cell bodies (C, arrows) in the granule cell layer (near the Purkinje cell layer border) in symptomatic Thy1-A53T hαSyn tg mice, while the cerebellar cortex of age-matched non-tg littermates was blank for immunoreactivity (C inset). Cleaved caspase-3+ cells in the granule cell layer were large cells (D, arrows) compared to the predominating granule cells that were negative. Scale bars = 25 μm (C), 50 μm (C inset), 15μm (D). E. Counts of cleaved caspase-3+ cells in the granule cell layer of Thy1-A53T tg mice at 1, 8, and 12 months of age revealed significant elevations above age-matched non-tg control levels (*, p < 0.05; **, p < 0.01, ***, p < 0.001). Values are mean ± SD (n = 6 mice/group). F. Silver-stained sections of cerebellum from 1-month-old Thy1-A53T tg mice confirmed the isolated degeneration of cells in the granule cell layer (GL, arrow) and demonstrated punctate degeneration at the GL-Purkinje cell layer (PL) border. Occasional apoptotic cells were also seen in the molecular layer (ML, arrow). hαSyn immunohistochemistry identified hαSyn+ cells in the GL that were attritional and separating from the extracellular matrix (inset arrow), as well as hαSyn immunoreactivity enriched in putative mossy fibers rosettes and decorating the surfaces of granule cells (inset). Scale bars = 8 μm (F), 25 μm (F inset). G. Double tg mice expressing Thy1-A53T hαSyn and GlyT2-eGFP showed that cerebellar Golgi cells expressing hαSyn undergo apoptosis. Scale bar = 6 μm. H,I. Nissl staining of cerebellar sections from endstage Thy1-A53T tg mice (H) and age-matched non-tg littermates (I) revealed degeneration neurons in the interpositus deep nucleus in tg mice including apparent cell dropout , reactive cell nests (H inset hatched arrow), and cytoplasmic vacuolation of residual neurons (H inset open arrow). Scale bars = 20 μm (H, same for I), 10 μm (H inset). J. Counts of nucleus interpositus neurons revealed a significant loss (asterisk, p < 0.01) in Thy1-A53T tg mice at 12 months of age but not in tg mice at 1 month of age. Values are mean ± SD (n = 6 mice/group).
Thy1-A53T tg C57BL/6 mice at endstage disease also had prominent degeneration of the deep cerebellar nuclei. Nissl and silver staining gave indications of pathology in this region. The interposed nucleus contained neurons with prominent large vacuoles, apparent cell elimination, and nests of small inflammatory-like cells (Fig. 7H). The vacuolar type of neuronal pathology has been seen with chronic deafferentation (Ginsberg and Martin, 1998). Age-matched non-tg controls had normal appearing neurons in cerebellar deep nuclei (Fig. 7I). Counts of neurons in the interposed nucleus confirmed the impression of cell dropout by demonstrating severe (~80%) loss of neurons in endstage mice but a normal complement of neurons in young adult tg mice (Fig. 7J). The interposed and lateral deep cerebellar nuclei also had degeneration in silver-stained preparations (data not shown).
3.8 Red nucleus degeneration in Thy1-A53T tg mice
Attention was directed to the red nucleus (RN) in Thy1-A53T tg C57BL/6 mice because of the degeneration of the cerebellar interpositus nucleus (Fig. 7H), which projects to the contralateral RN through the superior cerebellar peduncle (Voogd et al., 1985), and because damage within the RN was suggested while examining midbrain sections for SNc pathology (Figs. 3A,B, 4A,B). Nissl staining indicated a loss of magnocellular neurons (Figs. 3A,B, 8A,B) in the RN at endstage disease. Cell counting showed a significant loss (~60%) of RN neurons in mice at endstage disease but not in young adult tg mice (Fig. 8D). Many of the remaining neurons in tg mice had marked chromatolytic changes (Fig. 8C) indicative of axonal injury (Martin and Liu, 2002) or accumulation of granulovacuole-like inclusions (Fig. 8E). Apoptotic cells were observed in the RN in tg mice (Fig. 8F). Silver staining demonstrated prominent axonal and punctate degeneration in the RN and intermingling superior cerebellar peduncle of tg mice (Figs. 4A,B, 8G,) while the RN in age-matched non-tg mice was blank for argyrophilic degeneration (Figs. 4C, 8G inset). Immunohistochemistry for LC3A was done to examine whether the granulovacuole-like inclusions reflected autophagy. LC3A-positive granule inclusions accumulated in subsets of remaining RN neurons in A53T mice (Fig. 8I) but not in control mouse RN neurons (Fig. 8H).
Figure 8.

Red nucleus (RN) degeneration in Thy1-A53T hαSyn tg mice. A–C. Nissl staining of midbrain sections from age-matched non-tg littermates (A) and endstage Thy1-A53T tg mice (B,C) revealed degeneration neurons in the RN in tg mice including apparent cell dropout and reactive cell changes (B,C). Many remaining neurons showed chromatolytic changes (C, arrows). Scale bar = 42 μm (A, same for B), 19 μm (C). D. Counts of RN neurons revealed a significant loss (asterisk, p < 0.01) in Thy1-A53T tg mice at 12 months of age but not in tg mice at 1 month of age. Values are mean ± SD (n = 6 mice/group). E. Some residual RN neurons (arrow) accumulated granulovacuole-like inclusions in their cytoplasm. Scale bar = 11 μm. F. Apoptotic profiles (arrow) were present in the RN of tg mice. Scale bar = 8 μm. G. Silver staining revealed severe degeneration of axons and puncta (black) in the red nucleus neuropil, while the RN in age-matched non-tg littermates was blank for argyrophilia (inset). Scale bar = 33 μm (same for inset). H,I. Immunohistochemistry for autophagy marker LC3A (brown) showed enhanced LC3A immunostaining of granules in the cytoplasm of RN neurons (I, arrows) in A53T tg mice compared to neurons in age-matched non-tg littermates (H). Scale bar (in H, same for I) = 17 μm.
