

Significance
ADP is best known as a constituent of nucleic acids and for its role in energy metabolism. ADP is also an important signaling molecule that activates cell surface receptors in a broad range of cells. ADP receptor antagonists are widely used to treat cardiovascular disease. We studied the signaling pathways activated by ADP receptors in endothelial cells, which form the lining of blood vessels. Vascular disease states cause abnormalities in endothelial function. We identified a critical role for the reactive oxygen species hydrogen peroxide (H2O2) in mediating cellular responses to ADP. We discovered that P2Y1 ADP receptors promote transactivation of the receptor tyrosine kinase Flt3, suggesting a new mechanism whereby cancer chemotherapy with receptor tyrosine kinase inhibitors cause vascular dysfunction.
Keywords: signal transduction, purinergic signaling, FRET
Abstract
ADP activates a family of cell surface receptors that modulate signaling pathways in a broad range of cells. ADP receptor antagonists are widely used to treat cardiovascular disease states. These studies identify a critical role for the stable reactive oxygen species hydrogen peroxide (H2O2) in mediating cellular responses activated by the G protein-coupled P2Y1 receptor for ADP. We found that ADP-dependent phosphorylation of key endothelial signaling proteins—including endothelial nitric oxide synthase, AMP-activated protein kinase, and the actin-binding MARCKS protein—was blocked by preincubation with PEG-catalase, which degrades H2O2. ADP treatment promoted the H2O2-dependent phosphorylation of c-Abl, a nonreceptor tyrosine kinase that modulates the actin cytoskeleton. Cellular imaging experiments using fluorescence resonance energy transfer-based biosensors revealed that ADP-stimulated activation of the cytoskeleton-associated small GTPase Rac1 was independent of H2O2. However, Rac1-dependent activation of AMP-activated protein kinase, the signaling phospholipid phosphatidylinositol-(4, 5)-bisphosphate, and the c-Abl–interacting protein CrkII are mediated by H2O2. We transfected endothelial cells with differentially targeted HyPer2 H2O2 biosensors and found that ADP promoted a marked increase in H2O2 levels in the cytosol and caveolae, and a smaller increase in mitochondria. We performed a screen for P2Y1 receptor-mediated receptor tyrosine kinase transactivation and discovered that ADP transactivates Fms-like tyrosine kinase 3 (Flt3), a receptor tyrosine kinase expressed in these cells. Our observation that P2Y1 receptor-mediated responses involve Flt3 transactivation may identify a unique mechanism whereby cancer chemotherapy with receptor tyrosine kinase inhibitors promotes vascular dysfunction. Taken together, these findings establish a critical role for endogenous H2O2 in control of ADP-mediated signaling responses in the vascular wall.
Beyond their established roles in intracellular energy flux and nucleic acid metabolism, purine nucleotides also serve as intercellular messenger molecules that regulate signal transduction pathways in a broad range of cells and tissues (1–3). The purine nucleotide ADP binds to G protein-coupled P2Y purinergic cell surface receptors, which are expressed in diverse mammalian cells, including blood platelets and vascular endothelial cells (reviewed in refs. 2 and 4). ADP is a critical determinant of platelet aggregation, blood vessel tone, and vascular wall integrity. Platelet granules contain high concentrations of ADP, which is released during platelet aggregation. The released ADP binds to P2Y12 and P2Y1 cell surface receptors for ADP on platelets and further potentiates platelet aggregation. P2Y receptor antagonists play a central role in cardiovascular therapeutics (2, 4): The P2Y12 blocker clopidogrel is one of the most commonly prescribed drugs in the United States, and other P2Y1 and P2Y12 blockers are being actively developed and tested for treatment of cardiovascular and cerebrovascular disease states. ADP also binds to P2Y1 receptors in vascular endothelial cells and rapidly activates endothelial nitric oxide synthase (eNOS) (5). Endothelium-generated nitric oxide (NO) inhibits platelet aggregation (3, 6) and provides an important feedback loop between endothelial cells and platelets that serves to attenuate the direct proaggregatory effects of ADP on platelets. ADP may also be released from the vascular endothelium and act in an autocrine or paracrine fashion to exert longer-term effects on vascular cell migration and barrier function (1, 3, 7–9). Clearly, a deeper understanding of P2Y receptor pharmacodynamics could inform current efforts in the development of novel purinergic antagonist drugs.
Purinergic receptors for ADP can be classified by their structure and mode of action into two distinct receptor families, P2X and P2Y. P2X receptors are ligand-gated ion channels, whereas members of the P2Y receptor family are G protein-coupled receptors. ADP signaling pathways in platelets have been extensively characterized, yet the roles of ADP in the modulation of endothelial responses are less well understood. The current studies have focused on exploring the signaling pathways activated by P2Y1 receptors in vascular endothelial cells. We have shown (6) that ADP acts via P2Y1 receptors to activate the endothelial isoform of nitric oxide synthase (eNOS) in cultured endothelial cells and also modulates the activation of key signaling protein kinases including the AMP-activated protein kinase (AMPK). We also found that ADP promotes the P2Y1 ADP receptor-dependent endothelial cell migration through activation of the small GTPase Rac1 (6, 10). Discovering the involvement of Rac1 provided an important clue to the mechanisms whereby ADP exerts its influence on endothelial cell responses.
