

Significance
Chronic granulomatous disease (CGD) has an immunodeficiency component and, in addition, an autoinflammatory component in which autophagy and inflammasome activation are linked and amenable to IL-1 blockade. This study provides a rationale to perform clinical trials to investigate the efficacy of blocking IL-1 in CGD colitis and expands the therapeutic potential of IL-1 antagonists to inflammatory diseases with defective autophagy.
Keywords: interleukin-1, S. aureus, LPS, Crohn disease, autoinflammatory disease
Abstract
Patients with chronic granulomatous disease (CGD) have a mutated NADPH complex resulting in defective production of reactive oxygen species; these patients can develop severe colitis and are highly susceptible to invasive fungal infection. In NADPH oxidase-deficient mice, autophagy is defective but inflammasome activation is present despite lack of reactive oxygen species production. However, whether these processes are mutually regulated in CGD and whether defective autophagy is clinically relevant in patients with CGD is unknown. Here, we demonstrate that macrophages from CGD mice and blood monocytes from CGD patients display minimal recruitment of microtubule-associated protein 1 light chain 3 (LC3) to phagosomes. This defect in autophagy results in increased IL-1β release. Blocking IL-1 with the receptor antagonist (anakinra) decreases neutrophil recruitment and T helper 17 responses and protects CGD mice from colitis and also from invasive aspergillosis. In addition to decreased inflammasome activation, anakinra restored autophagy in CGD mice in vivo, with increased Aspergillus-induced LC3 recruitment and increased expression of autophagy genes. Anakinra also increased Aspergillus-induced LC3 recruitment from 23% to 51% (P < 0.01) in vitro in monocytes from CGD patients. The clinical relevance of these findings was assessed by treating CGD patients who had severe colitis with IL-1 receptor blockade using anakinra. Anakinra treatment resulted in a rapid and sustained improvement in colitis. Thus, inflammation in CGD is due to IL-1–dependent mechanisms, such as decreased autophagy and increased inflammasome activation, which are linked pathological conditions in CGD that can be restored by IL-1 receptor blockade.
Chronic granulomatous disease (CGD) is an immunodeficiency characterized by defective production of reactive oxygen species (ROS) (1) due to mutations in the proteins forming the NADPH complex (2, 3). The most frequent form of CGD is hereditary and X-linked, and is caused by a mutation in the gene CYBB, which encodes the protein gp91phox, the catalytic subunit of the NADPH oxidase complex. In autosomal and recessive forms of CGD, the mutations affect the genes encoding p22phox, p67phox, p40phox, or p47phox, which are all part of the NADPH complex, resulting in a defective NADPH oxidase complex. As a result, patients who have CGD have defective microbial killing by phagocytic cells and an increased susceptibility to infections, especially Staphylococcus aureus and Aspergillus spp. (4, 5). Paradoxically, ROS deficiency in patients with CGD results in a hyperinflammatory state (6), and one-third of the patients develop an inflammatory colitis indistinguishable from Crohn disease (7–10).
The hyperinflammatory state in CGD is linked to inflammasome activation (11–13). Studies in mice and humans reveal that autophagy is crucial for IL-1β transcription (14) and processing of pro–IL-1β (15, 16); defects in autophagy result in increased secretion of IL-1β. ROS production is commonly believed to be necessary for autophagy (17), and mice deficient in autophagy (ATG161−/−) can develop severe colitis and exhibit increased production of IL-1β (18).
In the present study, we aimed to decipher the link between autophagy and inflammasome activation in CGD. ROS deficiency in CGD mice and patients with CGD resulted in defective autophagy, eventually leading to increased IL-1β secretion and IL-1–dependent inflammation. Blocking the IL-1 receptor (IL-1R) with anakinra (the recombinant form of the naturally occurring IL-1R antagonist) not only limited inflammasome activation but restored protective autophagy in CGD.
Results
ROS Deficiency Results in Defective Autophagy.
