

Significance
Cytoskeletal filaments drive many dynamic cellular processes, such as the regulation of shape by actin networks in eukaryotes and by the actin homolog MreB in rod-shaped bacteria. Here, we use all-atom molecular dynamics simulations to demonstrate close parallels between the conformational dynamics of actin and MreB, in which polymerization induces flattening of MreB subunits that restructures the ATP binding pocket to promote hydrolysis. We also find that ATP-bound MreB filaments are substantially more curved than ADP-bound filaments, and this bending is highly correlated with the degree of flattening of the subunits. Our results provide molecular-scale insight into the diverse structural states of MreB during its assembly process, revealing properties that may be general to the broad actin family.
Keywords: bacterial cytoskeleton, polymer mechanics, actin superfamily, filament assembly, cell shape control
Abstract
The assembly of protein filaments drives many cellular processes, from nucleoid segregation, growth, and division in single cells to muscle contraction in animals. In eukaryotes, shape and motility are regulated through cycles of polymerization and depolymerization of actin cytoskeletal networks. In bacteria, the actin homolog MreB forms filaments that coordinate the cell-wall synthesis machinery to regulate rod-shaped growth and contribute to cellular stiffness through unknown mechanisms. Like actin, MreB is an ATPase and requires ATP to polymerize, and polymerization promotes nucleotide hydrolysis. However, it is unclear whether other similarities exist between MreB and actin because the two proteins share low sequence identity and have distinct cellular roles. Here, we use all-atom molecular dynamics simulations to reveal surprising parallels between MreB and actin structural dynamics. We observe that MreB exhibits actin-like polymerization-dependent structural changes, wherein polymerization induces flattening of MreB subunits, which restructures the nucleotide-binding pocket to favor hydrolysis. MreB filaments exhibited nucleotide-dependent intersubunit bending, with hydrolyzed polymers favoring a straighter conformation. We use steered simulations to demonstrate a coupling between intersubunit bending and the degree of flattening of each subunit, suggesting cooperative bending along a filament. Taken together, our results provide molecular-scale insight into the diversity of structural states of MreB and the relationships among polymerization, hydrolysis, and filament properties, which may be applicable to other members of the broad actin family.
The cytoskeletal protein MreB, a structural homolog of eukaryotic actin (1), is essential for cell-shape determination in many rod-shaped bacteria. Inhibition of MreB causes uncontrolled cell-wall insertion and eventual cell lysis (2, 3). Dozens of single-residue substitutions in MreB have been found to confer subtle shape changes, such as precise tuning of cell width, without negatively affecting growth rate or viability (4–6). Because emerging evidence suggests that MreB guides the patterning of cell-wall synthesis (7–9), it has become increasingly important to uncover the molecular factors that govern the structural and mechanical properties of MreB. Like actin, MreB is an ATPase, with four subdomains surrounding a nucleotide-binding pocket (1). However, MreB and actin share only 15% residue identity, mostly between conserved ATPase motifs common to the actin superfamily (10). Although both actin and MreB regulate cell shape, actin forms dynamic, branched cytosolic networks (11), whereas MreB forms distinct, stable filaments in vitro (12) and associates with the membrane in vivo to coordinate cell-wall elongation in many rod-shaped bacteria (13). Thus, the validity of inferring MreB filament properties from the extensive structural knowledge of actin is unclear.
Although ample experimental efforts have shed light on the biochemical and cellular properties of MreB, some seemingly conflicting data and the lack of molecular-level knowledge of MreB structural dynamics impede a full understanding of the mechanism by which cell shape is controlled. In vitro, MreB polymerizes into filaments that can bind liposomes and cause inward indentations indicative of stiff, preferentially curved filaments (14). However, other studies have observed long, straight filaments (15) or a combination of straight and curved filaments (1, 12, 16, 17), suggesting that experimental conditions may affect filament curvature, or that MreB filaments might adopt multiple conformations. In vivo, MreB binds to the cytoplasmic face of the inner membrane as distinct diffraction-limited puncta (18) or filaments (19) of unknown structure.
Understanding the structural dynamics of MreB filaments requires a method with atomic resolution that can capture molecular behavior. Here, we examine the conformational dynamics of MreB assembly from monomer to dimer in various nucleotide-binding states using all-atom molecular dynamics (MD) simulations. MD simulations have previously elucidated the polymerization- and hydrolysis-dependent structural transitions of various prokaryotic and eukaryotic cytoskeletal proteins (20–25). For the bacterial tubulin homolog FtsZ, MD simulations predicted a hydrolysis-dependent filament bending (24) that was later validated by X-ray crystallography (22). In the case of actin, MD simulations have shed light on the structural transition between ATP-bound “globular” actin (G-actin) and models of polymerized ADP-bound “filamentous” or “fibrous” actin (F-actin) (26).
In our simulations, we found that the structural dynamics of MreB polymer assembly exhibits remarkable parallels with actin polymerization. Specifically, simulated MreB monomers adopt conformations that deviate significantly from crystallized protofilaments, and the subsequent polymerization introduces flattening of the polymer subunits, a process also observed in actin. As with actin, MreB polymerization restructures the nucleotide-binding pocket into a configuration that promotes nucleotide hydrolysis. MreB dimers also exhibit nucleotide-dependent mechanical properties, wherein ATP-bound dimers are substantially more curved than ADP-bound dimers. We demonstrate that intersubunit bending is coupled to the flattening of MreB subunits, suggesting that polymer bending may be highly cooperative along a filament. These results provide a simple interpretation to reconcile the different MreB filament structures, straight and highly bent, obtained from in vitro experiments (1, 12, 15, 17). Furthermore, our results provide a molecular-scale framework for interpreting the mechanisms underlying cell-shape changes owing to perturbations of MreB structure. Finally, the high degree of parallels in the assembly of MreB and actin suggest that the structural dynamics of actin family proteins may be broadly conserved.
