

Abstract
Pre-existing immunity (PEI) to human adenovirus serotype 5 (Ad5) worldwide is the primary limitation to routine clinical use of Ad5-based vectors in immunization platforms. Using systemic and mucosal PEI induction models in rodents (mice and guinea pigs), we assessed the influence of PEI on the type of adaptive immune response elicited by an Ad5-based vaccine for Ebola with respect to immunization route. Splenocytes isolated from vaccinated animals revealed that immunization by the same route in which PEI was induced significantly compromised Ebola Zaire glycoprotein (ZGP)-specific IFN-γ+ CD8+ T cells and ZGP-specific multifunctional CD8+ T cell populations. ZGP-specific IgG1 antibody levels were also significantly reduced and a sharp increase in serum anti-Ad5 neutralizing antibody (NAB) titers noted following immunization. These immune parameters correlated with poor survival after lethal challenge with rodent-adapted Ebola Zaire virus (ZEBOV). Although the number of IFN-γ+ CD8+ T cells was reduced in animals given the vaccine by a different route from that used for PEI induction, the multifunctional CD8+ T cell response was not compromised. Survival rates in these groups were higher than when PEI was induced by the same route as immunization. These results suggest that antigen-specific multi-functional CD8+ T cell and Th2 type antibody responses compromised by PEI to Ad5 are required for protection from Ebola. They also illustrate that methods for induction of PEI used in pre-clinical studies must be carefully evaluated for successful development of novel Ad5-based vaccines.
Keywords: Pre-existing immunity, Adenovirus serotype 5, Zaire Ebola virus, Vaccine, Mouse, Guinea pig, Multifunctional CD8+ T cell response
Introduction
Recombinant human adenovirus serotype 5 (Ad5) is currently one of the most potent vaccine platforms that can induce strong adaptive immune responses against encoded antigens1–3. Therefore, Ad5 vectors have been actively developed as vaccines for many pathogenic bacteria and viruses (anthrax, hepatitis C, malaria, botulism, rabies and Ebola) 4–9. Although pre-clinical results obtained with adenovirus-based vaccines are quite encouraging, pre-existing immunity (PEI) to Ad5 in the general population has prevented progression to clinical use10, 11. Approximately 45% of the healthy adult population in the United States and India, 72% in China and 80% in South Africa, Gambia and Thailand have serological evidence of PEI to Ad5 in the context of measurable anti-Ad5 neutralizing antibody (NAB) titers in the circulation 12–15. This may manifest as high levels of pro-inflammatory cytokines at the time the vaccine is given, a humoral antibody response that neutralizes the adenovirus, or a cellular immune response that targets and destroys cells expressing adenoviral antigens 11, 16.
Several different approaches have been taken to resolve issues associated with PEI to Ad5. Increasing the vaccine dose to override vector neutralization by NAB was the first and most obvious tactic 17, 18. Even though this can incrementally improve the potency of Ad5-based vaccines in those with PEI, the risk of severe side effects due to the large dose of virus required to achieve this effect outweighed the utility of this approach. Sophisticated methods to improve antigen expression through optimization of promoter and codon sequences and incorporation of protein adaptor sequences in antigen expression cassettes has allowed for notable antigen expression from the portion of the vaccine which is not neutralized in those with PEI 19, 20. Use of rare human adenovirus serotypes (Ad11, Ad12, Ad35) or those that infect other species (chimpanzee, porcine, bovine) as vaccine vectors alone or in combination with other Ad serotypes in prime-boost regimens represents another rational approach taken to improve vaccine efficacy in those with PEI to Ad5 21–25. However, many of these viruses are difficult to produce on a large scale and are less immunogenic than Ad5. In addition, timelines needed for prime-boost protocols are often not amenable to effective, widespread immunization campaigns in regions where transportation and access to medical care are limited.
Modification of Ad5 capsid proteins at the genetic level to create hexon-chimeric adenoviruses and chemical modification virus proteins with biocompatible polymers represent some of the most highly sophisticated efforts to protect vectors from recognition by the immune system in those with PEI to Ad5 26, 27. Unexpected changes in the natural in vivo transduction efficiency of the modified viruses and additional modifications required to maintain potency of these vaccines currently limit the feasibility of these platforms in a clinical setting 28. More recent studies have focused on eliminating the necessity of virus transduction for effective immunization through fusion of antigen to capsid proteins on the outer surface of intact virions or generation of hapten-conjugates with antigen and disrupted virus capsids 29, 30.
After careful review of the literature, it is evident that intramuscular (IM) injection currently remains the primary route of administration of many vaccines 31. This is not surprising since it is the most direct way to induce strong, systemic immune responses to antigens. Thus, early studies designed to test novel immunization platforms that circumvent PEI to Ad5 established PEI by IM injection of an unrelated Ad5 vector prior to immunization 10, 11, 16. Considering that Ad5 naturally infects humans through the respiratory tract 32, direct injection of virus into the systemic circulation of a given animal model in this manner does not resemble what naturally occurs in the human population. Even though there is a general consensus within the scientific community that PEI to vectors developed from viruses commonly found in the environment is a significant issue that must be addressed when developing a clinical therapeutic containing these agents 10, a standardized method for establishing PEI that closely reflects natural infection currently does not exist. In addition, very little is known about this particular immunological state in the context of how it influences the immune response to an antigen and other microbial pathogens.
One of the primary goals of our laboratory is to develop a potent, long-acting Ad5-based vaccine for Ebola Zaire that is highly effective in those with prior-exposure to Ad5. In order to meet this objective, we conducted the study summarized here to determine how PEI to Ad, when induced by either the systemic or mucosal route, influences the immune response to a model antigen, Ebola Zaire glycoprotein (ZGP). A secondary aim was to identify a method for induction of PEI to Ad5 which could be viewed as the most stringent test under which to evaluate novel formulation candidates designed to improve vaccine potency in those with PEI. Assays to evaluate quantitative and qualitative antigen-specific CD8+ T cell responses were performed on samples obtained from naïve animals and those with PEI established by different routes. Antibody responses to ZGP and to the Ad5 vector were also assessed. Survival after lethal challenge served as a final indicator of the stringency of each model of PEI. Data obtained from these studies assisted us in identifying specific types of immune responses that must be reconstituted through formulation or other modification of the vaccine in order to promote survival from Ebola in those with prior-exposure to Ad5.
Experimental Section
Adenovirus Production
The codon optimized full-length Zaire Ebola glycoprotein sequence (Genbank/NCBI; Mayinga strain 76, Gene accession number: AF086833) was cloned in an E1/E3-deleted adenovirus serotype 5 vector under the control of chicken-β-actin promoter (Ad-CAGoptZGP) and further amplified in HEK 293 cells (ATCC, CRL-1573) as previously described 19, 33. Virus was purified from cell lysates by two rounds of cesium chloride gradient ultracentrifugation. Virus bands were desalted by dialysis overnight in 100 mM potassium phosphate buffer (pH 7.4) and infectious titer was determined using the Adeno-X Rapid Titer Kit (Clontech, Mountain View, CA) according to the manufacturer’s instructions. Preparations with infectious to physical particle ratios below 1:200 were used in this study.
Animal Studies
All procedures were approved by the Institutional Animal Care and Use Committees at The University of Texas at Austin and the University of Texas Medical Branch in Galveston and are in accordance with the guidelines established by the National Institutes of Health for the humane treatment of animals. The time course for establishment of PEI, immunization, sample collection and analysis is illustrated in Figure 1.
Figure 1. Timeline for Establishment of Pre-Existing Immunity to Adenovirus, Immunization, Sample Collection and Challenge for (A) Mice and (B) Guinea Pigs.

PEI was established by administration of either 5×1010 (mice: n=12) or 1×1012 (guinea pigs: n=5) particles of adenovirus containing the β-galactosidase transgene by the intramuscular (IM) or intranasal (IN) route 28 days prior to vaccination. These groups are denoted as IM/systemic PEI or IN/mucosal PEI, respectively throughout the manuscript. Naïve animals and those with PEI to Ad5 were then given either 1×108 ivp (mice) or 1×107~1×109 ivp (guinea pigs) of our vaccine (Ad-CAGoptZGP) by various routes. Control, naïve mice were given saline (PBS). Mononuclear cells were collected from the spleen, bronchioalveolar lavage (BAL) fluid, mesenteric lymph nodes (MLNs) and submandibular lymph nodes (SMLNs) of mice 10 days post-immunization for characterization of the ZGP-specific T cell response. Serum was collected 24 (guinea pigs) and 42 (mice) days after immunization for assessment of circulating anti-ZGP antibodies. Animals were challenged with 1,000 pfu of either MA-ZEBOV (30,000 × L.D.50) or GPA-ZEBOV (100 × L.D.50) 28 days after immunization.
Immunization
Six-week-old male B10.Br mice (MHC H-2k) and male Hartley guinea pigs (body weight: 400 g) were obtained from the Jackson Laboratory (Bar Harbor, ME) and Charles River Laboratories (Wilmington, MA), respectively. Animals were housed in a temperature-controlled, 12-hour light-cycled facility at the Animal Research Center of The University of Texas at Austin and were given free access to standard rodent chow (Harlan Teklad, Indianapolis, IN) and tap water. Animals were anesthetized by a single intra-peritoneal injection of a 3.9:1 (mice) or 7.5:1 (guinea pigs) mixture of ketamine (100 mg/ml, Wyeth, Fort Dodge, Animal Health, Overland Park, KS) and xylazine (100 mg/ml, Sigma Aldrich, St. Louis, MO). Once deep plane anesthesia was achieved, animals were immunized with 1×108 (mice) or 1×107 – 1×109 (guinea pigs) infectious virus particles (ivp) of Ad-CAGoptZGP by various routes. Intramuscular injection involved direct injection of the preparation into each gastrocnemius muscle located on the hind limb (mice: 50 μl/muscle, guinea pigs: 100 μl/muscle). Nasal immunization was performed by slowly dripping the preparation into each nostril (mice: 10 μl/nostril, guinea pigs: 50 μl/nostril) using a standard micropipette (Gilson, Middleton, WI). For sublingual immunization, sterile forceps were placed under the tongue of the animal and 10 μl of the preparation slowly dispensed with a standard micropipette as described previously 33. Animals given the vaccine by the nasal and sublingual routes were maintained in an upright position for 30 minutes after immunization to minimize choking and accidental swallowing of the vaccine.