Rac1 is an actin-binding cytoskeletal regulatory protein and is a member of the Rho GTPase protein family. The activation of eNOS by P2Y1 receptors for ADP depends on Rac1 (1, 6). Rac1 has been identified as a critical determinant of endothelial cell migration and barrier function, at least in part by modulating the levels of intracellular NO and hydrogen peroxide (H2O2) (11–15). H2O2 is a stable reactive oxygen species (ROS) that has been identified in recent years as a physiologically important intracellular messenger molecule (11–14), belying the classical concept of ROS functioning solely as deleterious molecules responsible for pathological states such as aging and neurodegeneration (14, 16). We reported (17, 18) that endogenous H2O2 regulates endothelial cell migration via dynamic signaling pathways involving the MARCKS protein, a ubiquitous phosphoprotein that translocates from the cell membrane to the actin cytoskeleton. The MARCKS protein also reversibly sequesters the signaling phospholipid phosphatidylinositol-(4, 5)-bisphosphate (PIP2). PIP2 is an important activator of proteins that initiate actin nucleation, including the phosphoprotein c-Abl, a nonreceptor tyrosine kinase that has been implicated in the dynamic cytoskeletal rearrangements that modulate endothelial barrier function. Endogenous H2O2 induces changes in cellular phospholipid metabolism via the phosphorylation and translocation of MARCKS in endothelial cells, yet the connections between receptor activation and intracellular modulation of H2O2 levels are incompletely understood.
The roles of H2O2 as a physiological intracellular messenger molecule were initially discovered through studies of growth factor-dependent activation of their cognate receptor tyrosine kinases (19, 20), which then signal to redox-regulated phosphoprotein phosphatases via H2O2 (20). In contrast to the widespread involvement of H2O2 in receptor tyrosine kinase signaling, only a handful of G protein-coupled receptors have been shown to directly modulate H2O2 levels (11, 16, 21, 22). Indeed, the roles of H2O2 in modulation of physiological responses have not yet been clearly defined for G protein-coupled receptors. In these studies, we present observations that establish that the G protein-coupled P2Y1 receptor for ADP modulates key H2O2-dependent signaling responses in the vascular endothelium via transactivation of the receptor tyrosine kinase Flt3.
Materials and Methods
Reagents.
FBS was from HyClone Laboratories. All other cell culture reagents, media, and Lipofectamine 2000 were from Invitrogen. Antibodies directed against eNOS, phospho-eNOS (S1177), AMPK, phospho-AMPK (T175), MARCKS, phospho-MARCKS, and caveolin-1 were from Cell Signaling. Antibodies against vinculin and GAPDH were from Sigma. AlexaFluor-coupled secondary antibodies and phalloidin/AlexaFluor 568 were from Invitrogen. SuperSignal chemiluminescence detection reagents and secondary antibodies conjugated with HRP were from Pierce. Processing and analysis of treated cells for the receptor tyrosine kinase screen used the PathScan RTK Signaling Antibody Array (Cell Signaling) according to the manufacturer’s protocols. All EV-fluorescence resonance energy transfer (FRET) biosensor plasmids were kind gifts of Michiyuki Matsuda (Department of Tumor Virology, Research Institute for Microbial Diseases, Kyoto University, Japan) (23). The AMPK FRET sensor was the kind gift of Lewis Cantley (Harvard Medical School) (24). ADP and PEG-catalase and all other biochemical reagents were from Sigma. MRS2179, sunitinib, and bosutinib were from SellcheckChem. pCMV LiveAct-TagGFP2 was from Ibidi. Arterial preparations from human tissues were from discarded surgical specimens and were obtained under a protocol approved by the Partners Healthcare Institutional Review Board (protocol 1999P000740). Arterial preparation from mice was obtained under a protocol approved by the Harvard Medical Area Standing Committee on Animals (protocol 03082). The HyPer2 constructs targeted to cytosol, mitochondria, and nucleus were from Evrogen. The HyPer2 construct targeted to caveolae was created by fusing the HyPer2 coding sequence downstream of the N-terminal 500 amino acids of eNOS, a caveolae-targeted protein (25).
Cell Culture, Transfection, and Treatment.
Bovine aortic endothelial cells (BAEC) were obtained from Genlantis and were maintained in culture, transfected with plasmids, and treated as described (18). When used, PEG-catalase treatments (100 U/mL) were for 8 h, with 0.05% PEG as control. After drug treatments, lysates from BAEC were prepared and analyzed in immunoblots, as described in detail (18). For live-cell imaging, the cells were seeded onto individual glass-bottom dishes (Mattek) 24 h after transfection and analyzed 24 h later.
Fluorescence Microscopy.
Live cell imaging experiments were carried out as described (18) by using a GM-4000 on-stage incubator mounted on a fully motorized Olympus IX81 inverted microscope equipped with a Hamamatsu Orca ER cooled-CCD camera in conjunction with a zero-drift focus compensation system. For imaging, cells were maintained in complete medium [low-glucose DMEM/10% (vol/vol) FBS] without phenol red in a humidified atmosphere containing 5% CO2. HyPer2 fluorescence was excited with 420/40 nm and with 500/16 nm band-pass excitation filters; corresponding YFP emission was acquired every 5 s for 15 min by using a 535/30 band-pass emission filter. The HyPer2 ratio was quantitated in cells as described in detail (18). Microscopic F-actin was imaged by using the plasmid construct pCMV LiveAct-TagGfp2 using a standard GFP filter set (Semrock). Images were acquired by using a 20×, 40×, or 100× differential interference contrast oil immersion objective lens and analyzed by using MetaMorph software (Universal Imaging).
FRET Imaging.
