

Significance
Voltage-gated sodium channels are one of the most fundamental electrical components in the nervous system and are key targets for local anesthesia and therapeutics for neurological and cardiac disorders. We have used multimicrosecond simulations to provide molecular-level descriptions of sodium channel function. We describe an almost barrier-less three-ion conduction mechanism involving competing knock-on and “pass-by” processes, intimately linked to signature glutamate ring protonation and structural isomerizations. These simulations have uncovered a high degree of protein flexibility, with conformational fluctuations in the pore domain involving residues central to slow-type inactivation, leading to gate collapse, helix bending, filter disruption, and changes in lipid-facing fenestrations linked to Nav drug pathways.
Abstract
Voltage-gated Na+ channels play an essential role in electrical signaling in the nervous system and are key pharmacological targets for a range of disorders. The recent solution of X-ray structures for the bacterial channel NavAb has provided an opportunity to study functional mechanisms at the atomic level. This channel’s selectivity filter exhibits an EEEE ring sequence, characteristic of mammalian Ca2+, not Na+, channels. This raises the fundamentally important question: just what makes a Na+ channel conduct Na+ ions? Here we explore ion permeation on multimicrosecond timescales using the purpose-built Anton supercomputer. We isolate the likely protonation states of the EEEE ring and observe a striking flexibility of the filter that demonstrates the necessity for extended simulations to study conduction in this channel. We construct free energy maps to reveal complex multi-ion conduction via knock-on and “pass-by” mechanisms, involving concerted ion and glutamate side chain movements. Simulations in mixed ionic solutions reveal relative energetics for Na+, K+, and Ca2+ within the pore that are consistent with the modest selectivity seen experimentally. We have observed conformational changes in the pore domain leading to asymmetrical collapses of the activation gate, similar to proposed inactivated structures of NavAb, with helix bending involving conserved residues that are critical for slow inactivation. These structural changes are shown to regulate access to fenestrations suggested to be pathways for lipophilic drugs and provide deeper insight into the molecular mechanisms connecting drug activity and slow inactivation.
Voltage-gated Na+ (Nav) channels are responsible for the generation of action potentials in electrically excitable cells, allowing critical physiological functions (1). Their functional selectivity identifies them as effective targets in the treatments of a wide range of pathologies spanning all major therapeutic areas (2). However, a full molecular-level description of Nav function has yet to be provided and is essential for future developments in anticonvulsants, antiarrhythmics, and local anesthetics (3).
Mammalian Nav channels are complex proteins composed of more than 2,000 amino acids and have eluded structural determination for decades. The discovery of the bacterial ion channel family NavBac (4, 5), and subsequent solution of high-resolution structures (6, 7), has given us an opportunity to begin to understand Nav function at the molecular level. The homotetrameric NavBac structures are far simpler to study structurally and functionally and are modulated by drugs in similar ways to their mammalian counterparts (8), making them ideal systems to explore the molecular mechanisms of Nav function.
The structure of the bacterial NavAb channel from Arcobacter butzleri (6) is reminiscent of the voltage-gated K+ (Kv) channels, with four identical monomers comprised of a voltage sensor domain (VSD) and a pore domain (PD), symmetrically organized around a central pore (Fig. 1 A and B). In contrast to the narrow, ion-sized selectivity filter (SF) of the K+ channel, the NavAb SF is wider and lined not only by protein backbone (residues 175–178) but also by the side chains of a ring of four glutamates (E177) (Fig. 1C). Curiously, this ring is conserved within the NavBac family, but is indicative of a Ca2+-selective and not of a Na+-selective mammalian channel, which has a conserved DEKA locus (9). Despite the EEEE-lined SF, bacterial channels nominally favor Na+ over Ca2+ or K+, with relative permeabilities 5 < PNa/PK < 171 and 7 < PNa/PCa < 70 in NaChBac (4, 10, 11) (depending an order of magnitude on experimental conditions; 11). This Na+ selectivity emerging from a Glu-lined pore leads us to question the mechanism by which these channels optimally conduct Na+ ions. Simple physical models have proposed a charge/space competition theory to explain the variable Na+/Ca2+ selectivity of Glu-lined SFs, where the volume and electrostatic environment both play a role in the number of ions that the channels can bind and efficiently conduct (10, 12, 13). Selectivity between the monovalent cations Na+ and K+ may be interpreted in terms of ligand field strength (14), where amide carbonyls provide weaker fields that favor larger ions, like K+, whereas carboxylates create a high field strength site (SHFS) (6) favorable to Na+.
Fig. 1.

