

Significance
Previous studies have suggested that aldolase plays an essential role in parasite motility and host-cell invasion by connecting surface-adhesive proteins to the actin–myosin motor of the parasite. However, our studies show that Toxoplasma aldolase is critical for metabolism but not directly required for parasite motility or invasion. Aldolase-depleted T. gondii cells were sensitive to glucose but showed normal motility and host-cell invasion when grown without glucose, indicating that aldolase does not fulfill an essential role in these important aspects of parasite biology. This conclusion was also supported by studies from adhesin mutants with altered interactions with aldolase in vitro. Taken together, our results force a revision to the current model for host-cell invasion of apicomplexan parasites.
Keywords: micronemal adhesin, motor complex, glycolysis
Abstract
Gliding motility and host-cell invasion by apicomplexan parasites depend on cell-surface adhesins that are translocated via an actin–myosin motor beneath the membrane. The current model posits that fructose-1,6-bisphosphate aldolase (ALD) provides a critical link between the cytoplasmic tails of transmembrane adhesins and the actin–myosin motor. Here we tested this model using the Toxoplasma gondii apical membrane protein 1 (TgAMA1), which binds to aldolase in vitro. TgAMA1 cytoplasmic tail mutations that disrupt ALD binding in vitro showed no correlation with host-cell invasion, indicating this interaction is not essential. Furthermore, ALD-depleted parasites were impaired when grown in glucose, yet they showed normal gliding and invasion in glucose-free medium. Depletion of ALD in the presence of glucose led to accumulation of fructose-1,6-bisphosphate, which has been associated with toxicity in other systems. Finally, TgALD knockout parasites and an ALD mutant that specifically disrupts adhesin binding in vitro also supported normal invasion when cultured in glucose-free medium. Taken together, these results suggest that ALD is primarily important for energy metabolism rather than interacting with microneme adhesins, challenging the current model for apicomplexan motility and invasion.
The phylum Apicomplexa is a group of mostly intracellular parasites that contains a number of human pathogens, including Toxoplasma gondii and Plasmodium spp., the causative agent of malaria. As intracellular pathogens, efficient host-cell invasion is critical for survival, dissemination, and transmission of these parasites. Although they infect different types of host cells, apicomplexan parasites share a conserved mode of host-cell invasion that relies on regulated secretion of adhesive proteins and active motility that is powered by an actin–myosin motor complex (1, 2). According to the current model, motility and host-cell invasion depend on transmembrane adhesins that are secreted apically from the micronemes and translocated along the cell surface in a conveyor belt fashion, using the force generated by the motor complex beneath the parasite membrane (1, 2).
Micronemal adhesins contain a variety of extracellular adhesive domains, a transmembrane domain, and a short cytoplasmic tail that is rich in acidic residues and contains a tryptophan residue (Trp) at or near the extreme carboxyl terminus (3). These conserved features of the cytoplasmic tails are critical to their function, as shown by mutational studies and functional replacement of the thrombospondin-related adhesive protein (TRAP) tail (TRAPt) in P. berghei with the T. gondii Microneme Protein 2 (MIC2) tail (TgMIC2t) (4). The cytoplasmic tails of several micronemal adhesins are thought to be anchored to the actin–myosin motor through a bridging molecule, the glycolytic enzyme fructose-1,6-bisphosphate aldolase (ALD) (5, 6). Mutational analysis has shown that in vitro binding of TgMIC2t and PbTRAPt to ALD is mediated by the acidic residues and Trp residue in the adhesin tails (5–7). ALD has also been shown to interact with MIC2 and TRAP in coimmunoprecipitation experiments using parasite lysates (5, 6). Further support that this interaction plays a role in vivo comes from a conditional knockout (cKO) of TgALD, which shows impaired invasion and growth, consistent with a role in metabolism and/or bridging of adhesins (8). Evidence that TgALD plays a specific role in bridging to adhesins was provided by the TgALD mutant K41E-R42G, which dramatically reduces TgMIC2t binding in vitro whereas having a minimal effect on enzyme activity (8). When expressed in the conditional knockout strain of TgALD (ALD cKO), the K41E-R42G mutant has normal ATP levels, yet it shows decreased host-cell invasion (8).
The micronemal adhesin AMA1 is also important for host-cell invasion by T. gondii and Plasmodium spp. (9–11). AMA1 has similar topology with TRAP/MIC2 with its bulky ectodomain binding to rhoptry neck proteins (RONs) that are secreted from the rhoptries and inserted into host plasma membrane to mediate formation of a moving junction between the host and parasite membranes (12–15). The cytoplasmic tail of AMA1 (AMA1t) also binds rabbit ALD in vitro, and a TgAMA1 mutant with a pair of aromatic residues changed to alanines (i.e., F547A, W548A) disrupts aldolase binding in vitro and blocks host-cell invasion (16). Similarly located FW residues in P. falciparum AMA1t were also shown to mediate binding to rabbit aldolase in vitro, implying this interaction is conserved (17). These studies suggest that AMA1 binding to ALD may play a key role during parasite invasion, similar to that proposed for MIC2 (8) and TRAP (6).