Monitoring of FRET biosensors was performed by using methods described in detail (18, 23). In brief, endothelial cells were transfected with plasmids encoding FRET biosensors as indicated. After 24 h, the cells were seeded onto individual glass-bottom dishes (Mattek), then pretreated with inhibitors as indicated and subjected to excitation of CFP-PH at 425 ±10 nm; emission was collected at 475 ± 10 (CFP) and 540 ± 10 nm (YFP) by using the Semrock FRET-CFP/YFP-B 4-filter single-band set. A series of fluorescence images were taken at 30-s time intervals before and after drug treatments; visualization and analysis was performed by using the MetaMorph FRET module.
Electric Cell Impedance Sensing.
ADP-induced changes in endothelial resistance were analyzed by using an electric cell impedance sensing (ECIS) device (Applied Biophysics). Impedance arrays were coated with gelatin for 30 min. A total of 20,000 endothelial cells were seeded per well and grown for 48 h at 37 °C/5% CO2. The cell medium was changed before the array was connected to the ECIS device. Drugs were added as soon as impedance had stabilized, and resistance (Ω) was analyzed for 24 h.
Statistical Analyses.
All experiments were performed at least three times. Mean values for experiments are expressed as mean ± SE. Statistical differences were assessed by analysis of variance. A P value less than 0.05 was considered statistically significant.
Results
We first used immunohistochemical approaches to explore the expression of the P2Y1 ADP receptor in intact arterial preparations. We analyzed human arterioles from tissues that were incidentally obtained as discarded surgical specimens, which were then fixed and stained with antibodies directed against either the P2Y1 receptor or the endothelial marker protein von Willebrand factor (vWF). As shown in Fig. 1A, antibodies specific for the P2Y1 receptor reveal a robust signal in the endothelial cell layer; the P2Y1 staining in endothelium colocalizes with the staining pattern observed using antibodies directed against vWF. We next analyzed mouse arterial preparations and found that mouse aorta showed robust staining of P2Y1 in vascular endothelium and smooth muscle cells (Fig. 1A). We previously characterized agonist-stimulated H2O2-mediated phosphorylation responses in endothelial cells and cardiac myocytes (18, 26), and in the present studies, we extended these approaches to explore the role of H2O2 in ADP signaling in intact blood vessels and endothelial cells. Fig. 1B shows results of immunoblots analyzed in freshly isolated mouse aortae that were treated with ADP in the presence and absence of PEG-catalase (a H2O2-catabolizing cell-permeant enzyme) and probed with phosphospecific antibodies. Fig. 1B shows representative immunoblots; statistical analyses of pooled data from identically configured experiments are shown in Fig. S1 A and B. The ADP-promoted increase in phosphorylation of AMPK, MARCKS, or eNOS (at phosphoserine 1179) is blocked both by PEG-catalase and by the P2Y1 receptor antagonist MRS2179. These findings indicate that ADP-promoted phosphorylation responses in intact arterial preparations are mediated by the P2Y1 cell surface receptor for ADP and implicate H2O2-dependent signaling pathways in coupling receptor activation to the phosphorylation of MARCKS, AMPK, and eNOS. We next explored the role of H2O2 in ADP-modulated signaling responses in cultured vascular endothelial cells, which represent a more tractable model system for the analysis of cell signaling pathways. Just as we found in mouse arterial preparations, ADP-stimulated phosphorylation of AMPK, eNOS, and MARCKS in cultured bovine aortic endothelial cells (BAEC) was blocked by PEG-catalase and by the P2Y1 antagonist MRS2179 (Fig. 1B and Figs. S1B and S2 A and B).
Fig. 1.
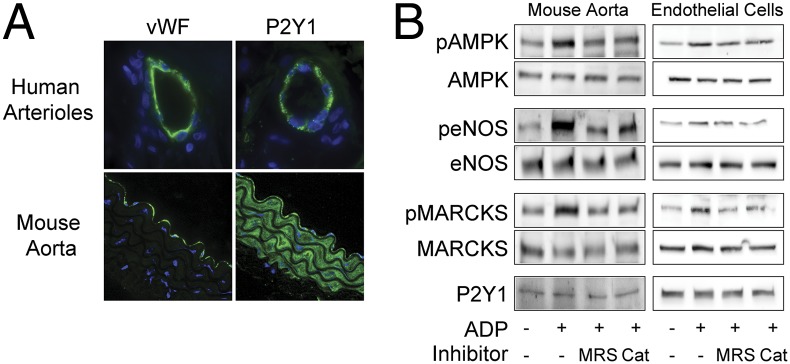
P2Y1 receptor expression and signaling responses in vascular preparations and cultured endothelial cells. A shows representative photomicrographs of human arterioles and murine aortic preparations that were fixed, paraffin-embedded, and stained with antibodies directed against the endothelium-specific marker vWF or the P2Y1 receptor, as indicated. Nuclei were stained with DAPI. Images were obtained by confocal imaging, as discussed in the text. In B, immunoblots were analyzed in murine aortic preparations or cultured endothelial cells that were incubated with ADP (50 μM, 30 min) in the presence or absence of the P2Y1-specific blocker MRS2179 (5 μM) or of the cell permeant H2O2-catabolizing enzyme PEG-catalase (100 U/mL). Membranes were probed with total and phosphospecific antibodies directed against eNOS, AMPK, and MARCKS; P2Y1 served as loading control. Fig. S1 A and B show statistical analyses of pooled data from four identical immunoblot experiments that yielded similar results.