(A) NavAb channel embedded in a hydrated DPPC bilayer (light blue sticks), surrounded by water (red and white sticks) with Na+ and Cl− ions (yellow and cyan balls). One of the four monomers shows the VSD in purple/blue and the PD in green/yellow/red. (B) Zoomed view of two monomers of the PD showing S5 (green) and S6 (red), and pore helices P1 (yellow) and P2 (orange) for one of them. The SF and key S6 residues are represented by sticks and labels. (C) Top view of the pore showing the EEEE ring.
Permeation of ions through the filter is expected to be modulated by the protonation states of ion-coordinating ligands. Although exhibiting the different DEKA sequence, mammalian Nav channels exhibit reduced conductances at lower extracellular pH (15)—interpreted as proton block (16). Importantly, external pH has been observed to have the same effect on Ca2+ channels and is significantly altered by mutation of the EEEE locus (17). This effect in mammalian channels is of particular interest, because pH is known to be modulated by neuronal activity (18) and to drop substantially when ischemia occurs in the brain or the heart (19). Although the influence of pH has not yet been characterized in bacterial Nav, the binding of protons to the EEEE ring is expected to be a critical factor in its ion conduction mechanism.
In this study, we carry out extensive molecular dynamics (MD) simulations, using the fully atomistic system shown in Fig. 1, to describe Na+ permeation and competitive binding of Na+, K+, and Ca2+ ions. Although the channel has been solved in a preopen state, with active voltage sensors and closed gate (6), the pathway through the SF and hydrophobic cavity (Fig. 1B) is in a conducting state, allowing for studies of multiple ion movements. We examine the effects of EEEE protonation and reveal extensive protein fluctuations, unseen in previous simulations on shorter timescales (11, 20–22), but suggested to play a role in a recent study (23). We will demonstrate that protein fluctuations away from the published crystal structure are critical for describing ion conduction mechanisms.
Another important feature of Nav function is its ability to terminate conductance under sustained depolarization via inactivation (24, 25). Inactivation is associated with numerous neuronal and cardiac pathologies (26). Bacterial channels display slow inactivation reminiscent of C-type inactivation in Kv channels (27), involving its PD (28, 29), and is known to be modulated by local anesthetics (8). Interestingly, there is recent evidence that mammalian channels are also affected by this process (30–32), despite a historical focus on fast inactivation (33). Our multimicrosecond simulations reveal conformational fluctuations in common with proposed inactivated crystal structures (34), which allow us to begin to describe the molecular mechanisms of inactivation and its relation to Nav drug inhibition.
Results
Ion Occupation and the Role of Protonation States.
The E177 ring lining the SF produces a high charge density that attracts cations into the pore. Although the aqueous pKa of Glu is 4.4 (35), the interactions between these side chains, ions, and a nonaqueous environment will lead to unknown pKa shifts. Fig. 2 shows that protonation states dramatically influence the distribution of Na+ ions in the channel. For protonation states PS0 (no E177 protonated) and PS1 (one protonated), ion densities are continuous, reflecting a substantial exchange between the SF and bulk. For PS0 and PS1, there are 2–3 ions in the SF (SI Appendix, Fig. S1), suggesting a mechanism involving interconversion between two and three ions, whereas this number decreases to just one ion for other protonation states (PS2, PS3, and PS4 with 2–4 protonated), where a reduction in ion density appears at the level of the EEEE ring, preventing conduction via intrapore proton block (16, 17).
Fig. 2.

Radial-axial Na+ free energy surfaces for protonation states PS0 to PS4, (with PS2A and PS2B averaged; breakdown provided in SI Appendix, Fig. S3), with 1D projections and initial SF structure shown to the right (binding sites SHFS, SCEN, or SIN indicated with horizontal lines). Dashed curves indicate incomplete sampling for PS3 and PS4. Representative conformations are shown beneath with Na+ and Cl− ions as yellow and cyan balls (for clarity only three monomers shown, with all four shown for PS2B to highlight distortion).
The 1D free energy projection (Fig. 2, Right) for PS0 is consistent with the proposed binding site locations in the NavAb crystal structure (6). These sites are shifted slightly in PS1, with a double well in the SHFS site due to E177 rotational isomerizations, resulting in an almost barrier-less profile. Only SIN (inner site) is common to all protonation states, with shifts owing to reduced electrostatic attraction to E177 in PS3 and PS4. Furthermore, Cl− ions enter the SF for both PS3 and PS4 on numerous occasions, being stable for tens of nanoseconds in SHFS for PS4 (Fig. 2, Lower). In the case of PS2, although still attractive for Na+, pairs of protonated/deprotonated E177 form H bonds that close the SF (Fig. 2, Lower and SI Appendix, Figs. S2 and S3). Therefore, PS0 and PS1 are the protonation states likely to be involved during channel conduction.