The autophagosome-associated protein light chain 3 (LC3) is recruited to macrophage phagosomes upon engulfment of bacteria (19). To test whether p40phox, encoding the p40phox subunit of NADPH oxidase, is required for LC3 recruitment to the autophagosomes, peritoneal macrophages were isolated from LC3-GFP transgenic and p40phox−/−mice crossed with LC3-GFP mice and exposed to adherent and invasive Escherichia coli (AIEC) strain LF82. This Crohn disease-associated strain of E. coli has previously been shown to accumulate preferentially in epithelial cells deficient in autophagy (20). LC3-GFP is recruited to phagocytized AIEC within macrophages obtained from WT mice expressing LC3-GFP within 30 min of infection (Fig. 1A and Fig. S1A). However, in p40phox−/−mice, there is minimal recruitment of LC3 to internalized bacteria. Quantification of the fraction of internalized bacteria surrounded by LC3-GFP revealed significantly more recruitment in WT compared with p40phox−/− macrophages (P < 0.001) (Fig. 1A). Thus, p40phox, which is necessary for NADPH-dependent ROS production, is required for LC3 recruitment to engulfed bacteria.
Fig. 1.
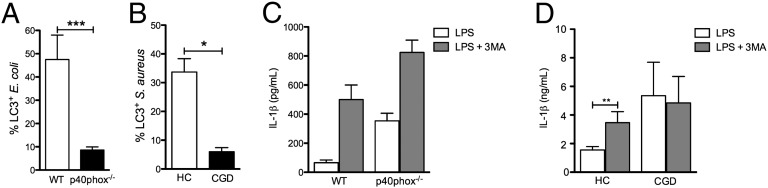
Effects of ROS deficiency on autophagy and IL-1β release from murine macrophages and human monocytes. (A) Mean ± SEM percentage of colocalization of LC3 with E. coli (strain LF82) in peritoneal macrophages from WT and p40phox−/−mice after 30 min of exposure. (B) Mean ± SEM percentage of LC3 colocalized with S. aureus in monocytes from HCs and patients with CGD following a 1-h exposure to FITC-labeled S. aureus. Data are representative of one of two separate observations in two patients with CGD. (C) Mean ± SEM. IL-1β was secreted from BMMs of WT and p40phox−/−mice stimulated with 100 ng/mL LPS in the absence (open bars) or presence (filled bars) of 10 nM 3MA. (D) IL-1β released from PBMCs of four patients with CGD and four HCs under the same conditions described in C. *P < 0.05; **P < 0.01; ***P < 0.001.
To investigate whether NADPH-dependent ROS in humans is important for LC3 recruitment to the phagosome, we assessed LC3 recruitment to the phagosome in cells isolated from patients with CGD and healthy controls (HCs). The cells were exposed to FITC-labeled S. aureus, a prominent pathogen in CGD (5), and were subsequently stained for LC3. The percentage of colocalization of S. aureus-FITC with labeled LC3 was determined (Fig. 1B and Fig. S1B). Monocytes from patients with CGD showed significantly less colocalization between LC3 and phagocytized S. aureus compared with monocytes from HCs (Fig. 1B). Therefore, the NADPH component p47phox in humans is required for LC3 recruitment upon engulfment of microorganisms.
Defects in Autophagy in p40phox−/− Mice and Patients with CGD Result in Increased IL-1β Production.
Because autophagy inhibits IL-1β production (15, 16), we assessed whether defective autophagy in ROS-deficient mice is responsible for the increased IL-1β secretion. Bone marrow-derived macrophages (BMMs) in the presence (WT) or absence (p40phox−/−) of ROS were stimulated with LPS. As shown in Fig. 1C, LPS treatment induced IL-1β in both WT and p40phox−/− BMMs, but p40phox−/− BMMs produced nearly sixfold greater levels of the cytokine after stimulation (WT: 61 ± 20 pg/mL and p40phox−/−: 354 ± 50 pg/mL; P < 0.001). Blocking autophagy with 3-methyladenine (3MA) pretreatment enhanced LPS-induced IL-1β secretion in both WT and p40phox−/−BMMs (WT: 502 ± 100 pg/mL and p40phox−/−: 828 ± 86 pg/mL; P < 0.01; Fig. 1C). However, the IL-1β production due to inhibition of autophagy by 3MA was increased twofold in p40phox−/−BMMs compared with 8.5-fold increased in WT BMMs (Fig. 1C). Next, we investigated whether the same differences are present in patients with CGD. In cells of HCs, autophagy inhibition by 3MA significantly increases LPS-induced IL-1β production (P < 0.01) (Fig. 1D). In contrast, 3MA did not increase IL-1β secretion in LPS-stimulated peripheral blood mononuclear cells (PBMCs) from patients with CGD (Fig. 1D). Collectively, these data provide evidence that a defect in autophagy is responsible for the increased IL-1β production in patients with CGD.