H2O2 has been implicated in the modulation of vascular wall integrity (14), and our next experiments explored the involvement of H2O2 in modulating the effects of ADP on endothelial permeability. We performed cell impedance measurements of cultured endothelial monolayers as an assay for endothelial barrier integrity (27). As shown in Fig. 2A, ADP treatment significantly increases the impedance of the endothelial monolayer, indicating an increase in barrier integrity; this ADP response is completely blocked by MRS2179 and is partially, but significantly, blocked by PEG-catalase treatment. Because of the importance of the actin cytoskeleton in control of the endothelial barrier, we performed pursuing cell imaging studies with FRET-based biosensors to explore the role of H2O2 in signaling pathways activated by ADP. We previously reported (6) that ADP promotes the P2Y1 receptor-dependent activation of the cytoskeleton-associated small GTPase Rac1 in endothelial cells. Fig. 2B shows the results of experiments in endothelial cells transfected with the Rac1 biosensor Raichu-RacEV (23) and analyzed by using FRET time-lapse imaging following addition of ADP. We found that the ADP-promoted increase in Rac1 activity was unaffected by PEG-catalase, indicating that ADP signaling to Rac1 is independent of changes in intracellular H2O2. We next prepared lysates from endothelial cells after treatment with ADP, and analyzed immunoblots probed with phosphospecific antibodies directed against the nonreceptor tyrosine kinase c-Abl or against MARCKS phosphoproteins, both of which are known to be involved in actin assembly and phospholipid signaling in these cells (1, 18, 28, 29). ADP treatment led to the robust phosphorylation of each of these proteins; these phosphorylation responses were blocked both by the P2Y1 antagonist MRS2179, PEG-catalase, and the specific c-Abl tyrosine kinase inhibitor bosutinib (Fig. 2 C and D).
Fig. 2.

ADP- and H2O2-mediated changes in endothelial cell impedance, Rac1 activation, and phosphorylation responses. A shows representative tracings of endothelial cells analyzed in impedance measurements in the presence or absence of ADP (50 μM), the P2Y1 receptor blocker MRS2179 (5 μM), or the H2O2 scavenger PEG-catalase (100 U/mL). The findings shown are representative of three identical experiments that yielded similar results. B shows representative photomicrographs of endothelial cells transfected with a plasmid encoding a Rac1 FRET biosensor and then analyzed by quantitative time-lapse microscopy before and 5 min after the addition of ADP in the presence or absence of MRS2179 (MRS) or PEG-catalase (Cat); pooled data are shown from four identical experiments, presenting the slope of the fluorescence increase following the addition of ADP, measured 5 min after adding ADP in the presence or absence of MRS2179 or PEG-catalase. C shows representative immunoblots of cultured endothelial cells incubated with ADP in the presence or absence of MRS2179, the c-Abl inhibitor bosutinib, or PEG-catalase as indicated, probed with antibodies as shown. D shows statistical analyses of pooled data from three identical experiments that yielded similar results; *P < 0.05 (ANOVA).
We next analyzed the involvement of H2O2 in the ADP-dependent modulation of the signaling phospholipid PIP2, the AMPK, and the c-Abl–interacting protein CrkII by using highly sensitive FRET biosensors. We first studied ADP-dependent modulation of PIP2, which we implicated in MARCKS-dependent regulation of endothelial cell motility (17, 18). We transfected endothelial cells with the PIP2-specific biosensor PiPi (23) and analyzed the fluorescence signal in response to ADP in the presence or absence of PEG-catalase. As shown in Fig. 3A, treatment with ADP leads to an increase in the PIP2 signal that is abrogated by PEG-catalase. We next explored the role of H2O2 in ADP-dependent activation of AMPK by using a recently developed FRET-based AMPK biosensor (24). Endothelial cells transfected with a plasmid encoding this biosensor were treated with ADP in the presence and absence of PEG-catalase. As shown in Fig. 3B, ADP-stimulated AMPK activation was completely blocked by PEG-catalase. We found similar effects on PEG-catalase on the ADP-promoted activation of the actin-regulatory protein CrkII, which is known to be a downstream target of c-Abl (18). We transfected endothelial cells with a plasmid by expressing a well-characterized CrkII FRET biosensor (23) and performed cellular imaging analyses after exposing the cells to ADP in the presence and absence of PEG-catalase. As shown in Fig. 3C, ADP treatment led to a significant activation of CrkII that was entirely blocked by PEG-catalase.
Fig. 3.

Effects of PEG-catalase on ADP-dependent modulation of PIP2, AMPK, and c-Abl. Endothelial cells were transfected with plasmids encoding FRET biosensors specific for PIP2 (A), AMPK (B), and CrKII (C) and then analyzed by quantitative FRET microscopy before and after the addition of ADP (50 μM, 5 min) in the presence and absence of PEG-catalase. A–C Left show representative photomicrographs; A–C Right show representative tracings of FRET ratios as well as statistical analysis of the ADP-promoted FRET slope change, as pooled and plotted from four independent experiments; *P < 0.05 (ANOVA).
The well-established involvement of Rac1 in modulation of the endothelial cytoskeleton led us to extend these cell imaging approaches to directly explore the direct effects of ADP and H2O2 on cytoskeletal fibers. We transfected endothelial cells with a plasmid construct expressing the actin-binding protein F-tractin fused to eGFP (pCMV LiveAct-TagGFP2) and analyzed the cells by time-lapse fluorescence microscopy. As shown in Fig. 4, ADP treatment markedly enhances actin polymerization and induces cell spreading. These effects of ADP on actin polymerization are entirely blocked by the P2Y1 inhibitor MRS2179 and by PEG-catalase.
Fig. 4.