Results
Polymerization Causes Flattening and Closing of the MreB Monomer.
To interrogate the first step in protofilament assembly of MreB monomers, we performed all-atom MD simulations of MreB as a monomer and a dimer bound to ATP (Fig. 1 A and B; simulations 1 and 2, Table S1; and Methods). Simulations were initialized either from the crystal structure of the MreB protofilament from Thermotoga maritima (PDB ID code 1JCG) (1) or from equilibrium structures obtained in our simulations starting with the same crystal structure. All unconstrained simulations were performed until an equilibrium structure was reached (55–130 ns; Methods). By analogy with actin, we refer to our MreB monomer and dimer structures as G-MreB and F-MreB, respectively, and distinguish the two subunits in a dimeric F-MreB as the (+) and (−) ends (Fig. 1B). The four subdomains of MreB subunits are defined by convention (Fig. 1C and Table S2).
Fig. 1.

Polymerization and hydrolysis cause flattening of the MreB polymer. (A and B) MD simulation of an ATP-bound monomeric G-MreB (A) and a dimeric F-MreB (B), with the top and bottom monomer referred to as the (+) and (−) subunits, respectively. (C) The MreB subunit, described by its four subdomains, quickly adopts an open and unflattened conformation in monomeric G-MreB simulations. (D) A polymerization-dependent transition in flatness can be described by the dihedral angle Φ. (E) Starting from the MreB crystal structure, ATP-bound G-MreB unflattens to a new state with a larger dihedral angle (solid curve is the smoothed version of the light curve); after this unflattened monomer is placed back on the (+) end of a protofilament subunit, it reflattens (blue curve) to a value close to that of the monomer to which it is bound (red line) and to the MreB crystal structure (horizontal dashed line). (F) Distributions of the dihedral angle over the last 30 ns of each simulation in E.
In all simulations of monomeric G-MreB, the IB and IIB subdomains rapidly hinged away from each other and formed a stable conformation with an open and sheared structure (Fig. 1 C and D and Movie S1). The structural changes within a monomeric MreB can be quantified by measuring the center-of-mass positions of the four subdomains, allowing for the definitions of two inner opening angles and a dihedral angle Φ (Fig. 1 C and D) (25). In our simulations, ATP-bound G-MreB stabilized at a dihedral angle of Φ ≈ 30° at the end of a 55-ns simulation (Fig. 1E, orange trace), an angle substantially larger than that seen in the MreB protofilament crystal structure (11°). In our simulations with dimeric F-MreB bound to ATP (Fig. 1B), both subunits maintained a smaller dihedral angle (Φ ≈ 22°; Fig. 1F and Fig. S1), indicating that F-MreB is inherently flatter than G-MreB.
By comparison, actin also exhibits a well-characterized polymerization-induced shift in its dihedral angle (25, 26). Specifically, G-actin structures display a dihedral angle of 21–24° and flatten to 4–10° upon polymerization into F-actin (25). Given the large difference in dihedral angles between G-MreB and F-MreB, we hypothesized that, like actin, polymerization of a newly incorporated G-MreB monomer would lead to a decrease in its dihedral angle to a flatter F-MreB configuration. To test this hypothesis, we performed a simulation mimicking the polymerization process by placing an equilibrium structure of G-MreB onto the (+) end of an MreB subunit initialized from the protofilament crystal structure. Despite the large difference in initial structures, the two proteins remained attached for the full simulation duration of 66 ns, and the (+) subunit that was initially in a G-MreB configuration rapidly flattened back to the dihedral angle of the (−) subunit at ∼17° (Fig. 1E, blue trace and simulation 3, Table S1). The (−) subunit remained in an F-MreB configuration with its dihedral angle also fluctuating around 17° (Fig. 1E, red trace). Thus, our simulations reveal that, like actin, MreB undergoes reversible monomer flattening upon polymerization.
Although the adsorbed subunit flattened rapidly over the time course of this simulation, it maintained the G-MreB open configuration, as measured by the average of the two opening angles (Fig. S2 C and D). Interestingly, in one repeat simulation of ATP-bound F-MreB, the (+) subunit transiently adopted a G-MreB–like open conformation before returning to a stable, closed conformation. In all simulations of F-MreB initialized from the MreB crystal structure, MreB subunits equilibrated to a closed state, suggesting that the adsorbed subunit would close in an extended simulation. Taken together, our simulations suggest the existence of distinct open and closed MreB conformations, wherein MreB maintains an open state as a monomer and a closed state upon polymerization.
MreB Requires a Chelating Mg2+ Ion for Stability of the Bound Nucleotide.
A divalent cation (Mg2+ or Ca2+) is required for the in vitro polymerization of MreB (1, 12, 15–17) and actin (11) and has been hypothesized to stabilize the bound nucleotide (27). To determine the effects of Mg2+ on the structural dynamics of MreB monomers, we performed simulations of MreB as a monomer or dimer bound to ATP with the chelating Mg2+ ion removed (Fig. 2 and simulations 4 and 5, Table S1). Whereas the γ-phosphate of ATP remained coordinated with the Mg2+ ion throughout the simulations in which Mg2+ was present (Fig. 2A), the rmsd of the ATP γ-phosphate increased by approximately eightfold in the absence of Mg2+ (Fig. 2B). In one dimer simulation performed in the absence of Mg2+, an ATP molecule fully dissociated from the binding pocket (Fig. S3 and Movie S2). These data reinforce the notion that a divalent cation such as Mg2+ or Ca2+ plays an important structural role in stabilizing ATP in the nucleotide-binding pocket of MreB.