3.9 Motor thalamus degeneration in Thy1-A53T tg mice
The thalamus receives cerebellar input (Voogd et al., 1985; Bostan and Strick, 2010), and because of the degeneration of the nucleus interpositus of cerebellum and superior cerebellar peduncle near the RN, our attention was directed to the motor thalamus. Silver staining offered the first indication of degeneration in this region (Fig. 9). Highly selective nuclear-specific degeneration was seen in the ventral anterior (VA) and ventral lateral (VL) thalamic nuclei in Thy1-A53T tg mice at endstage disease (Fig. 9B), while the thalamus in age-matched non-tg mice was negative for argyrophilia (Fig. 9A). The central medial, central lateral, and paracentral thalamic nuclei were also affected (data not shown). Closer examination showed the presence of degeneration cell bodies, axons, and puncta in these regions (Fig. 9C,D). Nissl staining suggested reactive changes in response to neurodegeneration in the VA/VL of Thy1-A53T tg mice (Fig. 9F,H), while controls gave no indication of neurodegeneration and small cell changes (Fig. 9E,G). Apoptotic profiles were encountered in the VA/VL of Thy1-A53T tg mice at endstage disease (Fig. 9H). Neuronal cell counting revealed a significant loss of neurons (~40%) in the VA/VL of 12 month old tg mice but not in 1 month old tg mice (Fig. 9I).
Figure 9.

Motor thalamus degeneration in Thy1-A53T hαSyn tg mice. A–D. Silver-stained sections through the diencephalon of endstage Thy1-A53T tg mice displayed selective robust degeneration (black staining) of the ventrolateral nucleus (VL) of thalamus (B,C), while the thalamus in age-matched non-tg littermates was blank for argyrophilia (A). Some of the territory designated as VL might include the the ventral anterior (VA) thalamic nucleus. Reticular thalamic nucleus (Rt), globus pallidus (GP), internal capsule (ic). Degenerating axons (D, open arrow), cell bodies (D, hatched arrow), and puncta were abundant in the VL of A53T tg mice. Scale bars = 150 μm (A), 200 μm (B), 100 μm (C), 40 μm (D). E–H. Nissl-stained sections through diencephalon viewed at low magnification (E,F) revealed dilation of the lateral ventricle (lv) and 3rd ventricular cavities of the cerebroventricular system and dark-staining enhancement in VL of Thy1-A53T hαSyn tg mice. At higher magnification, the VL thalamus contained numerous apoptotic profiles (H, arrows) and chromatolytic neurons (H, open arrows), and apparent small cell infiltrates. Scale bars = 200 μm (E, same for F), 25 μm (G, same for H). I. Counts of VL neurons revealed a significant loss (asterisk, p < 0.01) in Thy1-A53T tg mice at 12 months of age but not in tg mice at 1 month of age. Values are mean ± SD (n = 6 mice/group).
3.10 Mitochondrial permeability transition pore (mPTP) involvement in disease mechanisms in Thy1-A53T tg mice
Mitochondria have been implicated in the pathogenesis of PD in humans (Olanow and Tatton, 1999; Beal, 2005; Martin, 2010; Reddy and Reddy, 2011; Pilsl and Winklhofer, 2012; Csokun et al., 2012) and in A53T-hαSyn tg mice (Martin et al., 2006). The mPTP may have particular importance in this regard (Martin, 2010). We attempted to establish direct cause-effect relationships between the mPTP and disease mechanisms in PD using tg mice. Putative components or regulators of the mPTP are expressed in mouse SNc neurons (Fig. 10A,B). CyPD (Fig. 10A) and the ANT (Fig. 10B) are present in mitochondria marked by SOD2. Western blotting for mPTP components revealed a modest increase in CyPD levels in brainstem, striatum and cortex of early and late symptomatic Thy1-A53T tg mice, but ANT and VDAC levels were not different from age-matched non-tg mice (Fig. 10C,D). Co-immunoprecipitation showed that A53T-hαSyn avidly binds VDAC in brainstem, striatum and cortex of early and late symptomatic Thy1-A53T tg mice, but only slightly binds CyPD in cortex in vivo (Fig. 10E). A53T-hαSyn associates with neuronal mitochondria that appear aggregated and swollen in Thy1-A53T tg mouse SNc neurons (Fig. 10F). Reducing the levels of CyPD by genetic ablation significantly delayed disease onset and extended the lifespan of Thy1-A53T tg mice (Fig. 10G).
Figure 10.

The mitochondrial permeability transition pore (mPTP) contributes to disease mechanisms in Thy1-A53T hαSyn tg mice. A,B. Putative components of the mPTP are enriched in substantia nigra compacta (SNc) neurons compared to substantia nigra reticulata (SNr) neurons as shown by immunofluorescence for cyclophilin D (CyPD, A, red) and adenine nucleotide translocator (ANT, B, red) and co-labeling with the mitochondrial marker superoxide dismutase-2 (SOD2, A and B, green). Co-localization is seen as yellow. Nuclei were stained with DAPI (blue). Scale bars = 12.5 μm (A), 4 μm (A inset), 10 μm (B), 5 μm (B inset). C. Western blots for putative components of the mPTP, including ANT, voltage-dependent anion channel (VDAC), and CyPD, in the brainstem, striatum, and cerebral cortex of early and late symptomatic Thy1-A53T hαSyn tg mice and non-tg mice. A representative Ponceau S-stained membrane shows protein loading. D. Quantification of immunoreactivity by densitometry showed a significant increase (*, p < 0.05) in CyPD levels in brainstem of early symptomatic Thy1-A53T hαSyn tg mice compared to non-tg controls. Similar elevations in CyPD were seen in brainstem, striatum, and cortex of early and late symptomatic mice. E. hαSyn interacts strongly with VDAC (all brain regions) and slightly with CyPD (cortex) as shown by co-immunoprecipitation. F. hαSyn associates with mitochondria as shown by the immunofluorescent co-localization of hαSyn (red) and the mitochondrial marker SOD2 (green) in SNc neurons. Co-localization is seen as yellow. Some mitochondria positive for hαSyn appear swollen (arrow). Scale bar = 4 μm. G. Genetic deletion of CyPD delayed disease onset and extended lifespan in Thy1-A53T hαSyn tg mice.
4. Discussion
This study provides new information on the pathogenesis of PD using a tg mouse model. This mouse model expresses the human A53T mutant variant of αSyn driven by a Thy1 promoter. The original tg mouse line was generated in the Sudhof laboratory on a hybrid background (Chandra et al., 2005), and we backcrossed this original tg mouse line over many generations into a pure C57BL/6 background. These Thy1-A53T-hαSyn C57BL/6 tg mice develop robust neurological and neuropathological phenotypes and a fully penetrant fatal disease consistent with PD. They show age-related ataxia, bradykinesia, postural instability, and rigidity (see supplemental Videos 1 and 2) and have motor system-preferential neuropathology involving synucleinopathy and interneuron degeneration early in the disease. HαSyn-A53T mutant protein associates with putative components of the mPTP in these mice and genetic inactivation of the ppif gene encoding CyPD, a major regulator of the mPTP (Baines et al., 2005; Basso et al., 2005), in Thy1-A53T-α-Syn C57BL/6 tg mice delays disease onset and extends lifespan. These results demonstrate for the first time that Thy1-A53T-α-Syn C57BL/6 tg mice are a useful model that faithfully simulates neurological and neuropathological aspects of human PD and that the mPTP is involved directly in the disease mechanisms in these mice.