In the present study, we undertook a broader analysis of TgAMA1t mutants that disrupt binding to TgALD in vitro to determine the role of this interaction during host-cell invasion. Unexpectedly, our results indicate that the TgALD–TgAMA1 interaction is not required for parasite motility or invasion. Moreover, we found that the previously described role for TgALD during invasion is alleviated by the absence of glucose. Taken together, these results suggest that ALD is primarily important for energy metabolism but does not play an essential role in coupling adhesins to the motor complex during invasion.
Results
Key Residues in the Tail of TgAMA1 Mediate Binding to TgALD in Vitro.
The cytoplasmic tails of AMA1 from T. gondii and P. falciparum contain a pair of hydrophobic (i.e., FW) residues (Fig. 1A) that are important for binding rabbit muscle aldolase in vitro (16, 17). To test whether TgAMA1t also interacts with aldolase from T. gondii, we used a pull-down assay to examine the ability of recombinant His-TgALD to bind GST-TgAMA1t expressed and purified from Escherichia coli. Consistent with previous reports (16), TgALD bound efficiently to GST-TgAMA1t but not to GST alone (Fig. 1B). To define the residues important for this interaction, we introduced alanine mutations into a number of residues in TgAMA1t and examined their interaction with TgALD using the pull-down assay (Fig. 1 A and B). We identified the residues critical for TgALD binding as W548 (corresponding to W520 in a previous gene model) (16) and the four negatively charged residues immediately downstream (Fig. 1 A and B). The double mutant F547A-W548A showed decreased binding to TgALD. However, the individual single mutants contributed differently to this phenotype: W548A was dramatically impaired, whereas F547A only showed a modest decrease in binding (Fig. 1 A and B). The residues E566 and D568 at the extreme C terminus were also important for this interaction, whereas the terminal tyrosine (Y569) played little if any role (Fig. 1 A and B and Fig. S1A).
Fig. 1.

Interaction between TgAMA1t and TgALD in vitro. (A) Amino acid sequence of the cytoplasmic tail of TgAMA1. When mutated to alanine, the residues colored in green had no effect on TgALD binding, whereas residues in blue increased binding and residues in red decreased binding. (B) Pull-down assay between recombinant TgALD and WT or mutant alleles of TgAMA1t fused to GST (GST-TgAMA1t), resolved by SDS/PAGE, and stained with Coomassie blue. (C) Binding between TgALD and TgAMA1t mutants measured by ELISA. Expressed as % of maximum binding to WT TgAMA1t. Means ± SD (n = 3) from one representative of three independent experiments.
The in vitro binding of TgAMA1t mutants to TgALD was also examined using a semiquantitative ELISA (Fig. 1C and Table 1). The double mutant E552A-E553A had the most dramatic effect on binding (∼45-fold increase in observed Kd; Kobs), whereas other mutants showed changes in Kobs that ranged from 7- to 35-fold higher than WT or were essentially unchanged (F547A) (Table 1). The specificity of the TgALD–TgAMA1t interaction in vitro was also confirmed using the previously characterized mutant TgALD K41E-R42G, which has decreased binding to TgMIC2t in vitro (8). This TgALD mutant displayed dramatically reduced interaction with TgAMA1t in the pull-down assay (Fig. S1B), suggesting that TgAMA1t and TgMIC2t rely on the same positively charged surface on TgALD for binding. Although the TgALD–TgAMA1t interaction was readily established in vitro, we were unable to demonstrate this interaction in vivo, although the moving junction component TgRON4 was easily detected in complex with TgAMA1 by coimmunoprecipitation (co-IP) (Fig. S2). These results suggest that despite robust binding between TgAMA1 and ALD in vitro, conditions are less favorable for this interaction to occur in vivo.
Table 1.
Summary of in vitro TgALD binding and host-cell invasion properties of TgAMA1 mutants
| TgAMA1 alleles | Kobs of TgALD binding, nM* | Host-cell invasion, %† |
| WT | 27.28 ± 1.41 | 100.0 ± 11.0 |
| F547A-W548A | 494.60 ± 27.79 | 9.8 ± 2.0 |
| F547A | 41.54 ± 3.14 | 12.8 ± 3.9 |
| W548A | 288.00 ± 10.03 | 10.5 ± 2.6 |
| D549A-E550A | 920.00 ± 68.65 | 83.2 ± 10.1 |
| E552A-E553A | 1251.00 ± 85.75 | 106.3 ± 6.6 |
| E566A-D568A | 186.00 ± 13.95 | 83.7 ± 9.6 |
In Vivo Characterization of TgAMA1 Mutants.
To examine the role of mutations in TgAMA1 in vivo, we expressed FLAG-tagged mutants under the endogenous promoter in the conditional knockout strain (AMA1 cKO) (9), which expresses a myc-tagged copy of TgAMA1 that is suppressed by addition of anhydrotetracycline (ATc) (Fig. 2A). Stable clones were selected based on similar expression levels of TgAMA1 (Fig. 2B) and efficient suppression of the regulatable copy of TgAMA1-myc by ATc (Fig. S3). The WT and mutant alleles of TgAMA1 were all correctly localized to micronemes at the apical end of the parasite, as shown by colocalization with MIC2-associated protein (M2AP) (Fig. S4).