Blocking IL-1 Is Beneficial in p47phox−/− Mice with Colitis.
Colitis is a severe manifestation of CGD (21). To investigate whether the deficiency of NADPH-dependent ROS leading to defective autophagy and subsequently increased IL-1β production is relevant in vivo, we evaluated the effects of blocking IL-1 with anakinra in WT and p47phox−/− mice with 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis. We observed that TNBS-treated p47phox−/− mice have significantly more weight loss compared with WT mice (more than 20% loss of their initial body weight on day 5) upon TNBS treatment (Fig. 2A) and display severe inflammatory histopathology (Fig. 2B and Fig. S1A) and inflammatory cytokine responses (Fig. S1B). By contrast, TNBS-treated mice p47phox−/− mice rapidly regained weight after anakinra treatment, histological examination of colonic tissues and blinded histological scoring of colitis improved, and inflammatory cytokines were significantly lower (Fig. 2 A and B and Fig. S1 A and B). Thus, anakinra has significant beneficial effects on the outcome of colitis in CGD mice.
Fig. 2.

Blocking IL-1R in p47phox−/−mice with colitis and invasive aspergillosis. C57BL/6 or p47phox−/−mice received 2.5 mg of TNBS dissolved in 50% (vol/vol) ethanol given intrarectally and were treated i.p. with daily anakinra (10 mg/kg). Control mice received 50% ethanol. (A) Mean ± SEM weight change in two experiments. dpi, days postinfection. (B) Mean ± SEM histological score of the colon from one of two experiments. (C and D) C57BL/6 or p47phox−/−mice were infected intranasally with live A. fumigatus conidia and treated with daily anakinra (1 or 10 mg/kg). (C) Mean ± SEM percentage of survival. (D) Mean ± SEM log10 cfu in the lungs of infected mice (day 7). (E) Mean ± SEM cytokine levels in lung homogenates. (F) Mean ± SEM fold difference in MPO mRNA levels in the lungs at day 7. Data are representative of three experiments with four to eight mice per group. *P < 0.05; **P < 0.01; ***P < 0.001.
Blocking IL-1 Protects p47phox−/− Mice from Invasive Aspergillosis.
A prominent complication of CGD is the increased susceptibility to pulmonary aspergillosis that is associated with a marked inflammatory response (22). To investigate whether blocking IL-1R would decrease detrimental inflammatory responses in CGD mice with aspergillosis, WT and p47phox−/− mice were infected with Aspergillus fumigatus and treated with anakinra daily. Mice were monitored for survival, local fungal growth, inflammatory cell recruitment in the bronchoalveolar lavage (BAL), and lung histopathology. In contrast to WT mice, the majority of p47phox−/− mice died of the infection (Fig. 2C), were unable to restrict fungal growth in the lung (Fig. 2D), and showed mycotic pneumonia at necropsy with more than half of the pulmonary parenchyma being involved (Fig. S2A), as well as BAL neutrophilia (Fig. S2A, Inset). Anakinra at 10 mg/kg, a dose that is known to be pharmacologically active in mice and mimics human therapeutic dosages (23), significantly increased survival (Fig. 2C), reduced fungal growth (Fig. 2D), decreased IL-1β (Fig. 2E), decreased BAL neutrophilia, decreased levels of MPO (Fig. 2F and Fig. S2A, Inset), and ameliorated lung pathology (Fig. S2A) in p47phox−/− mice. The extent of granuloma formation was also significantly reduced by anakinra in CGD mice, as observed by gross pathology. These data demonstrate that anakinra restores immunocompetence in CGD by inhibiting excess neutrophil influx in the lung during infection and restraining fungal growth in p47phox−/− mice.