Many tg mouse lines have been engineered to express hαSyn controlled by different promoters. These mice have varying neurological and pathological characteristics, some of which may be present in human PD, but most hαSyn tg mouse models are imperfect for modeling human PD (Chesselet and Richter, 2011; Antony et al., 2011). Mice expressing wildtype hαSyn driven by a human platelet-derived growth factor-β promoter displayed a mild motor phenotype, developed amorphous nonfilamentous intranuclear inclusions, and reduced striatal TH immunoreactivity, but did not have SNc neuron loss or fatal disease (Masliah et al., 2000). Mice expressing wildtype or mutant (A53T or A30P) hαSyn driven by the rat TH promoter did not develop clinical or neuropathological phenotypes (Matsukoka et al., 2001), but, mice expressing doubly mutant (G88C and G209A) hαSyn driven by a rat TH promoter showed reduced locomotor activity, mild loss of striatal dopamine, and moderate reduction of TH immunostained SNc neurons, but no fatal disease (Richfield et al., 2002; Thiruchelvam et al., 2004). Tg mice expressing truncated hαSyn driven by a rat TH promoter developed a mild motor phenotype, mild loss of striatal dopamine, and non-progressive loss (~50%) of SNc neurons, but no fatal disease (Wakamatsu et al., 2008). Tg mice expressing hαSyn (wildtype, A53T and A30P variants) under the control of the murine Thy-1 promoter developed pronounced motor deficits on the rotating rod task, intraneuronal hαSyn accumulation, axonal degeneration in spinal roots, and evidence for muscle denervation, but no reported loss of SNc neurons or fatal disease (van der Putten et al., 2000). In contrast, using the mouse prion protein (PrP) promoter, Giasson et al engineered mice expressing A53T mutant hαSyn and found that these mice become paralyzed and develop hαSyn inclusions, primarily in brainstem and spinal cord, and axonal degeneration in spinal roots (Giasson et al., 2002). Similarly, Lee et al generated tg mice expressing hαSyn wildtype, A53T, and A30P variants driven by a murine PrP promoter and showed A53T mice with a fatal paralytic disease phenotype (Lee et al., 2002). These mice also showed mitochondrially-associated A53T hαSyn, mitochondrial morphology and biochemical abnormalities, and neuronal cell death in neocortex, brainstem, and spinal cord (Martin et al., 2006). However, no evidence of neurodegeneration and neuronal cell death in the SNc of PrP hαSyn tg mice has been reported (Giasson et al., 2002; Lee et al., 2002; Martin et al., 2006). Recently, tg mice with conditional expression of hαSyn-A53T in midbrain dopaminergic neurons using a tetracycline-regulated inducible PITX3 promoter failed to develop major neurological disease but were reported to have deficits in striatal dopamine release and early onset loss of dopaminergic neurons in SNc and VTA (Lin et al., 2012). The distinguishing observations that set apart our study from these earlier studies are the findings that human Thy1- driven A53T-hαSyn C57BL/6 tg mice show cardinal neurological and motor system-preferential pathological abnormalities and fatal disease consistent with PD. The pronounced clinical neurological features of these mice were rigidity/spasticity, postural instability, bradykinesia, and akinesia (Videos 1 and 2). The robust neuropathological features in these mice, that are also cardinal pathologies in PD, were major age-related loss of SNc neurons, denervation of striatum, neocortical degeneration, and formation of intraneuronal hαSyn+ inclusions. Additional pathological changes in these mice, which are not examined commonly in human PD, include cerebellar and red nucleus degeneration and loss of striatal and cerebellar cortical inhibitory interneurons.
PD is a multisystem neural circuit disorder (DeLong and Wichmann, 2007; Jellinger 2012). We show that A53T-hαSyn tg mice have neuropathology consistent with a neural circuit degenerative disorder organized by connectivity. This is best illustrated by the degeneration in the SNc and nigrostriatal pathway and in the known network formed by the DCN, RN, VA/VL thalamus, and motor-sensory neocortex. The striking selective degeneration of VA/VL regions of thalamus highlights a probable transynaptic mechanism related to basal ganglia and cerebellar degeneration in these mice. Most investigations of PD focus on the basal ganglia; in contrast, the cerebellum has received little attention, but a cerebellar role in PD is now realized (Helmich et al., 2012; Wu and Hallett, 2013). fMRI studies have found hyperactivation of the cerebellum in human PD (Rascol et al., 1997; Yu et al., 2007). Cerebellar abnormalities may contribute directly to manifestation of resting tremor in PD oscillatory activity (Bostan and Strick, 2010; Helmich et al., 2012). Our work on a tg mouse model of PD reinforces the idea that be cerebellum may have a role in the pathophysiology of PD. However, the cerebellum, red nucleus, globus pallidus, and thalamus in human PD are generally thought spared from neuropathology (Dickson, 2012), but these regions, as well as the SNc and striatum, are affected human progressive supranuclear palsy (PSP) (Dickson, 2012). Like PD, PSP is characterized clinically by postural instability and rigidity (Dickson, 2012). Thus, it is possible that this A53T-hαSyn C57BL/6 tg mouse is a model of PSP, although this interpretation is contingent on a characterization of the tau pathology in these mice.
The early degeneration of cerebellar Golgi interneurons and striatal parvalbumin interneurons in A53T-hαSyn tg mice is instructive by demonstrating that 1) interneurons are involved in the disease process in PD mice, and 2) interneuron disease emerges early in life, but the overt clinical manifestations appear much later in life. Epidemiological accounts suggest that individuals with PD may have a preclinical phase spanning decades (Savica et al., 2010). A clinical study of the orbicularis oculi reflex in PD patients found indications of brainstem interneuron dysfunction (Kimura, 1973). The 6-hydroxydopamine model of PD has also revealed evidence for interneuron abnormalities. Striatal interneuron innervation of striatopallidal and striatonigral neurons undergoes differential structural (Salin et al., 2009) and functional (Gittis et al., 2011) remodeling of synapses after toxin exposure. Electrophysiological studies support the idea that inhibitory interneurons may be vulnerable in hαSyn tg mice. Patch-clamp recording shows deficits in GABAergic inhibitory transmission in frontal cortex of these mice (Ito et al., 2012). Early presymptomatic degeneration or dysfunction of interneurons has also been found in mouse models of amyotrophic lateral sclerosis (Martin and Chang, 2012) and Huntington’s disease (Dougherty et al., 2014). The disease within the cerebello-rubro-thalamo-cortical circuit in our tg mice possibly starts with the degeneration of hαSyn+ Golgi cell interneurons in cerebellar cortex because we found these neurons degenerating in 1-month old mice. In human Thy1-driven A53T-hαSyn C57BL/6 tg mice, hαSyn protein levels are generally higher in hindbrain structures than in forebrain/diencephalic structures early in disease, but, later in disease, hαSyn protein is present more globally throughout the brain. A cerebellum to midbrain to diencephalon to forebrain spread of pathology would be consistent with a prion-like neuron-to-neuron transmission of hαSyn-mediated disease (Desplats et al., 2009).