Fig. 2.
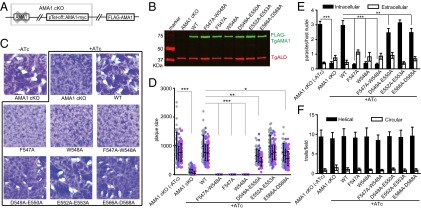
Characterization of TgAMA1 mutants with altered TgALD binding. (A) Diagram of the AMA1 cKO strain complemented with FLAG-tagged TgAMA1 (FLAG-AMA1). The AMA1 cKO strain contains an anhydrotetracycline regulatable copy of TgAMA1 (pTet-off::AMA1-myc), whereas the endogenous TgAMA1 is deleted. (B) FLAG-TgAMA1 mutants were expressed in the AMA1 cKO at similar levels as the WT complement. Western blot using mouse anti-FLAG and rabbit anti-TgALD as a loading control. Primary antibodies were detected using IRDye-conjugated secondary antibodies and imaged using the LI-COR Odyssey imaging system. (C) Parasites were grown ±0.5 μg/mL ATc for 8 d to form plaques on HFF monolayers. (D) Average plaque size in C. Mutants that formed no plaques are shown as having a size of zero (i.e., F547A-W548A, F547A, and W548A). Means ± SD from three independent experiments each with triplicate wells. Each dot represents one plaque; separate experiments are indicated by a different color. Two-way ANOVA with Bonferroni posttest comparisons. Differences were significant in all three experiments at the indicated P values of 0.05 (*), 0.01 (**), or <0.001 (***). (E) Host-cell invasion of TgAMA1 mutants evaluated by a two-color assay to distinguish intracellular from extracellular parasites, with a 20-min infection. Student t test, **P ≤ 0.005, ***P ≤ 0.0001. Means ± SD from three independent experiments each in triplicate (n = 9). (F) Gliding motility of TgAMA1 mutants as determined by SAG1 trail formation. Means ± SD from three independent experiments each in triplicate (n = 9). For both E and F, parasites were cultured in 0.5 μg/mL ATc for 48 h before evaluation. The AMA1 cKO strain grown without ATc served as a control.
TgAMA1 is necessary for parasite growth on fibroblast monolayers, as shown by the inability of the AMA1 cKO strain to form plaques in the presence of ATc, in contrast to the robust growth seen in its absence (Fig. 2C). Complementing the AMA1 cKO strain with WT FLAG-TgAMA1 fully restored the growth in the presence of ATc, as evident by normal plaquing (Fig. 2C). To test the function of TgAMA1 mutants in supporting parasite growth, we compared their ability to complement the formation of plaques in the presence of ATc. Of the six mutants tested, F547A, W548A, and the combined F547A-W548A did not make any visible plaques, indicating that these two residues are critical for TgAMA1 function (Fig. 2 C and D). In contrast, the mutants D549A-E550A and E566A-D568A produced similar numbers of plaques as the WT complement, although the plaques were slightly smaller (Fig. 2 C and D). The plaquing phenotype of the E552A-E553A mutant was indistinguishable from that of WT parasites (Fig. 2 C and D).
In comparing the various mutants, there was no correlation between TgALD binding activity in vitro and the ability to support growth in vivo. Mutants such as W548A and F547A-W548A had reduced TgALD binding and failed to complement the AMA1 cKO strain. However, the mutant F547A showed only a modest drop in TgALD binding yet it did not complement growth, and mutant E552A-E553A, which had the least TgALD binding activity among all the mutants, was fully functional for growth.
Binding of TgAMA1t to TgALD Is Neither Sufficient Nor Necessary for Host-Cell Invasion.
To examine the potential role of TgALD binding for TgAMA1 during host-cell invasion, we compared the invasion efficiency of the above TgAMA1 mutants using a quantitative invasion assay. Consistent with previous studies (9), depletion of AMA1 in the AMA1 cKO strain led to impaired invasion, which was rescued in the WT-complemented line (Fig. 2E). Invasion by strains complemented with the single mutants F547A and W548A, as well as the double mutant F547A-W548A, was reduced to levels similar to that of TgAMA1-depleted cells (AMA1 cKO, +ATc) (Fig. 2E). In contrast, mutants D549A-E550A and E566A-D568A showed a mild (∼20%) reduction in invasion, whereas E552A-E553A invaded as efficiently as the WT complement (Fig. 2E). Although the invasion and plaquing phenotypes of the TgAMA1 mutants were similar, there was again no correlation between TgALD binding and host-cell invasion, suggesting that TgALD–TgAMA1 interactions may not be required for invasion.
Recently, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was reported to bind the cytoplasmic tails of several merozoite Duffy binding-like (DBL) and reticulocyte homology (RH) ligands in P. falciparum (18). We tested whether TgAMA1 might also bind to GAPDH, as this might provide an alternative means to connect adhesins to the motor, thus potentially masking defects in aldolase binding. We detected an interaction between TgAMA1t and rabbit muscle GAPDH in vitro, and binding was significantly reduced in some TgAMA1t mutants (Fig. S5). However, the interaction of TgAMA1t mutants with GAPDH was not correlated with observed invasion phenotypes. In particular, mutants D549A-E550A, E552A-E553A, and E566A-D568A, which all showed reduced binding to ALD (Fig. 1) and GAPDH (Fig. S5) in vitro, displayed normal or nearly normal invasion (Fig. 2).
