

Abstract
Many secretory proteins are targeted by signal sequences to a protein-conducting channel, formed by prokaryotic SecY- or eukaryotic Sec61-complexes, and are translocated across the membrane during their synthesis1,2. Crystal structures of the inactive channel show that the SecY subunit of the heterotrimeric complex consists of two halves that form an hourglass-shaped pore with a constriction in the middle of the membrane and a lateral gate that faces the lipid phase3-5. The closed channel has an empty cytoplasmic funnel and an extracellular funnel that is filled with a small helical domain, called the plug. During initiation of translocation, a ribosome–nascent chain complex binds to the SecY/Sec61 complex, resulting in insertion of the nascent chain. However, the mechanism of channel opening during translocation is unclear. Here, we have addressed this question by determining structures of inactive and active ribosome–channel complexes with cryo-electron microscopy. Non-translating ribosome–SecY channel complexes derived from Methanococcus jannaschii or Escherichia coli show the channel in its closed state, and indicate that ribosome binding per se causes only minor changes. The structure of an active E. coli ribosome–channel complex demonstrates that the nascent chain opens the channel, causing mostly rigid body movements of the N- and C-terminal halves of SecY. In this early translocation intermediate, the polypeptide inserts as a loop into the SecY channel with the hydrophobic signal sequence intercalated into the open lateral gate. The nascent chain also forms a loop on the cytoplasmic surface of SecY rather than directly entering the channel.
Opening of the SecY channel during initiation of translocation involves two events: binding of the ribosome and insertion of the nascent chain. To analyze how ribosome binding per se affects the structure of a translocation channel, we first determined the structure of complexes lacking a nascent chain. Initial experiments were performed with complexes from M. jannaschii, because this allows a direct comparison with a crystal structure of SecY3. Purified M. jannaschii ribosomes were incubated with an excess of SecY complex, and complexes were imaged by cryo-electron microscopy (cryo-EM). A total of ~37,000 particles were analyzed, resulting in an electron density map with a resolution of 9.0 Å for the ribosome and ~12.7 Å for the channel (Supplementary Table 1).
A ribosome model from Pyrococcus furiosus6, a species related to M. jannaschii, was fit into the density map, allowing the identification of essentially all RNA helices and many helical features of ribosomal proteins (Fig. 1a and Supplementary Fig. 1). A crystal structure of the M. jannaschii SecY complex could be docked into density for the SecY channel (Fig. 1b; Supplementary Fig. 2), and Molecular Dynamics Flexible Fitting (MDFF)7 resulted in only small changes (Fig. 1c). All trans-membrane segments (TMs), including the 10 TMs of SecY, and the single TMs of the SecE and Secβ subunits, could be accounted for in the map. Several TM helices and the extracellular loop between TMs 5 and 6 were partially resolved (Supplementary Fig. 3). A comparison with the crystal structure shows that, with the exception of some adjustments in the cytoplasmic helix of SecE, membrane-embedded domains remained essentially unaltered (Fig. 1c). As previously observed with other species8-11, loops between TMs 6 and 7 (6/7 loop) and TMs 8 and 9 (8/9 loop) of SecY, as well as the cytoplasmic helix of SecE (Fig. 1b), all interact with components of the large ribosomal subunit at the tunnel exit (Supplementary Figs. 4a–c). These interactions clearly do not induce major structural changes in the SecY channel and leave the lateral gate closed.
Figure 1. Structures of non-translating ribosome–channel complexes.
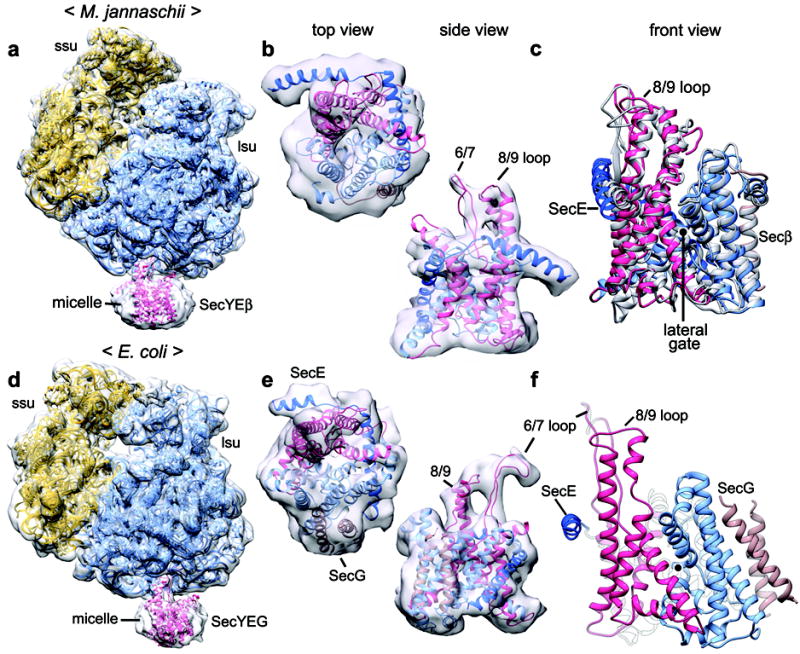
a, Density map for the M. jannaschii complex. Models for ribosomal RNA and proteins of the small and large ribosomal subunits (ssu and lsu; in gold and blue, respectively) and of the SecY complex (in red) were docked into the map. b, Fit of the M. jannaschii SecY complex into the segmented density map, as viewed from the cytoplasm (top view) and from the side. The N- and C-terminal halves of SecY are in light blue and red, respectively. SecE is in dark blue and Secβ in brown. c, Comparison between the crystal structure of an M. jannaschii SecY complex (grey) and the EM structure (in color), as viewed facing the lateral gate (front view). d-e, As in a and b, but for the E. coli complex. SecG, the bacterial equivalent of Secβ, is in brown. f, A model for the E. coli channel in a front view.