The increased susceptibility of p47phox−/− mice to Aspergillus infection is associated with failure to activate protective T helper (Th) 1 and regulatory T-cell responses and the occurrence of inflammatory Th17 cells (22). Therefore, we also evaluated parameters of adaptive Th immunity in WT and p47phox−/− mice. Blocking the IL-1R with anakinra greatly reduced IL-17A (Fig. 2E) and increased IFN-γ and IL-10 production in p47phox−/−-infected mice (Fig. S2 B and C). Notably, treatment of anakinra in WT mice with invasive aspergillosis resulted in decreased IFN-γ production without affecting IL-17A and IL-10 production. Anakinra inhibited IL-4 in both WT and CGD mice (Fig. S2 B and C). Thus, blocking the IL-1R modulates innate and adaptive antifungal immune responses in murine CGD.
Blocking IL-1 Inhibits Increased Inflammasome Activation in p47phox−/− Mice.
To investigate whether blocking IL-1 could modulate increased inflammasome activation, which has been described in patients with CGD (11–13), we evaluated inflammasome activation in p47phox−/− and WT mice with and without anakinra treatment during invasive aspergillosis. Consistent with previous reports, we observed increased inflammasome activation in p47phox−/− compared with WT mice (Fig. 3 A and B). Treatment with anakinra during invasive aspergillosis resulted in decreased processing of caspase-1 in p47phox−/− mice during infection (Fig. 3 A and B).
Fig. 3.

Anakinra inhibits inflammasome activation and restores autophagy in p47phox−/−mice with invasive aspergillosis. (A) Western blotting of pro- and processed caspase-1 in lung homogenates on day 20 following infection with live A. fumigatus in untreated (None) and anakinra-treated mice compared with uninfected mice (Naive). β-tub, β-tubulin. (B) Density of bands from A. Data are representative of three experiments. (C) Mean percentage of LC3+ cells in EGFP-LC3–transfected RAW 264.7 cells exposed to A. fumigatus SC in the absence (None) or presence of anakinra (1 or 10 μg/mL) or rapamycin (50 μM) for 4 h (n = 3). **P < 0.01 (anakinra- or rapamycin-treated vs. untreated cells). (D) Western blot of LC3b and p62 in homogenates of lungs of naive, Aspergillus-infected untreated, or Aspergillus-infected anakinra-treated (10 mg/kg) mice on day 7 postinfection. (E) Density of bands from D. (F) LC3 staining in alveolar macrophages from C57BL/6 and p47phox−/−mice exposed to 20 μg/mL poly(I:C) (Sigma–Aldrich), A. fumigatus SC, and/or 10 μg/mL anakinra for 2 h. (Magnification: 100×.) DAPI was used to detect nuclei. One experiment representative of two experiments is shown.
Autophagy in p47phox−/− Mice Is Restored by Inhibiting the IL-1R Pathway.
Because autophagy and inflammasome activation are reciprocally regulated (24), we assessed the effects of inhibiting the IL-1 pathway on autophagy. We investigated autophagy in response to Aspergillus in vitro and in vivo in the presence of anakinra. We transiently transfected RAW 264.7 cells with the EGFP-LC3 plasmid and cultured the macrophages with live swollen conidia (SC) (25). Rapamycin, a known inducer of autophagy, was used as a positive control. Similar to rapamycin, blocking IL-1 dose-dependently increased autophagy in response to SC, as indicated by the increased number of cells with punctate dots containing EGFP-LC3 (Fig. 3C and Fig. S3A); the increased ratio of LC3-II/I (Fig. S3B); and the decreased p62, a ubiquitin-binding protein that is selectively degraded by autophagy (Fig. S3B). Furthermore, blocking IL-1 decreased caspase-1 cleavage in macrophages exposed to Aspergillus SC (Fig. S3C), which is similar to findings from the in vivo experiments. In vivo, blocking IL-1 promoted autophagy in WT and p47phox−/− mice with invasive aspergillosis, as revealed by LC3-II/I and p62 immunoblotting (Fig. 3 D and E) and steady-state mRNA levels of selective autophagy genes, such as LC3a, LC3b, Bcn1, Atg4a, Atg4b, and Atg5 (Fig. S3E). LC3 immunofluorescence in ex vivo purified macrophages from p47phox−/− mice demonstrated that blocking IL-1 restores defective autophagy to Aspergillus SC in CGD (Fig. 3F).