We also examined gliding motility of the TgAMA1 mutants using staining of surface antigen 1 (SAG1), which is deposited in the trails when parasites glide on substrate. Consistent with previous studies (9), TgAMA1 was not required for gliding motility because the AMA1 cKO displayed very similar gliding properties in the presence and absence of ATc (Fig. 2F). Similarly, none of the TgAMA1 mutants displayed an obvious change in motility compared with the WT complement (Fig. 2F), indicating that they do not have a dominant-negative phenotype for gliding.
Glucose Inhibits the Growth of TgALD-Depleted Parasites.
The lack of correlation between aldolase binding to TgAMA1 in vitro and invasion in vivo led us to consider other models to explain the previously reported requirement for aldolase. In other microorganisms, aldolase deficiency leads to growth arrest only when cells are cultured with glucose or related sugars (19, 20), likely as a consequence of a toxic intermediate(s) that accumulates due to interrupted glycolysis. To test whether aldolase depletion in T. gondii led to growth arrest in a nutrient-dependent manner, we grew the ALD cKO strain and the WT complement (ALD cKO/ALD) with or without glucose in medium containing glutamine and pyruvate. Parasite growth was monitored by the lysis of host-cell monolayers, as described previously (21). When expression of TgALD was suppressed by ATc treatment in the ALD cKO strain, parasite growth was inhibited only when glucose was present in the medium, whereas the WT TgALD-complemented strain grew well under both conditions (Fig. S6). To confirm this phenotype under more stringent suppressive conditions, we pretreated the ALD cKO strain with ATc for 36 h in glucose-free medium, resulting in suppression of the regulatable copy of TgALD to less than 2% of the endogenous level (Fig. 3A). TgALD-depleted parasites were then used to infect fresh monolayers in the presence or absence of glucose plus ATc, and parasite growth was monitored by monolayer lysis. Depletion of TgALD in ALD cKO treated with ATc prevented lysis of the host monolayer in the presence of glucose at any inoculation density, yet TgALD-depleted parasites cultured without glucose grew similar to WT TgALD-complemented cells (Fig. 3B). We also compared the growth of TgALD-depleted parasite in fructose vs. glucose using similar procedures. Although fructose also decreased the growth of TgALD-depleted cells at high concentrations, it was substantially less toxic than glucose, which completely blocked the growth at ≥800 mg/L (Fig. 3C).
Fig. 3.
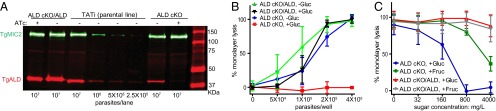
Growth inhibition of TgALD-depleted parasites by glucose or fructose. (A) Depletion of TgALD in the ALD cKO strain. The ALD cKO strain and WT complement (ALD cKO/ALD) were grown in glucose-free medium ±0.5 μg/mL ATc for 36 h before analysis. TgALD was detected using rabbit anti-TgALD. TgMIC2 was detected using mAb 6D10 as a loading control. Primary antibodies were detected as above. Dilutions of the parental TATi line (tet-transactivator) were used to estimate levels of TgALD. (B) Host monolayer lysis by parasite growth for 48 h in media ±4,500 mg/L glucose (containing 0.5 μg/mL ATc). (C) Host monolayer lysis by growth of parasites for 48 h in media (with 0.5 μg/mL ATc) containing glucose or fructose (4 × 105 parasites per well). (B and C) Parasites were pretreated with 0.5 μg/mL ATc for 36 h in glucose-free medium before analysis. Means ± SD combined from two independent experiments (each in triplicate) for B (n = 6) or three for C (n = 9).
Impaired Motility, Invasion, and Replication of TgALD-Depleted Parasites Are Dependent on Growth in Glucose.
Previous studies on the ALD cKO strain indicated that depletion of TgALD inhibited parasite motility and invasion; however, these studies were conducted in glucose-rich medium (i.e., 4,500 mg/L) (8). Because the above findings clearly show that glucose has a toxic effect on TgALD-depleted parasites, we wanted to reexamine the cellular phenotypes of the ALD cKO strain grown under glucose-replete vs. -depleted conditions. In the presence of ATc, the ALD cKO strain displayed a dramatic reduction in host-cell invasion when grown in glucose-containing medium, compared with the same strain grown in the absence of ATc (Fig. 4A). This invasion defect was fully reverted in the WT TgALD-complemented strain (Fig. 4A). In contrast, depletion of TgALD had no effect on invasion when parasites were grown in glucose-depleted medium (Fig. 4A). Similarly, gliding motility was impaired only when ALD cKO was grown in the presence of ATc and glucose, but not in glucose-free medium (Fig. 4B). TgALD-depleted parasites showed significantly slower replication than control parasites when grown in the presence of glucose (Fig. 4C). However, when glucose was excluded from the medium, replication of TgALD-depleted parasites was normal (Fig. 4C). In other systems, disruption of aldolase leads to accumulation of fructose-1,6-bisphosphate (FBP) (22, 23). Consistent with this, we observed a dramatic increase in the concentration of FBP in TgALD cKO parasites grown in the presence of ATc and glucose, and this was largely eliminated when parasites were grown in the absence of glucose (Fig. 4D). Taken together, these results indicate that the impairment of TgALD-depleted parasites is only apparent when cells are grown in glucose, likely as the result of accumulation of toxic metabolites such as FBP.