Next we determined the structure of a non-translating ribosome–channel complex from E. coli, with a larger dataset than used previously8. A total of ~39,000 particles were analyzed, resulting in a density map with a resolution of ~9.5 Å for the ribosome and ~14 Å for the channel (Supplementary Table 1). Models for ribosomal subunits11,12 were docked into the density map (Fig. 1d) and all RNA helices were visible, as well as some partially resolved helices of ribosomal proteins (Supplementary Fig. 5). Because there is no crystal structure of the E. coli SecY complex, we generated a homology model based on crystal structures of Thermus thermophilus and Thermotoga maritima complexes4,13 (Supplementary Figs. 6 and 7). This model was subjected to MDFF using the entire density map of the ribosomal large subunit and channel as a restraint. This resulted in movements of cytoplasmic loops, while membrane-embedded domains remained essentially unchanged (Supplementary Fig. 8). Many features of the channel are clearly visible in a segmented map (Figs. 1e and Supplementary Fig. 9, 10), including cytoplasmic loops of SecY, two helices of SecE, two TMs of SecG (the bacterial equivalent of archaeal Secβ), and some partially resolved TMs of SecY. Connections between the channel and ribosome were similar to those in the M. jannaschii complex, with the exception of the longer 6/7 loop of SecY, which is repositioned between RNA helices 6 and 7 (Supplementary Figs. 4d–f). Importantly, the ribosome alone does not induce major changes in the channel structure, so the lateral gate remains closed (Fig. 1f).
To determine the structure of an active E. coli ribosome–channel complex, we used a new strategy. Previous attempts to obtain a structure of an active translocation channel showed that a translating ribosome was bound to the channel, but there was little biochemical evidence that a nascent chain was inserted in the channel and no clear electron density was visible for the polypeptide10,11. These studies employed small amounts of ribosome-nascent chain complexes (RNCs) that were formed in vitro and subsequently added to purified channel. To obtain a more physiological sample, we generated an early translocation intermediate of a secretory protein in living E. coli cells by expressing a polypeptide with 100 amino acids from an inducible promoter14,15. The polypeptide has an N-terminal signal sequence derived from DsbA, which targets the protein to the co-translational translocation pathway16, and a C-terminal SecM-stalling sequence, which arrests translation of the ribosome17 (Fig. 2a). We also expressed the endoribonuclease MazF from an inducible promoter to cleave mRNAs between ribosomes, which results in the depletion of nascent chains associated with non-stalled ribosomes18. To generate a stable complex between the SecM-stalled RNC and the channel, we used disulfide crosslinking. The nascent chain contained a cysteine at position 19 of the signal sequence, which can be crosslinked to a cysteine at position 68 in the SecY plug14. Disulfide bond formation was achieved by adding an oxidant to the E. coli culture, resulting in 70% of nascent chains being linked to SecY.
Figure 2. Purification of a ribosome–nascent chain–channel complex.
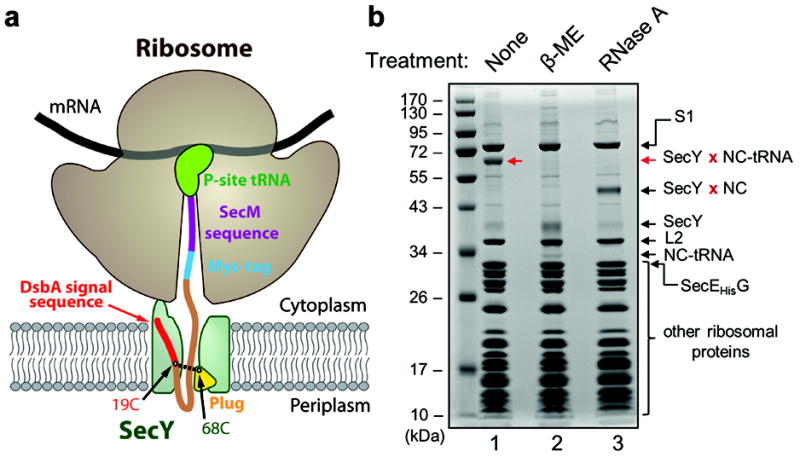
a, The complex was generated in living E. coli cells by expressing a nascent chain (NC) of 100 amino acids with a signal sequence and SecM-stalling sequence. The NC also contains a Myc-tag. A cysteine at position 19 of the NC (19C) was disulfide-crosslinked to a cysteine in the plug of SecY (68C). b, Coomassie-stained SDS-gel of the ribosome-NC (RNC)–channel complex (lane 1). The red arrow indicates the crosslinked product of SecY and the NC-tRNA adduct. This band disappears after treatment with β-mercaptoethanol (β-ME) or RNaseA (lanes 2 and 3). Ribosomal proteins (including S1) and the fusion between SecE and SecG are indicated.
To purify the RNC–channel complex, we replaced the endogenous ribosomal protein L12 with a Strep-tagged version, allowing the enrichment of ribosomes on a Strep-Tactin column. This purification step was performed at high salt concentration to remove SecY complexes lacking a nascent chain (Supplementary Fig. 11a). A second purification step exploited a His-tag attached to a fusion between SecE and SecG, and allowed the enrichment of channel-containing complexes by Co2+-affinity chromatography. Finally, the sample was subjected to gel filtration. The purified RNC–channel complex eluted as a homogeneous peak at the position of monosomes (Supplementary Fig. 11b). On a Coomassie-stained SDS gel, the SecY–nascent chain–tRNA species was the only major band besides those from ribosomal proteins (Fig. 2b, lane 1). As expected, the band disappeared when the sample was treated with a reducing agent to remove the disulfide bridge or with RNase A to degrade the tRNA (Fig. 2b, lanes 2 and 3). We found that the previous protocol of adding purified RNCs to SecY complex, either in detergent or in nanodiscs10,11, resulted in inefficient insertion of the nascent chain into the channel (Supplementary Fig. 12). Also, when RNC–channel complexes were generated in vivo and crosslinked after purification, crosslinks between different nascent chain molecules and between the nascent chain and unidentified proteins were observed (Supplementary Fig. 13). Hence, crosslinking in vivo is required to maintain the nascent chain in the channel.
Purified RNC–channel complexes were frozen over holes on EM grids, as the channel was lost when complexes were placed on a carbon film. A total of ~167,000 individual particles were used, of which ~50% contained the channel. Additional sorting for the best signal-to-noise ratio identified ~53,000 particles for structure determination and resulted in a density map at ~10 Å resolution for the ribosome and ~11 Å for the channel (Fig. 3a; Supplementary Table 1). Ribosomal RNAs and proteins were clearly visible in the density map (Supplementary Fig. 14), including A- and P-site tRNAs, as expected for a SecM-stalled ribosome19 (Supplementary Fig. 15a). Moreover, there was density for mRNA underneath the anticodon regions of tRNAs (Supplementary Fig. 15b). We also observed density for ribosomal protein S1, which was more extensive than seen before20 (Fig. 3a and Supplementary Figs. 15c-e).