Inhibiting IL-1 Restores Autophagy in Human CGD Cells.
We investigated whether inhibition of IL-1 could also restore defective autophagy in human CGD cells. Monocytes isolated from patients with CGD were exposed to Aspergillus SC or spores in the absence or presence of anakinra. Monocytes from patients with CGD show significantly less colocalization between LC3 and phagocytized Aspergillus spores or SC compared with monocytes isolated from HCs (Fig. 4A). However, when the IL-1R was blocked, the capacity of monocytes from patients with CGD to recruit LC3 upon engulfment of Aspergillus increased to levels comparable to those with LC3 recruitment in monocytes from healthy subjects (Fig. 4B). Thus, blocking the IL-1 activity not only decreases inflammasome activation but restores defective autophagy in murine and human CGD cells.
Fig. 4.

Anakinra restores autophagy in human CGD cells and reduces disease severity in patients with CGD colitis. (A) Percentage of colocalization of LC3 with resting conidia (RC) or SC in PBMCs isolated from two patients with CGD and two HCs. (B) Percentage of colocalization of LC3 with RC or SC in the same PBMCs isolated from two patients with CGD in the absence or presence of anakinra (10 μg/mL). Data are representative of two separate experiments performed in PBMCs from two patients with CGD. *P < 0.05; **P < 0.01. (C) Two patients with CGD (P1 and P2) with active colitis were treated with anakinra at a dosage of 100 mg daily for 3 mo. C-reactive protein (CRP) (milligrams per liter) and the number of stools per day are shown. (D) Patient 2 (P2). The number of perirectal abscesses during a 3-mo period of treatment with anakinra is shown.
Improved Clinical Outcome in Patients With CGD Who Had Colitis Treated with Anakinra.
Two patients with CGD who had refractory colitis were treated s.c. with anakinra at a dosage of 100 mg daily for 3 mo. The first patient (p47phox-deficient) suffered from colitis, with 15–20 loose stools per day; over the 3-mo period with anakinra, there was progressive improvement, with a reduction in frequency to eight to 10 stools per day (Fig. 4C). The second patient (gp91phox-deficient), described by van de Veerdonk et al. (26), suffered from perirectal granulomas and abscesses, which were refractory to corticosteroid therapy. During the 3-mo treatment period, the inflammatory parameters improved and the patient showed a good clinical response (Fig. 4C), with resolution of the perirectal abscesses (Fig. 4D). After stopping anakinra, he was disease-free for several months; however, his colitis flared, and anakinra was restarted for a period of 4 wk after which he remained free of symptoms. Notably, although the first patient previously had a history of invasive pulmonary fungal disease with Exophiala dermatitidis and the second patient had multiple severe S. aureus infections (pneumonia and liver abscesses), no infections were observed during treatment with anakinra in these patients with CGD.
Discussion
Using murine and human cells, we demonstrate here that CGD is characterized by defective autophagy resulting in increased release of IL-1β. Although CGD is characterized by increased inflammasome activation (11–13), the present study expands on those findings by demonstrating that blocking IL-1 itself decreased IL-1β secretion and restored defective autophagy in CGD in in vitro and in vivo settings. Clinically, we report here the beneficial effects observed in two patients suffering from CGD colitis treated with the IL-1R antagonist anakinra at the approved daily dose of 100 mg.
Autophagy defects resulting in inflammation appear to be a key feature in the pathogenesis of Crohn colitis, a disease that is indistinguishable from CGD colitis (27, 28). Given the suggested role for ROS in autophagy (29, 30), a defective autophagic process responsible for hyperproduction of IL-1β is consistent with NADPH deficiency. Indeed, defective autophagy (25) was observed in macrophages isolated from p40phox−/− mice, mimicking the intestinal inflammation of CGD (31). The defect in autophagy demonstrated here was accompanied by significantly higher secretion of IL-1β in the p40phox−/− macrophages compared with cells from WT mice and by increased secretion of IL-1β in PBMCs isolated from patients with CGD. The human data that defective autophagy contributes to the uncontrolled production of IL-1β are also observed in macrophages from Atg16L1-deficient mice, which produce greater levels of IL-1β compared with WT mice (18). Moreover, loss-of-function polymorphisms in the autophagy genes ATG16L1 and IRGM increase susceptibility to Crohn disease (32, 33), and the risk allele is associated with increased production of IL-1β (34).