Fig. 2.

Lack of chelation with Mg2+ ions disrupts ATP binding. (A) In the absence of Mg2+, the ATP loses its coordination with the nucleotide-binding pocket (red) compared with the initial structure (gray). (B) Distribution of the rmsd values of γ-phosphate with and without Mg2+. The γ-phosphate exhibits a several-fold increase in rmsd in the absence of Mg2+.
Polymerization Restructures the Nucleotide-Binding Pocket to Promote Hydrolysis.
Given the large conformation changes of G-MreB upon polymerization, we next queried how these changes affect other aspects of MreB filament assembly. In vitro polymerization assays have demonstrated that the ATPase activity of MreB occurs only after polymerization (12, 15–17). In actin, polymerization leads to a 40,000-fold increase in the rate of hydrolysis (28). Previous MD simulations of F-actin indicated that polymerization-induced flattening of the protein promotes hydrolysis by coordinating water molecules and catalytic side chains with the ATP γ-phosphate, and by stabilizing the ATP β-phosphate, which acts as the leaving group during hydrolysis (25, 29). Given the large number of conserved hydrolytic residues between actin and MreB (1), we compared catalytic water coordination and β-phosphate stability in our simulations of G- and F-MreB. We observed a similar restructuring of catalytic residues around ATP in F-MreB simulations (Fig. 3A), including E131, a highly conserved residue in MreB equivalent to Q137 in actin that has been implicated in hydrolysis promotion (26). This restructuring was coincident with the stabilization of a network of catalytic water molecules around the chelating Mg2+ ion (Fig. 3A). In contrast, water did not form a well-defined network in simulations of G-MreB bound to ATP (Fig. 3B). The rmsd values of the β-phosphate were larger in the case of G-MreB compared with F-MreB and also exhibited a wider distribution (Fig. 3C). Our results therefore suggest that the promotion of ATP hydrolysis upon MreB polymerization follows molecular restructuring similar to that proposed for actin.
Fig. 3.
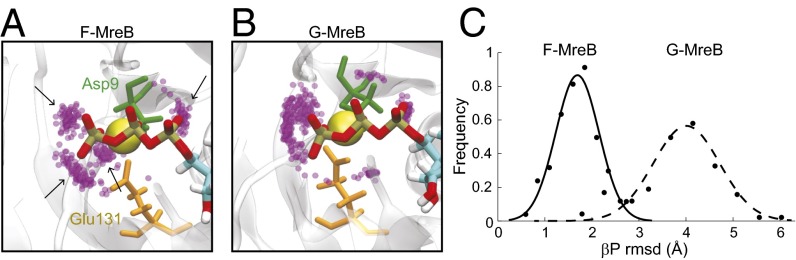
Polymerization restructures the ATP binding pocket, promoting ATP hydrolysis through coordination of a catalytic water network around the γ-phosphate of ATP. (A and B) Simulation snapshot of the ATP-binding pocket, depicting the Mg2+ ion (yellow), the conserved catalytic residues Asp9 (green) and Glu131 (orange), and the ATP tail (red and gold). Positions of nearby water oxygens (within 4 Å, purple) are shown for each frame in the last 5 ns of each simulation (each frame is 0.1 ns). Stable catalytic water networks (arrows) form in F-MreB (A) but not G-MreB (B). (C) The β-phosphate of ATP is associated with a lower and more stable rmsd in F-MreB than in G-MreB; a stabilized hydrolytic leaving group can help promote ATP hydrolysis upon polymerization.
Bending of MreB Dimers Is Nucleotide-Dependent.
Nucleotide identity regulates the structural properties of actin filaments, with ADP-bound filaments exhibiting lower stiffness than ATP-bound filaments (30). To probe the effects of nucleotide identity on F-MreB structures, we performed MreB monomer and dimer simulations with each subunit bound to either ATP or ADP. The replacement of ATP with ADP resulted in flattening in G-MreB and both F-MreB subunits (Fig. S1 and simulations 6 and 7, Table S1). We also observed intermolecular bending between F-MreB subunits, which was more substantial in the ATP-bound case than in the ADP-bound case (Fig. 4A). To quantify the mechanical properties of MreB dimers, we measured the relative orientation between the (+) and (−) subunits using three Euler angles, which can be interpreted as two orthogonal bending directions (θ1 and θ2) and twist along the axis of the filament (θ3) (Methods and Supporting Information). We define zero bending and twisting to be the configuration adopted by the crystal structure (1). Most of the bending in our simulations occurred around a single axis (θ2; Table S3). The intrinsic curvature of the MreB dimer measured by θ2 was nucleotide-dependent, with ATP-bound F-MreB bent at ∼26.0° (corresponding to a radius of curvature R = 10 nm, because each MreB is ∼5 nm along the filament axis), whereas the ADP-bound F-MreB was bent at ∼8.6° (R = 30 nm; Fig. 4B).
Fig. 4.