Figure 3. Structure of the active SecY channel.
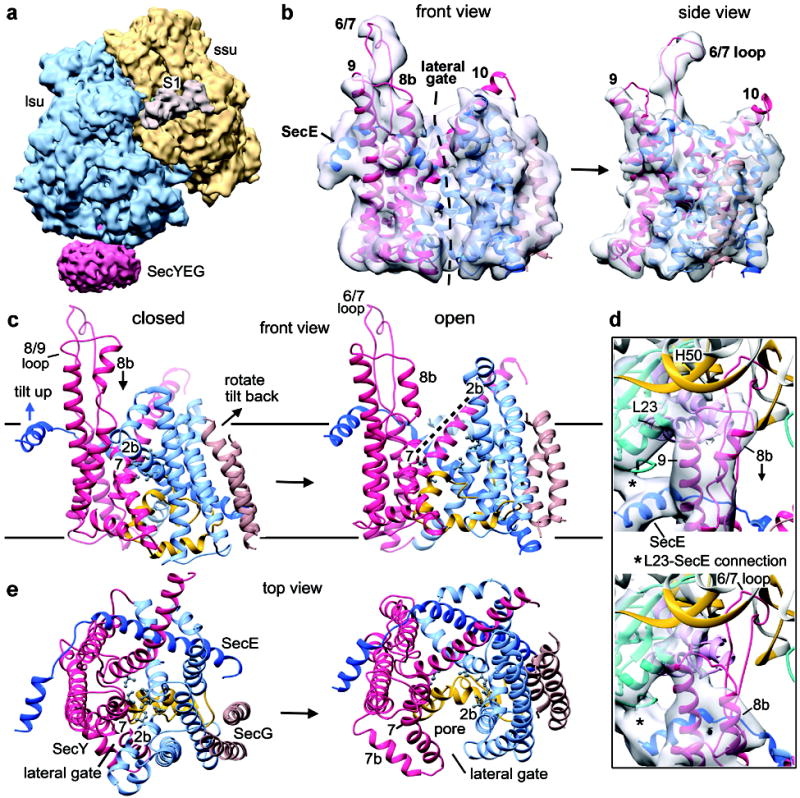
a, Structure of the E. coli RNC–SecY channel complex, with large and small ribosomal subunits in blue and gold, respectively, the SecY complex in red, and ribosomal protein S1 in tan. b, Front and side views of the channel fit into the segmented density map (grey). The nascent chain was omitted for clarity. The N-terminal half of SecY is in light blue, the C-terminal half in red, SecE in dark blue, and SecG in brown. c, Comparison of front views of the closed and open E. coli SecY channels with the approximate position of the membrane indicated by solid horizontal lines. The N-terminal half of SecY is in light blue, the C-terminal half in red, SecE in dark blue, SecG in brown, and the plug in yellow. Some movements during channel opening are indicated, such as the rotation and tilting of the N-terminal half of SecY, the tilting of SecE, and the movement of helix 8b. Labels for helices 2b and 7 are placed at the same position in the closed and open channel. Pore residues forming the constriction in the closed channel are indicated with grey balls and sticks. d, Connections of the ribosome with the 8/9 loop of SecY and the cytoplasmic helix of SecE in the closed and open channels (upper and lower panels, respectively). Note the large movement of helix 8b towards the membrane. e, As in c, but viewed from the top.
To generate a model for the active channel, we created an E. coli homology model based on a crystal structure of the SecY complex from P. furiosus5, which has the most open lateral gate among known crystal structures (Supplementary Fig. 16), and used MDFF to adjust the model to the experimental density map. The 6/7 loop and TM9 of SecY were well resolved (Fig. 3b), and ribosomal components interacting with the channel were the same as with the non-translating complex. The cytoplasmic helix of SecE and TM10 of SecY were clearly visible, and there was good density for SecG (Supplementary Figs. 17 and 18). In addition, many TMs were partially resolved, with only occasional density breaks in the helices. Density for the nascent chain was clearly identifiable without segmentation of the density map. Specifically, additional density for a helix was visible in the cytoplasmic part of the lateral gate (see below), explaining why a channel with a fully open lateral gate could be fitted into the density map. In fact, the lateral gate is more open than in the P. furiosus crystal structure5 (Supplementary Table 2). Calculated cross correlation coefficients showed that the model for the open SecY channel is a significantly better fit in the density map than the model for the closed channel (Supplementary Table 1).
The modeled conformational change of the E. coli channel is supported by the fact that the conversion from a closed to an open channel involves mostly rigid body movements of the N- and C-terminal halves of SecY (Supplementary Fig. 19). To open the lateral gate, the N-terminal half of SecY undergoes a significant rotation and tilt, while the C-terminal half moves less in the opposite direction (Fig. 3c; see also Supplementary Video 1). SecE undergoes a tilting motion to accommodate movements of SecY, and SecG moves with the N-terminal half of SecY. These conformational changes would maintain the hydrophobic belt of the SecY complex within the lipid environment. In addition to rigid body movements, there are changes in the 5/6 loop which connects the two halves of SecY to accommodate the large opening motion. There are also movements in TM8 and the lower part of TM7. One particularly large change occurs in the upper part of TM8 (helix 8b), which undergoes a large displacement towards the membrane surface (Fig. 3d). The 6/7 loop and TM9, as well as preceding loop residues, including a conserved arginine (Arg357), do not move appreciably (Fig. 3d), consistent with their role in tethering the channel to the ribosome. The plug domain moves only a small distance, probably because it is restrained by the disulfide bridge to the signal sequence. However, the plug does not have to move much to allow translocation21. When viewed from the cytoplasmic side, these conformational changes open a pore adjacent to the lateral gate (Fig. 3e; see Supplementary Video 2). Overall, the changes are more pronounced than seen previously10,11.