Fig. 4.
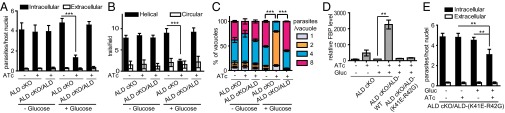
Effect of glucose on host-cell invasion, gliding motility, and intracellular replication. (A) Host-cell invasion as evaluated by a two-color assay to distinguish intracellular from extracellular parasites, with a 20-min infection. (B) Gliding of parasites on BSA-coated coverslips as detected by trails stained with SAG1. In both A and B, parasites were pretreated by growth in ±ATc, ±glucose as indicated for 36 h before analysis. Independent experiments (each in triplicate) were combined for three experiments in A (n = 9) and two for B (n = 6). Means ± SD, Student t test, ***P ≤ 0.0001. (C) Parasites pretreated by growth in ±ATc, ±glucose for 36 h were used to infect fresh host monolayers. Invaded parasites were allowed to replicate for 18 h under the corresponding pretreatment conditions, and the number of vacuoles containing one, two, four, or eight parasites was determined by epifluorescence microscopy after SAG1 staining. Means ± SD combined from two independent experiments each in triplicate (n = 6). Two-way ANOVA, ***P ≤ 0.0001. (D) LC-MS/MS analysis of FBP levels in parasite extracts. Samples were normalized to the ALD cKO +Gluc –ATc sample that was set at 100%. Means ± SD from three experiments each with a single sample (n = 3). **P < 0.01. (E) Host-cell invasion (as described above) of the TgALD-K41E-R42G mutant after growth for 36 h under the indicated conditions. Means ± SD combined from two independent experiments each in triplicate (n = 6). Student t test, **P ≤ 0.001.
TgALD Mutant Defective in Adhesin Binding Showed Normal Invasion When Cultured in Glucose-Free Medium.
The complete reversal of the phenotypes of TgALD-depleted parasites by growth in the absence of glucose calls into question the adhesin-bridging role proposed previously (5, 8). Key support for this model comes from the mutant TgALD K41E-R42G, which has a partial invasion defect but also reduced enzymatic activity despite normal cellular ATP levels (8). Consistent with this previous report (8), we found that TgALD K41E-R42G had dramatically reduced TgMIC2t and TgAMA1t binding in vitro (Fig. S1). We reasoned that if the decrease in adhesin binding was responsible for the decreased invasion, the phenotype of the TgALD K41E-R42G mutant should be independent of growth in glucose. However, when cultured in glucose-free medium, the partial invasion defect of the TgALD K41E-R42G mutant seen in the presence of glucose was reverted to normal levels (Fig. 4E). Although we did not detect accumulation of FBP in this mutant when grown in the presence of glucose (Fig. 4D), the defect in this mutant is likely due to a metabolic insufficiency, as invasion was normal when grown in the absence of glucose (Fig. 4E).
TgALD Knockout Parasites Invade Normally When Grown in Glucose-Free Medium.
To further confirm the independence of host-cell invasion of aldolase, we constructed a loxP-TgALD strain and subsequently used Cre recombinase to delete TgALD, as described (24) (Fig. 5 A and B). Following Cre transfection and growth for 48 h, ∼25% of replicating parasites were YFP+ and lacked detectable TgALD expression (Fig. 5C). After FACS sorting (Fig. 5D), we grew aldolase-deleted (YFP+ TgALD−) vs. wild-type (YFP− TgALD+) parasites in glucose-free medium for another 48 h (approximately eight generations) to ensure complete depletion of the protein (Fig. S7). Subsequent invasion assays showed that YFP+ TgALD− cells had similar invasion efficiency to YFP− TgALD+ parasites (Fig. 5E), confirming that host-cell invasion does not require aldolase. Collectively, these findings suggest a revision to the bridging model, as discussed further below.
Fig. 5.

Host-cell invasion of TgALD knockout parasites grown in glucose-free medium. (A) Diagram of loxP-ALD strain generation. (B) Diagnostic PCR used to confirm the loxP-ALD strain. The asterisks denote nonspecific amplification. (C) Immunofluorescence staining for TgALD and YFP expression in loxP-ALD cells 48 h post Cre transfection and growth in glucose-free medium. TgALD was detected with rabbit anti-TgALD and YFP by mouse anti-GFP, followed by Alexa Fluor-conjugated secondary antibodies. (D) Following natural egress, parasites were FACS-sorted based on YFP expression and cultured for another 48 h in glucose-free medium. (E) Host-cell invasion of parasites collected and expanded from D as described above. Representative of three independent experiments (C and D); E contains data from three experiments combined (each in triplicate, means ± SD, n = 9).