One unexpected finding in this report is that blocking the IL-1R with anakinra reduced IL-1β production and restored autophagy in vivo (24). Indeed, anakinra restored LC3 recruitment to near-normal levels in both mice and human NADPH oxidase-deficient cells in vitro, as well as in vivo in mice with invasive pulmonary aspergillosis, with both suggesting that inhibiting IL-1 is beneficial in CGD not only by dampening IL-1–mediated inflammation but by restoring defective LC3 recruitment. Although ROS deficiency leads to impaired pathogen killing (35, 36), the defect in autophagy observed in this study suggests a significant role for autophagy in the host defense mechanism against Aspergillus infections in CGD. Whereas promotion of autophagy restores CD8+ T-cell memory in CGD mice with aspergillosis (25), blocking autophagy in WT mice infected with Aspergillus resulted in increased inflammation similar to that of p47phox−/− mice (Fig. S4). Not surprisingly, impaired autophagy results in impaired killing of A. fumigatus (37). Thus, the beneficial effects of anakinra observed in CGD mice with invasive aspergillosis is not only due to the reduction of neutrophil influx and IL-17 production but to the restoration of autophagy, which increases the killing of A. fumigatus by host phagocytes.
With the autophagy defect and the IL-1β hyperproduction being fundamental to the inflammatory features of CGD, the management of the disease appears to have advanced, as shown in the two patients with CGD colitis treated with anakinra. Anakinra dampened the inflammatory reaction and improved the clinical condition in patients with CGD colitis, providing a proof of principle for this therapeutic approach in the management of CGD. Although anti-TNF antibodies improved CGD colitis, the treatment was accompanied by life-threatening infectious complications (38). Anakinra is relatively safe (39), its short t1/2 provides an exit in the event of an infectious process, and blocking IL-1 deserves a trial in a larger cohort of patients with CGD.
Not only can we explain the increased IL-1β production but we can demonstrate that CGD is a disease associated with abnormal states of IL-1–dependent mechanisms, such as decreased autophagy and increased inflammasome activation, that cannot be viewed as separate pathological conditions in CGD. However, we cannot conclude that IL-1 alone explains the pathology of CGD because, downstream, there is also IL-18. In human heart tissue, IL-1 drives caspase-1 activation and increases active IL-18 (40). Support for this concept that IL-18 plays a role in IL-1–dependent disease can be found in a report using IL-18–deficient mice with inflammasome-dependent high levels of IL-1β (41). The earlier reports demonstrating that IL-1 can induce IL-1β through a positive feed-forward loop (42) explain the observation that IL-1 regulatory systems are dependent on the IL-1R signaling pathway. In this regard, CGD can no longer be regarded as an immunodeficiency but rather as a severe autoinflammatory disorder in which autophagy and inflammasomes are linked and amenable to IL-1 blockade.
Materials and Methods
Ethics Statement.
Patients and healthy volunteers gave written consent to participate as approved by the Radboud University Institutional Review Board. Experiments were performed according to the Italian Approved Animal Welfare Assurance A-3143-01. The Subcommittee on Research Animal Care approved the studies in Boston.
Patients and HCs.
For cytokine production, PBMCs were isolated and stimulated as previously described (43) from HCs and patients with CGD harboring homozygous mutations in the NCF1 gene (p47phox). To induce autophagy, cells were incubated for 4 h in Earle's Balanced Salt Solution starvation medium. After 4 h, IL-1β mRNA was assessed by quantitative RT-PCR. To inhibit autophagy, cells were incubated with 10 mM 3MA for 24 h.
Mice.
C57BL6 WT mice were purchased from Jackson Laboratories. Homozygous p47phox−/− mice on C57BL6 background were purchased (Harlan) and bred under specific pathogen-free conditions. The p40phox−/− mice were generated previously (31). The p40phox−/− × LC3-GFP mice were generated by crossing p40 phox−/− mice with LC3-GFP transgenic mice for two generations (44) (SI Materials and Methods).
In Vitro Production of IL-1β in Mice.