Density for the nascent chain was seen inside the ribosomal tunnel, on the cytoplasmic surface of the SecY complex, inside the channel, and on its periplasmic side (Fig. 4a; Supplementary Fig. 20). Based upon biochemical data14, an approximate model for the nascent chain in the RNC–channel complex was built into the density. The last ~40 amino acids are located inside the ribosome, as cysteines introduced into this segment are inaccessible to a bulky modification reagent. In addition, cysteines at positions 19 to 34 are most favored to form a disulfide bridge with a cysteine in the plug. Finally, the position of the end of the signal sequence in our structure is constrained by the disulfide crosslink between position 19 of the nascent chain and position 68 of the plug.
Figure 4. Path of the nascent chain.
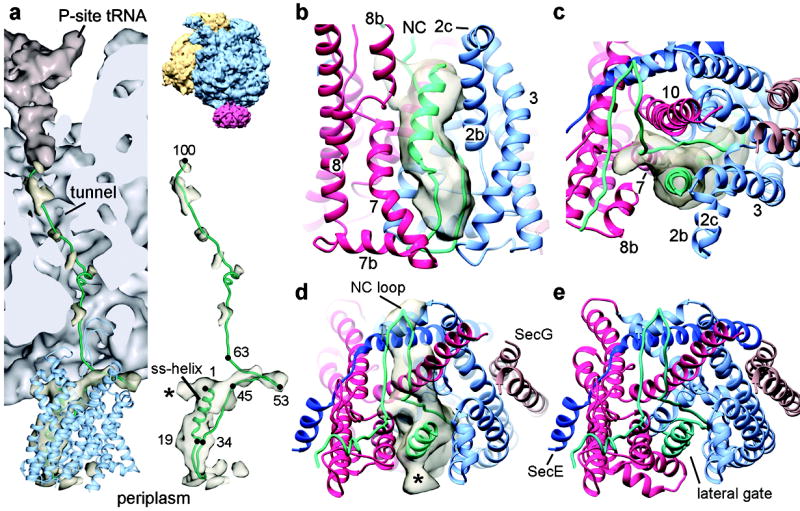
a, Density (in light gold) and model (green line) for the nascent chain (NC) in the RNC-channel complex. The P-site tRNA is in brown, the ribosome in grey, and the channel in blue. The right upper panel shows the entire RNC-channel complex from the same viewing angle. The right lower panel shows the density and model for the NC, with ribosome and channel omitted. The asterisk indicates density for an alternative orientation of the NC loop on the cytoplasmic side of the channel (see also d). b, Side view of the signal sequence (ss) helix in the lateral gate. Density for the NC on the cytoplasmic surface was removed for clarity. c, As in b, but viewed from the top along the axis of the signal sequence helix. d, As in c, but from a slightly different angle of view with NC density on the cytoplasmic surface included. e, As in d, but without the density map.
The resulting model shows that the hydrophobic core of the signal sequence forms a helix in the lateral gate (residues 1 to 15) (Figs. 4b–d and Supplementary Fig. 21), consistent with crosslinking data obtained with the yeast Sec61 complex22. The signal sequence helix is contacted by TM2b, helix 8b, and TM7 of SecY (Fig. 4b). In a lipid bilayer, much of the signal sequence, including parts that follow the hydrophobic region, would be exposed to the hydrocarbon chains of phospholipids, again in agreement with crosslinking experiments22. Additional density below and adjacent to the signal sequence helix can account for the other side of the nascent chain loop. The pore through which the mature region of the nascent chain would move into the extracellular funnel is not exactly in the center of the channel, but the translocating polypeptide may still be surrounded by pore ring residues that form a constriction in the closed channel (Supplementary Video 2). It is possible that crosslinking to the nascent chain may restrain the plug, keeping it in the center of the channel. However, there is still room for the nascent chain to form a loop in the pore.
We modeled density on the cytoplasmic surface of the channel as a loop that extends parallel to the surface and towards the back of the channel (residues ~45 to 63) (Figs. 4a and e). This part of the nascent chain lies in a V-shaped groove, which is framed by the base of the 6/7 loop and TM10 of SecY (Supplementary Fig. 22 and Video 3). However, the nascent chain may adopt an alternative orientation with a loop that extends above the lateral gate (marked with an asterisk in Figs. 4a and e). The nascent chain may also slide up and down the axis of the channel to some extent, as there is density on the periplasmic side that is not fully accounted for in our model.
In summary, our structures show that ribosome binding alone does not induce major changes in the SecY channel, although it may cause transient opening23. Rather, stable opening of the channel requires loop insertion of the nascent chain24. As predicted3,22, the hydrophobic part of the signal sequence forms a helix that occupies the open lateral gate. The signal sequence would thus become part of the channel wall, thereby increasing the size of the pore through which the polypeptide moves across the membrane. At later stages of translocation, the signal sequence is cleaved from the nascent chain and released from the lateral gate, which may result in a narrower pore. It is also possible that the signal sequence leaves the lateral gate before cleavage. This hypothesis would be consistent with a two-dimensional crystal structure of the SecY complex that showed a synthetic signal peptide bound to the outside of an essentially closed channel25
Our results also indicate that most nascent chains form a loop on the cytoplasmic surface of SecY, rather than adopting a fully extended conformation between the ribosome and channel. Although the observed looping of the nascent chain at the cytoplasmic surface of the channel needs to be confirmed with other substrates, it seems possible that a pulling force or ratcheting mechanism26,27 may be required to achieve efficient translocation. SecDF could use a proton gradient across the membrane together with movements of a periplasmic domain to pull on the nascent chain28. In addition, polypeptide chain folding or the binding of periplasmic chaperones may help to move the polypeptide chain across the membrane.