Establishment of Pre-Existing Immunity to Adenovirus
First generation adenovirus expressing β-galactosidase under the control of the cytomegalovirus promoter (AdlacZ) was used to establish PEI in mice and guinea pigs 33, 34. Twenty-eight days prior to immunization, systemic PEI (IM PEI) or mucosal PEI (IN PEI) was induced by administration of 5×1010 (mice) or 1×1012 (guinea pigs) virus particles (vp) to the muscle of each hind limb or in the nasal cavity as described above. Blood was collected via the saphenous (mice) or cephalic (guinea pigs) vein and serum screened for anti-Ad5 neutralizing antibodies (NAB). At the time of vaccination, animals had an average anti-Ad5 circulating NAB titer of 1:171 ± 48 (IM PEI) or 1:103 ± 26 (IN PEI), which falls within the lower range of average values reported in humans after natural infection 12–15.
Challenge with Ebola Zaire
All challenge experiments were performed under biosafety level 4 (BSL-4) conditions in an AAALAC accredited animal facility at the Robert E. Shope BSL-4 Laboratory at The University of Texas Medical Branch in Galveston, Texas. Twenty-four days post-immunization, vaccinated mice were transferred to the BSL-4 lab where they were challenged on day 28 by intraperitoneal injection with 1,000 pfu of mouse (MA-ZEBOV; 30,000 × L.D.50) 35 or guinea pig adapted Ebola Zaire (GPA-ZEBOV; 100 × L.D.50) 36. Animals were monitored for clinical signs of disease and weighed daily. At the end of the study, survivors were bled and serum samples gamma-irradiated (5 Mrad) prior to removal from the BSL-4 lab for cytokine and transaminase analysis.
ELISPOT
IFN-γ ELISPOT assays were performed using the ELISpot Mouse Set (BD Pharmingen) according to the manufacturer’s instructions. Briefly, a 96-well ELISPOT plate was pre-coated with 5 μg/ml anti-mouse IFN-γ capture antibody overnight at 4 °C. Coated wells were blocked with complete Dulbecco’s Modified Eagle’s Medium (DMEM, Mediatech) for 2 hours at room temperature. Cells were isolated from the spleen, lung, mesenteric lymph nodes (MLNs) and submandibular lymph nodes (SMLNs) as described previously 33, washed twice with complete DMEM and added to each well (5×105 cells/well) with the TELRTFSI peptide (0.5 μg/well, New England Peptide, Gardner, MA) that bears the ZGP immunodominant MHC class I epitope for mice with the H-2k haplotype (B10.Br) 37. Plates were placed at 37°C for 20 hours with 5% CO2. Wells were washed twice with deionized water, washed with buffer (0.05% Tween-20 in PBS, pH 7.4) and subsequently incubated with biotinylated anti-mouse IFN-γ antibody at a concentration of 2 μg/ml for 2 hours at room temperature. Following three consecutive washes with buffer, wells were incubated with 5 μg/ml of horseradish peroxidase (HRP)-conjugated streptavidin antibody for an additional hour. Wells were then washed and plates developed with AEC substrate (Sigma). Spots were counted using an automated ELISpot reader (CTL-ImmunoSpot® S5 Micro Analyzer, Cellular Technology Ltd., Shaker Heights, OH).
Intracellular cytokine staining
Splenocytes isolated from mice were washed three times with complete DMEM containing 50 μM β-mercaptoethanol (Sigma), penicillin (10,000 I.U./ml)/streptomycin (10,000 μg/ml) (Gibco, Invitrogen, Grand Island, NY), L-glutamine (1mM, Hyclone, Salt Lake City, UT), 1 mM sodium pyruvate (Lonza, Walkersville, MD), 1 mM non-essential amino acids (Lonza) and stimulated with TELRTFSI peptide (0.5 μg/well) in the presence of Brefeldin A at 37°C for 6 hours. Following stimulation, cells were surface stained with fluorescein isothiocyanate (FITC)-labeled anti-mouse CD8α antibody (1:150 in PBS, BD Pharmingen, San Diego, CA) for 30 minutes at 4 °C and treated with Cyto-fix/Cytoperm (BD Pharmingen) for 20 minutes at 4 °C. For intracellular cytokine staining, cells were labeled with PE-labeled anti-mouse IFN-γ, PerCP-Cy5.5-labeled anti-mouse TNF-α and APC-labeled anti-mouse IL-2 antibodies (1:150 in Permwash, BD Pharmingen) for 30 minutes at 4 °C. Cells were washed and positive cells counted using four-color flow cytometry (FACS Fortessa, BD Biosciences, Palo Alto, CA) in The University of Texas at Austin Flow Cytometry Core Facility. Over 500,000 events were captured per sample. Data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Characterization of Ebola Zaire glycoprotein-specific antibodies
Full length Ebola Zaire glycoprotein was produced using the Zaire GP33-637ΔTM-HA construct according to published methods 38. Sub-confluent HEK 293T cells were transfected with the plasmid using a standard calcium phosphate transfection protocol. Four days later, supernatant was collected, concentrated by centrifugation (Centricon Plus-100, Millipore, Billerica, MA) and purified by immunoaffinity chromatography (Anti-HA Affinity Matrix, Roche, Mannheim, Germany). To assess anti-ZGP immunoglobulin levels in serum, Immulon 2HB plates (Fisher Scientific, Pittsburgh, PA) were coated with purified Zaire GP33-637ΔTM-HA (3 μg/well) in PBS (pH 7.4) overnight at 4°C. Plates were washed 4 times with PBS containing 0.05% Tween 20 and blocked in PBS containing 1% BSA (Sigma) for 1 hour at room temperature. Heat-inactivated serum samples were diluted 1:20 in sterile PBS. One hundred microliters of each dilution was added to the antigen-coated plates for 2 hours at room temperature. Plates were washed 4 times with PBS containing 0.05% Tween 20 and incubated with HRP-conjugated goat anti-mouse IgG, IgG1, IgG2a, IgG2b, IgM (1:2,000, Southern Biotechnology Associates, Birmingham, AL) or HRP-conjugated goat anti-guinea pig IgG, IgG1, IgG2, IgM antibodies (1:2,000, Bethyl Laboratories, Inc., Montgomery, TX) in separate wells for 1 hour at room temperature. Plates were washed and 200 μl of substrate solution (0.4 mg/ml o-phenylenediamine (Sigma) in 50 mM phosphate-citrate buffer, pH 5.0 with 0.03 % (v/v) hydrogen peroxide) were added to each well. The plate was incubated at room temperature for 10 minutes and optical densities read at 450 nm on a microplate reader (GloMax-Multi+ Detection System, Promega, Madison, WI).
Neutralizing antibody assay
Anti-Ad5 neutralizing antibody (NAB) titers were assessed in serum samples from mice and guinea pigs prior to and after immunization according to established methods 33, 34, 39. Heat-inactivated serum was diluted in DMEM in twofold increments starting from a 1:20 dilution. Each dilution was mixed with AdlacZ for 1 hour at 37 °C and added to HeLa cells grown on 96-well plates. Two hours later, 100 microliters of DMEM supplemented with 20% FBS were added to each well. Cells were incubated additional 24 hours and β-galactosidase expression visualized by histochemical staining. For each sample, the serum dilution that corresponded to a 50% reduction in transgene expression was obtained by the method of Reed and Muench as described previously 39. The absence of neutralization in samples containing medium only (negative control) and FBS (serum control) and an average titer of 1:1,280 ± 210 read from an internally generated positive control stock serum were criteria for qualification of each assay.
Serum Cytokines and Transaminases
Cytokines (IL-6, IL-12, TNF-α, IL-2 and IL-10) were quantitated with commercially available ELISA kits (Invitrogen, BioSource International, Camarillo CA). Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were determined using Vitros AST/SGOT and ALT/SGPT DT slides on a Vitros DTSC autoanalyzer (Ortho-Clinical Diagnostics, Rochester, NY).
Statistical Analysis
Data were analyzed for statistical significance using SigmaStat (Systat Software Inc., San Jose, CA) by performing a one-way analysis of variance (ANOVA) between control and experimental groups, followed by a Bonferroni/Dunn post-hoc test when appropriate. Differences in the raw values among treatment groups were considered statistically significant when p<0.05.
Results
Pre-Existing Immunity and the Magnitude of the T Cell Response in Mice
In order to determine how establishment of PEI by either the systemic (IM) or mucosal (IN) route influenced the antigen-specific T cell response, mononuclear cells were collected 10 days after immunization. Ad-CAGoptZGP induced the strongest responses in the systemic and mucosal compartments of naïve animals than in those with PEI, regardless of immunization route (Figure 2). Prior exposure to the virus through IM injection followed by vaccination by the same route reduced the number of activated cells in the spleen to a much greater degree than when PEI was established by the nasal route (IM PEI/IM: 44.0% reduction with respect to naïve animals vs. 25.8% reduction IN PEI/IM, p<0.001, Figure 2A). Establishment of PEI by instillation of virus in the nasal cavity had the most profound effect on ZGP-specific IFN-γ secreting mononuclear cells (MNCs) in the spleen of animals also immunized by the IN route (IN PEI/IN, 55.3% reduction). Establishment of PEI by the nasal route had a similar effect on the systemic antigen-specific T cell response after sublingual immunization. Prior exposure to Ad by the IN route suppressed production of these cells by 56.5% while establishment of PEI by the IM route reduced cell numbers by 19.5% (Figure 2A).