There is little doubt about the loss of SNc dopamine neurons in PD (Rudow et al., 2008), yet an important unresolved basic question is how SNc neurons degenerate and are eventually eliminated. Cells can die by different ways through the involvement of different molecular mechanisms. Apoptosis, necrosis, autophagy, and hybrids of apoptosis and necrosis are forms of cell death (Martin et al., 1998; Klionsky and Emr, 2000; Martin, 2010). The reports on the incidence of dying cells in the PD SNc with DNA fragmentation based on in situ end labeling methods are conflicting. Some studies report no labeling (Jellinger, 1999; Jellinger, 2000), but other studies have found nuclear DNA fragmentation (Tompkins et al., 1997; Tatton et al., 1998; Martin, 2010). Some studies report evidence for apoptosis in PD (Anglade et al., 1997; Tatton et al., 1998; Hartmann et al., 2000), but other studies reveal little evidence of apoptosis (Burke and Kholodilov, 1998; Wullner et al., 1999). Some of the confusion might be due to the use of DNA fragmentation detection systems that do not distinguish between DNA strand breaks associated with apoptosis or necrosis and to arbitrary morphological interpretations of nuclear morphology and chromatin condensation (Martin et al., 1998; Jellinger, 1999). In human PD, despite the reported presence of TUNEL+ and cleaved caspase-3+ neurons in the SNc (Hartmann et al., 2000; Martin, 2010), the morphology of degenerating SNc neurons appears different from classical apoptosis (Martin, 2001, 2010). In contrast, in A53T-hαSyn tg mice we find that SNc neuron death appears to be consistent with a caspase-3-driven classically apoptotic form of degeneration. The activation of p53 in SNc neurons suggested DNA damage induced by mutant hαSyn as a possible upstream mechanism of SNc neuron apoptosis. Apoptosis and cleaved caspase-3 immunoreactivity was also observed in the neocortex, striatum, thalamus, red nucleus, and cerebellar cortex in these mice, while evidence for necrotic-like cell death was found in the deep cerebellar nuclei. The absence of neurodegenerative phenotype that fully manifests as classical apoptosis has been seen also in human ALS (Martin, 1999; Martin, 2010). It is unknown why rodent experimental systems appear to more readily engage classically apoptotic forms of neuronal cell death in mature CNS compared to adult human CNS. It is possible that the molecular regulations of neuronal aging, compensation, and cell death are nuanced in human and rodent neurons because of differences in DNA damage and repair mechanisms, DNA methylation, gene promoter and transcriptome activity, senescence, caspase activation and utilization, and mitochondrial biology (Panov et al., 2007; Horvath et al., 2007; Odom et al., 2007; Loerch et al., 2008; Banuelos et al., 2008; Westerman, 2010; Bell et al., 2012). Therefore, the value of this tg mouse as a translational model for testing therapies for neuroprotection in PD warrants exploration.
In addition to the new clinical and neuropathological description of this tg mouse model of PD, a novel contribution of this work relates to the identification of the role of the mPTP in the disease process in these mice. Mitochondrial permeability transition is a mitochondrial state in which the proton-motive force is disrupted (Crompton, 1999; van Gurp et al., 2003; Bernadri et al., 2006; Leung and Halestrap, 2008; Bernardi, 2013). This disruption involves the mPTP that functions as a voltage, thiol, and Ca2+ sensor. Conditions of mitochondrial Ca2+ overload, excessive oxidative stress, and decreased electrochemical gradient (ΔP), ADP, and ATP can favor mitochondrial permeability transition. The mPTP is believed to be a transmembrane channel formed at the contact sites between the inner mitochondrial membrane (IMM) and the outer mitochondrial membrane (OMM). The components of the mPTP are still controversial. Dimers of the F0F1 ATP synthase (complex V) form a channel that appears electrophysiologically equivalent to the mPTP (Bernardi, 2013). The VDAC in the OMM, the ANT in the IMM, and CyPD in the matrix are also believed to have some involvement (Crompton, 1999; van Gurp et al., 2003; Bernardi et al., 2006), but they are not obligatory for mPTP formation (Bernardi et al., 2006; Bernardi, 2013). The VDAC adopts an open conformation at low or zero membrane potential and a closed conformation at potentials above 30–40 mV making the OMM permeable to most small hydrophilic molecules up to 1.3 kDa for free exchange of respiratory chain substrates (Rostovtseva et al., 2005). The ANT is a multi-pass membrane protein, with odd-numbered transmembrane helices having kinks because of proline residues, which mediates the exchange of cytosolic ADP for mitochondrial ATP (Graham et al., 1997). During normal mitochondrial function the OMM and the IMM are separated by the intermembrane space, and the VDAC and the ANT do not interact (Crompton, 1999). Permeability transition is activated by the formation of the mPTP; the IMM loses its integrity and the ANT changes its conformation from its native state into a non-selective pore (Crompton, 2003). This process is catalyzed by CyPD which is a peptidylprolyl isomerase (PPIase) that functions in protein cis-trans isomerization and chaperoning (Waldmeier et al., 2003). The ANT and CyPD interact directly (Woodfield et al., 1998). The molar concentration of CyPD (in heart mitochondria) is much less (>5%) than ANT; thus, under normal conditions only a minor fraction of the ANT can be in a complex with CyPD (Johnson et al., 1999; Leung and Halestrap, 2008). When this occurs, small ions and metabolites permeate freely across the IMM and oxidation of metabolites by O2 proceeds with electron flux not coupled to proton pumping, resulting in collapse of ΔP, dissipation of ATP production, production of ROS, equilibration of ions between the matrix and cytosol, matrix volume increases, and mitochondrial swelling (van Gurp et al., 2003; Rostovtseva et al., 2005). We demonstrate that putative components or regulators of the mPTP are present in mouse SNc neurons. We also show that A53T-hαSyn associates with neuronal mitochondria and interacts with VDAC and CyPD in vivo. Mitochondria positive for A53T-hαSyn undergo prominent morphological changes in SNc neurons in these mice. Other groups have shown that hαSyn is imported into mitochondria in cultured dopaminergic neurons and in human PD brain neurons (Devi et al., 2008) and that this impairs complex I activity (Devi et al., 2008; Liu et al., 2009). Mitochondrial membrane associated A53T-hαSyn can cause morphological changes in HeLa cell mitochondria (Nakamura et al., 2011). Moreover, hαSyn interacts with the ANT in rat midbrain (Zhu et al., 2011). Finally, we show that the genetic deletion of a major mPTP regulator, CyPD, has robust effects in PD mice by delaying disease onset and extending survival. Our finding that the mPTP has a role in the development of PD in A53T-hαSyn mice is consistent with observations made in cell culture showing that loss of PINK1 and DJ-1 function mutations result in mPTP opening (Giaime et al., 2012; Gautier et al., 2012). Noteworthy in this regard is the finding that CyPD deletion protects interneurons from apoptosis in the adult CNS (Martin, 2011). This work shows for the first time a direct causal role of a mitochondrial mechanism (i.e., the mPTP) in the pathobiology of a genetic mouse model of PD.