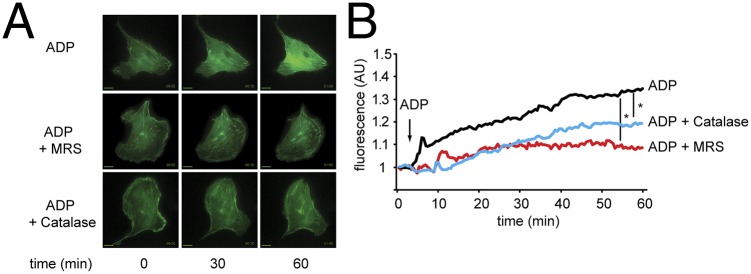
ADP-dependent modulation of actin polymerization. A shows representative photomicrographs of endothelial cells that were transfected with GFP-labeled F-tractin, and then treated with PBS or ADP (50 µM), with or without MRS2179 (5 μM) or PEG-catalase. (Magnification: 100×.) (Scale bars: 5 µm.) B shows individual tracings of cellular fluorescence over time; the slope of the fluorescence increase before and 60 min after addition of ADP was determined for each experiment. The asterisk notes P < 0.05, showing a significant effect of both PEG-catalase and MRS2179 on ADP-promoted F-tractin fluorescence.
To further characterize the intracellular sources of H2O2 that are modulated by ADP, we used differentially targeted constructs expressing the H2O2 biosensor HyPer2. Endothelial cells were transfected with HyPer2 constructs targeted to various subcellular locales: the mitochondrial membrane, caveolae, nucleus, or cytosol (Fig. 5 A and B). After addition of ADP, there was a marked increase in the HyPer2 signal in cells transfected with the cytosolic and the caveolae-targeted HyPer2 constructs; a smaller ADP-stimulated H2O2 response was observed for the HyPer2 variant targeted to the mitochondria, and no ADP-stimulated H2O2 response was seen for the HyPer2 variant targeted to the nucleus. All these differentially targeted HyPer2 constructs responded to exogenous H2O2 (Fig. S3). The NADPH oxidase inhibitor VAS2870 completely blocked the effect of ADP on AMPK, eNOS, and MARCKS phosphorylation (Fig. 5C and Figs. S1C and S2C) and blocking the HyPer2 response (Fig. S2D). In contrast, the mitochondrial inhibitor rotenone had no effect on ADP-modulated phosphorylation of MARCKS or AMPK, yet completely blocked ADP-stimulated eNOS phosphorylation (Fig. 5C). A more detailed time course showing the effects of these inhibitors on ADP-modulated phosphorylation responses is shown in Fig. S2.
Fig. 5.

Detection of ADP-promoted changes in intracellular H2O2 levels. A shows representative photomicrographs of endothelial cells transfected with differentially targeted variants of the hydrogen peroxide specific biosensor HyPer2 expressed in cytosol (Cyto), caveolae (Cav), nucleus (Nuc), or mitochondria (Mito), as indicated, and then treated with ADP. B shows representative tracings for the individual differentially targeted HyPer2 constructs; ADP addition is indicated by an arrow. C shows immunoblots of cultured endothelial cells incubated with ADP in the presence or absence of MRS2179 (“MRS”; 5 μM), PEG-catalase (“Cat”; 100 U/mL), the NADPH oxidase inhibitor VAS2870 (“VAS”; 10 μM), or the inhibitor of mitochondrial respiration rotenone (“Rot”; 10 μM) as indicated. Membranes were probed with total and phosphospecific antibodies for AMPK, eNOS, and MARCKS. The results shown are representative of three identical experiments that yielded similar results; statistical analyses of pooled data are presented in Fig. S1C.
We next explored the possibility that ADP leads to transactivation of receptor tyrosine kinases, which have been linked to H2O2-mediated signaling responses (11–14). We screened for ADP-dependent activation of receptor tyrosine kinases in endothelial cells by using a chip-based receptor tyrosine kinase phosphorylation assay that allowed us to screen 39 different tyrosine kinases, along with a series of positive and negative controls. We used this chip assay to analyze the time course of ADP-simulated phosphorylation responses in the absence and presence of the P2Y1 inhibitor MRS2179. As shown in Fig. 6A, ADP treatment led to the reversible and robust phosphorylation of Flt3, and had a smaller and more transient effect on phosphorylation of fibroblast growth factor receptor-4 (FGF4). We performed standard immunoblot analyses and confirmed that ADP-dependent Flt3 and AMPK phosphorylation responses are completely blocked by the nonselective receptor tyrosine kinase inhibitor sunitinib, yet sunitinib had no substantive effect on ADP-modulated eNOS phosphorylation (Fig. 6B and Fig. S1D). Sunitinib also entirely abrogated ADP-induced changes in the endothelial cytoskeleton (Fig. 6C).
Fig. 6.

ADP induces actin rearrangement via Flt3 transactivation. A shows results from a receptor tyrosine kinase activation screen that permits quantitation of phosphorylation responses for a panel of 39 different receptor tyrosine kinases. Endothelial cells were treated with ADP (50 µM) for the indicated time points in the presence or absence of MRS2179 (5 μM), and then harvested and lysed. Whole-cell lysates were subjected to a RTK PathScan antibody array (Cell Signaling) and analyzed by densitometry. The arrows note a significant time-dependent increase and decrease in phosphorylation of the receptor tyrosine kinase Flt3. The receptor tyrosine kinase FGF4 also showed an increase in phosphorylation, but only at the 15-min time point. B shows immunoblots of cultured endothelial cells incubated with ADP in the presence or absence of MRS2179 or the receptor tyrosine kinase inhibitor sunitinib, as indicated. Membranes were probed for phosphospecific and total AMPK, eNOS, and Flt3, as shown in these representative immunoblots. C shows representative photomicrographs of endothelial cells transfected with GFP-labeled F-tractin treated with ADP (50 µM) with or without sunitinib. (Magnification: 100×.) (Scale bars: 5 µm.) B and C show individual tracings of cellular fluorescence over time; the asterisk notes a significant difference in the slope of the fluorescence signal measured before and 30 min after the addition of ADP, as analyzed in pooled data from four independent experiments. D shows a schematic of the signaling pathways being probed in these studies; see text for details.