Figure 2. The Manner by which PEI is Induced Significantly Affects the Magnitude of the CD8+ T Cell Response Against Ebola Zaire Glycoprotein in the Systemic and Mucosal Compartments of Mice Immunized by Various Routes.
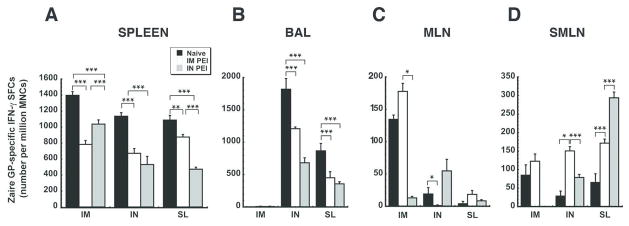
The number of IFN-γ secreting mononuclear cells was quantitated in isolates taken from the spleen (Panel A), bronchioalveolar lavage (BAL) fluid (Panel B), mesenteric lymph nodes (MLN, Panel C) and submandibular lymph nodes (SMLN, Panel D) from naïve mice and those with PEI to Ad5 10 days after immunization by ELISPOT. Results are expressed as average values ± the standard error of the mean and are representative of data collected over the course of three separate experiments. *p<0.05, **p<0.01, ***p<0.001, one-way ANOVA, Bonferroni/Dunn post-hoc analysis.
Mucosal PEI significantly reduced the average number of ZGP-specific IFN-γ+ MNCs in BAL fluid of mice immunized by the IN route to a much greater degree than when PEI was established by IM injection (IN PEI/IN: 62.5% reduction with respect to naïve animals vs. 33.5% reduction IM PEI/IN, p<0.001, Figure 2B). A similar trend was noted in BAL of mice immunized by the SL route (IN PEI/SL: 58.8% reduction; IM PEI/SL: 47.9% reduction, p<0.001). In sharp contrast, prior exposure to Ad5 significantly enhanced the number of activated cells in MLNs and SMLNs with respect to naïve animals when the vaccine was given by either the IN or SL route (Figures 2C and 2D). The most prominent increases were found in samples from SMLNs of mice immunized by IN route in the presence of systemic PEI (85 ± 27.83 SFCs/million MNCs: Naive/IN vs. 122.5 ± 19.6 SFCs/million MNCs: IM PEI/IN, p<0.05) and mice immunized by the SL route in the presence of mucosal PEI (Naïve/SL: 65.5 ± 23.46 SFCs/million MNCs, IN PEI/SL: 294 ± 15.64 SFC/million MNCs; p<0.001; Figure 2D).
Pre-Existing Immunity and Multifunctional CD8+ T cell Responses in Mice
Previously, we have shown that, even though a single dose of vaccine given by the IN route to naïve animals induced significantly less ZGP-specific IFN-γ+ CD8+ T cells than immunization by IM injection, it could afford full protection from lethal challenge with Ebola Zaire 33, 34. When PEI was established by IM injection, animals immunized by the IN route survived while those immunized by the IM route succumbed to infection. Further analysis revealed that the amount of ZGP-specific IFN-γ+ CD8+T cells produced in animals with systemic PEI given the vaccine nasally was substantially lower than that found in naïve mice immunized by IM injection, yet similar to that seen in naïve animals vaccinated by the IN route. Collectively, these data suggest that the magnitude of the antigen-specific IFN-γ response is not fully predictive of vaccine-elicited protection from ZEBOV challenge in mice with PEI to the vaccine carrier.
To further characterize the influence of PEI induced by systemic or nasal route on antigen-specific CD8+ T cell responses, we performed a comprehensive functional analysis using multiparameter flow cytometry 40, 41. With this strategy, we were able to delineate seven distinct cytokine-producing cell populations and characterize them at the single-cell level based on IFN-γ, IL-2 and TNF-α secretion patterns. The relative frequency of these distinct populations defines the quality of the vaccine-induced CD8+ response. Complete analysis of IFN-γ producing cells identified four distinct populations: IFN-γ+, IFN-γ+IL-2+, IFN-γ+TNF-α+ and IFN-γ+IL-2+TNF-α+. This analysis further revealed a correlation between the frequency of robust, multifunctional CD8+ T cells (IFN-γ+IL-2+TNF-α+), which are most effective at responding to a given antigen, and the manner by which PEI was induced in mice immunized by IM, IN or SL routes. Ten days after immunization, systemic PEI not only suppressed the total number of Zaire GP-specific IFN-γ producing CD8+ T cells in the spleen of mice given the vaccine by intramuscular injection (5.30%: Naïve/IM vs. 1.82%: IM PEI/IM; Figure 3A), but also reduced ZGP-specific multifunctional CD8+ T cells (triple producers: IFN-γ+IL-2+TNF-α+; 35.4 ± 0.37 %: Naive/IM vs. 24.29 ± 2.01 %: IM PEI/IM, p<0.001; red arcs Figure 3G) with a concurrent increase in single cytokine producers (blue arcs Figure 3G). When PEI was established by the nasal route, the number of IFN-γ+ CD8+ T cells was reduced to a lesser degree (5.30%: Naïve/IM vs. 3.18%: IN PEI/IM; Figure 3A) and the multifunctional CD8+ T cell population was not significantly compromised (35.4 ± 0.37 %, Naive/IM vs. 37.88 ± 0.83 %, IN PEI/IM, Figure 3G).
Figure 3. Prior Exposure to Adenovirus Profoundly Affects the Ebola Zaire Glycoprotein-Specific Multifunctional CD8+ T cell Response Elicited After Immunization by Various Routes.

Naïve B10.Br mice and those with PEI established by the IM (systemic) or IN (mucosal) route (10/group) were given 1 × 108 ivp of Ad-CAGoptZGP by the IM, IN or sublingual (SL) route. Mononuclear cells were harvested from the spleen 10 days later and the multifunctional T cell response assessed by FACS. Panels A–C. Representative FACS analysis of cell populations secreting individual cytokines. Numbers written in the upper right corner of each scatter plot represent the percentage of CD8+ T cells secreting the cytokine listed at the far left of each row after TELRTFSI peptide stimulation. Panels D–F. Quantitative analysis of cell populations secreting individual and combinations of cytokines in response to antigen stimulation. Each positively responding cell was assigned to one of 7 possible categories reflecting the production of IFN-γ, IL-2 and TNF-α alone or in combination. Average values and standard errors were calculated for each treatment group for statistical evaluation. Panels G–I. Depiction of Zaire glycoprotein-specific multifunctional CD8+ T cells in pie chart format. The relative frequency of triple producers defines the quality of the vaccine-induced CD8+ T cell response. Triple (IFN-γ+IL-2+TNF-α+) and single cytokine producers are highlighted in the red and blue arcs, respectively. Numbers in the pie chart denote the percentage of triple (red) or single (black) cytokine producers in a given population. Results are reported as the mean ± the standard error of the mean. *, p<0.05, **, p<0.01, ***, p<0.001, one-way ANOVA, Bonferroni/Dunn post-hoc analysis.
The next series of experiments was designed primarily to determine how nasal administration of our Ad5-based Ebola vaccine was able to bypass PEI induced by IM injection 34. In this context, mucosal PEI suppressed the number of IFN-γ+ CD8+ T cells to a larger degree than systemic PEI (2.58%: Naïve/IN vs. 0.89%: IM PEI/IN vs. 0.77%: IN PEI/IN, Figure 3B). A similar trend was also noted in the multifunctional CD8+ T cell response (triple producers: 41.11 ± 3.59 %: Naive/IN vs. 30.74 ± 1.30 %, IM PEI/IN, p<0.05; 41.11 ± 3.59 %: Naive/IN vs. 23.52 ± 4.22 %, IM PEI/IN, p<0.01; red arcs; Figure 3H). These parameters were also evaluated after SL immunization. The total frequency of IFN-γ+ CD8+ T cells was reduced to a larger degree in mice with PEI induced by the mucosal route than in mice with systemic PEI (0.21%: IN PEI/SL and 0.66%: IM PEI/SL, respectively; Figure 3C). Despite this notable reduction in the number of IFN-γ+ CD8+ T cells, a significant increase in multifunctional CD8+ T cells was observed (triple producers: 24.20 ± 0.91 %: Naive/SL vs. 37.04 ± 1.91 %: IM PEI/SL, p<0.01; red arcs; Figure 3I). The multifunctional CD8+ T cell population was not affected by mucosal PEI (24.20 ± 0.91 %: Naive/SL vs. 20.93 ± 4.92 %: IN PEI/SL, p>0.05; red arcs; Figure 3I).
Pre-Existing Immunity and Antibody responses to Zaire GP and Ad5 in Mice
To characterize the impact of systemic and mucosal PEI on B cell-mediated antibody responses, anti-ZGP-specific immunoglobulin isotypes and adenovirus serotype 5-specific neutralizing antibody (NAB) levels were assessed in samples collected 42 days after immunization. Induction of PEI by the IM route reduced serum anti-ZGP-specific total IgG levels by 63.6% with respect to levels of naïve mice vaccinated by IM injection (Figure 4A). Of the serotypes tested in this group, IgG1 was the most profoundly affected by PEI (90.8% reduction). IgG2a and IgG2b were reduced by 78.8 and 78.4%, respectively. When PEI was established by the mucosal route, a significant decrease in the production of IgG1 was observed with respect to naïve animals immunized by instillation of the vaccine in the nasal cavity (IgG1: 88.99% reduction; p<0.05; Figure 4B). Aside from this, none of the other anti-ZGP-specific isotypes were affected by PEI induced by either route in animals immunized by the IN route. Mucosal PEI also did not significantly affect anti-ZGP-specific IgG isotype levels in mice given the vaccine by the SL route (Figure 4C). Systemic induction of PEI, however, did significantly reduce ZGP-specific total IgG (66.3%), IgG1 (77.5%), IgG2a (80.6%) and IgM (80.5%) levels with respect to those seen in naïve mice (Figure 4C).