Hydrolysis decreases filament bending. (A) Snapshot of ATP-bound F-MreB, illustrating the three possible intersubunit bending and twisting directions (Supporting Information). Bending occurs primarily around the θ2 axis (dashed box). (B) Distribution of θ2 bending angles of ATP- and ADP-bound MreB dimers (solid and dashed, respectively). The histogram of bending from the last 30 ns of each simulation is shown as solid circles and Gaussian fits are shown as continuous curves, with the means and SDs of θ2 bending given in degrees (Table S3).
Both nucleotide conditions in the MreB dimer simulations led to approximately equal bending stiffness as characterized by the width of the θ2 spectrum, corresponding to a spring constant for bending of k = kBT/σ2 = 196 kBT/rad2 at T = 310 K. As a complementary estimate of dimer stiffness, we calculated the Young’s modulus for filament stretching from the distribution of distances between the center-of-mass positions of the two subunits. We found nearly identical values of 1.7 GPa for ATP- and ADP-bound F-MreB dimers (Fig. S4 and Supporting Information), a value comparable to that of actin double filaments (1.9 GPa) (31) and energetically sufficient to orient membrane-bound filaments along the circumference of the cell according to a theoretical study (32). We note that stretching would likely be a dominant contributor to the bending stiffness of higher-order MreB structures such as double filaments.
To test whether the observed change in dimer filament structure was caused by differences in the stability of the monomer–monomer interface, we compared the subunit interface contact area. Specifically, we calculated the buried solvent-accessible surface area (Methods and Supporting Information) for both nucleotide states of F-MreB and found that they were similar in value and variation (Fig. S4). Therefore, we propose that nucleotide hydrolysis causes further subunit flattening and straightens the polymer without destabilizing the monomer–monomer interface.
Subunit Flattening Is Inversely Correlated with Bending of the MreB Dimer.
Closer inspection of trajectories from our F-MreB dimer simulations revealed a strong correlation between the dihedral motion of the (−) subunit and the bending of the dimer (Fig. 5A and Fig. S5, Pearson’s P = 0.76, highly significant), suggesting that the two motions may be inherently coupled. To test this hypothesis, we performed MD simulations in which the dihedral angle of the (−) subunit was steered to different values, starting from the equilibrium value of the dihedral angle observed for ATP-bound F-MreB (Φ = 23°) (simulations 8–10, Table S1). When the dihedral angle of the (−) subunit was steered from Φ = 23° to either Φ = 16° or 11° over 1 ns, the bending of the dimer quickly attained a new value from θ2 = 24° to θ2 = 13° and 2° in simulations 8 and 9, respectively (Fig. 5B, green and red traces and Fig. S6 and Movie S3). By contrast, when the dihedral angle of the (−) subunit was instead constrained to its initial value (simulation 10, Table S1), the bending remained around 24° (Fig. 5B, blue trace). These results indicate that the intersubunit bending motion can be inversely modulated by the dihedral motion of the (−) subunit.
Fig. 5.
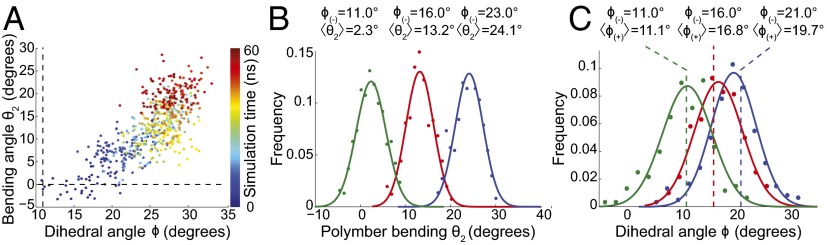
Intersubunit bending of ATP-bound F-MreB is coupled to subunit flatness. (A) Dihedral angle of the (+) subunit is highly correlated with the bending angle (Pearson’s P = 0.76, highly significant) in unconstrained simulations of F-MreB. (B) Continuously moving (“steering”) the dihedral angle of the (−) subunit from 23° to either 16° or 11° results in a reduction in the intersubunit bending of the dimer, whereas constraining the (−) dihedral angle at 23° maintained the initial bending angle, demonstrating inverse coupling between the flatness of the (−) subunit and bending of the dimer. The steered value of the (−) subunit and mean dimer bending are given above each bending distribution. (C) In simulations where an equilibrium monomer structure of MreB was placed on a subunit with a constrained dihedral angle of 11° (green dashed line), 15° (red dashed line), or 21° (blue dashed line), the dihedral angle of the adsorbed (+) subunit was biased to a similar value (respective solid distributions). The constrained dihedral value of the (−) subunit and the average dihedral value of the (+) subunit are given above each curve.
As we reported above, when an equilibrium G-MreB structure was adsorbed onto a flat subunit, both subunits adopted comparable dihedral angles (Fig. 1F and simulation 3, Table S1). To test whether this behavior was more general, we repeated this simulation while constraining the (−) subunit to three different dihedral angle values (Φ = 11°, 16°, and 21°; simulations 11–13, Table S1). In each case, the equilibrium dihedral angle of the (+) subunit adopted the value of the (−) subunit (Fig. 5C). Given this observed bias of the (+) subunit dihedral angle by the (−) subunit, if a third subunit is added onto the (+) end of the dimer, its dihedral angle should be biased by the flatness of the (−) end monomer. More generally, our results suggest that subunit dihedral angle, which is coupled to intersubunit bending (Fig. 5 A and B), can cooperatively propagate from the (−) subunit to the (+) subunit in an MreB filament. Cooperative changes in stiffness have been observed in F-actin (11), although the molecular mechanism of transduction along the filament is unknown. Taken together, our MreB dimer simulations suggest that nucleotide identity (ATP or ADP) affects the dihedral angle of the MreB subunits, and this intramolecular conformation determines the properties of an MreB polymer by biasing both the intersubunit bending and the dihedral angles of subsequent subunits.