FULL METHODS
Construction of plasmids and E. coli strains
Plasmids used in this study are listed and described in Supplementary Table 3. PCR reactions were performed with Phusion polymerase (New England Biolabs) or KOD polymerase (Novagen). E. coli DH5α strain was used for all cloning procedures.
pBAD(MazF)-NC100, a plasmid expressing a SecM-stalled nascent chain under an arabinose-inducible (ara) promoter, and the MazF endoribonuclease under a tetracycline-inducible (tet) promoter, has been described15. Briefly, a DNA sequence coding for a 100-amino acid nascent chain was placed after the ara promoter of pBAD His/C (Invitrogen). The nascent chain contains an N-terminal signal sequence derived from E. coli DsbA, a Myc-tag, and a C-terminal translational arrest sequence from E. coli SecM. The SecM nucleotide sequence contains three ‘aca’ sites (ttc agc aca ccc gtc tgg ata tca caa gca caa ggc atc cgt gct ggc cct); MazF will cleave the mRNA at these positions and convert polysomes into monosomes. To keep MazF uninduced, a TetR repressor was expressed. The tetR gene from Tn10 was cloned and inserted immediately downstream of the β-lactamase gene of the plasmid (for bicistronic expression). A DNA sequence for a tet promoter followed by E. coli MazF was cloned and placed between tetR and the replication origin of the plasmid. pACYC EhG/Y(68C), expressing a SecE–SecG fusion protein and SecY(68C) from a constitutive promoter, was constructed as follows. DNA sequences coding for E. coli SecE (residues 2 to 127) and SecG (residues 2 to 110) were fused with a sequence coding for a His-tag linker (GGSDGHHGHHHHGHHGDSGG). The fusion construct also contains an N-terminal calmodulin-binding peptide (CBP) tag (MGSRWKKNFIAVSAANRFKKISGGG). The resulting (CBP-tag)–SecE–(His-tag)–SecG fusion construct was ligated into pACYC-SecYEG14, replacing the original SecE segment. Subsequently, the original SecG coding sequence from pACYC-SecYEG was removed by restriction enzyme digestion and re-ligation. For information on other plasmids, see Supplementary Table 3.
E. coli strains containing chromosome modifications were generated using standard λRed recombination techniques31. To construct an E. coli strain (EP71; BW25113 Δrmf ΔompT rplL-strep::aadA(StrR)) in which ribosomal protein L12 (rplL) is C-terminally tagged with a Strep-tag (WSHPQFEK), we first synthesized a ‘rplL-strep-RBS-aadA’ DNA cassette, containing the C-terminal part of the rplL gene followed by the Strep-tag, a stop codon, a ribosome binding site (RBS), the coding sequence of a streptomycin resistance gene (aadA), and a short sequence downstream of the rplL gene. This cassette was amplified by PCR and electroporated into Δrmf ΔompT cells (EP51)15 expressing λRed recombinase from the pKD46 plasmid. The resulting cells were selected on agar medium containing 25 μg/mL streptomycin. Incorporation of the cassette into the chromosome was verified by PCR and immunoblotting using Strep-tag antibodies (Novagen). To delete the chromosomal secY gene (strain EP72), EP71 cells were first transformed with pKD46 and pACYC EhG/Y(68C). After induction of λRed recombinase, the cells were electroporated with a PCR product containing a hygromycin resistance gene (hph), flanked by short sequences homologous to the chromosomal secY locus (so that the secY coding sequence is replaced by the hph coding sequence). Deletion of chromosomal secY was verified by PCR.
Preparation of SecY complex and ribosomes
All protein purification procedures were performed at 4°C unless otherwise indicated. M. jannaschii and E. coli SecY complexes and E. coli 70S ribosomes were purified as previously described3,8. M. jannaschii cells were obtained from the University of Georgia Bioexpression and Fermentation Facility. M. jannaschii 70S ribosomes were purified by multiple ultracentrifugation steps as follows. Cells were homogenized in buffer containing 50 mM HEPES-NaOH pH 7.5, 100 mM KCl, 10 mM MgCl2, and 1 mM dithiothreitol (DTT), using a French press. After removing cell debris by centrifugation (SS34 rotor; 1 h at 16,000 rpm), the cell homogenate was loaded onto a sucrose cushion (containing 50 mM HEPES pH 7.5, 1 M NH4Cl, 10 mM MgCl2, 1 mM DTT, and 30% w/v sucrose), and ribosomes were pelleted by ultracentrifugation at 45,000 rpm for 5 h (Beckman Ti50.2 rotor). The pelleted ribosomes were resuspended in buffer containing 50 mM HEPES pH 7.5, 1 M NH4Cl, 5 mM MgCl2, and 1 mM DTT, and then sedimented by ultracentrifugation (SW-28 rotor, 24,000 rpm, 12 h) through a linear sucrose gradient (10–40% w/v sucrose in the resuspension buffer). Fractions containing the 30S and 50S ribosomal subunits were collected separately and concentrated. The buffer was exchanged to 50 mM HEPES pH 7.5, 100 mM NH4Cl, 50 mM MgCl2, and 1 mM DTT using a 100-kDa cut-off AmiconUltra (GE Healthcare) device. 30S and 50S subunits were mixed at a molar ratio of 2:1. To purify 70S ribosomes from excess 30S subunits, the complexes were subjected to centrifugation (SW-28 rotor, 24,000 rpm, 12 h) through a 10–40% sucrose gradient in 50 mM HEPES pH 7.5, 100 mM NH4Cl, 50 mM MgCl2, 1 mM DTT. Fractions containing the 70S ribosomes were pooled, concentrated, and dialyzed against buffer containing 50 mM HEPES pH 7.5, 100 mM NH4Cl, 10 mM MgCl2, and 1 mM DTT. It should be noted that the resulting specimen contained an E-site tRNA at high occupancy.