Figure 4. Establishment of Pre-Existing Immunity to Adenovirus by the Same Route as that used for Immunization Significantly Impairs Ebola Zaire Glycoprotein-Specific Antibody Production and Exacerbates the Anti-Adenovirus Antibody Response.

Serum was collected from naïve B10.Br mice and those with PEI established by the IM (systemic) or IN (mucosal) route 4 days prior to and 42 days after immunization. Panels A–C. Characterization of the Antigen-Specific Response. Individual samples were evaluated for ZGP-specific IgM and IgG isotypes by ELISA. The average optical density read from samples obtained from each treatment group are presented to serve as a measure of relative antibody concentration. Panel D. Characterization of Anti-Adenovirus Antibody Production After Induction of PEI (PEI only) and Post-Immunization. Neutralization was assessed by serial dilution of each sample and incubation with a fixed concentration of adenovirus expressing beta-galactosidase (AdlacZ) prior to infection of HeLa cells. The reciprocal dilution plotted for each treatment group reflects the dilution at which the ability of the AdlacZ vector to infect target cells was reduced by 50%. In each panel, results are expressed as average values ± the standard error of the mean and are representative of three separate experiments each containing 12 mice per immunization route. *p<0.05, **p<0.01, ***p<0.001, one-way ANOVA, Bonferroni/Dunn post-hoc analysis.
In order to understand how PEI affects the B cell-mediated immune response against the adenoviral vector itself, circulating anti-Ad5 NAB levels were evaluated in serum samples collected 42 days after immunization. Systemic PEI greatly increased the amount of circulating anti-Ad5 NABs after IM immunization as levels rose by a factor of 5 from an average of 1:171 ± 48 prior to vaccination to 1:925± 213 after treatment (p<0.05, Figure 4D). However, the anti-Ad5 NAB titers found in mice with IM PEI after IN or SL immunization were not significantly higher than levels measured prior to vaccination (1:171 ± 40 (IM PEI only), 1:184 ± 40 (IM PEI/IN), 1:314 ± 186 (IM PEI/SL); p>0.05; Figure 4D). A similar trend was found in samples from mice immunized by IM or IN route in the presence of mucosal PEI.
Pre-Existing Immunity and Survival of Mice after Lethal Challenge
The most direct way of evaluating the impact of PEI on vaccine potency is to assess protection from a lethal dose of pathogen. We did this by monitoring survival rate, weight loss and toxicity after exposure to mouse-adapted ZEBOV (MA-ZEBOV). The virus was uniformly lethal in control mice given saline (PBS, Figure 5A). In contrast, all naive mice immunized by IM injection survived without notable loss of body weight (Figure 5A, D). As seen previously 33, 34, systemic PEI significantly compromised the efficacy of the vaccine when it was given by the IM route with only 20% survival observed (IM PEI/IM; p<0.01; Figure 5A). Chemical analysis of samples taken from mice in this treatment group (IM PEI/IM) post-challenge revealed serum ALT levels that were 47.5 times that of naïve, immunized animals (IM) and AST levels that were increased by a factor of 27, indicative of severe liver damage from Zaire Ebola infection (p<0.001; Figure 5G). As is commonly seen in samples taken from non-survivors of Ebola infection 42–44, samples from these animals also contained elevated levels of cytokines (IL-6: 597.72 ± 577.32 pg/ml, IL-10: 1383.8 ± 143.96 pg/ml, IFN-γ: 635.5 ± 294.65 pg/ml; Figure 5J). As routinely found in survivors of Ebola hemorrhagic fever, samples from the Naïve/IM group contained only trace amounts of each compound (IL-6: not detected, IL-10: 17.49 ± 3.02 pg/ml, IFN-γ: not detected, Figure 5J).
Figure 5. Prior Exposure to Adenovirus by a Route Different from that of Immunization Affords Better Protection from Lethal Challenge than when the Same Route is used for Both Procedures in Mice.

Naïve B10.Br mice and those with prior exposure to adenovirus 5 were challenged with a lethal dose of MA-ZEBOV 28 days after immunization by the IM, IN or SL route. Control, naïve mice were given saline (PBS). Panels A–C. Kaplan–Meier survival curves. In each panel, asterisks indicate a significant difference with respect to naïve, immunized animals. Panels D–F. Body Weight Profiles after Challenge. No significant change in body weight was noted in animals that survived challenge. Panels G–I. Post-challenge serum alanine (ALT) and aspartate (AST) aminotransferase levels. Samples were taken from survivors at the termination of the study and from non-survivors at time of death. Asterisks indicate a significant difference with respect to naïve, immunized animals. Panels J–L. Serum Cytokines Levels After Challenge. Samples were taken from survivors 14 days after challenge and from non-survivors at time of death. In all panels, data reflect average values ± the standard error of the mean for 9–10 mice per group. *p<0.05, **p<0.01, ***p<0.001, one-way ANOVA, Bonferroni/Dunn post-hoc analysis.
Interestingly, 90 % of the mice given the vaccine by the IM route in the presence of mucosal PEI survived (IN PEI/IM, Figure 5A) without a notable increase in Ebola-induced liver toxicity (Figure 5G). To fully define how PEI affects protective efficacy following nasal immunization, naïve mice and those with systemic or mucosal PEI were also given a lethal dose of MA-ZEBOV. Consistent with previous studies 34, naïve mice (Naïve/IN) and those with systemic PEI (IM PEI/IN) survived without notable changes in body weight (Figures 5B and 5E). However, 60% of the animals with mucosal PEI survived challenge (IN PEI/IN, Figure 5B). As seen with animals immunized by IM injection, samples taken from this group also revealed sharp elevations in serum ALT (768.75 ± 337.48 U/L) and AST (IN PEI/IN: 1086 ± 584.97 U/L, p < 0.001, Figure 5H) with respect to naïve animals immunized by the same route that survived challenge (Naive/IN: 144 ± 30.19 (ALT) and 330 ± 102.94 U/L (AST) respectively)). They also contained significant amounts of IL-10 and IFN-γ with respect to those from naïve immunized mice (p<0.05, Figure 5K).
Naïve mice and those with systemic or mucosal PEI vaccinated by the SL route were also challenged with MA-ZEBOV 28 days after vaccination. Eighty percent of naïve mice and 100% of those with systemic PEI to Ad5 survived (Figure 5C). Interestingly, mucosal PEI did not significantly compromise the efficacy of the vaccine when given by the SL route with 87.5% survival observed in this treatment group (IN PEI/SL, Figure 5C). Serum samples taken from this group post-challenge revealed moderate increases in ALT (118.75 ± 24.09 U/L (p<0.05)) and IFN-γ (1,384.4 ± 222.15 pg/ml (p<0.001)) with respect to naïve, immunized animals (Figures 5I and 5L).
Pre-Existing Immunity and the Zaire GP-specific Antibody Response in Guinea Pigs
Several animal models have been developed to study the pathogenesis of Ebola virus infection, assess the safety and efficacy of vaccine candidates and identify the immune mechanisms necessary for optimal protection against lethal challenge. Currently, the progression and pathogenesis of infection in guinea pigs and non-human primates most closely resembles that of the human disease 36, 45. Thus, in order to determine if our observations in mice would translate to that which occurs in humans, systemic or mucosal PEI was established in male Hartley guinea pigs 28 days prior to immunization with doses of vaccine ranging from 1 ×107 to 1 ×109 ivp as outlined in Figure 1B. Due to the lack of commercially available reagents to perform T cell assays in guinea pigs, we were not able to characterize the ZGP-specific T cell responses as was done in mice.
Systemic PEI reduced serum anti-ZGP-specific total IgG levels by 67.4% in all animals immunized by IM injection (p<0.05, Figure 6A). IgG1 was also suppressed (~ 95.2% reduction) without regard to vaccine dose. At the highest vaccine dose tested (1 ×109 ivp), IgG1 was the only isotype affected by mucosal PEI in animals given the vaccine nasally (82.2% reduction, p<0.05, Figure 6B). Although antibody production was significantly blocked in animals given the lowest dose of vaccine nasally regardless of the method by which PEI was induced, mucosal PEI held the most prominent effect (Total IgG: 92.1%, IgG1: 97.3%, IgG2: 84.7 and IgM: 99.2% reduction p<0.01, Figure 6B). There was no significant difference in the amount of each ZGP-specific IgG isotype produced after SL immunization of naïve animals and those with mucosal or systemic PEI (p >0.05, Figure 6C).
Figure 6. Pre-Existing Immunity to Adenovirus Significantly Reduces Ebola Zaire Glycoprotein-Specific Antibody Response and Exacerbates the Anti-Adenovirus Antibody Response in Guinea Pigs in a Dose-Dependent Manner.

Serum was collected from naïve guinea pigs and those with PEI established by the IM (systemic) or IN (mucosal) route 4 days prior to and 24 days after immunization. Panels A–C. Characterization of the Ebola-Zaire Glycoprotein-Specific Response. Characterization of the Antigen-Specific Response. Individual samples were evaluated for ZGP-specific IgM and IgG isotypes by ELISA. D. The Anti-Adenovirus Response. The value plotted for each treatment group reflects the dilution at which the ability of an AdlacZ vector to infect target cells was reduced by 50%. In each panel, results are expressed as average values ± the standard error of the mean and are representative of three separate experiments each containing 5 guinea pigs per treatment. *In each panel, indicates a significant difference with respect to naïve, immunized animals. *p<0.05, **p<0.01, one-way ANOVA, Bonferroni/Dunn post-hoc analysis.