To generalize our observations to higher-order MreB polymers, we conducted a simulation of an ATP-bound F-MreB trimer initialized from the protofilament crystal structure (simulation 14, Table S1). The trimer remained stable over the time course of the simulation and also exhibited coupling of monomer unflattening and intersubunit bending, consistent with our dimer simulations (Fig. S7), suggesting that the filament dynamics that we have observed in dimers exist in higher-order structures of MreB.
Discussion
For the challenge of understanding the role of the prokaryotic actin homolog MreB in coordinating cell-wall synthesis in many rod-shaped bacteria, our simulations provide key insight into the structural properties of MreB at the molecular and polymer length scales. Our simulations elucidate important rules in the first steps of the MreB assembly process: Polymerization introduces conformational changes in MreB subunits that drive hydrolysis, and the subsequent change in nucleotide identity leads to a shift in polymer orientation through coupled motions between subunit flattening and intersubunit bending (Fig. 6). Our results are generally applicable for different species of MreB and are relevant to its cellular roles for three reasons. First, higher-order assembly of MreB is preceded by dimerization of subunits. Second, our results derived from dimer simulations are recapitulated in a simulation of an F-MreB trimer (Figs. S2 and S7). Third, while our simulations were based on the structure of Thermotoga maritima MreB, its interfacial and nucleotide-proximal residues most relevant to our observations are highly conserved in MreB from other Gram-negative model organisms (Supporting Information).
Fig. 6.

Model of MreB assembly. (A) G-MreB subunits require Mg2+ and ATP to polymerize onto the F-MreB protofilament. Polymerization promotes flattening of the monomer (blue arrows) and restructuring of the nucleotide-binding pocket. (B and C) ATP-bound (B) and ADP-bound (C) MreB filaments adopt more and less curved conformations, respectively (orange and green arrows, respectively).
The different degree of bending between ADP- and ATP-bound dimers (Fig. 4B) suggests a simple model of MreB assembly (Fig. 6) that potentially reconciles the varied MreB filament architectures reported in in vitro studies. Initial studies of MreB architecture using electron microscopy described a combination of straight filaments, planar sheets, and tightly curved arcs (1, 12, 16, 17). Hydrolysis did not occur immediately in these reports, with incomplete hydrolysis reactions later attributed to inhibitory salt conditions (15). Our simulations suggest that this mixed population of structures represents the two nucleotide states of straight, ADP-bound and curved, ATP-bound filaments. Moreover, the extended straight and curved regions within filaments from these studies support the prediction of our simulations that the bending of a filament is cooperatively driven by coupling between the subunit dihedral angles (Fig. 5C). Consistent with our hypothesis of hydrolysis-driven polymer straightening, more recent studies in which hydrolysis occurred to stoichiometric completion reported only straight MreB filaments (15); in contrast, when bound to nonhydrolyzable nucleotide analogs such as adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP), MreB filaments are exclusively bent or tightly wound, with no straight filaments reported (14, 17). Taken together, our simulations suggest that any subpopulation of curved filaments observed via in vitro electron microscopy is composed of segments within the polymers that have not hydrolyzed.
Although MreB has been observed to form micrometer-long polymeric structures in vivo on the inner membrane (9, 19) and in the cytoplasm (14), other studies reported only diffraction-limited puncta (7, 8), and the native structure of MreB in the cell remains unknown. The strong conformational dependence of F-MreB on the identity of the bound nucleotide in our simulations indicates that in vitro assays using nonhydrolyzable ATP analogs might provide an incomplete picture of how MreB mechanically stiffens the cell (13, 33) and deforms membranes (14), especially considering that MreB may be predominantly bound to ADP in the cell (15). Our results suggest that the hydrolysis state of MreB must also be considered; for example, binding of a curved, ATP-bound MreB filament to the relatively flat membrane could straighten the filament and promote hydrolysis or drive preferential binding of MreB to regions of specific membrane curvature (32). Nucleotide state might also affect the affinity of MreB for other proteins, as observed between actin and actin-binding proteins (34). Ultimately, cell shape is determined by a complex set of interactions among the cell envelope, the wall-synthesis machinery, and MreB (9, 35, 36). Our results outlining the critical first step of MreB assembly provide a molecular framework for understanding how MreB and MreB-associated proteins determine cell shape. Future studies can incorporate MreB-associated proteins such as RodZ (37, 38) to understand how they interact with MreB filament mechanics and contribute to cell shape maintenance.
Previously identified MreB mutants that lead to varied MreB localization phenotypes and cell morphologies can be used to demonstrate the utility of our results for interpreting MreB behavior in cells. For example, the Caulobacter crescentus MreBD16G mutant likely has impaired hydrolysis activity based on the importance of D16 as a conserved hydrolytic residue. In these cells, MreB mislocalizes at the cell poles, which have a higher degree of curvature than the rest of the cell body (4). This observation is consistent with our prediction that ATP-bound MreB filaments have higher intrinsic curvature and hence prefer a more curved surface (Fig. 4B). In addition, a recent study has found that fluorescent-protein fusions to Escherichia coli wild-type MreB localize in a curvature-dependent manner, targeting growth to regions of low mean curvature to straighten bent regions of the cell (39). Finally, given that mutations in the nucleotide-binding pocket (40) have been shown to alter cell width in E. coli, we speculate that altered MreB filament properties such as curvature and stiffness may be directly responsible for transducing these changes in cell morphology, for example by changing MreB localization and the subsequent patterning of cell-wall growth in the cell (32, 39). How the pattern of cell-wall synthesis determines and maintains cell shape remains to be understood.