Purification of disulfide-crosslinked E. coli RNC–SecY complexes
EP72 (Δrmf ΔompTrplL-strep∷aadA ΔsecY∷hph pACYC-EhG/Y(68C)) cells harboring pBAD(MazF)-NC100 were grown to logarithmic phase in a medium containing 5g/L trypton, 2.5g/L yeast extract, 10g/L casamino acids, and 5g/L NaCl. The expression of the nascent chain was induced by addition of 0.06% arabinose for 2 h at 37°C, followed by E. coli MazF induction with 100 ng/mL anhydrotetracycline for 30 min at 30°C. Disulfide crosslinking between NC100(19C) and SecY(68C) was then induced by addition of 1mM 5,5’-dithiobis-(2-nitrobenzoic acid) (DTNB) to the culture medium for 20 min. DTNB facilitates disulfide bond formation between SecY and the nascent chain as efficiently as CuPh315. The cells were pelleted, washed once with buffer containing 50 mM Tris-HCl pH 7.2, 5 mM Mg(OAc)2, 150 mM KCl, and frozen. RNC–SecY complexes were purified as follows. The cells were resuspended in buffer containing 50 mM Tris-acetate pH 7.2, 25 mM Mg(OAc)2, 0.3 M NH4Cl and homogenized with a French press. One percent dodecyl maltoside (DDM) was added to the cell lysate for 1 h to solubilize membranes. After centrifugation (SS-34 rotor, 13,000 rpm, 30 min), ribosomes containing Strep-tagged L12 were purified by applying the lysate to a Strep-Tactin Sepharose column (IBA). The column was washed with 8 column volumes (CV) of buffer containing 50 mM Tris-acetate pH 7.2, 25 mM Mg(OAc)2, 0.4 M NH4Cl, 0.03% DDM, and then with 2 CV of buffer (TMP200) containing 50 mM Tris-acetate pH 7.2, 25 mM Mg(OAc)2, 0.2 M KOAc, 0.03% DDM. Ribosomes were eluted from the column with 4 CV of the TMP200 buffer containing 4 mM desthiobiotin. To enrich for channel-bound RNCs containing His-tagged SecE-SecG fusion protein, the eluate was incubated with Dynal-Talon beads (Invitrogen) for 30 min. The beads were washed 3 times with TMP200 buffer, and bound complexes were eluted with TMP200 buffer containing 120 mM imidazole. The complexes were further purified by gel filtration on a Superose 6 column (GE Healthcare) equilibrated with buffer containing 50 mM Tris-acetate pH 7.2, 10 mM Mg(OAc)2, 80 mM KOAc, 0.03% DDM. Monomeric ribosome fractions were collected and concentrated to 8–9 mg/mL.
Test for in vitro reconstitution of the RNC-SecY complex
For the experiments shown in Supplementary Fig. 12, RNCs containing the DsbA108His or NC100 nascent chain were isolated as follows. pBAD-DsbA108His(19C) or pBAD-NC100(19C) were transformed into Δrmf ΔompT cells (EP51) harboring the pRARE2 plasmid. Cells were grown to log phase in 2xYT medium supplemented with 100 μg/mL ampicillin and 40 μg/mL chloramphenicol. Nascent chain expression was induced by addition of 0.4% arabinose for 3 h. The cells were resuspended in buffer (TMA750) containing 50 mM Tris-acetate pH 7.2, 25 mM Mg(OAc)2, 0.75 M NH4Cl, and 1.5 mM DTT and homogenized in a French press. To solubilize the membranes, 1% DDM was added to the cell extract. The extract was cleared by centrifugation at 13,000 rpm for 1 h. The ribosomes were sedimented through a sucrose cushion (TMA750, 30% sucrose, 0.03% DDM), and resuspended in TMA750. The buffer was exchanged on a PD-10 desalting column (GE Healthcare) to buffer TMP100 (50mM Tris-acetate pH 7.2, 25mM Mg(OAc)2, 0.1 M KOAc). To purify RNCs containing monosomes, the ribosomes (OD260nm=500–1000) in TMP750 were briefly incubated with 20 μg/mL RNase A at room temperature and immediately injected into a Superose 6 gel-filtration column (GE Healthcare) equilibrated with TMP100 containing 50 mM Tris-acetate pH 7.2, 25 mM Mg(OAc)2, and 100 mM KOAc. Fractions containing monomeric ribosomes were collected.
DsbA108His- or NC100-containing RNCs (0.27 μM total ribosomes) were mixed with a 15-fold excess (4.1 μM) of the SecY(68C) complex in TMP100 containing 0.03% DDM. When SecY-nanodiscs were used instead of SecY-detergent complexes, 0.138 μM of RNCs were mixed with a 5-fold (0.7 μM) excess of SecY–nanodiscs in the same buffer lacking detergent. After incubating solutions at 4°C for 1 h or at 30°C for 30 min, disulfide bridge formation was induced by addition of 0.1 mM Cu-phenanthroline (CuPh3) for 20 min at room temperature. The reaction was stopped by addition of 20 mM N-ethyl maleimide (NEM) for 30 min at 4°C. The samples were subjected to non-reducing SDS-PAGE and analyzed by immunoblotting with Myc- and SecY-antibodies.
Nanodiscs containing SecY(68C) complex were generated as previously described32 using the scaffold protein MSP1D133. Briefly, SecY(68C) complexes, MSP1D1, and deoxyBigChap-solubilized E. coli polar lipid (Avanti Polar Lipids) were mixed in a molar ratio of 1:4:100 in 50 mM Tris-acetate pH 7.2, 150 mM KOAc. After removal of the detergent with Biobeads (Bio-Rad), the sample was injected into a Superdex 200 column equilibrated with buffer TMP100. Fractions containing the SecY–nanodiscs complex were pooled and concentrated with an Amicon Ultra device (100-kDa cut-off).
SDS-PAGE and immunoblotting
SDS-PAGE was performed using 4–12% Bis-Tris gels (Bio-Rad) with either MES-SDS or MOPS-SDS running buffer (Invitrogen). Images of immunoblots were recorded with a CCD-based device (Fujifilm LAS-3000) and a standard ECL reagent. Antibodies against the C-terminus of SecY were described previously34. Anti-Myc and anti-CBP antibodies were obtained from Sigma and Genscript, respectively.
Cryo-electron microscopy and 3D image processing
M. jannaschii ribosomes were mixed with a 5-fold excess of M. jannaschii SecYEβ in 100 mM NH4Cl, 30 mM MgCl2, 20 mM HEPES-KOH pH 7.5, 6 mM β-mercaptoethanol and ~0.1% DDM. Samples were added to 400 mesh Cu grids with a holey carbon film (Quantafoil 2/1; ~2 ul/grid at an OD260 of 60-120) or diluted and added to 400 mesh grids with a thin continuous carbon film. After blotting, samples were plunge frozen into liquid ethane with a Vitrobot Mark 3 (FEI). Grids were mounted on an Oxford cold holder and imaged at 200 kV on a Tecnai F20. Data were collected manually on Kodak SO163 film at 50,000x with a defocus range of -1.0 to -2.5 um. Micrographs were scanned on Zeiss SCAI and Creoscitex EVERSMART scanners and particles selected with EMAN boxer35 were binned and scaled to 2.73 Å/pixel. In total, ~59,000 particles were CTF corrected with EMAN2 and classified with a supervised multi-reference refinement into groups, with and without channel, to give a dataset with ~37,000 particles that contained the channel. Three-dimensional reconstructions from six EMAN2 refinements carried out with different parameters and estimated resolutions of 9.2-9.5Å (based on half-data set comparisons), were aligned in Chimera and averaged to obtain a final 3D density map.