Anti-Ad5 NAB levels were evaluated in serum collected 24 days after immunization. Systemic PEI greatly increased the amount of circulating anti-Ad5 NABs after IM immunization by a factor of 10 from an average of 1:212 ± 79 prior to vaccination to ~1:1,985± 363 after treatment for all vaccine doses tested (p<0.01, Figure 6D). Mucosal PEI also boosted circulating anti-Ad5 NABs after IN administration of 1 ×108 and 1 × 109 ivp of vaccine by a factor of 4 (p<0.01) while there was no significant difference between anti-Ad5 NAB levels of naïve guinea pigs given 1 × 107 ivp of vaccine and those with mucosal PEI immunized in the same manner (p=0.08). Systemic PEI increased anti-Ad5 NAB levels in guinea pigs immunized by the SL route fourfold while mucosal PEI increased NAB levels fivefold (p<0.01).
Pre-Existing Immunity and Survival of Guinea Pigs After Challenge
In a final effort to determine how changes in antibody production in response to PEI impacts survival, guinea pigs were challenged with 1,000 pfu (~100 × LD50) of guinea pig-adapted Ebola Zaire (GPA-ZEBOV). Unimmunized guinea pigs (negative control, PBS) experienced significant weight loss starting from day 5 post-challenge that progressed until death on days 6 through 9 (Figure 7A). Naïve guinea pigs given the vaccine by the IM route (dose: 1 × 109 ivp) were fully protected. Consistent with mouse challenge results, PEI established by the IM route significantly decreased protective efficacy in a dose dependent manner (IM PEI/IM (1 × 109), 80% survival; IM PEI/IM (1 × 108), 40% survival, p < 0.01, Figure 7A). Survival was reduced by 60% when PEI was established by the mucosal route and the vaccine given by IM injection (IN PEI/IM (1 × 108)). Samples taken after challenge revealed that serum AST levels were significantly elevated in guinea pigs with PEI established by either route with respect to naïve, immunized animals (Naïve/IM,104 ± 37.1 U/L) by a factor of 2.4 (IM PEI/IM: 1 × 109), 6.2 (IM PEI/IM: 1 × 108) and 5.8 (IN PEI/IM: 1 × 108, Figure 7G).
Figure 7. Protective Efficacy of Various Doses of an Ad-Based Ebola Vaccine in Guinea Pigs with Pre-Existing Immunity to Ad5.

Naïve guinea pigs and those prior exposure to adenovirus were challenged with a lethal dose of GPA-ZEBOV 28 days after immunization with 1×107 ~ 1×109 ivp of Ad-CAGoptZGP by the IM, IN or SL route. Panels A–C. Survival curves. In each panel, asterisks indicate a significant difference with respect to naïve, immunized animals. Panels D–F. Body Weight Profiles after Challenge. Significant changes in body weight were only noted in animals that did not survive challenge. Panels G–I. Serum alanine (ALT) and aspartate (AST) aminotra nsferase levels. Samples were taken from survivors at the termination of the study and from non-survivors at time of death. Asterisks indicate a significant difference with respect to naïve, immunized animals. In all panels, data reflect average values ± the standard error of the mean for 5 guinea pigs per treatment. *p<0.05, **p<0.01, ***p<0.001, one-way ANOVA, Bonferroni/Dunn post-hoc analysis.
PEI induced by the nasal route also compromised the protective efficacy of the vaccine after IN immunization in a dose dependent manner (Figure 7B). A high vaccine dose (1×109 ivp) fully protected immunized animals with mucosal PEI (IN PEI/IN (1 × 109)). However, the cha llenge was uniformly lethal when the vaccine dose was reduced to 1×107 ivp regardless of the manner by which PEI was induced (IM PEI/IN (1 × 107), IN PEI/IN (1 × 107): no survivors, Figure 7B). Samples taken from guinea pigs with systemic or mucosal PEI after challenge revealed that serum ALT (IM PEI/IN (1 × 107): 229 ± 6.78 U/L, IN PEI/IN (1 × 107): 323 ± 157.72 U/L) and AST (IM PEI/IN (1 × 107): 1,525 ± 123.64 U/L, IN PEI/IN (1 × 107): 2,192 ± 726.29 U/L) levels were significantly elevated in those with PEI established by either route with respect to naïve, immunized animals (naïve/IN: ALT: 37 ± 4.85 U/L, AST: 74.4 ± 13.78 U/L, p < 0.001, Figure 7H).
The protective efficacy of sublingual (SL) immunization in guinea pigs in the presence of systemic or mucosal PEI was also evaluated only with the highest dose of the vaccine (1×109 ivp, Figure 7C). Consistent with the mouse data, 80% of naïve guinea pigs and those with systemic PEI vaccinated by the SL route survived without notable loss of body weight (Figure 7C, 7F). However, when PEI was induced by the IN route, the efficacy of the SL vaccine was significantly compromised with only 40% survival observed in this treatment group (IN PEI/SL, p < 0.05, Figure 7C). None of the samples taken post-challenge from guinea pigs immunized by the SL route contained significantly elevated serum ALT and AST from baseline values (Figure 7I).
Discussion
Ebola hemorrhagic fever is a severe, often-fatal disease caused by a single stranded RNA virus of the Filoviridae family 46. Four of the five known species of Ebola are infectious to humans with case fatality rates up to 90% 47. Lack of remedies, unpredictability of outbreaks and its potential use as a bioweapon highlight the necessity for an effective strategy to prevent Ebola infection. Although there are currently no licensed vaccines or post-exposure treatments available for preventing or managing Ebola infection, several promising vaccine platforms that employ recombinant viruses to deliver genetic sequences for Ebola proteins have been developed 48–53. Among these, recombinant Ad5 and human parainfluenza virus type 3 (HIPV3) vectors, generated from common respiratory tract viruses are currently the most efficacious in animal models. It is not clear, however, how either of these platforms will perform in those with prior exposure by natural infection.
Natural transmission of Ebola involves either direct contact with body fluids of infected individuals or aerosol transmission of virus particles to the respiratory mucosa 54–56. Therefore, fully protective vaccines against the virus require strong systemic and mucosal antigen-specific adaptive immune responses to prevent or substantially limit infection 57, 58. Administration of a vaccine by different routes can have profoundly different effects on the type and strength of the immune response generated against a given antigen 59, 60. This is illustrated by the fact that IM injection of Ad5-based vaccines induces strong, systemic compartment-restricted immune responses while mucosal immunization (nasal, sublingual) induces both systemic and mucosal immune responses. Even though mucosal immunization often generates low-grade antigen-specific CD8+ T cell responses in comparison to those generated by IM injection, it fully protects naïve animals and those with prior exposure to Ad5 from lethal Ebola infection 33, 34, 61. In this regard, mucosal administration of adenovirus-based vaccines has long been thought to be a promising strategy to bypass PEI. However, specific immunological parameters altered by PEI to Ad5 and the mechanisms to explain poor survival following systemic injection of an Ad5-based Ebola vaccine in animals with PEI are not fully delineated.
The initial paradigm to explain how PEI to Ad dampens the adaptive immune response to an encoded antigen identified neutralization of virus particles to block transduction of target cells by anti-Ad antibodies and killing of Ad infected cells by cytotoxic T cells as primary events that limit the utility of Ad-based vaccines 62–65. We believe that this supports our observation that establishment of PEI by the same route significantly compromises the antigen-specific multifunctional CD8+ T cell response. It does not, however, fully explain why establishing PEI by a route different from that of immunization has little to no effect on this facet of the immune response. Although a few early reports suggested that Ad5 might transduce and activate dendritic cells (DCs), the role of transduced DCs in priming T cell responses was not clear 66, 67. The recent observation that neutralizing antibodies in the systemic circulation block uptake of Ad and subsequent transgene expression in CD11c+ DCs with a corresponding reduction in the antigen-specific CD8+ T cell response in vivo represents an important shift in the PEI model 41. Given this information, we believe that there is a minimal threshold and/or requirement for the number and types of DCs and other antigen presenting cells (APCs) needed to induce Th1 and CD8+ T cell responses that varies according to the route of exposure (for PEI) or administration (for vaccination). This minimum is clearly not met when PEI is established by the same route as that of immunization while differing routes stimulate a diverse array of APCs to support production of antigen-specific multifunctional T cells sufficient to protect animals from lethal challenge 68. This may also explain why PEI appeared to strengthen the antigen-specific CD8+ T cell response in SMLNs and MLNs after IN or SL immunization as different types of local, resident DCs and other migratory APCs recruited to the area in response to local inflammation processed antigen within these regions to magnify the immune response 69. Additional studies to identify and characterize the types of APCs recruited to immunization sites and their distribution patterns are necessary to validate this model. Another very important role of DCs, cross-presentation of antigen to promote inhibitory CD8+ T cell responses was not addressed in the studies outlined here 70, 71. Additional studies to evaluate the activation status of CD4+CD25+ regulatory T cells (Treg) in naïve animals and those with PEI are currently underway in our laboratories to further characterize the influence of PEI on the immunogenicity of our Ad5-based vaccine.
A controversy exists regarding which specific immune parameters correlate with protection against Ebola virus infection. A recent report showed that the antigen-specific CD8+ T cell response is unequivocally required for protection in non-human primates 57. Others have provided evidence suggesting that amount of antigen-specific IgG is the immune parameter that correlates with survival58, 72, 73. This is the first report in which multi-parameter analysis of T cell isolates has been performed in rodents infected with and immunized against Ebola. Identification of changes in cell populations that have enhanced antigen-specific CTL activity (IFN-γ+ and TNF-α+) as well as those that foster memory T cell differentiation (IFN-γ+, TNF-α+, IL-2+) allowed us to determine that the quality of the CD8+ T cell response more closely predicts survival rather than the quantitative measure of this response 74–77. We also found that the antigen-specific antibody response is highly dependent on the manner by which PEI is induced and the immunization route. Since T cells play a key role in B cell activation for antibody production, we believe that the suboptimal activation of the multifunctional CD8+ T cell response in conjunction with reduced level of serum ZGP-specific IgG isotypes, especially IgG1, were collectively responsible for poor survival. Even though we could not characterize the T cell response elicited by the vaccine in the guinea pig model, it was clear that the antigen-specific antibody response alone was not sufficient for protection from lethal challenge. Studies in naïve primates and those with prior exposure to Ad5 in which multifunctional analysis of both antigen-specific T and B cell subsets produced by our vaccine are evaluated are currently underway and will provide additional information about requirements for protection from Ebola.