The structural dynamics and mechanics of MreB polymerization seem to have close parallels to its eukaryotic counterpart, actin. Like actin, MreB polymerization induces monomeric flattening (Fig. 1E), followed by the molecular reorganization of the nucleotide-binding pocket to promote hydrolysis (Fig. 3) (25, 26). Our observation of well-defined open and closed states of monomeric MreB (Fig. 1C and Fig. S2) parallels a growing consensus in the actin field that stable open states exist in actin even without interaction with actin-binding proteins, and that these states might play important physiological roles (41). It will be intriguing to probe whether our observation of direct coupling between subunit flatness and polymer bending (Fig. 4B) also exists in F-actin filaments. The similarities between MreB and actin revealed in our study encourage the pursuit of further illumination of MreB structural dynamics through the existing breadth of actin knowledge.
A previous cryo-electron microscopy study has suggested that MreB can form short double protofilaments that seem to be relatively untwisted (14), unlike the interwoven structure of actin filaments (26, 42). Our F-MreB simulations exhibited small twist angles (Table S3), suggesting that longer MreB double filaments might form slightly twisted protofilaments, as has been observed in vitro for E. coli MreB (43). With further description of the structure of an MreB double protofilament, the mechanical parameters extracted from our dimer simulations can be used to infer the properties of higher-order MreB assemblies, and how those properties might change upon genetic perturbation of the MreB subunits.
Beyond MreB, at least seven bacterial actin-related proteins are known to polymerize into filaments, and assembly properties of bacterial actin-related proteins seem to be conserved even when amino acid sequences are highly disparate (44). Although the roles of these proteins vary widely, including cell-wall synthesis (MreB), plasmid segregation (ParM) (45), and organization of membrane invaginations (MamK) (46), our results open the possibility that structural principles of polymerization are well conserved across the actin family. Interestingly, many of the conformational dynamics of the highly conserved division protein FtsZ, a tubulin homolog and GTPase, are in sharp contrast with those of MreB. For example, there is little nucleotide-dependent monomer structural variation in FtsZ, and, like tubulin, nucleotide hydrolysis induces bending in FtsZ rather than straightening for the case of MreB (22, 24). In general, the actin and tubulin families serve complementary cellular roles in both prokaryotes and eukaryotes, perhaps by invoking orthogonal molecular principles. Ultimately, our study reinforces the importance of computational studies in revealing potentially conserved structural dynamics across protein families.
Methods
Equilibrium MD.
All simulations were performed using the MD package NAMD (47) with the CHARMM27 force field (48, 49). Water molecules were described with the TIP3P model (50). Long-range electrostatic forces were evaluated by means of the particle-mesh Ewald summation approach with a grid spacing of <1 Å. An integration time step of 2 fs was used (51). Bonded terms and short-range, nonbonded terms were evaluated at every time step, and long-range electrostatics were evaluated at every other time step. Constant temperature (T = 310 K) was maintained using Langevin dynamics, with a damping coefficient of 1.0 ps−1. A constant pressure of 1 atm was enforced using the Langevin piston algorithm (52) with a decay period of 200 fs and a time constant of 50 fs. Steering of the dihedral angle was achieved by introducing collective forces to constrain the dihedral angle to a defined value through the collective variables functionality of NAMD (47).
Simulated Systems.
Fourteen MD simulations were performed as described in Table S1. For simulations initialized from an MreB crystal structure, the crystallographic structure of T. maritima MreB bound to AMP-PNP (PDB ID code 1JCG) (1) was used, and the bound AMP-PNP was replaced with ATP or ADP. Water and neutralizing ions were added around each monomer, dimer, or trimer, resulting in final simulation sizes of up to ∼140,000 atoms. All unconstrained simulations were performed for ∼55–130 ns, except in simulations without Mg2+, in which the nucleotide was unstable. All steered and constrained simulations were run until equilibrium was reached. Setup, analysis, and rendering of the simulation systems were performed with the software VMD (53). For average values and distributions of measurements, only the last 30 ns of each of the simulation trajectories were used. To ensure that the simulations had reached equilibrium, measurement distributions were fit to a Gaussian. A satisfactory fit implies that the system is located within an energy well approximated by a harmonic potential. All simulation systems except the trimer were repeated at least twice, and repeat simulations gave similar results.
Determination of Filament Mechanical Properties.
Relative bending orientations of dimer subunits were calculated by determining the rotational transformation required to align the subunits (Supporting Information). The elastic moduli of dimer simulations were measured from the distribution of bending orientations or the distance between the centers-of-mass (Supporting Information). As a measure of the size of a monomer–monomer interface, we calculate buried solvent-accessible surface area (SASA), which is defined as half the difference between the SASA of both subunits alone and the SASA of the entire dimer (24).
Supplementary Material
Acknowledgments
The authors thank members of the K.C.H. lab for insightful discussions and Leigh Harris, Nikolay Ouzounov, Zemer Gitai, and Zev Bryant for careful readings of the manuscript. This work was supported by a Stanford Graduate Fellowship (to A.C.), National Institutes of Health Ruth L. Kirschstein National Research Service Award 1F32GM100677 (to J.H.), a Stanford School of Medicine Dean’s Postdoctoral Fellowship (to J.H.), National Science Foundation CAREER Award MCB-1149328 (to K.C.H.), National Institutes of Health (NIH) Director’s New Innovator Award DP2OD006466 (to K.C.H.), and the Simbios NIH Center for Biomedical Computation under Award U54 GM072970. All simulations were performed with computer time provided by the Extreme Science and Engineering Discovery Environment, which is supported by National Science Foundation Grant OCI-1053575, with allocation number TG-MCB110056 (to J.H. and K.C.H.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317061111/-/DCSupplemental.