Bone marrow was harvested, and cells were cultured for 5 d in M-CSF to drive macrophage differentiation (SI Materials and Methods). On day 6, cells were harvested, counted, replated for 3 h, and incubated with 3MA for 1 h before LPS stimulation.
Mouse Macrophage Infection and Induction of Autophagy.
Peritoneal macrophages were harvested and allowed to adhere to the coverslips (SI Materials and Methods).
Human Monocyte Infection and Induction of Autophagy.
Monocytes were isolated from PBMCs on anti-CD14–coated beads (MACS Miltenyi) and allowed to adhere to glass coverslips for 1 h, after which they were exposed to pathogens. The coverslips were then washed, fixed in cold methanol, and examined by immunofluorescence (SI Materials and Methods).
TNBS-Induced Colitis.
As reported previously, mice received 2.5 mg of TNBS and were concomitantly treated i.p. with anakinra (10 mg/kg) daily. Weight changes were recorded daily, and on day 5, mice were killed and tissues were collected for histology, RNA analysis, and cytokine analysis. Colonic sections were stained with H&E, and histology was scored as described elsewhere (45).
Experimental Invasive Pulmonary Aspergillosis in Mice.
Details on this model using viable conidia (>95%) from the A. fumigatus Af293 strain are described in SI Materials and Methods. Different doses of anakinra were administered i.p. daily until the end of the experiment.
Cell Line Cultures, Transfection, and Autophagy.
RAW 264.7 cells were transiently transfected with the EGFP-LC3 plasmid (Addgene) for 48 h and exposed to A. fumigatus SC at a cell/fungus ratio of 1:1 in the absence or presence of different dosages of anakinra or rapamycin as a positive control. LC3 staining, LC3b and p62 blotting, and gene transcription of autophagy genes were used to investigate autophagy (SI Materials and Methods).
Supplementary Material
Acknowledgments
This study was supported by a Veni grant of the Netherlands Organization for Scientific Research (to F.L.v.d.V.) and a Vici grant (to M.G.N.). It was also supported by the European Union’s Seventh Framework Programme Agreement ERC-2011-AdG-293714 [metabolomics of fungal diseases: a systems biology approach for biomarkers discovery and therapy (FUNMETA)] (to L.R.) and by National Institutes of Health (NIH) Grants DK-83756, AI-62773, DK-60049, and DK-43351 (to R.J.X.) and NIH Grant AI-15614 (to C.A.D.).
Footnotes
The authors declare no conflict of interest.
3L.R. and F.L.v.d.V. share senior authorship.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1322831111/-/DCSupplemental.
References
- 1.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000;79(3):170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Holmes B, Page AR, Good RA. Studies of the metabolic activity of leukocytes from patients with a genetic abnormality of phagocytic function. J Clin Invest. 1967;46(9):1422–1432. doi: 10.1172/JCI105634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnston RB, Jr, et al. The role of superoxide anion generation in phagocytic bactericidal activity. Studies with normal and chronic granulomatous disease leukocytes. J Clin Invest. 1975;55(6):1357–1372. doi: 10.1172/JCI108055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antachopoulos C. Invasive fungal infections in congenital immunodeficiencies. Clin Microbiol Infect. 2010;16(9):1335–1342. doi: 10.1111/j.1469-0691.2010.03289.x. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Ari J, Wolach O, Gavrieli R, Wolach B. Infections associated with chronic granulomatous disease: Linking genetics to phenotypic expression. Expert Rev Anti Infect Ther. 2012;10(8):881–894. doi: 10.1586/eri.12.77. [DOI] [PubMed] [Google Scholar]
- 6.Rieber N, Hector A, Kuijpers T, Roos D, Hartl D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin Dev Immunol. 2012;2012:252460. doi: 10.1155/2012/252460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitomi H, et al. Colitis in chronic granulomatous disease resembling Crohn’s disease: Comparative analysis of CD68-positive cells between two disease entities. Dig Dis Sci. 1999;44(2):452–456. doi: 10.1023/a:1026643609944. [DOI] [PubMed] [Google Scholar]
- 8.Marciano BE, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114(2):462–468. doi: 10.1542/peds.114.2.462. [DOI] [PubMed] [Google Scholar]
- 9.Huang JS, et al. Chronic granulomatous disease caused by a deficiency in p47(phox) mimicking Crohn’s disease. Clin Gastroenterol Hepatol. 2004;2(8):690–695. doi: 10.1016/s1542-3565(04)00292-7. [DOI] [PubMed] [Google Scholar]
- 10.Rahman FZ, et al. Phagocyte dysfunction and inflammatory bowel disease. Inflamm Bowel Dis. 2008;14(10):1443–1452. doi: 10.1002/ibd.20449. [DOI] [PubMed] [Google Scholar]
- 11.Meissner F, et al. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood. 2010;116(9):1570–1573. doi: 10.1182/blood-2010-01-264218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Bruggen R, et al. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood. 2010;115(26):5398–5400. doi: 10.1182/blood-2009-10-250803. [DOI] [PubMed] [Google Scholar]