One critical issue not addressed by this report and many others evaluating the role of PEI to Ad5 on in vivo performance of vaccine candidates is that the viruses used to establish PEI do not replicate as the wild type virus does during the natural course of human infection. In fact, replication of wild-type Ad5 obtained from human isolates is restricted in most rodent animal models and non-human primates 78. Thus, we realize that our PEI model may not fully reflect the immunological state induced by replication competent Ad5 in humans after natural exposure. Very recently, a host range mutant adenovirus, capable of replicating in non-human primates has been developed and used in studies where PEI to Ad5 is required 79, 80. Several pilot studies using this virus in primates are currently planned to determine how virus replication alters the immune response to our vaccine and parameters required for protection from lethal Ebola challenge.
One of the most important findings described in this manuscript is that the ZGP-specific multifunctional T cell response induced by our vaccine is more predictive of survival from lethal Ebola challenge than the quantitative T cell response in rodents. This cannot be considered alone but in tandem with the strength of the antibody response in larger animal models of Ebola infection. We also found that induction of pre-existing immunity to Ad5 through the nasal mucosa, which closely models natural exposure, may be limiting for nasal vaccine platforms using adenoviral vectors but not for those that plan to administer the vaccine by other routes. Thus, establishing PEI in this manner during pre-clinical study of nasal vaccine candidates will provide the most stringent test for evaluating novel formulations or other strategies designed to bypass PEI. Lastly, we believe that formulations that can restore and strengthen components of the adaptive immune response impaired by PEI to Ad5 and which are necessary for protection from Ebola will greatly improve vaccine potency in naïve individuals and those with PEI.
Acknowledgments
The authors thank Jennifer Smith, Tatyana Yun and Bersabeh Tigabu of the UTMB BSL-4 facility for their help with Ebola challenge studies. We would also like to thank Dr. Erica Ollmann Saphire and Marnie Fucso of The Scripps Research Institute for providing Zaire Ebola GP33-637 TM-HA plasmid and assistance with glycoprotein purification. This work was funded by the National Institutes of Health NIAID Grant U01AI078045 (MAC).
Cited Literature
- 1.Kron MW, Kreppel F. Adenovirus Vectors and Subviral Particles for Protein and Peptide Delivery. Curr Gene Ther. 2012;12(5):362–373. doi: 10.2174/156652312802762563. [DOI] [PubMed] [Google Scholar]
- 2.Rollier CS, Reyes-Sandoval A, Cottingham MG, Ewer K, Hill AV. Viral Vectors as Vaccine Platforms: Deployment in Sight. Curr Opin Immunol. 2011;23(3):377–382. doi: 10.1016/j.coi.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Bassett JD, Swift SL, Bramson JL. Optimizing Vaccine-Induced CD8(+) T-Cell Immunity: Focus on Recombinant Adenovirus Vectors. Expert Rev Vaccines. 2011;10(9):1307–1319. doi: 10.1586/erv.11.88. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Jex E, Feng T, Sivko GS, Baillie LW, Goldman S, Van Kampen KR, Tang DC. An Adenovirus-Vectored Nasal Vaccine Confers Rapid and Sustained Protection against Anthrax in a Single-Dose Regimen. Clin Vaccine Immunol. 2013;20(1):1–8. doi: 10.1128/CVI.00280-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnes E, Folgori A, Capone S, Swadling L, Aston S, Kurioka A, Meyer J, Huddart R, Smith K, Townsend R, Brown A, Antrobus R, Ammendola V, Naddeo M, O’hara G, Willberg C, Harrison A, Grazioli F, Esposito ML, Siani L, Traboni C, Oo Y, Adams D, Hill A, Colloca S, Nicosia A, Cortese R, Klenerman P. Novel Adenovirus-Based Vaccines Induce Broad and Sustained T Cell Responses to HCV in Man. Sci Transl Med. 2012;4(115):115ra1. doi: 10.1126/scitranslmed.3003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuldt NJ, And Amalfitano A. Malaria Vaccines: Focus on Adenovirus Based Vectors. Vaccine. 2012;30(35):5191–5198. doi: 10.1016/j.vaccine.2012.05.048. [DOI] [PubMed] [Google Scholar]
- 7.Fehlner-Gardiner C, Rudd R, Donovan D, Slate D, Kempf L, Badcock J. Comparing ONRAB® and RABORAL V-RG® Oral Rabies Vaccine Field Performance in Raccoons and Striped Skunks, New Brunswick, Canada, and Maine, USA. J Wildl Dis. 2012;48(1):157–167. doi: 10.7589/0090-3558-48.1.157. [DOI] [PubMed] [Google Scholar]
- 8.Grant-Klein RJ, Altamura LA, Schmaljohn CS. Progress in Recombinant DNA-Derived Vaccines for Lassa Virus and Filoviruses. Virus Res. 2011;162(1–2):148–161. doi: 10.1016/j.virusres.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Xu Q, Pichichero ME, Simpson LL, Elias M, Smith LA, Zeng M. An Adenoviral Vector-Based Mucosal Vaccine is Effective in Protection Against Botulism. Gene Ther. 2009;16(3):367–375. doi: 10.1038/gt.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saxena M, Van TT, Baird FJ, Coloe PJ, Smooker PM. Pre-Existing Immunity against Vaccine Vectors - Friend or Foe? Microbiology. 2013;159(Pt 1):1–11. doi: 10.1099/mic.0.049601-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaiss AK, Machado HB, Herschman HR. The Influence of Innate and Pre-Existing Immunity on Adenovirus Therapy. J Cell Biochem. 2009;108(4):778–790. doi: 10.1002/jcb.22328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu B, Zhou Y, Wu H, Wang Z, Zhan Y, Feng X, Geng R, Wu Y, Kong W, Yu X. Seroprevalence of Neutralizing Antibodies to Human Adenovirus Type 5 in Healthy Adults in China. J Med Virol. 2012;84(9):1408–1414. doi: 10.1002/jmv.23325. [DOI] [PubMed] [Google Scholar]
- 13.Pilankatta R, Chawla T, Khanna N, Swaminathan S. The Prevalence of Antibodies to Adenovirus Serotype 5 in an Adult Indian Population and Implications for Adenovirus Vector Vaccines. J Med Virol. 2010;82(3):407–414. doi: 10.1002/jmv.21721. [DOI] [PubMed] [Google Scholar]
- 14.Mast TC, Kierstead L, Gupta SB, Nikas AA, Kallas EG, Novitsky V, Mbewe B, Pitisuttithum P, Schechter M, Vardas E, Wolfe ND, Aste-Amezaga M, Casimiro DR, Coplan P, Straus WL, Shiver JW. International Epidemiology of Human Pre-Existing Adenovirus (Ad) Type-5, Type-6, Type-26 and Type-36 Neutralizing Antibodies: Correlates of High Ad5 Titers and Implications for Potential HIV Vaccine Trials. Vaccine. 2010;28(4):950–957. doi: 10.1016/j.vaccine.2009.10.145. [DOI] [PubMed] [Google Scholar]
- 15.Nwanegbo E, Vardas E, Gao W, Whittle H, Sun H, Rowe D, Robbins PD, Gambotto A. Prevalence of Neutralizing Antibodies to Adenoviral Serotypes 5 and 35 in the Adult Populations of the Gambia, South Africa, and the United States. Clin Diagn Lab Immunol. 2004;11(2):351–357. doi: 10.1128/CDLI.11.2.351-357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aldhamen YA, Seregin SS, Amalfitano A. Immune Recognition of Gene Transfer Vectors: Focus on Adenovirus as a Paradigm. Front Immunol. 2011;2:40. doi: 10.3389/fimmu.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varnavski AN, Calcedo R, Bove M, Gao G, Wilson JM. Evaluation of Toxicity from High-Dose Systemic Administration of Recombinant Adenovirus Vector in Vector-Naive and Pre-Immunized Mice. Gene Ther. 2005;12(5):427–436. doi: 10.1038/sj.gt.3302347. [DOI] [PubMed] [Google Scholar]