Discussion
Purinergic receptors modulate a broad range of cellular responses in cardiovascular cells and tissues. Blood platelets express G protein-coupled P2Y1 and P2Y12 receptors for ADP (1, 4, 6, 8, 30); both receptor subtypes are linked to platelet aggregation (30). The P2Y12 ADP receptor antagonist clopidogrel is one of the most frequently prescribed cardiovascular drugs, yet clopidogrel has limited potency in blocking P2Y1 receptors (6). Because of frequent treatment failures with clopidogrel, there are ongoing efforts to develop new ADP antagonists that block P2Y1 and P2Y12 receptors. Signal transduction pathways modulated by platelet P2Y12 receptors have been extensively characterized, whereas our knowledge of P2Y1 receptor responses remains less complete. In contrast to blood platelets, P2Y1 receptors represent the predominant isoform found in vascular endothelial cells (6) and modulate ADP-dependent eNOS activation, leading to vascular relaxation. Because ADP receptor antagonists may affect ADP signaling pathways in the vascular wall, it is essential to develop a more comprehensive understanding of P2Y1 receptor-mediated responses. The principal discoveries in the present studies are twofold: First, our observation that ADP-modulated signaling responses in endothelial cells are modulated by H2O2-regulated pathways, and second, our discovery of ADP-dependent transactivation of the receptor tyrosine kinase Flt3.
We have shown that ADP stimulates G protein-coupled P2Y1 cell surface receptor in endothelial cells and leads to the rapid activation of diverse protein kinase pathways, while also modulating endothelial cell migration (6, 10). The present studies have documented a central role for H2O2 in modulating cellular responses activated by the P2Y1 ADP receptor in blood vessels and in vascular endothelial cells (Fig. 1). The inhibition of key ADP-dependent phosphorylation responses—AMPK, MARCKS, and eNOS—by the cell-permeant H2O2-catabolizing reagent PEG-catalase provides strong evidence that H2O2 is a critical intermediate both in intact arteries and in cultured endothelial cells. However, as shown in Fig. 2A, PEG-catalase provides only a partial (but statistically significant) inhibition of the ADP-promoted increase in cell impedance (a marker of endothelial barrier function). The fact that the P2Y1 receptor antagonist MRS2179 is more effective than PEG-catalase in blocking these and several other ADP-stimulated responses analyzed in these cells likely reflects the fact that PEG-catalase is less effective at fully degrading all intracellular H2O2 than is the high-affinity receptor antagonist MRS2179 in fully blocking P2Y1-mediated responses. It is also likely that H2O2-independent pathways are involved in the control of cell impedance (27).
We used a series of informative FRET-based biosensors to further probe the involvement of H2O2 in signaling pathways stimulated by ADP in these cells. As shown in Fig. 3, PEG-catalase entirely abrogates the ADP-stimulated increase in PIP2, a key signaling phospholipid that reversibly binds MARCKS, which we showed also to be regulated by ADP in a H2O2-dependent fashion (Fig. 3). ADP-dependent activation of the c-Abl–binding protein CrkII is similarly blocked by PEG-catalase, consistent with our observation that ADP-stimulated c-Abl phosphorylation is blocked by PEG-catalase. ADP-dependent activation of the “energy gauge” kinase AMPK—which we have shown to be modulated by redox-sensitive upstream kinases (6, 31)—is also blocked by PEG-catalase (Fig. 3). These results help to establish the critical role of H2O2 in modulating the response to ADP.
These studies provide multiple lines of evidence establishing a key role for H2O2 in modulating ADP signaling to the cytoskeleton. PEG-catalase completely blocked ADP-stimulated phosphorylation of c-Abl and MARCKS (Fig. 2), both of which are reversibly targeted to the actin cytoskeleton. The P2Y1 receptor-modulated formation of actin stress fibers is similarly attenuated by PEG-catalase (Fig. 4). We sought to complement these approaches with PEG-catalase by directly detecting intracellular H2O2 using the biosensor HyPer2 (18), which we have previously studied in these cells. Using differentially targeted HyPer2 constructs, we found that ADP promoted a robust increase in H2O2 levels in endothelial cells transfected with cytosolic or caveolae-targeted HyPer2 constructs, whereas cells transfected with mitochondria-targeted HyPer2 variants showed a smaller response; the nuclear-targeted HyPer2 showed no response whatsoever (Fig. 5). This finding suggested that mitochondria—which are a quantitatively important source of H2O2 in these (and in most other) cells (11)—are not the major source of increased H2O2 levels seen after ADP treatment. Differential utilization of intracellular H2O2 sources in different signaling pathways is consistent with our observation (Fig. 5C) that the mitochondrial inhibitor rotenone completely blocks ADP signaling to eNOS, while having no effect on ADP-stimulated phosphorylation of MARCKS or AMPK. These observations represent a point of convergence with features of the angiotensin-II AT1 receptor, in which the increase in intracellular ROS seen after AT1 receptor activation is due to an increase in NADPH oxidase activity (16). However, in contrast to the pathophysiological consequences of ROS generation typically seen in response to angiotensin-II (13, 16), the increase in H2O2 in response to ADP observed in the present study instead appears to reflect the activation of a physiological signaling pathway.