In summary we characterized the neurological abnormalities, neuropathology, and mechanisms of disease in Thy1-hαSyn-A53T C57BL/6 tg mice. We found that these mice develop a profound movement disorder with many PD-like features and develop a motor circuit neuropathology and synucleinopathy involving the nigrostriatal and cerebellar pathways. Interneurons degeneration is a prominent early feature of the disease in these mice suggesting that interneuron disease emerging early in life may have neurological consequences later in life. Genetic deletion of the mPTP regulator CyPD had major disease modifying effects. We conclude that the mPTP actively participates in the mechanisms of neurodegeneration in this PD mouse model. This work defines new cellular and mitochondrial mechanisms for neurodegeneration in PD that might be useful for preclinical translation studies and the identification of molecular mechanism-based therapies for treating this debilitating fatal disease.
Supplementary Material
Thy1-A53T hαSyn C57BL/6 tg mouse at early symptomatic stage of disease with postural instability, slight tremor, and jerking movements.
Thy1-A53T hαSyn C57BL/6 tg mouse at near endstage disease with sever rigidity and postural instability.
Acknowledgments
The authors thank Ann Price and Yan Pan for their outstanding technical work and Isabella Martin for her editorial assistance. This work was supported by grant NS034100 and NS052098 from the NIH-NINDS and AG016282 from the NIH-NIA.
Footnotes
Disclosure Statement: All authors have no actual or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marguez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Antony PMA, Diederich NJ, Balling R. Parkinson’s disease mouse models in translational research. Mamm Genome. 2011;22:401–19. doi: 10.1007/s00335-011-9330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascherio A, Chen H, Weisskopf MG, O’Reilly E, McCullough ML, Calle EE, Schwarzchild MA, Thun MJ. Pesticide exposure and risk for Parkinson’s disease. Ann Neurol. 2006;60:197–203. doi: 10.1002/ana.20904. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair HS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Bañuelos CA, Banáth JP, MacPhail SH, Zhao J, Eaves CA, O’Connor MD, Lansdorp PM, Olive PL. Mouse but not human embryonic stem cells are deficient in rejoining of ionizing radiation-induced DNA double-strand breaks. DNA Repair. 2008;7:1471–83. doi: 10.1016/j.dnarep.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Fortes MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem. 2005;280:18558–61. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- Bell CG, Wilson GA, Butcher LM, Roos C, Walter L, Beck S. Human-specific CpG “beacons” identify loci associated with human-specific traits and disease. Epigenetics. 2012;7:1188–99. doi: 10.4161/epi.22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol. 2013;4:95. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Krauskopf A, Basso E, Petronilli V, Blalchy-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–99. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- Bindoff LA, Birch-Machin M, Cartlidge NEF, Parker WD, Turnbull DM. Mitochondrial function in Parkinson’s disease. Lancet. 1989;2:49. doi: 10.1016/s0140-6736(89)90291-2. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Bostan A, Strick PL. The cerebellum and basal ganglia are interconnected. Neuropsychol Rev. 2010;20:261–70. doi: 10.1007/s11065-010-9143-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, Kholodilov NG. Programmed cell death: does it play a role in Parkinson’s disease? Ann Neurol. 1998;44 (3 Suppl 1):S1126–133. doi: 10.1002/ana.410440719. [DOI] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudholf TC. α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell. 2005;123:383–96. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chesselet MF, Richter F. Modeling of Parkinson’s disease in mice. Lancet. 2011;10:1108–18. doi: 10.1016/S1474-4422(11)70227-7. [DOI] [PubMed] [Google Scholar]
- Cleeter MWJ, Cooper JM, Schapira AHV. Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridine: evidence for free radical involvement. J Neurochem. 1992;58:786–9. doi: 10.1111/j.1471-4159.1992.tb09789.x. [DOI] [PubMed] [Google Scholar]
- Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, LaFerla F, Wallace DC. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophysica Acta. 2012;1820:553–64. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook J, Hendrickson A, Robinson FR. Co-localization of glycine and GABA immunoreactivity in interneurons in Macaca monkey cerebellar cortex. Neuroscience. 2006;141:1951–9. doi: 10.1016/j.neuroscience.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–49. [PMC free article] [PubMed] [Google Scholar]
- Crompton M. Mitochondria and aging: a role for the permeability transition? Aging Cell. 2004;3:3–6. doi: 10.1046/j.1474-9728.2003.00073.x. [DOI] [PubMed] [Google Scholar]
- Daher JPL, Pletnikova O, Biskup S, Musso A, Gellhaar S, Galter D, Troncoso JC, Lee MK, Dawson TM, Dawson VL, Moore DJ. Neurodegenerative phenotypes in an A53T α-synuclein transgenic mouse model are independent of LRRK2. Human Mol Genet. 2012;21:2420–31. doi: 10.1093/hmg/dds057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol. 2007;64:20–4. doi: 10.1001/archneur.64.1.20. [DOI] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci USA. 2009;106:13010–5. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW. Parkinson’s disease and parksonism: neuropathology. Cold Spring Harb Perspect Med. 2012;2:a009258. doi: 10.1101/cshperspect.a009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderow FA, Tanner CM. Projected number of people with Parkinson disease in the most populous nation, 2005 through 2030. Neurology. 2007;68:384–6. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- Dougherty SE, Hollimon JJ, McMeekin LJ, Bohannom AS, West AB, Lesort M, Hablitz JJ, Cowell RM. Hyperactivity and cortical disinhibition in mice with restricted expression of mutant huntingtin to parvablumin-positive cells. Neurobiol Dis. 2014;62:160–71. doi: 10.1016/j.nbd.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon JH, Loughlin SE. Substantia nigra. In: Paxinos G, editor. The Rat Nervous System. Vol. 1. Academic Press; Australia: 1985. pp. 353–74. [Google Scholar]
- Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. Neural activity control the synaptic accumulation of α-synuclein. J Neurosci. 2005;25:10913–21. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1997. [Google Scholar]