- 18.Pandey A, Singh N, Vemula SV, Couëtil L, Katz JM, Donis R, Sambhara S, Mittal SK. Impact of Preexisting Adenovirus Vector Immunity on Immunogenicity and Protection Conferred with an Adenovirus-Based H5N1 Influenza Vaccine. PLoS One. 2012;7(3):e33428. doi: 10.1371/journal.pone.0033428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richardson JS, Yao MK, Tran KN, Croyle MA, Strong JE, Feldmann H, Kobinger GP. Enhanced Protection Against Ebola Virus Mediated by an Improved Adenovirus-Based Vaccine. PLoS One. 2009;4(4):e5308. doi: 10.1371/journal.pone.0005308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shoji M, Yoshizaki S, Mizuguchi H, Okuda K, Shimada M. Immunogenic Comparison of Chimeric Adenovirus 5/35 Vector Carrying Optimized Human Immunodeficiency Virus Clade C Genes and Various Promoters. PLoS One. 2012;7(1):e30302. doi: 10.1371/journal.pone.0030302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teigler JE, Iampietro MJ, Barouch DH. Vaccination with Adenovirus Serotypes 35, 26, and 48 Elicits Higher Levels of Innate Cytokine Responses Than Adenovirus Serotype 5 in Rhesus Monkeys. J Virol. 2012;86(18):9590–9598. doi: 10.1128/JVI.00740-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colloca S, Barnes E, Folgori A, Ammendola V, Capone S, Cirillo A, Siani L, Naddeo M, Grazioli F, Esposito ML, Ambrosio M, Sparacino A, Bartiromo M, Meola A, Smith K, Kurioka A, O’hara GA, Ewer KJ, Anagnostou N, Bliss C, Hill AV, Traboni C, Klenerman P, Cortese R, Nicosia A. Vaccine Vectors Derived from a Large Collection of Simian Adenoviruses Induce Potent Cellular Immunity across Multiple Species. Sci Transl Med. 2012;4(115):115ra2. doi: 10.1126/scitranslmed.3002925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel A, Tikoo S, Kobinger G. A Porcine Adenovirus with Low Human Seroprevalence Is a Promising Alternative Vaccine Vector to Human Adenovirus 5 in an H5N1 Virus Disease Model. PLoS One. 2010;5(12):e15301. doi: 10.1371/journal.pone.0015301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du E, Tikoo SK. Efficient Replication and Generation of Recombinant Bovine Adenovirus-3 in Non-Bovine Cotton Rat Lung Cells Expressing I-Scel Endonuclease. J Gene Med. 2010;12(10):840–847. doi: 10.1002/jgm.1505. [DOI] [PubMed] [Google Scholar]
- 25.Abbink P, Lemckert AA, Ewald BA, Lynch DM, Denholtz M, Smits S, Holterman L, Damen I, Vogels R, Thorner AR, O’brien KL, Carville A, Mansfield KG, Goudsmit J, Havenga MJ, Barouch DH. Comparative Seroprevalence and Immunogenicity of Six Rare Serotype Recombinant Adenovirus Vaccine Vectors from Subgroups B and D. J Virol. 2007;81(9):4654–63. doi: 10.1128/JVI.02696-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaufmann JK, And Nettelbeck DM. Virus Chimeras for Gene Therapy, Vaccination, and Oncolysis: Adenoviruses and Beyond. Trends Mol Med. 2012;18(7):365–376. doi: 10.1016/j.molmed.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 27.Wortmann A, Vöhringer S, Engler T, Corjon S, Schirmbeck R, Reimann J, Kochanek S, Kreppel F. Fully Detargeted Polyethylene Glycol-Coated Adenovirus Vectors are Potent Genetic Vaccines and Escape from Pre-Existing Anti-Adenovirus Antibodies. Mol Ther. 2008;16(1):154–162. doi: 10.1038/sj.mt.6300306. [DOI] [PubMed] [Google Scholar]
- 28.Khare R, Chen CY, Weaver EA, Barry MA. Advances and Future Challenges in Adenoviral Vector Pharmacology and Targeting. Curr Gene Ther. 2011;11(4):241–258. doi: 10.2174/156652311796150363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matthews QL. Capsid-Incorporation of Antigens into Adenovirus Capsid Proteins for a Vaccine Approach. Mol Pharm. 2010;8(1):3–11. doi: 10.1021/mp100214b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De BP, Pagovich OE, Hicks MJ, Rosenberg JB, Moreno AY, Janda KD, Koob GF, Worgall S, Kaminsky SM, Sondhi D, Crystal RG. Disrupted Adenovirus-Based Vaccines against Small Addictive Molecules Circumvent Anti-Adenovirus Immunity. Hum Gene Ther. 2013;24(1):58–66. doi: 10.1089/hum.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scherliess R. Delivery of Antigens Used for Vaccination: Recent Advances and Challenges. Ther Deliv. 2011;2(10):1351–1368. doi: 10.4155/tde.11.80. [DOI] [PubMed] [Google Scholar]
- 32.Lynch JP, Fishbein M, Echavarria M. Adenovirus. Semin Respir Crit Care Med. 2011;32(4):494–511. doi: 10.1055/s-0031-1283287. [DOI] [PubMed] [Google Scholar]
- 33.Choi JH, Schafer SC, Zhang L, Kobinger GP, Juelich T, Freiberg AN, Croyle MA. A Single Sublingual Dose of an Adenovirus-Based Vaccine Protects against Lethal Ebola Challenge in Mice and Guinea Pigs. Mol Pharm. 2012;9(1):156–167. doi: 10.1021/mp200392g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Croyle MA, Patel A, Tran KN, Gray M, Zhang Y, Strong JE, Feldmann H, Kobinger GP. Nasal Delivery of an Adenovirus-Based Vaccine Bypasses Pre-Existing Immunity to the Vaccine Carrier and Improves the Immune Response in Mice. PLoS One. 2008;3(10):e3548. doi: 10.1371/journal.pone.0003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. A Mouse Model for Evaluation of Prophylaxis and Therapy of Ebola Hemorrhagic Fever. J Infect Dis. 1998;178(3):651–661. doi: 10.1086/515386. [DOI] [PubMed] [Google Scholar]
- 36.Connolly BM, Steele KE, Davis KJ, Geisbert TW, Kell WM, Jaax NK, Jahrling PB. Pathogenesis of Experimental Ebola Virus Infection in Guinea Pigs. J Infect Dis. 1999;179 (Suppl 1):S203–S217. doi: 10.1086/514305. [DOI] [PubMed] [Google Scholar]
- 37.Rao M, Matyas GR, Grieder F, Anderson K, Jahrling PB, Alving CR. Cytotoxic T Lymphocytes to Ebola Zaire Virus are Induced in Mice by Immunization with Liposomes Containing Lipid A. Vaccine. 1999;17(23–24):2991–2998. doi: 10.1016/s0264-410x(99)00170-x. [DOI] [PubMed] [Google Scholar]
- 38.Lee JE, Fusco ML, Ollman-Saphire E. An Efficient Platform for Screening Expression and Crystallization of Glycoproteins Produced in Human Cells. Nat Protoc. 2009;4(4):592–604. doi: 10.1038/nprot.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi JH, Dekker J, Schafer SC, John J, Whitfill CE, Petty CS, Haddad EE, Croyle MA. Optimized Adenovirus-Antibody Complexes Stimulate Strong Cellular and Humoral Immune Responses against an Encoded Antigen in Naive Mice and Those with Pre-Existing Immunity. Clin Vaccine Immunol. 2012;19(1):84–95. doi: 10.1128/CVI.05319-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL, Roederer M, Seder RA. Multifunctional Th1 Cells Define a Correlate of Vaccine-Mediated Protection against Leishmania Major. Nat Med. 2007;13(7):843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 41.Lindsay RW, Darrah PA, Quinn KM, Wille-Reece U, Mattei LM, Iwasaki A, Kasturi SP, Pulendran B, Gall JG, Spies AG, Seder RA. CD8+ T Cell Responses Following Replication-Defective Adenovirus Serotype 5 Immunization Are Dependent on CD11c+ Dendritic Cells but Show Redundancy in Their Requirement of TLR and Nucleotide-Binding Oligomerization Domain-Like Receptor Signaling. J Immunol. 2010;185(3):1513–1521. doi: 10.4049/jimmunol.1000338. [DOI] [PubMed] [Google Scholar]
- 42.Paessler S, Walker DH. Pathogenesis of the Viral Hemorrhagic Fevers. Annu Rev Pathol. 2013;8:411–440. doi: 10.1146/annurev-pathol-020712-164041. [DOI] [PubMed] [Google Scholar]
- 43.Hutchinson KL, Rollin PE. Cytokine and Chemokine Expression in Humans Infected with Sudan Ebola Virus. J Infect Dis. 2007;196(Suppl 2):S357–S363. doi: 10.1086/520611. [DOI] [PubMed] [Google Scholar]
- 44.Villinger F, Rollin PE, Brar SS, Chikkala NF, Winter J, Sundstrom JB, Zaki SR, Swanepoel R, Ansari AA, Peters CJ. Markedly Elevated Levels of Interferon (IFN)-Gamma, IFN-Alpha, Interleukin (IL)-2, IL-10, and Tumor Necrosis Factor-Alpha Associated with Fatal Ebola Virus Infection. J Infect Dis. 1999;179(Suppl 1):S188–S191. doi: 10.1086/514283. [DOI] [PubMed] [Google Scholar]
- 45.Bray M, Hatfill S, Hensley L, Huggins JW. Haematological, Biochemical and Coagulation Changes in Mice, Guinea-Pigs and Monkeys Infected with a Mouse-Adapted Variant of Ebola Zaire Virus. J Comp Pathol. 2001;125(4):243–253. doi: 10.1053/jcpa.2001.0503. [DOI] [PubMed] [Google Scholar]
- 46.Fausther-Bovendo H, Mulangu S, Sullivan NJ. Ebolavirus Vaccines for Humans and Apes. Curr Opin Virol. 2012;2(3):324–329. doi: 10.1016/j.coviro.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoenen T, Groseth A, Feldmann H. Current Ebola Vaccines. Expert Opin Biol Ther. 2012;12(7):859–872. doi: 10.1517/14712598.2012.685152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bukreyev AA, Dinapoli JM, Yang L, Murphy BR, Collins PL. Mucosal Parainfluenza Virus-Vectored Vaccine against Ebola Virus Replicates in the Respiratory Tract of Vector-Immune Monkeys and is Immunogenic. Virology. 2010;399(2):290–298. doi: 10.1016/j.virol.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geisbert TW, Feldmann H. Recombinant Vesicular Stomatitis Virus-Based Vaccines against Ebola and Marburg Virus Infections. J Infect Dis. 2011;204(Suppl 3):S1075–S1081. doi: 10.1093/infdis/jir349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warfield KL, Aman MJ. Advances in Virus-Like Particle Vaccines for Filoviruses. J Infect Dis. 2011;204(Suppl 3):S1053–S1059. doi: 10.1093/infdis/jir346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kobinger GP, Feldmann H, Zhi Y, Schumer G, Gao G, Feldmann F, Jones S, Wilson JM. Chimpanzee Adenovirus Vaccine Protects against Zaire Ebola Virus. Virology. 2005;346(2):394–401. doi: 10.1016/j.virol.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 52.Geisbert TW, Bailey M, Hensley L, Asiedu C, Geisbert J, Stanley D, Honko A, Johnson J, Mulangu S, Pau MG, Custers J, Vellinga J, Hendriks J, Jahrling P, Roederer M, Goudsmit J, Koup R, Sullivan NJ. Recombinant Adenovirus Serotype 26 (Ad26) and Ad35 Vaccine Vectors Bypass Immunity to Ad5 and Protect Non-Human Primates against Ebolavirus Challenge. J Virol. 2011;85(9):4222–4233. doi: 10.1128/JVI.02407-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sullivan NJ, Sanchez A, Rollin PE, Yang ZY, Nabel GJ. Development of a Preventive Vaccine for Ebola Virus Infection in Primates. Nature. 2000;408(6812):605–609. doi: 10.1038/35046108. [DOI] [PubMed] [Google Scholar]