References
- 1.van den Ent F, Amos LA, Löwe J. Prokaryotic origin of the actin cytoskeleton. Nature. 2001;413(6851):39–44. doi: 10.1038/35092500. [DOI] [PubMed] [Google Scholar]
- 2.Bean GJ, et al. A22 disrupts the bacterial actin cytoskeleton by directly binding and inducing a low-affinity state in MreB. Biochemistry. 2009;48(22):4852–4857. doi: 10.1021/bi900014d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gitai Z. The new bacterial cell biology: Moving parts and subcellular architecture. Cell. 2005;120(5):577–586. doi: 10.1016/j.cell.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 4.Dye NA, Pincus Z, Fisher IC, Shapiro L, Theriot JA. Mutations in the nucleotide binding pocket of MreB can alter cell curvature and polar morphology in Caulobacter. Mol Microbiol. 2011;81(2):368–394. doi: 10.1111/j.1365-2958.2011.07698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kruse T, Møller-Jensen J, Løbner-Olesen A, Gerdes K. Dysfunctional MreB inhibits chromosome segregation in Escherichia coli. EMBO J. 2003;22(19):5283–5292. doi: 10.1093/emboj/cdg504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones LJ, Carballido-López R, Errington J. Control of cell shape in bacteria: Helical, actin-like filaments in Bacillus subtilis. Cell. 2001;104(6):913–922. doi: 10.1016/s0092-8674(01)00287-2. [DOI] [PubMed] [Google Scholar]
- 7.Domínguez-Escobar J, et al. Processive movement of MreB-associated cell wall biosynthetic complexes in bacteria. Science. 2011;333(6039):225–228. doi: 10.1126/science.1203466. [DOI] [PubMed] [Google Scholar]
- 8.Garner EC, et al. Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science. 2011;333(6039):222–225. doi: 10.1126/science.1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Teeffelen S, et al. The bacterial actin MreB rotates, and rotation depends on cell-wall assembly. Proc Natl Acad Sci USA. 2011;108(38):15822–15827. doi: 10.1073/pnas.1108999108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kabsch W, Holmes KC. The actin fold. FASEB J. 1995;9(2):167–174. doi: 10.1096/fasebj.9.2.7781919. [DOI] [PubMed] [Google Scholar]
- 11.Sheterline P, Clayton J, Sparrow JC. Actin. Prot Profile. 1998;4:1–272. [PubMed] [Google Scholar]
- 12.Esue O, Cordero M, Wirtz D, Tseng Y. The assembly of MreB, a prokaryotic homolog of actin. J Biol Chem. 2005;280(4):2628–2635. doi: 10.1074/jbc.M410298200. [DOI] [PubMed] [Google Scholar]
- 13.Shaevitz JW, Gitai Z. The structure and function of bacterial actin homologs. Cold Spring Harb Perspect Biol. 2010;2(9):a000364. doi: 10.1101/cshperspect.a000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salje J, van den Ent F, de Boer P, Löwe J. Direct membrane binding by bacterial actin MreB. Mol Cell. 2011;43(3):478–487. doi: 10.1016/j.molcel.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bean GJ, Amann KJ. Polymerization properties of the Thermotoga maritima actin MreB: Roles of temperature, nucleotides, and ions. Biochemistry. 2008;47(2):826–835. doi: 10.1021/bi701538e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esue O, Wirtz D, Tseng Y. GTPase activity, structure, and mechanical properties of filaments assembled from bacterial cytoskeleton protein MreB. J Bacteriol. 2006;188(3):968–976. doi: 10.1128/JB.188.3.968-976.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Popp D, et al. Filament structure, organization, and dynamics in MreB sheets. J Biol Chem. 2010;285(21):15858–15865. doi: 10.1074/jbc.M109.095901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gitai Z, Dye N, Shapiro L. An actin-like gene can determine cell polarity in bacteria. Proc Natl Acad Sci USA. 2004;101(23):8643–8648. doi: 10.1073/pnas.0402638101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reimold C, Defeu Soufo HJ, Dempwolff F, Graumann PL. Motion of variable-length MreB filaments at the bacterial cell membrane influences cell morphology. Mol Biol Cell. 2013;24(15):2340–2349. doi: 10.1091/mbc.E12-10-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu JW, Voth GA. Allostery of actin filaments: Molecular dynamics simulations and coarse-grained analysis. Proc Natl Acad Sci USA. 2005;102(37):13111–13116. doi: 10.1073/pnas.0503732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gebremichael Y, Chu JW, Voth GA. Intrinsic bending and structural rearrangement of tubulin dimer: molecular dynamics simulations and coarse-grained analysis. Biophys J. 2008;95(5):2487–2499. doi: 10.1529/biophysj.108.129072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, et al. FtsZ protofilaments use a hinge-opening mechanism for constrictive force generation. Science. 2013;341(6144):392–395. doi: 10.1126/science.1239248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wells DB, Aksimentiev A. Mechanical properties of a complete microtubule revealed through molecular dynamics simulation. Biophys J. 2010;99(2):629–637. doi: 10.1016/j.bpj.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsin J, Gopinathan A, Huang KC. Nucleotide-dependent conformations of FtsZ dimers and force generation observed through molecular dynamics simulations. Proc Natl Acad Sci USA. 2012;109(24):9432–9437. doi: 10.1073/pnas.1120761109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfaendtner J, Lyman E, Pollard TD, Voth GA. Structure and dynamics of the actin filament. J Mol Biol. 2010;396(2):252–263. doi: 10.1016/j.jmb.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oda T, Iwasa M, Aihara T, Maéda Y, Narita A. The nature of the globular- to fibrous-actin transition. Nature. 2009;457(7228):441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- 27.Kinosian HJ, Selden LA, Estes JE, Gershman LC. Nucleotide binding to actin. Cation dependence of nucleotide dissociation and exchange rates. J Biol Chem. 1993;268(12):8683–8691. [PubMed] [Google Scholar]