Non-programmed E. coli ribosome-channel complexes were prepared for cryo-EM and imaged at 50,000x with a Gatan (626-DH) cold holder at 200 kV, as described previously8. After identifying and removing complexes without channels, ~39,000 particles were processed with EMAN135 at a pixel size of 2.73 Å (for details see ref. 8). Aliquots of E. coli ribosome-nascent chain complexes with SecYEG (OD260 = 120–160 in ~0.06–0.1% DDM) were thawed and kept on ice. Samples were applied to 300 mesh Cu grids with a holey support film (Quantafoil 2/1 for imaging at 42,000x) and 400 mesh grids (Quantafoil 1.2/1.3 for imaging at 50,000x). The holey grids had a very thin layer of carbon freshly applied by evaporation and were airglow discharged prior to use. A Vitrobot or a manual plunger was used to plunge-freeze grids after blotting into liquid ethane, with the chamber at room temperature and a relative humidity of ~95-100%. Samples were loaded onto an Oxford cold holder and images obtained at 160 kV on a 4096 × 4096 CCD (TVIPS) with a semi-automated, single-particle collection program in EMtools (TVIPS) on a TF-20. Particle images were selected using e2boxer and further processed with EMAN229.
The CTF correction was based on all particles from each ccd frame (~450,000 from ~3500 frames), including ribosome-nascent chain-channel complexes that formed aggregates, after scaling data collected at 50,000x to 2.12 Å/pixel. Subsequently, multiple cycles of reference free classification in EMAN2 were used to extract ~167,000 single particles without close nearest neighbors for final processing. A ribosome at 25Å resolution, with and without the channel, was used as a starting model. The program e2refinemulti.py was used to separate the data set into two groups, which were refined separately to a resolution of ~11–12Å. A final supervised classification with e2refinemulti.py at an angular step size appropriate for 14 Å resolution was then carried out with the full data set, using 3D references with and without the channel filtered to 14Å. This step used the FRC comparator and provided an improved separation of the data set. At this stage ~83,000 particles with channels from the supervised classification were sorted further with e2ligandclassify.py, based on their signal-to-noise ratio, to give a final data set of ~53,000 particles. Two separate structure refinements were then done, starting with either the best 3D reference from the original low-resolution ribosome model or using a 6.8Å resolution E. coli ribosome map (EMDB id: 5036) scaled to 2.12 Å/pixel. After convergence, the four best maps (two from each structure path calculated with different refinement parameters) were then aligned in Chimera and averaged to give the final 3D map.
Molecular modeling and docking
Maps from M. jannaschii and active E. coli ribosome-channel complexes were subjected to a local normalization in EMAN2 to allow densities for ribosomal proteins, RNA, channel and micelle to be displayed and analyzed using a single density cutoff. Maps were segmented with Chimera using Zone and difference map options (vop subtract)30. Small and large ribosomal subunit models were fit into the ribosome-channel density maps using Chimera fit in map option30 and MDFF7 with runs of 500,000 steps (0.5 ns). Since no model was available for the M. jannaschii ribosome, we used a model of the related complex from P. furiosus (ref. 6, PDB ID: 3J20, 3J21 and 3J2L). Extra copies of ribosomal proteins and rRNA loops from the P. furiosus model that are absent in M. jannaschii were omitted. For E. coli ribosome-channel complexes, a nearly complete model of the large ribosomal subunit based on EM modeling and a crystal structure (ref. 11, PDB ID: 3J01; ref. 12, PDB ID: 2I2T) were used, along with a crystal structure of the small subunit (ref. 12, PDB ID: 2I2P). Models for tRNAs and mRNA were obtained from a crystal structure of a programmed T. thermophilus ribosome (ref. 36, PDB ID: 3I8G).
The global resolution in experimental density maps was determined separately for the ribosome and channel in each structure using Fourier Shell Correlation (FSC) in EMAN2, with reference maps calculated from Protein Databank files of docked models. Reference maps were calculated with pdb2mrc in EMAN at 7Å resolution and aligned in Chimera to the appropriate experimental map, then saved with vop resample onGrid. Experimental maps of ribosomes, as part of their cognate ribosome-channel complex, had a soft mask applied after calculation in EMAN2. Density maps for channels were created by segmentation in Chimera which also effectively created a mask. However, no masks were created for reference maps to prevent spurious correlations between similar masks in the FSC calculations between the two volumes being compared. The 0.5 criterion was used in all cases to identify the resolution.
Models for closed and open E. coli SecYEG channels were constructed as follows. SecY in the closed channel was based on individual structural elements (helices and turns) from the crystal structure of T. thermophilus SecY4. These segments were docked onto the closed crystal structure of SecY from M. jannaschii3 in Chimera, based on sequence alignments between the three organisms. Loops were then regularized and additional residues added as needed in Coot37. SecE and SecG subunits were taken from the crystal structure of T. maritima SecYEG13. The structural model was then mutated to E. coli sequences, energy minimized with NAMD38 and fit into the map with Chimera30 and MDFF7. A model for the open E. coli channel was constructed in a similar way, based on a crystal structure of a partially open SecYE channel from P. furiosus5. SecY models were positioned initially in the maps by docking the 6/7 and 8/9 loops into their density with Rosetta39. All MDFF runs with these components were done with segmented maps that contained the large ribosomal subunit and complete density for the channel and micelle. Models for the large subunit and channel were minimized together. Importantly, the partially open channel model moved into correct density, to reveal the signal sequence helix and associated density for the nascent chain. Finally, no density was observed for the first two TMs of E. coli SecE, which are connected by an extended linker to the surface helix and C-terminal helix and thus, may be flexible.
Supplementary Material
Acknowledgments
We thank K. Matlack and T. Guettler for reading the manuscript. This work was supported by the National Institute of Health grants GM067887 to J.C.G., GM080139 to S.J.L., GM052586 to T.A.R., and GM45377 to C.W.A. T.A.R. is a Howard Hughes Institute investigator.