Discussion
The current model for gliding motility and host-cell invasion by apicomplexan parasites postulates the need for a mechanical connection between surface adhesins and the actin–myosin motor to generate power (1, 2). For the past decade, this connection was thought to be mediated via aldolase bridging between the adhesin tails and F-actin (5). There are a number of features that make this an attractive model. ALD is tetrameric, with four binding sites, such that it can bind to and cross-link actin filaments (25). The binding sites important for this interaction are also conserved in parasite actins (26) and aldolases (27). The cytoplasmic tails of many transmembrane adhesins bind to ALD in vitro, including MIC2 (5), MIC6 (28), and AMA1 (16) in T. gondii, as well as RH1, RH2B, RH4 (18), TRAP (6), and AMA1 (17) in Plasmodium spp. The molecular details of the interaction between ALD and TRAPt and MIC2t have been probed by structural (29) as well as mutational studies (6, 8). Together, these prior studies reveal that the binding site is mediated by a positively charged groove in aldolase that interacts with negatively charged residues in the adhesin tails, combined with hydrophobic interactions. Moreover, ALD, actin, and TgMIC2 or PbTRAP can be coprecipitated from cell lysates of T. gondii (5) or P. berghei (6), suggesting these interactions occur in vivo. However, the significance of such interactions was largely untested, with the exception of TgALD mutants made in T. gondii (8).
Although initially established from studies in the related adhesins MIC2 and TRAP (5, 6), recent attention has focused on other adhesins that may also interact with ALD, broadening interest in the role of this interaction during host-cell invasion (16, 17). For example, TgAMA1t was shown to bind rabbit muscle ALD in vitro, and a TgAMA1 mutant (F547A-W548A) defective in ALD binding lost the ability to invade host cells, suggesting this interaction was critical for function (16). To explore this interaction further, we generated additional TgAMA1 mutants and tested their ability to bind TgALD in vitro and the ability to complement the AMA1 depletion strain for growth and invasion. There was no correlation between the ability of TgAMA1 mutants to bind TgALD in vitro and their ability to support host-cell invasion. In addition, we were unable to detect the TgAMA1–TgALD interaction by co-IP, implying that it is of low affinity or absent in vivo. Together, these results suggest that if binding betweenTgAMA1 and TgALD occurs in vivo, it is not essential for invasion.
The lack of a requirement for binding between TgAMA1 and TgALD during invasion led us to reexamine the proposed role for aldolase in bridging to adhesins during host-cell invasion. We discovered that growth of TgALD-depleted parasites was only inhibited when high levels of glucose or fructose were present in the medium. In contrast, when TgALD-depleted parasites were cultured in medium containing pyruvate and glutamine, growth and host-cell invasion were normal. Furthermore, the reversal of the invasion defect of the TgALD K41E-R42G mutant in the absence of glucose suggests that the defect in this mutant is not due to the decreased binding to adhesins but rather to metabolic deficiency as a result of its lower enzyme activity (8). In light of the present findings, we conclude that if ALD–adhesin interactions occur in vivo, they do not play an essential role in invasion but rather aldolase is primarily important in energy metabolism. Our finding that aldolase is dispensable in the absence of glucose is consistent with the observation that growth is normal following deletion of the major glucose transporters in T. gondii, as long as glutamine is present (30). Whether ALD is required for long-term growth of T. gondii under these conditions is uncertain, although it might be necessary to participate in gluconeogenesis, a pathway that is complete in the genome (www.toxodb.org).
The dramatic inhibition of ALD-depleted parasites in the presence of glucose/fructose mirrors the growth inhibition of temperature sensitive ALD mutants of bacteria and yeast (19, 20) grown in glucose and related sugars, as well as fructose intolerance caused by aldolase B (an isoform preferentially expressed in the liver) deficiency in humans (22). The basis for the latter defects is thought to be the toxic effects of accumulated FBP or fructose-1-phosphate (22, 23). A similar mechanism may explain the growth inhibition of TgALD-depleted parasites cultured in glucose, because we observed a dramatic increase in the concentration of FBP when grown in the presence of glucose. Hence, it is likely that most of the previous effects of aldolase depletion can be attributed to the accumulation of toxic intermediates. Aldolase is also likely to be essential in vivo, because human serum has ∼1,000 mg glucose/L, levels that would completely block the growth of TgALD-depleted parasites.
Although ALD is important for sugar metabolism in T. gondii, it is apparently not required to link microneme adhesins to the motor during host-cell invasion. It has been suggested that other adaptors may contribute to this function, including GAPDH that binds to several Plasmodium adhesin tails in vitro (18) and TgAMA1 as shown here. Our data suggest that binding to GAPDH may also not be required for AMA1 function because mutants that were defective in binding to both GAPDH and ALD (i.e., E552A-E553A) showed nearly normal invasion. Also, the phenotype of mutants such as TgAMA1t-F547A, which still binds aldolase and GAPDH although is unable to support invasion, suggests that neither interaction is sufficient for invasion. Rather, it is possible that adhesin tails serve other functions, as mutational studies clearly demonstrate critical roles for conserved residues in TgAMA1 (16), TgMIC2 (7), PbTRAP (4), and others. As gliding motility and cell invasion are critical for parasite survival, defining these functions remains an important focus for future study.