- 28.Blanchoin L, Pollard TD. Hydrolysis of ATP by polymerized actin depends on the bound divalent cation but not profilin. Biochemistry. 2002;41(2):597–602. doi: 10.1021/bi011214b. [DOI] [PubMed] [Google Scholar]
- 29.Saunders MG, Voth GA. Water molecules in the nucleotide binding cleft of actin: Effects on subunit conformation and implications for ATP hydrolysis. J Mol Biol. 2011;413(1):279–291. doi: 10.1016/j.jmb.2011.07.068. [DOI] [PubMed] [Google Scholar]
- 30.Isambert H, et al. Flexibility of actin filaments derived from thermal fluctuations. Effect of bound nucleotide, phalloidin, and muscle regulatory proteins. J Biol Chem. 1995;270(19):11437–11444. doi: 10.1074/jbc.270.19.11437. [DOI] [PubMed] [Google Scholar]
- 31.Kojima H, Ishijima A, Yanagida T. Direct measurement of stiffness of single actin filaments with and without tropomyosin by in vitro nanomanipulation. Proc Natl Acad Sci USA. 1994;91(26):12962–12966. doi: 10.1073/pnas.91.26.12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang S, Wingreen NS. Cell shape can mediate the spatial organization of the bacterial cytoskeleton. Biophys J. 2013;104(3):541–552. doi: 10.1016/j.bpj.2012.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuson HH, et al. Measuring the stiffness of bacterial cells from growth rates in hydrogels of tunable elasticity. Mol Microbiol. 2012;84(5):874–891. doi: 10.1111/j.1365-2958.2012.08063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.dos Remedios CG, et al. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83(2):433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- 35.Furchtgott L, Wingreen NS, Huang KC. Mechanisms for maintaining cell shape in rod-shaped Gram-negative bacteria. Mol Microbiol. 2011;81(2):340–353. doi: 10.1111/j.1365-2958.2011.07616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Furchtgott L, Huang KC, Shaevitz JW. Helical insertion of peptidoglycan produces chiral ordering of the bacterial cell wall. Proc Natl Acad Sci USA. 2012;109(10):E595–E604. doi: 10.1073/pnas.1117132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bendezú FO, Hale CA, Bernhardt TG, de Boer PA. RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. coli. EMBO J. 2009;28(3):193–204. doi: 10.1038/emboj.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiomi D, Sakai M, Niki H. Determination of bacterial rod shape by a novel cytoskeletal membrane protein. EMBO J. 2008;27(23):3081–3091. doi: 10.1038/emboj.2008.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ursell TS, et al. Rod-like bacterial shape is maintained by feedback between cell curvature and cytoskeletal localization. Proc Natl Acad Sci USA. 2014 doi: 10.1073/pnas.1317174111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kruse T, Bork-Jensen J, Gerdes K. The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Mol Microbiol. 2005;55(1):78–89. doi: 10.1111/j.1365-2958.2004.04367.x. [DOI] [PubMed] [Google Scholar]
- 41.Klenchin VA, Khaitlina SY, Rayment I. Crystal structure of polymerization-competent actin. J Mol Biol. 2006;362(1):140–150. doi: 10.1016/j.jmb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Holmes KC, Popp D, Gebhard W, Kabsch W. Atomic model of the actin filament. Nature. 1990;347(6288):44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- 43.Nurse P, Marians KJ. Purification and characterization of Escherichia coli MreB protein. J Biol Chem. 2013;288(5):3469–3475. doi: 10.1074/jbc.M112.413708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rivera CR, Kollman JM, Polka JK, Agard DA, Mullins RD. Architecture and assembly of a divergent member of the ParM family of bacterial actin-like proteins. J Biol Chem. 2011;286(16):14282–14290. doi: 10.1074/jbc.M110.203828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garner EC, Campbell CS, Mullins RD. Dynamic instability in a DNA-segregating prokaryotic actin homolog. Science. 2004;306(5698):1021–1025. doi: 10.1126/science.1101313. [DOI] [PubMed] [Google Scholar]
- 46.Komeili A, Li Z, Newman DK, Jensen GJ. Magnetosomes are cell membrane invaginations organized by the actin-like protein MamK. Science. 2006;311(5758):242–245. doi: 10.1126/science.1123231. [DOI] [PubMed] [Google Scholar]
- 47.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacKerell AD, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 49.Foloppe N, MacKerell JAD. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J Comput Chem. 2000;21(2):86–104. [Google Scholar]
- 50.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 51.Tuckerman M, Berne BJ, Martyna GJ. Reversible multiple time scale molecular dynamics. J Chem Phys. 1992;97(3):1990–2001. [Google Scholar]
- 52.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103(11):4613–4621. [Google Scholar]
- 53.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.