The signal detected by the caveolae-targeted HyPer2 construct provides suggestive evidence that NADPH oxidases may represent an important ADP-modulated source of H2O2. NADPH oxidases are a family of membrane-associated multisubunit enzymes that synthesize superoxide or H2O2 in response to a broad range of extracellular stimuli. Rac1 is an essential component of some NADPH oxidase isoforms, and the critical dependence of ADP signaling on Rac1 (ref. 6 and Fig. 2) provides additional evidence for the involvement of NADPH oxidase in the P2Y1 receptor-mediated response. The fact that ADP-dependent Rac1 activation is not blocked by PEG-catalase (Fig. 2) indicates that H2O2 is “downstream” of Rac1 activation. The NADPH oxidase inhibitor VAS2180 completely blocked ADP-stimulated phosphorylation responses, whereas the mitochondrial inhibitor rotenone blocked only eNOS phosphorylation while having no effect on AMPK or MARCKS phosphorylation (Fig. 5C). These findings suggest that different signaling responses elicited by P2Y1 receptors for ADP may differentially modulate specific intracellular H2O2 sources. FRET imaging of the ADP-dependent activation of Rac1 reveals that most of the Rac1 activation is taking place at cell membranes. This observation is consistent with the subcellular distribution of NADPH oxidase isoforms, which typically are membrane-targeted proteins (13, 16). Taken together, ADP-dependent activation of NADPH oxidase appears to be a key determinant of P2Y1-mediated responses in these cells.
The critical role of H2O2 in receptor-dependent cell signaling has been extensively characterized for receptor tyrosine kinases. Many G protein-coupled receptors “transactivate” receptor tyrosine kinases as a fundamental component of their signal transduction pathways, yet a role for H2O2 in directly mediating physiological G protein-coupled receptor signal transduction has been observed for only a handful of receptors (11). These previous observations on the interactions between G protein-coupled receptors and receptor tyrosine kinases inspired us to explore whether the P2Y1 receptor for ADP might follow a similar signaling paradigm. We therefore explored whether ADP might activate receptor tyrosine kinase(s) in these cells by using a chip-based receptor tyrosine kinase screen, which revealed that ADP treatment promoted the robust time-dependent reversible phosphorylation of Flt3, and a transient increase in FGF4 phosphorylation (Fig. 6). ADP-dependent phosphorylation of both receptor tyrosine kinases was entirely blocked by MRS2179, indicating that these responses are mediated by the P2Y1 receptor. Fig. 6D shows a schematic model that synthesizes our key findings. We found that the ADP-dependent phosphorylation of AMPK and ADP-modulated cytoskeleton reorganization was entirely blocked by the highly selective Flt3 inhibitor sunitinib. Activation of the endothelial cell P2Y1 receptor leads to the transactivation of the receptor tyrosine kinase Flt3, which leads to the H2O2-dependent phosphorylation of the nonreceptor tyrosine kinase c-Abl, which then promotes phosphorylation of MARCKS and leads to an increase in endothelial barrier integrity. This pathway appears to involve NADPH oxidase and is independent of mitochondria-derived H2O2. In contrast, the ADP/P2Y1 signaling pathway leading to eNOS activation is independent of Flt3 and appears to involve H2O2 derived from mitochondrial sources, providing a clear point of departure between P2Y1 signaling to eNOS vs. AMPK and MARCKS. These observations indicate that P2Y1-mediated ADP-dependent transactivation of the Flt3 receptor tyrosine kinase differentially modulates downstream signaling responses in endothelial cells.
These studies establish a role for Flt3 in endothelial signaling and may help to explain the frequent occurrence of vascular dysfunction in patients treated with sunitinib and other receptor tyrosine kinases inhibitors used in cancer chemotherapy (32). Because ADP modulates vascular wall integrity, the abrogation of ADP responses by blockade of Flt3 might represent one mechanism whereby sunitinib leads to dose-limiting vascular dysfunction. In the process of explicating unique features of H2O2 in ADP signaling, these studies have identified a mechanism for receptor transactivation that may have implications for cancer chemotherapy, revealing the complex interplay between oxidant-modulated signaling and vascular homeostasis.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants R01HL46457 and P01 HL48743 (to T.M.) and R01 HL104006 (to C.V.C.). H.K. was supported by an American Heart Association Postdoctoral Fellowship and by Postdoctoral Fellowship PDR-09-002 from the Fonds National de la Recherche (Luxembourg), and N.R. was supported by a Pew-Latin American Trust Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320854111/-/DCSupplemental.