- Gautier CA, Giaime E, Caballero E, Núńez L, Song Z, Chan D, Villalobos C, Shen J. Regulation of mitochondrial permeability transition pore by PINK1. Mol Neurodegener. 2012;7:22. doi: 10.1186/1750-1326-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaime E, Yamaguchi H, Gautier CA, Kitada T, Shen J. Loss of DJ-1 does not affect mitochondrial respiration but increases ROS production and mitochondrial permeability transition pore opening. PLoS One. 2012;7(7):e40501. doi: 10.1371/journal.pone.0040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IVJ, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VMY. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science. 2000a;290:985–9. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VMY. A panel of epitope-specific antibodies detects protein domains distributed throughout human α-synuclein in lewy bodies of Parkinson’s disease. J Neurosci Res. 2000b;59:528–33. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee WMY. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Martin LJ. Ultrastructural analysis of the progression of neurodegeneration in the septum following fimbria-fornix transection. Neuroscience. 1998;86:1259–72. doi: 10.1016/s0306-4522(98)00136-5. [DOI] [PubMed] [Google Scholar]
- Gispert S, Del Turco D, Garrett L, Chen A, Bernard DJ, Hamm-Clement J, Korf HW, Deller T, Braak H, Auburger G, Nussbaum RL. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol Cell Neurosci. 2003;24:419–29. doi: 10.1016/s1044-7431(03)00198-2. [DOI] [PubMed] [Google Scholar]
- Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, Voos W, Leuner K, Mueller WE, Kudin AP, Kunz WS, Zimmermann A, Roeper J, Wenzel D, Jendrach M, Garcia-Arenciba M, Fernandez-Ruiz J, Huber L, Rohrer H, Barrera M, Reichert AS, Rub U, Chen A, Nussbaum RL, Auburger G. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One. 2009;4(6):e5777. doi: 10.1371/journal.pone.0005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Hang GB, LaDow ES, Shoenfeld LR, Atallah BV, Finkbeiner S, Kreitzer AC. Rapid target-specific remodeling of fast-spiking inhibitory circuits after loss of dopamine. Neuron. 2011;71:858–68. doi: 10.1016/j.neuron.2011.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T, Irizarry MC, Mariash A, Cheung B, Soto O, Schrump S, Sondel J, Kotilinek L, Day J, Schwarzschild MA, Cha JHJ, Newell K, Miller DW, Ueda K, Young AB, Hyman BT, Ashe KH. Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2003;24:245–58. doi: 10.1016/s0197-4580(02)00091-x. [DOI] [PubMed] [Google Scholar]
- Graham BH, Waymire KG, Cotrell B, Trounce IA, MacGregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet. 1997;16:226–34. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–31. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu CS, Ng AR, Rosales PL, Shimizu N, Toda T, Mizuno Y, Hattori N. Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol. 2004;56:424–7. doi: 10.1002/ana.20251. [DOI] [PubMed] [Google Scholar]
- Hartmann A, Michel PP, Troadec JD, Mouatt-Prignet A, Faucheux BA, Ruberg M, Agid Y, Hirsch EC. Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease? J Neurochem. 2001;76:1785–93. doi: 10.1046/j.1471-4159.2001.00160.x. [DOI] [PubMed] [Google Scholar]
- Helmich RC, Hallett M, Deuschl G, Toni I, Bloem BR. Cerebral causes and consequences of parkinsonian resting tremor: a tale of two circuits. Brain. 2012;135:3206–26. doi: 10.1093/brain/aws023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath MM, Wang X, Resnick MA, Bell DA. Divergent evolution of human p53 binding sites: cell cycle versus apoptosis. PLoS ONE. 2007;3(7):e127. doi: 10.1371/journal.pgen.0030127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–10. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Nakayama K, Jin C, Suzuki Y, Yazawa I. α-Synuclein accumulation reduces GABAergic inhibitory transmission in a model of multiple system atrophy. Biochem Biophys Res Comm. 2012;428:348–53. doi: 10.1016/j.bbrc.2012.10.057. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79:368–76. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Is there apoptosis in Lewy body disease? Acta Neuropathol. 1999;97:413–415. doi: 10.1007/s004010051006. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Cell death mechanisms in Parkinson’s disease. J Neural Transm. 2000;107:1–29. doi: 10.1007/s007020050001. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov Disord. 2012;27:8–30. doi: 10.1002/mds.23795. [DOI] [PubMed] [Google Scholar]
- Johnson N, Khan A, Virji S, Ward JM, Crompton M. Import and processing of heart mitochondrial cyclophilin D. Eur J Biochem. 1999;263:353–9. doi: 10.1046/j.1432-1327.1999.00490.x. [DOI] [PubMed] [Google Scholar]
- Kimura J. Disorder of interneurons in parkinsonism. The orbicularis oculi reflex to paired stimuli. Brain. 1973;96:87–96. doi: 10.1093/brain/96.1.87. [DOI] [PubMed] [Google Scholar]
- Kita H, Kossak T, Heizmann CW. Parvalbumin-immunoreactive neurons in the rat neostriatum: a light and electron microscopic study. Brain Res. 1990;536:1–15. doi: 10.1016/0006-8993(90)90002-s. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Klein C, Schlossmacher MG. Parkinson disease, 10 years after its genetic revolution. Neurology. 2007;69:2093–104. doi: 10.1212/01.wnl.0000271880.27321.a7. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degeneration. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler J, Hattori K, Sawada M, DeVries AC, Martin LJ, Hurn PD, Traystman RJ. Histopathological and behavioral characterization of a novel model of cardiac arrest and cardiopulmonary resuscitation in mice. J Neurosci Meth. 2004;136:33–44. doi: 10.1016/j.jneumeth.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Langston JM, Ballard P. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–80. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA. 2002;99:8968–73. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Browstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451–2. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- Lesuisse C, Martin LJ. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol. 2001;51:9–23. doi: 10.1002/neu.10037. [DOI] [PubMed] [Google Scholar]
- Leung AWC, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777:946–52. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, Shim H, Gu XL, Luo J, Long CX, Ding J, Mateo Y, Sullivan PH, Wu LG, Goldstein DS, Lovinger D, Cai H. Conditional expression of Parkinson’s disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci. 2012;32:9248–64. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhang C, Yin J, Li X, Cheng F, Li Y, Yang H, Ueda K, Chan P, Yu S. α-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett. 2009;454:187–92. doi: 10.1016/j.neulet.2009.02.056. [DOI] [PubMed] [Google Scholar]