Materials and Methods
Parasite Strains and Growth Conditions.
T. gondii tachyzoites were maintained by growth in human foreskin fibroblasts (HFFs) cultured as described previously (8) (SI Materials and Methods). The AMA1 cKO strain (9) was kindly provided by Gary Ward (University of Vermont, Burlington, VT), and complementing mutants were constructed in the plasmid pDHFR-AMA1 and used to obtain transgenic parasites (SI Materials and Methods). The TgALD cKO and complementing (WT and K41E-R42G) strains were described previously (8). Anhydrotetracycline (0.5 μg/mL) (Clontech Laboratories) was used to treat parasites for 36 or 48 h unless otherwise indicated. Plaque assays were performed on HFF monolayers as described in SI Materials and Methods. Motility, invasion, and replication assays were done as previously described (8), with modifications described in SI Materials and Methods.
Mass Spectrometry.
Parasites were grown under the indicated conditions for 36 h, harvested, purified, washed with PBS, extracted, and processed for LC-MS/MS as described in SI Materials and Methods. Samples were analyzed using a Shimadzu LC system interfaced with an AB Sciex 4000 QTRAP mass spectrometer. Pipes [piperazine-N,N′-bis(2-ethanesulfonic acid)] was spiked into the samples before extraction and used as an internal standard to normalize the extraction efficiency, as described in SI Materials and Methods.
Protein Purification, Pull-Down, and ELISA.
His-TgALD and GST-TgAMA1t were purified from E. coli BL21(DE3) containing pET16b-TgALD (8) and pGEX3X-TgAMA1t, respectively, as described previously (7). Pull-down assays were conducted as described (7). Western blotting was performed as described in SI Materials and Methods. Quantitative ELISA was done as previously described (7), with modifications as described in SI Materials and Methods. Samples were tested in triplicate, and the Kobs was determined using the nonlinear regression curve-fitting function in Prism version 5 (GraphPad Software).
Statistics.
Statistical comparisons were conducted in Prism (GraphPad Software) using two-tailed Student t test with unpaired samples with equal variance. For comparing multiple datasets or more than two groups, ANOVA statistical tests were conducted in Prism.
Supplementary Material
Acknowledgments
We are grateful to Gary Ward for providing the TgAMA1 conditional knockout strain and for helpful discussions, Vern Carruthers and John Boothroyd for providing antibodies, Markus Meissner and Ke Hu for plasmids, members of the L.D.S. laboratory, Jurgen Sygusch, and Lou Miller for advice, Sophie Alvarez (Donald Danforth Plant Science Center) for mass spectrometry analysis, and Jennifer Barks for technical assistance. This work was supported by a grant from the National Institutes of Health (AI034036).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315156111/-/DCSupplemental.
References
- 1.Cowman AF, Berry D, Baum J. The cellular and molecular basis for malaria parasite invasion of the human red blood cell. J Cell Biol. 2012;198(6):961–971. doi: 10.1083/jcb.201206112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sibley LD. How apicomplexan parasites move in and out of cells. Curr Opin Biotechnol. 2010;21(5):592–598. doi: 10.1016/j.copbio.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carruthers VB, Tomley FM. Microneme proteins in apicomplexans. Subcell Biochem. 2008;47:33–45. doi: 10.1007/978-0-387-78267-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kappe S, et al. Conservation of a gliding motility and cell invasion machinery in Apicomplexan parasites. J Cell Biol. 1999;147(5):937–944. doi: 10.1083/jcb.147.5.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jewett TJ, Sibley LD. Aldolase forms a bridge between cell surface adhesins and the actin cytoskeleton in apicomplexan parasites. Mol Cell. 2003;11(4):885–894. doi: 10.1016/s1097-2765(03)00113-8. [DOI] [PubMed] [Google Scholar]
- 6.Buscaglia CA, Coppens I, Hol WGJ, Nussenzweig V. Sites of interaction between aldolase and thrombospondin-related anonymous protein in Plasmodium. Mol Biol Cell. 2003;14(12):4947–4957. doi: 10.1091/mbc.E03-06-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starnes GL, Jewett TJ, Carruthers VB, Sibley LD. Two separate, conserved acidic amino acid domains within the Toxoplasma gondii MIC2 cytoplasmic tail are required for parasite survival. J Biol Chem. 2006;281(41):30745–30754. doi: 10.1074/jbc.M606523200. [DOI] [PubMed] [Google Scholar]
- 8.Starnes GL, Coincon M, Sygusch J, Sibley LD. Aldolase is essential for energy production and bridging adhesin-actin cytoskeletal interactions during parasite invasion of host cells. Cell Host Microbe. 2009;5(4):353–364. doi: 10.1016/j.chom.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mital J, Meissner M, Soldati D, Ward GE. Conditional expression of Toxoplasma gondii apical membrane antigen-1 (TgAMA1) demonstrates that TgAMA1 plays a critical role in host cell invasion. Mol Biol Cell. 2005;16(9):4341–4349. doi: 10.1091/mbc.E05-04-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Triglia T, et al. Apical membrane antigen 1 plays a central role in erythrocyte invasion by Plasmodium species. Mol Microbiol. 2000;38(4):706–718. doi: 10.1046/j.1365-2958.2000.02175.x. [DOI] [PubMed] [Google Scholar]