- 13.van de Veerdonk FL, et al. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci USA. 2010;107(7):3030–3033. doi: 10.1073/pnas.0914795107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crişan TO, et al. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS ONE. 2011;6(4):e18666. doi: 10.1371/journal.pone.0018666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris J, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286(11):9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi CS, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA. 2009;106(15):6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 19.Sanjuan MA, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 20.Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010;12(1):99–113. doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winkelstein JA, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000;79(3):155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Romani L, et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451(7175):211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- 23.Petrasek J, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122(10):3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodgers MA, Bowman JW, Liang Q, Jung JU. Regulation where autophagy intersects the inflammasome. Antioxidants & redox signaling. 2014;3:495–506. doi: 10.1089/ars.2013.5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Luca A, et al. CD4(+) T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J Clin Invest. 2012;122(5):1816–1831. doi: 10.1172/JCI60862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van de Veerdonk FL, Netea MG, Dinarello CA, van der Meer JW. Anakinra for the inflammatory complications of chronic granulomatous disease. Neth J Med. 2011;69(2):95. [PubMed] [Google Scholar]
- 27.Xavier RJ, Huett A, Rioux JD. Autophagy as an important process in gut homeostasis and Crohn’s disease pathogenesis. Gut. 2008;57(6):717–720. doi: 10.1136/gut.2007.134254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahida YR, Wu K, Jewell DP. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut. 1989;30(6):835–838. doi: 10.1136/gut.30.6.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J, Brumell JH. NADPH oxidases contribute to autophagy regulation. Autophagy. 2009;5(6):887–889. doi: 10.4161/auto.9125. [DOI] [PubMed] [Google Scholar]
- 30.Scherz-Shouval R, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellson CD, et al. Neutrophils from p40phox-/- mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med. 2006;203(8):1927–1937. doi: 10.1084/jem.20052069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hampe J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 33.Parkes M, et al. Wellcome Trust Case Control Consortium Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39(7):830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plantinga TS, et al. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut. 2011;60(9):1229–1235. doi: 10.1136/gut.2010.228908. [DOI] [PubMed] [Google Scholar]
- 35.Kuijpers T, Lutter R. Inflammation and repeated infections in CGD: Two sides of a coin. Cell Mol Life Sci. 2012;69(1):7–15. doi: 10.1007/s00018-011-0834-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nathan C, Cunningham-Bussel A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13(5):349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kyrmizi I, et al. Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting dectin-1/Syk kinase signaling. J Immunol. 2013;191(3):1287–1299. doi: 10.4049/jimmunol.1300132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uzel G, et al. Complications of tumor necrosis factor-α blockade in chronic granulomatous disease-related colitis. Clin Infect Dis. 2010;51(12):1429–1434. doi: 10.1086/657308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci USA. 2001;98(5):2871–2876. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brydges SD, et al. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest. 2013;123(11):4695–4705. doi: 10.1172/JCI71543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dinarello CA. Interleukin-1beta and the autoinflammatory diseases. N Engl J Med. 2009;360(23):2467–2470. doi: 10.1056/NEJMe0811014. [DOI] [PubMed] [Google Scholar]
- 43.van de Veerdonk FL, et al. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe. 2009;5(4):329–340. doi: 10.1016/j.chom.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 44.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15(3):1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takedatsu H, et al. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135(2):552–567. doi: 10.1053/j.gastro.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.