- Loerch PM, Lu T, Dakin KA, Vann JM, Issacs A, Geula C, Wang J, Pan Y, Gabuzda DH, Li C, Prolla TA, Yankner BA. Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE. 2008;3(10):e3329. doi: 10.1371/journal.pone.0003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J, Lennox G, Leigh PN. Disorders of movement and system degeneration. In: Graham DI, Lantos PL, editors. Greenfield’s Neuropathology. Arnold; London: 1997. pp. 281–366. [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminals. J Neurosci. 1988;8:2804–15. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58:459–71. doi: 10.1097/00005072-199905000-00005. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Neuronal cell death in nervous system development, disease, and injury. Int J Mol Med. 2001;7:455–78. [PubMed] [Google Scholar]
- Martin LJ. Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2006;65:1103–10. doi: 10.1097/01.jnen.0000248541.05552.c4. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Mitochondrial and cell death mechanisms in neurodegenerative disease. Pharmaceuticals. 2010;3:839–915. doi: 10.3390/ph3040839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ. An approach to experimental synaptic pathology using green fluorescent protein-transgenic mice and gene knockout mice to show mitochondrial permeability transition pore-driven excitotoxicity in interneurons and motoneurons. Toxicol Pathol. 2011;39:220–33. doi: 10.1177/0192623310389475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Chang Q. Inhibitory synaptic regulation of motoneurons: a new target of disease mechanisms in amyotrophic lateral sclerosis. Mol Neurobiol. 2012;45:30–42. doi: 10.1007/s12035-011-8217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z. Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and bax-dependent. J Neurobiol. 2002;5:181–97. doi: 10.1002/neu.10026. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Liu Z. Opportunities for neuroprotection in ALS using cell death mechanism rationales. Drug Discovery Today: Disease Models. 2004;1:135–43. [Google Scholar]
- Martin LJ, Chen K, Liu Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci. 2005;25:6449–59. doi: 10.1523/JNEUROSCI.0911-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Adams NA, Pan Y, Price A, Wong M. The mitochondrial permeability transition pore regulates nitric oxide-mediated apoptosis of neurons induced by target deprivation. J Neurosci. 2011;31:359–70. doi: 10.1523/JNEUROSCI.2225-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46:281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp Neurol. 2009;218:333–46. doi: 10.1016/j.expneurol.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Price AC, McClendon KB, Al-Abdulla NA, Subramaniam JR, Wong PC, Liu Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J Neurochem. 2003;85:234–47. doi: 10.1046/j.1471-4159.2003.01659.x. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, Golden WC. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007;500:20–46. doi: 10.1002/cne.21160. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–9. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowen E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA. Lack of nigral pathology in transgenic mice expressing alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–9. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VMY. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–20. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulated some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng Y, Finkbeiner S, Nussbaum PL, Masliah E, Edwards RH. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J Biol Chem. 2011;286:20710–26. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6,-tetrahydropyridine. Life Sci. 1985;36:2503–8. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- Odom DT, Dowell RD, Jacobsen ES, Gordon W, Danford TW, MacIssac KD, Rolfe PA, Conboy CM, Gifford DK, Fraenkel E. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet. 2007;39:730–2. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson’s disease. Ann Rev Neurosci. 1999;22:123–44. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Whitney Evans E, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Martínez-Gil A, Khan N, Johnson J, Ruiz Martinez J, Nicholl D, Marti Carrera I, Saénz Peňa A, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Panov A, Dikalov S, Shalbuyeva N, Hemendinger R, Greenamyre JT, Rosenfeld J. Species- and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am J Physiol Cell Physiol. 2007;292:C708–18. doi: 10.1152/ajpcell.00202.2006. [DOI] [PubMed] [Google Scholar]
- Pilsl A, Winklhofer KF. Parkin, PINK1 and mitochondrial integrity: emerging concepts of mitochondrial dysfunction in Parkinson’s disease. Acta Neuropathol. 2012;123:173–88. doi: 10.1007/s00401-011-0902-3. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau CP, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal death in adult brain are morphologically distinct: further evidence for an apoptosis necrosis continuum. J Comp Neurol. 1997;378:88–104. [PubMed] [Google Scholar]
- Ramsay RR, Salach JI, Dadgar J, Singer TP. Inhibition of mitochondrial NADH dehydrogenase by pyridine derivatives and its possible relation to experimental and idiopathic parkinsonism. Biochem Biophys Res Comm. 1986;135:269–75. doi: 10.1016/0006-291x(86)90972-1. [DOI] [PubMed] [Google Scholar]
- Rascol O, Fabre SN, Brefel C, Loubinoux I, Celsis P, Senard JM, Montastruc JL, Chollet F. The ipsilateral cerebellar hemisphere is overactive during hand movements in akinetic parkinsonian patients. Brain. 1997;120:103–10. doi: 10.1093/brain/120.1.103. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Reddy TP. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Cur Alzheimer Res. 2011;8:393–409. doi: 10.2174/156720511795745401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuetzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol. 2002;175:35–48. doi: 10.1006/exnr.2002.7882. [DOI] [PubMed] [Google Scholar]
- Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr. 2005;37:129–42. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- Rudow G, O’Brien R, Savonenko AV, Resnick SM, Zonderman AB, Pletnikova O, Marsh C, Dawson TM, Crain BT, West MT, Troncoso JC. Morphometry of the human substantia nigra in ageing and Parkinson’s disease. Acta Neuropathol. 2008;115:461–70. doi: 10.1007/s00401-008-0352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin P, López IP, Kachidian P, Barroso-Chinea P, Rico AJ, Gómez-Bautista V, Coulon P, Kerkerian-Le Goff L, Lanciego JL. Changes to interneuron-driven striatal microcircuits in a rat model of Parkinson’s disease. Neurobiol Dis. 2009;34:545–52. doi: 10.1016/j.nbd.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Savica R, Rocca WA, Ahlskog JE. When does Parkinson disease start? Arch Neurol. 2010;67:798–801. doi: 10.1001/archneurol.2010.135. [DOI] [PubMed] [Google Scholar]