- 11.Hehl AB, et al. Toxoplasma gondii homologue of Plasmodium apical membrane antigen 1 is involved in invasion of host cells. Infect Immun. 2000;68(12):7078–7086. doi: 10.1128/iai.68.12.7078-7086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexander DL, Mital J, Ward GE, Bradley PJ, Boothroyd JC. Identification of the moving junction complex of Toxoplasma gondii: A collaboration between distinct secretory organelles. PLoS Pathog. 2005;1(2):e17. doi: 10.1371/journal.ppat.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamarque M, et al. The RON2-AMA1 interaction is a critical step in moving junction-dependent invasion by apicomplexan parasites. PLoS Pathog. 2011;7(2):e1001276. doi: 10.1371/journal.ppat.1001276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tonkin ML, et al. Host cell invasion by apicomplexan parasites: Insights from the co-structure of AMA1 with a RON2 peptide. Science. 2011;333(6041):463–467. doi: 10.1126/science.1204988. [DOI] [PubMed] [Google Scholar]
- 15.Richard D, et al. Interaction between Plasmodium falciparum apical membrane antigen 1 and the rhoptry neck protein complex defines a key step in the erythrocyte invasion process of malaria parasites. J Biol Chem. 2010;285(19):14815–14822. doi: 10.1074/jbc.M109.080770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheiner L, et al. Toxoplasma gondii transmembrane microneme proteins and their modular design. Mol Microbiol. 2010;77(4):912–929. doi: 10.1111/j.1365-2958.2010.07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srinivasan P, et al. Binding of Plasmodium merozoite proteins RON2 and AMA1 triggers commitment to invasion. Proc Natl Acad Sci USA. 2011;108(32):13275–13280. doi: 10.1073/pnas.1110303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pal-Bhowmick I, et al. Binding of aldolase and glyceraldehyde-3-phosphate dehydrogenase to the cytoplasmic tails of Plasmodium falciparum merozoite Duffy binding-like and reticulocyte homology ligands. MBio. 2012;3(5):e00292-12. doi: 10.1128/mBio.00292-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Böck A, Neidhardt FC. Properties of a mutant of Escherichia coli with a temperature-sensitive fructose-1,6-diphosphate aldolase. J Bacteriol. 1966;92(2):470–476. doi: 10.1128/jb.92.2.470-476.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lobo Z. Saccharomyces cerevisiae aldolase mutants. J Bacteriol. 1984;160(1):222–226. doi: 10.1128/jb.160.1.222-226.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buguliskis JS, Brossier F, Shuman J, Sibley LD. Rhomboid 4 (ROM4) affects the processing of surface adhesins and facilitates host cell invasion by Toxoplasma gondii. PLoS Pathog. 2010;6(4):e1000858. doi: 10.1371/journal.ppat.1000858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouteldja N, Timson DJ. The biochemical basis of hereditary fructose intolerance. J Inherit Metab Dis. 2010;33(2):105–112. doi: 10.1007/s10545-010-9053-2. [DOI] [PubMed] [Google Scholar]
- 23.Schreyer R, Böck A. Phenotypic suppression of a fructose-1,6-diphosphate aldolase mutation in Escherichia coli. J Bacteriol. 1973;115(1):268–276. doi: 10.1128/jb.115.1.268-276.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andenmatten N, et al. Conditional genome engineering in Toxoplasma gondii uncovers alternative invasion mechanisms. Nat Methods. 2013;10(2):125–127. doi: 10.1038/nmeth.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Morris AJ, Tolan DR, Pagliaro L. The molecular nature of the F-actin binding activity of aldolase revealed with site-directed mutants. J Biol Chem. 1996;271(12):6861–6865. [PubMed] [Google Scholar]
- 26.Skillman KM, et al. Evolutionarily divergent, unstable filamentous actin is essential for gliding motility in apicomplexan parasites. PLoS Pathog. 2011;7(10):e1002280. doi: 10.1371/journal.ppat.1002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HC, Certa U, Döbeli H, Jakob P, Hol WGJ. Crystal structure of fructose-1,6-bisphosphate aldolase from the human malaria parasite Plasmodium falciparum. Biochemistry. 1998;37(13):4388–4396. doi: 10.1021/bi972233h. [DOI] [PubMed] [Google Scholar]
- 28.Zheng B, et al. MIC6 associates with aldolase in host cell invasion by Toxoplasma gondii. Parasitol Res. 2009;105(2):441–445. doi: 10.1007/s00436-009-1401-5. [DOI] [PubMed] [Google Scholar]
- 29.Bosch J, et al. Aldolase provides an unusual binding site for thrombospondin-related anonymous protein in the invasion machinery of the malaria parasite. Proc Natl Acad Sci USA. 2007;104(17):7015–7020. doi: 10.1073/pnas.0605301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blume M, et al. Host-derived glucose and its transporter in the obligate intracellular pathogen Toxoplasma gondii are dispensable by glutaminolysis. Proc Natl Acad Sci USA. 2009;106(31):12998–13003. doi: 10.1073/pnas.0903831106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.