References
- 1.Soulet C, et al. A differential role of the platelet ADP receptors P2Y1 and P2Y12 in Rac activation. J Thromb Haemost. 2005;3(10):2296–2306. doi: 10.1111/j.1538-7836.2005.01588.x. [DOI] [PubMed] [Google Scholar]
- 2.Hechler B, Gachet C. P2 receptors and platelet function. Purinergic Signal. 2011;7(3):293–303. doi: 10.1007/s11302-011-9247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zemskov E, Lucas R, Verin AD, Umapathy NS. P2Y receptors as regulators of lung endothelial barrier integrity. J Cardiovasc Dis Res. 2011;2(1):14–22. doi: 10.4103/0975-3583.78582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollopeter G, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409(6817):202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 5.Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res. 1987;61(6):866–879. doi: 10.1161/01.res.61.6.866. [DOI] [PubMed] [Google Scholar]
- 6.Hess CN, Kou R, Johnson RP, Li GK, Michel T. ADP signaling in vascular endothelial cells: ADP-dependent activation of the endothelial isoform of nitric-oxide synthase requires the expression but not the kinase activity of AMP-activated protein kinase. J Biol Chem. 2009;284(47):32209–32224. doi: 10.1074/jbc.M109.032656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buvinic S, Bravo-Zehnder M, Boyer JL, Huidobro-Toro JP, González A. Nucleotide P2Y1 receptor regulates EGF receptor mitogenic signaling and expression in epithelial cells. J Cell Sci. 2007;120(Pt 24):4289–4301. doi: 10.1242/jcs.03490. [DOI] [PubMed] [Google Scholar]
- 8.Shen J, DiCorleto PE. ADP stimulates human endothelial cell migration via P2Y1 nucleotide receptor-mediated mitogen-activated protein kinase pathways. Circ Res. 2008;102(4):448–456. doi: 10.1161/CIRCRESAHA.107.165795. [DOI] [PubMed] [Google Scholar]
- 9.Uehara K, Uehara A. P2Y1, P2Y6, and P2Y12 receptors in rat splenic sinus endothelial cells: An immunohistochemical and ultrastructural study. Histochem Cell Biol. 2011;136(5):557–567. doi: 10.1007/s00418-011-0859-2. [DOI] [PubMed] [Google Scholar]
- 10.Bretón-Romero R, Kalwa H, Lamas S, Michel T. Role of PTEN in modulation of ADP-dependent signaling pathways in vascular endothelial cells. Biochim Biophys Acta. 2013;1833(12):2586–2595. doi: 10.1016/j.bbamcr.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finkel T. Reactive oxygen species and signal transduction. IUBMB Life. 2001;52(1-2):3–6. doi: 10.1080/15216540252774694. [DOI] [PubMed] [Google Scholar]
- 12.Nathan C. Specificity of a third kind: Reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest. 2003;111(6):769–778. doi: 10.1172/JCI18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandes RP, Kreuzer J. Vascular NADPH oxidases: Molecular mechanisms of activation. Cardiovasc Res. 2005;65(1):16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Cai H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc Res. 2005;68(1):26–36. doi: 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 15.Seifert R, Burde R, Schultz G. Activation of NADPH oxidase by purine and pyrimidine nucleotides involves G proteins and is potentiated by chemotactic peptides. Biochem J. 1989;259(3):813–819. doi: 10.1042/bj2590813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehta PK, Griendling KK. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292(1):C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 17.Kalwa H, Michel T. The MARCKS protein plays a critical role in phosphatidylinositol 4,5-bisphosphate metabolism and directed cell movement in vascular endothelial cells. J Biol Chem. 2011;286(3):2320–2330. doi: 10.1074/jbc.M110.196022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalwa H, Sartoretto JL, Sartoretto SM, Michel T. Angiotensin-II and MARCKS: A hydrogen peroxide- and RAC1-dependent signaling pathway in vascular endothelium. J Biol Chem. 2012;287(34):29147–29158. doi: 10.1074/jbc.M112.381517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi MH, et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435(7040):347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 20.Pescatore LA, et al. Protein disulfide isomerase is required for platelet-derived growth factor-induced vascular smooth muscle cell migration, Nox1 NADPH oxidase expression, and RhoGTPase activation. J Biol Chem. 2012;287(35):29290–29300. doi: 10.1074/jbc.M112.394551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moniri NH, Daaka Y. Agonist-stimulated reactive oxygen species formation regulates beta2-adrenergic receptor signal transduction. Biochem Pharmacol. 2007;74(1):64–73. doi: 10.1016/j.bcp.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Pendyala S, Usatyuk PV, Gorshkova IA, Garcia JG, Natarajan V. Regulation of NADPH oxidase in vascular endothelium: The role of phospholipases, protein kinases, and cytoskeletal proteins. Antioxid Redox Signal. 2009;11(4):841–860. doi: 10.1089/ars.2008.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komatsu N, et al. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22(23):4647–4656. doi: 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsou P, Zheng B, Hsu CH, Sasaki AT, Cantley LC. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011;13(4):476–486. doi: 10.1016/j.cmet.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaul PW, et al. Acylation targets emdothelial nitric-oxide synthase to plasmalemmal caveolae. J Biol Chem. 1996;271(11):6518–6522. doi: 10.1074/jbc.271.11.6518. [DOI] [PubMed] [Google Scholar]
- 26.Sartoretto JL, Kalwa H, Pluth MD, Lippard SJ, Michel T. Hydrogen peroxide differentially modulates cardiac myocyte nitric oxide synthesis. Proc Natl Acad Sci USA. 2011;108(38):15792–15797. doi: 10.1073/pnas.1111331108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: Assessment of endothelial barrier function. Proc Natl Acad Sci USA. 1992;89(17):7919–7923. doi: 10.1073/pnas.89.17.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103(1):40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- 29.El Jamali A, Valente AJ, Clark RA. Regulation of phagocyte NADPH oxidase by hydrogen peroxide through a Ca(2+)/c-Abl signaling pathway. Free Radic Biol Med. 2010;48(6):798–810. doi: 10.1016/j.freeradbiomed.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leon C, Ravanat C, Freund M, Cazenave JP, Gachet C. Differential involvement of the P2Y1 and P2Y12 receptors in platelet procoagulant activity. Arterioscler Thromb Vasc Biol. 2003;23(10):1941–1947. doi: 10.1161/01.ATV.0000092127.16125.E6. [DOI] [PubMed] [Google Scholar]
- 31.Koh H, Chung J. AMPK links energy status to cell structure and mitosis. Biochem Biophys Res Commun. 2007;362(4):789–792. doi: 10.1016/j.bbrc.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 32.Ky B, Vejpongsa P, Yeh ET, Force T, Moslehi JJ. Emerging paradigms in cardiomyopathies associated with cancer therapies. Circ Res. 2013;113(6):754–764. doi: 10.1161/CIRCRESAHA.113.300218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.