- Schapira AHV. Etiology of Parkinson’s disease. Neurology. 2006a;66 (Suppl 4):S10–S23. doi: 10.1212/wnl.66.10_suppl_4.s10. [DOI] [PubMed] [Google Scholar]
- Schapira AHV. Mitochondrial disease. Lancet. 2006b;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- Schapira AHV. Mitochondrial diseases. Lancet. 2012;379:1825–34. doi: 10.1016/S0140-6736(11)61305-6. [DOI] [PubMed] [Google Scholar]
- Schapira AHV, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA. 1997;94:4103–8. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S, Swada H, Kitamura Y, Taniguchi T. Disease model: Parkinson’s disease. Trends Mol Med. 2003;9:360–5. doi: 10.1016/s1471-4914(03)00117-5. [DOI] [PubMed] [Google Scholar]
- Sillitoe RV, Chung SH, Fritschy JM, Hoy M, Hawkes R. Golgi cell dendrites are restricted by Purkinje cell stripe boundaries in the adult mouse cerebellar cortex. J Neurosci. 2008;28:2820–6. doi: 10.1523/JNEUROSCI.4145-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simat M, Ambrosetti L, Lardi-Studler B, Fritschy JM. GABAergic synaptogenesis marks the onset of differentiation of basket and stellate cells in mouse cerebellum. Eur J Neurosci. 2007;26:2239–56. doi: 10.1111/j.1460-9568.2007.05846.x. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Souza JM, Giasson BI, Chen Q, Lee VMY, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable α-synuclein polymers. J Biol Chem. 2000;265:18344–9. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, Comyns K, Batber Richards M, Meng C, Priestly B, Fernandez HH, Cambi F, Umbach DM, Blair A, Sandler DP, Langston W. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119(6):866–72. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton NA, MaClean-Fraser A, Tatton WG, Perl DP, Olnanow CW. A fluorescent double-labeling method to detect and confirm apoptotic nuclei in Parkinson’s disease. Ann Neurol. 1998;44:S142–S148. doi: 10.1002/ana.410440721. [DOI] [PubMed] [Google Scholar]
- Thiruchelvam MJ, Powers JM, Cory-Slechta DA, Richfield EK. Risk factors for dopaminergic neuron loss in human α-synuclein transgenic mice. Eur J Neurosci. 2004;19:845–54. doi: 10.1111/j.0953-816x.2004.03139.x. [DOI] [PubMed] [Google Scholar]
- Tompkins MM, Basgall EJ, Zamrini E, Hill WD. Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigra neurons. Am J Pathol. 1997;150:119–131. [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert A, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, Nelson LM. Incidence of Parkinson’s disease: variations by age, gender and race/ethnicity. Am J Epidemiol. 2003;157:1015–22. doi: 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WPJM, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G. Neuropathology in mice expressing human α-synuclein. J Neurosci. 2000;20:6021–9. doi: 10.1523/JNEUROSCI.20-16-06021.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gurp M, Festjens N, van Loo G, Saeleus X, Vandenabeele P. Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Comm. 2003;304:487–97. doi: 10.1016/s0006-291x(03)00621-1. [DOI] [PubMed] [Google Scholar]
- Vilkki J, Savontaus ML, Kalimo H, Nikoskelainen EK. Mitochondrial DNA polymorphism in Finish families with Leber’s hereditary optic neuroretinopathy. Hum Genet. 1989;82:208–12. doi: 10.1007/BF00291155. [DOI] [PubMed] [Google Scholar]
- Voogd J, Gerrittis NM, Marani E. Cerebellum. In: Paxinos G, editor. The Rat Nervous System. Vol. 2. Academic Press; Australia: 1985. pp. 251–91. [Google Scholar]
- Wakamatsu M, Ishii A, Iwata S, Sakagami J, Ukai Y, Ono M, Kanbe D, Muramatsu SI, Kabayashi K, Iwatsubo T, Yoshimoto M. Selective loss of nigral dopamine neurons induced by overexpression of truncated human α-synuclein in mice. Neurobiol Aging. 2008;29:574–85. doi: 10.1016/j.neurobiolaging.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem. 2003;10:1485–506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- Weissman TA, Sanes JR, Lichtman JW, Livet J. Generation and imaging of brainbow mice. Cold Spring Harb Protoc. 2011;7:851–6. doi: 10.1101/pdb.prot5632. [DOI] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial dynamics in model organisms: what yeasts, worms and flies have taught us about fusion and fission of mitochondria. Semin Cell Dev Biol. 2010;21:542–9. doi: 10.1016/j.semcdb.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Woodfield K, Rück A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336:287–90. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Hallett M. The cerebellum in Parkinson’s disease. Brain. 2013;136:696–709. doi: 10.1093/brain/aws360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullner U, Kornhuber J, Weller M, Schulz JB, Loschmann PA, Riederer P, Klockgether T. Cell death and apoptosis regulating proteins in Parkinson’s disease- a cautionary note. Acta Neuropathol. 1999;97:408–12. doi: 10.1007/s004010051005. [DOI] [PubMed] [Google Scholar]
- Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VMY. Mouse model of multiple system atrophy α-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45:847–59. doi: 10.1016/j.neuron.2005.01.032. [DOI] [PubMed] [Google Scholar]
- Yu H, Sternad D, Corcos DM, Vaillancourt DE. Role of hyperactive cerebellum and motor cortex in Parkinson’s disease. NeuroImage. 2007;35:222–33. doi: 10.1016/j.neuroimage.2006.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Studler B, Arabadzisz D, Schweizer C, Ahmadi S, Layh B, Bosl MR, Fritschy J-M. Glycinergic neurons expressing enhanced green fluorescent protein in bacterial artificial chromosome transgenic mice. J Comp Neurol. 2005;482:123–41. doi: 10.1002/cne.20349. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Duan C, Li L, Gao LL, Zhao C, Yu S, Ueda K, Chan P, Yang H. α-Synuclein overexpression impairs mitochondrial function by associating with adenylate translocator. Int J Biochem Cell Biol. 2011;43:732–41. doi: 10.1016/j.biocel.2011.01.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Thy1-A53T hαSyn C57BL/6 tg mouse at early symptomatic stage of disease with postural instability, slight tremor, and jerking movements.
Thy1-A53T hαSyn C57BL/6 tg mouse at near endstage disease with sever rigidity and postural instability.