Footnotes
Author contributions
E.P. designed and purified RNC–channel complexes, J.-F.M. and C.W.A. obtained and analyzed the EM data, J.C.G. helped with MDFF and channel modeling, S.J.L. helped with data analysis, W.L. and A.W. purified M.j. components, and T.A.R., E.P., and C.W.A. wrote the paper.
Author Information
Electron density maps and PDB files have been submitted to EM (EMD-5691, 5692 and 5693) and RCSB databases (3J43, 3J44, 3J45, 3J46 and 1VVK).
References
- 1.Park E, Rapoport TA. Mechanisms of Sec61/SecY-Mediated Protein Translocation Across Membranes. Annu Rev Biophys. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- 2.Shao S, Hegde RS. Membrane protein insertion at the endoplasmic reticulum. Annual review of cell and developmental biology. 2011;27:25–56. doi: 10.1146/annurev-cellbio-092910-154125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van den Berg B, et al. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 4.Tsukazaki T, et al. Conformational transition of Sec machinery inferred from bacterial SecYE structures. Nature. 2008;455:988–991. doi: 10.1038/nature07421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egea PF, Stroud RM. Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17182–17187. doi: 10.1073/pnas.1012556107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armache JP, et al. Promiscuous behaviour of archaeal ribosomal proteins: implications for eukaryotic ribosome evolution. Nucleic acids research. 2013;41:1284–1293. doi: 10.1093/nar/gks1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menetret JF, et al. Ribosome binding of a single copy of the SecY complex: implications for protein translocation. Molecular cell. 2007;28:1083–1092. doi: 10.1016/j.molcel.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 9.Menetret JF, et al. Single copies of Sec61 and TRAP associate with a nontranslating mammalian ribosome. Structure. 2008;16:1126–1137. doi: 10.1016/j.str.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Becker T, et al. Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science. 2009;326:1369–1373. doi: 10.1126/science.1178535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frauenfeld J, et al. Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nature structural & molecular biology. 2011;18:614–621. doi: 10.1038/nsmb.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berk V, Zhang W, Pai RD, Cate JH. Structural basis for mRNA and tRNA positioning on the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15830–15834. doi: 10.1073/pnas.0607541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park E, Rapoport TA. Preserving the membrane barrier for small molecules during bacterial protein translocation. Nature. 2011;473:239–242. doi: 10.1038/nature10014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park E, Rapoport TA. Bacterial protein translocation requires only one copy of the SecY complex in vivo. The Journal of cell biology. 2012;198:881–893. doi: 10.1083/jcb.201205140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schierle CF, et al. The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway. Journal of bacteriology. 2003;185:5706–5713. doi: 10.1128/JB.185.19.5706-5713.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakatogawa H, Ito K. The ribosomal exit tunnel functions as a discriminating gate. Cell. 2002;108:629–636. doi: 10.1016/s0092-8674(02)00649-9. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, et al. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Molecular cell. 2003;12:913–923. doi: 10.1016/s1097-2765(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 19.Muto H, Nakatogawa H, Ito K. Genetically encoded but nonpolypeptide prolyl-tRNA functions in the A site for SecM-mediated ribosomal stall. Molecular cell. 2006;22:545–552. doi: 10.1016/j.molcel.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 20.Sengupta J, Agrawal RK, Frank J. Visualization of protein S1 within the 30S ribosomal subunit and its interaction with messenger RNA. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11991–11996. doi: 10.1073/pnas.211266898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lycklama ANJA, Bulacu M, Marrink SJ, Driessen AJ. Immobilization of the plug domain inside the SecY channel allows unrestricted protein translocation. The Journal of biological chemistry. 2010;285:23747–23754. doi: 10.1074/jbc.M110.124636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plath K, Mothes W, Wilkinson BM, Stirling CJ, Rapoport TA. Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell. 1998;94:795–807. doi: 10.1016/s0092-8674(00)81738-9. [DOI] [PubMed] [Google Scholar]
- 23.Knyazev DG, et al. The Bacterial Translocon SecYEG Opens upon Ribosome Binding. The Journal of biological chemistry. 2013;288:17941–17946. doi: 10.1074/jbc.M113.477893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw AS, Rottier PJ, Rose JK. Evidence for the loop model of signal-sequence insertion into the endoplasmic reticulum. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:7592–7596. doi: 10.1073/pnas.85.20.7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hizlan D, et al. Structure of the SecY complex unlocked by a preprotein mimic. Cell Rep. 2012;1:21–28. doi: 10.1016/j.celrep.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matlack KE, Misselwitz B, Plath K, Rapoport TA. BiP acts as a molecular ratchet during posttranslational transport of prepro-alpha factor across the ER membrane. Cell. 1999;97:553–564. doi: 10.1016/s0092-8674(00)80767-9. [DOI] [PubMed] [Google Scholar]
- 27.Nicchitta CV, Blobel G. Lumenal proteins of the mammalian endoplasmic reticulum are required to complete protein translocation. Cell. 1993;73:989–998. doi: 10.1016/0092-8674(93)90276-v. [DOI] [PubMed] [Google Scholar]
- 28.Tsukazaki T, et al. Structure and function of a membrane component SecDF that enhances protein export. Nature. 2011;474:235–238. doi: 10.1038/nature09980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang G, et al. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 31.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alami M, Dalal K, Lelj-Garolla B, Sligar SG, Duong F. Nanodiscs unravel the interaction between the SecYEG channel and its cytosolic partner SecA. The EMBO journal. 2007;26:1995–2004. doi: 10.1038/sj.emboj.7601661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nath A, Atkins WM, Sligar SG. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- 34.Cannon KS, Or E, Clemons WM, Jr, Shibata Y, Rapoport TA. Disulfide bridge formation between SecY and a translocating polypeptide localizes the translocation pore to the center of SecY. The Journal of cell biology. 2005;169:219–225. doi: 10.1083/jcb.200412019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 36.Jenner LB, Demeshkina N, Yusupova G, Yusupov M. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nature structural & molecular biology. 2010;17:555–560. doi: 10.1038/nsmb.1790. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DiMaio F, Tyka MD, Baker ML, Chiu W, Baker D. Refinement of protein structures into low-resolution density maps using rosetta. Journal of molecular biology. 2009;392:181–190. doi: 10.1016/j.jmb.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.