- 54.Weingartl HM, Embury-Hyatt C, Nfon C, Leung A, Smith G, Kobinger G. Transmission of Ebola Virus from Pigs to Non-Human Primates. Sci Rep. 2012;2(811) doi: 10.1038/srep00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kobinger GP, Leung A, Neufeld J, Richardson JS, Falzarano D, Smith G, Tierney K, Patel A, Weingartl HM. Replication, Pathogenicity, Shedding, and Transmission of Zaire Ebolavirus in Pigs. J Infect Dis 2011. 2011;204(2):200–208. doi: 10.1093/infdis/jir077. [DOI] [PubMed] [Google Scholar]
- 56.Feldmann H, And Geisbert TW. Ebola Haemorrhagic Fever. Lancet. 2011;377(9768):849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sullivan NJ, Hensley L, Asiedu C, Geisbert TW, Stanley D, Johnson J, Honko A, Olinger G, Bailey M, Geisbert JB, Reimann KA, Bao S, Rao S, Roederer M, Jahrling PB, Koup RA, Nabel GJ. CD8+ Cellular Immunity Mediates rAd5 Vaccine Protection against Ebola Virus Infection of Nonhuman Primates. Nat Med. 2011;17(9):1128–1131. doi: 10.1038/nm.2447. [DOI] [PubMed] [Google Scholar]
- 58.Wong G, Richardson JS, Pillet S, Patel A, Qiu X, Alimonti J, Hogan J, Zhang Y, Takada A, Feldmann H, Kobinger GP. Immune Parameters Correlate with Protection against Ebola Virus Infection in Rodents and Nonhuman Primates. Sci Transl Med. 2012;4(158):158ra146. doi: 10.1126/scitranslmed.3004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pavot V, Rochereau N, Genin C, Verrier B, Paul S. New Insights in Mucosal Vaccine Development. Vaccine. 2012;30(2):142–154. doi: 10.1016/j.vaccine.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 60.Czerkinsky C, Holmgren J. Mucosal Delivery Routes for Optimal Immunization: Targeting Immunity to the Right Tissues. Curr Top Microbiol Immunol. 2012;354:1–18. doi: 10.1007/82_2010_112. [DOI] [PubMed] [Google Scholar]
- 61.Patel A, Zhang Y, Croyle M, Tran K, Gray M, Strong J, Feldmann H, Wilson JM, Kobinger GP. Mucosal Delivery of Adenovirus-Based Vaccine Protects against Ebola Virus Infection in Mice. J Infect Dis. 2007;196(Suppl 2):S413–S420. doi: 10.1086/520603. [DOI] [PubMed] [Google Scholar]
- 62.Sumida SM, Truitt DM, Kishko MG, Arthur JC, Jackson SS, Gorgone DA, Lifton MA, Koudstaal W, Pau MG, Kostense S, Havenga MJ, Goudsmit J, Letvin NL, Barouch DH. Neutralizing Antibodies and CD8+ T Lymphocytes Both Contribute to Immunity to Adenovirus Serotype 5 Vaccine Vectors. J Virol. 2004;78(6):2666–2673. doi: 10.1128/JVI.78.6.2666-2673.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Y, And Wilson JM. Clearance of Adenovirus-Infected Hepatocytes by MHC Class I-Restricted CD4+ CTLs in Vivo. J Immunol. 1995;155(5):2564–2570. [PubMed] [Google Scholar]
- 64.Yang Y, Ertl HC, Wilson JM. MHC Class I-Restricted Cytotoxic T Lymphocytes to Viral Antigens Destroy Hepatocytes in Mice Infected with E1-Deleted Recombinant Adenoviruses. Immunity. 1994;1(5):433–442. doi: 10.1016/1074-7613(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 65.Yang Y, Xiang Z, Ertl HC, Wilson JM. Upregulation of Class I Major Histocompatibility Complex Antigens by Interferon Gamma Is Necessary for T-Cell-Mediated Elimination of Recombinant Adenovirus-Infected Hepatocytes in Vivo. Proc Natl Acad Sci U S A. 1995;92(16):7257–7261. doi: 10.1073/pnas.92.16.7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rozis G, De Silva S, Benlahrech A, Papagatsias T, Harris J, Gotch F, Dickson G, Patterson S. Langerhans Cells Are More Efficiently Transduced Than Dermal Dendritic Cells by Adenovirus Vectors Expressing Either Group C or Group B Fibre Protein: Implications for Mucosal Vaccines. Eur J Immunol. 2005;35(9):2617–2626. doi: 10.1002/eji.200425939. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Chirmule N, Gao GP, Qian R, Croyle M, Joshi B, Tazelaar J, Wilson JM. Acute Cytokine Response to Systemic Adenoviral Vectors in Mice Is Mediated by Dendritic Cells and Macrophages. Mol Ther. 2001;3(5 Pt 1):697–707. doi: 10.1006/mthe.2001.0329. [DOI] [PubMed] [Google Scholar]
- 68.Steffensen MA, Jensen BA, Holst PJ, Bassi MR, Christensen JP, Thomsen AR. Pre-Existing Vector Immunity Does Not Prevent Replication Deficient Adenovirus from Inducing Efficient CD8 T-Cell Memory and Recall Responses. PLoS One. 2012;7(4):e34884. doi: 10.1371/journal.pone.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kweon MN. Sublingual Mucosa: A New Vaccination Route for Systemic and Mucosal Immunity. Cytokine. 2011;54(1):1–5. doi: 10.1016/j.cyto.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 70.Kwekkeboom J. Modulation of Dendritic Cells and Regulatory T Cells by Naturally Occurring Antibodies. Adv Exp Med Biol. 2012;750:133–144. doi: 10.1007/978-1-4614-3461-0_10. [DOI] [PubMed] [Google Scholar]
- 71.Pletinckx K, Döhler A, Pavlovic V, Lutz MB. Role of Dendritic Cell Maturity/Costimulation for Generation, Homeostasis, and Suppressive Activity of Regulatory T Cells. Front Immunol. 2011;2(39) doi: 10.3389/fimmu.2011.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qiu X, Audet J, Wong G, Pillet S, Bello A, Cabral T, Strong JE, Plummer F, Corbett CR, Alimonti JB, Kobinger GP. Successful Treatment of Ebola Virus-Infected Cynomolgus Macaques with Monoclonal Antibodies. Sci Transl Med. 2012;4(138):138ra81. doi: 10.1126/scitranslmed.3003876. [DOI] [PubMed] [Google Scholar]
- 73.Qiu X, Fernando L, Melito PL, Audet J, Feldmann H, Kobinger G, Alimonti JB, Jones SM. Ebola GP-Specific Monoclonal Antibodies Protect Mice and Guinea Pigs from Lethal Ebola Virus Infection. PLoS Negl Trop Dis. 2012;6(3) doi: 10.1371/journal.pntd.0001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seder RA, Darrah PA, Roederer M. T-Cell Quality in Memory and Protection: Implications for Vaccine Design. Nat Rev Immunol. 2008;8(4):247–58. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 75.Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 Signals During Priming Are Required for Secondary Expansion of CD8+ Memory T Cells. Nature. 2006;441(7095):890–3. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lichterfeld M, Yu XG, Waring MT, Mui SK, Johnston MN, Cohen D, Addo MM, Zaunders J, Alter G, Pae E, Strick D, Allen TM, Rosenberg ES, Walker BD, Altfeld M. HIV-1-Specific Cytotoxicity Is Preferentially Mediated by a Subset of CD8(+) T Cells Producing Both Interferon-Gamma and Tumor Necrosis Factor-Alpha. Blood. 2004;104(2):487–94. doi: 10.1182/blood-2003-12-4341. [DOI] [PubMed] [Google Scholar]
- 77.Sandberg JK, Fast NM, Nixon DF. Functional Heterogeneity of Cytokines and Cytolytic Effector Molecules in Human CD8+ T Lymphocytes. J Immunol. 2001;167(1):181–7. doi: 10.4049/jimmunol.167.1.181. [DOI] [PubMed] [Google Scholar]
- 78.Hoeben RC, Uil TG. Adenovirus DNA Replication. Cold Spring Harb Perspect Biol. 2013;5(3):a013003. doi: 10.1101/cshperspect.a013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Demberg T, Florese RH, Heath MJ, Larsen K, Kalisz I, Kalyanaraman VS, Lee EM, Pal R, Venzon D, Grant R, Patterson LJ, Korioth-Schmitz B, Buzby A, Dombagoda D, Montefiori DC, Letvin NL, Cafaro A, Ensoli B, Robert-Guroff M. A Replication-Competent Adenovirus-Human Immunodeficiency Virus (Ad-HIV) Tat and Ad-HIV Env Priming/Tat and Envelope Protein Boosting Regimen Elicits Enhanced Protective Efficacy against Simian/Human Immunodeficiency Virus SHIV 89.6P Challenge in Rhesus Macaques. J Virol. 2007;81(7):3414–27. doi: 10.1128/JVI.02453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Masek-Hammerman K, Li H, Liu J, Abbink P, La Porte A, O’brien KL, Whitney JB, Carville A, Mansfield KG, Barouch DH. Mucosal Trafficking of Vector-Specific CD4+ T Lymphocytes Following Vaccination of Rhesus Monkeys with Adenovirus Serotype 5. J Virol. 2010;84(19):9810–6. doi: 10.1128/JVI.01157-10. [DOI] [PMC free article] [PubMed] [Google Scholar]