Na+ Conduction Mechanism.
With an average number of 2.3 ions in the PS0 SF (SI Appendix, Fig. S1), permeation of Na+ involves transitions between two- and three-ion states, from which we compute potential of mean force (PMF) maps. In the presence of two ions, the PMF (Fig. 3A) reveals three states: A (and A′ due to E177 mobility), B2, and C2. In each state, ion 2 (upper ion) is bound to E177 and occupies SHFS, whereas ion 1 (lower ion) moves more freely from SIN (A with ion 2 in high SHFS, A′ with ion 2 in low SHFS) to SHFS (C2) via SCEN (central site; B2). Although the highest energy barrier encountered is ∼1.5 kcal/mol (∼±0.5 kcal/mol; SI Appendix, SI Text), ions appear trapped in the absence of a third ion.
Fig. 3.

2D PMF projections for Na+ ions in PS0 (see SI Appendix, Fig. S6 for PS1 results), when two (A) or three ions (B and C) occupy the SF, with z23 corresponding to the COM of the upper ions (2, 3). Contouring is at 0.5 kcal/mol, with lowest free energy pathways as dashed curves. The permeation mechanism is shown on the right, with free energies relative to state A and barriers (indicated on the arrows) in kcal/mol (∼±0.5 kcal/mol; negative values implying no barriers).
In Fig. 3B, we show the three-ion PMF for PS0 as a function of the position of the bottom ion (ion 1) and the center of mass (COM) of the two upper ions (2, 3). Four states—A, B3, C3, and F—can be identified, with state C′3 related to C3. State A, with two ions in the SF (SHFS and SIN) and one approaching from the extracellular solution, is common to both two- and three-ion maps. In B3, ion 3, initially extracellular, moves down, whereas ion 1 remains in SIN (C3/C′3), before being pushed into the cavity, leading to state F.
The projection shown in Fig. 3B reveals the conduction of an ion, but does not detail the involvement of ions 2 and 3 in the upper part of the SF. An alternative projection, as a function of only the top two ions, is shown in Fig. 3C and reveals an interesting competition of conduction mechanisms. One possibility involves the upper ion entering the SF from above, joining the lower ion at the EEEE ring, and then knocking the bottom ion downward, whereas the alternative is for one ion to pass the other via state E (on the diagonal), coexisting at the EEEE ring before pushing the bottom ion into the cavity. These direct knock-on (36) and pass-by mechanisms encounter similar low (<1 kcal/mol) free energy barriers and would both contribute to conduction.
Ion permeation in NavAb, in its fully charged PS0 state, can therefore be described as a two-stage process, alternating between three- and two-ion occupancy states (Fig. 3, Right). During the entire conduction process, the largest free energy barrier encountered is ∼1.2 kcal/mol during the transition from C3 to F (perhaps slightly elevated due to the closed gate), allowing for rapid conduction. This three-ion mechanism is supported by previous studies of simplified Glu-lined pores (13), and by recent simulations (23), but not in other studies that focused on a two-ion process (21, 22).
During the simulation of PS1, the SF is occupied by two ions most of the time (SI Appendix, Fig. S1), with shorter appearances of a third. The details of the permeation process for the PS1 state are described in SI Appendix, SI Text. Inspection of SI Appendix, Fig. S6A reveals that, in the presence of two ions, the upper ion is bounded below z = 6 Å in this simulation, essentially ruling out a two-ion conduction mechanism. In contrast, the PS1 PMF for three ions reveals a complete conduction path (A–F; as seen for PS0) that is nearly barrier-less (SI Appendix, Fig. S6B). Thus, both protonation states PS0 and PS1 display a conduction mechanism involving transitions between two- and three-ion occupancy states. The main difference is the change in net SF charge, reducing the affinity of the pore for three ions in PS1. We suggest that optimal conduction requires greater three-ion occupancy, as occurs with a fully charged EEEE ring.
Competitive Na+, K+, and Ca2+ Ion Binding.
Equilibrium sampling of mixed electrolytes can reveal the competition for ion binding sites, coordination, and differences in free energy to help understand experimental relative permeabilities. Independent simulations with different ion placements led to exchanges of Na+ and K+ between the SF, bulk, and cavity (SI Appendix, Fig. S7). In the case of Ca2+, however, stronger E177 binding led to little exchange (SI Appendix, Fig. S7), but enough movement to examine its binding locations. Fig. 4 shows results for PS0 (see SI Appendix, Fig. S8 for PS1 and SI Appendix, Fig. S9 for convergence). Na+ ions occupy the same binding sites as in pure NaCl solution (SHFS, SCEN, and SIN; magenta curves), shifted slightly due to the other ions. K+ ions (green curves) bind to comparable sites, separated by similar barriers, but with marginally lower affinity (by <1 kcal/mol). The K+ ions interact with slightly more protein oxygen atoms than Na+ in SCEN and SIN and slightly less in the carboxylate-lined site SHFS (SI Appendix, Fig. S10), as anticipated. Results for PS1 (SI Appendix, Fig. S8) are similar, offering little evidence for discrimination against K+. Acknowledging kcal/mol statistical errors, and up to ∼2 kcal/mol error due to the choice of ion parameters (SI Appendix, SI Text and ref. 37), this result is consistent with lower experimental estimates of relative permeability, as well as recent biased sampling calculations (11).
Fig. 4.

PMFs for Na+ (magenta), K+ (green), and Ca2+ (blue) ions during simulations of PS0 (see SI Appendix, Fig. S8 for PS1) in mixtures of Na+/K+ (Left) and Na+/Ca2+ (Right), with two independent simulations shown as solid and dashed curves.
Ca2+ ions in PS0 (Fig. 4, blue curves) occupy a SHFS-like site, which we call SCa, formed by 5–6 E177 oxygens and just two water molecules (SI Appendix, Fig. S10). The SCa site consists of a broad free energy well, including two local minima, reaching down to SCEN (displacing nearby Na+ ions), corresponding to different E177 orientations that allow Ca2+ to remain always coordinated by carboxylates, and demonstrating poor affinity for other parts of the SF. In PS1 (SI Appendix, Fig. S8), these minima involve either two charged and one protonated Glu, with additional contribution from the V176 carbonyl (lower minimum), or by three charged Glu side chains (upper minimum). The stronger binding of Ca2+ ions in the presence of Na+ could help explain experimental measures of selectivity (4, 10, 38). In fact, it has been shown that additional negative protein charges are needed to favor Ca2+ (10), consistent with our inability to sample additional Ca2+ entry that would promote ion movements.
Conformational Flexibility of the PD.
With its wide pore and Glu-lined SF, subject to the rotational isomerizations, the NavAb pore displays more flexibility than that familiar in K+ channels. We have observed large movements of Glu side chains, which are closely correlated with ion translocation through the SF (Movie S1 and Movie S2), as also described in a recent study (23). Fig. 5A, Left shows the 2D rmsd maps comparing SF structures at two different times (SI Appendix, SI Methods) and represents a state map for the protein’s SF conformation. We see by comparing PS0 (top) and PS1 (bottom) that the protonation of a single residue can impact significantly on the flexibility of the protein. In the initial X-ray structure, E177 carboxylates form H bonds with S178 hydroxyls (SI Appendix, Figs. S4 and S5), orienting one oxygen per group inward to form SHFS. In PS0, this conformation is maintained for much of the first microsecond of simulation, but then the rupture of two of these H bonds allows two E177 side chains to orient downward (SI Appendix, Fig. S5), promoting the entrance of a third ion (with 2–3 ion exchanges continuing throughout the rest of the simulation; SI Appendix, Fig. S1). We note that previous studies (21, 22) relied on biased sampling on the time scale of just 0.1–0.5 ns per simulation, far too short to allow for the conformational changes observed here. The situation is different in PS1, where the SF displays a succession of short-lived states, with E177 side chains moving freely (SI Appendix, Fig. S4). Regardless of the protonation state, it is clear that the X-ray structure, or any short simulation based on that structure, provides just one snapshot of the protein, inadequate for describing ion conduction.
Fig. 5.

Fluctuations of the PD. (A) 2D rmsd maps for PS0 (Upper) and PS1 (Lower), considering the SF (backbone of residues 175–179, Left) and PD (backbone of residues 130–220, Right). The black dashed lines delimit blocks of stable conformations (labeled with roman numerals) with representative conformations as insets (SI Appendix, Figs. S5 and S13). Bottom views of the PD in the initial conformation (PDB ID code 3rvy) (B), after 2.5 µs in PS1 (C), and in the proposed inactivated conformation (PDB ID code 4ekw, chains AB) (D), aligned on the S6 helices, are displayed beneath.
The behavior of the SF is strongly correlated with the dynamics of the PD. This is evident in the comparison of state maps for the SF (Left) and PD (Right) in Fig. 5A. In PS0, two states of the SF are associated with two conformations of the PD, defined by bending of an S6 helix at the level of residues P200–F203, at the same time the SF exhibits some distortion (at t ∼ 1 µs). In PS1, five PD states (I to V) can be identified, associated with more extensive S6 bending (Movie S3), with all but state II having well-defined corresponding states in the SF.
Our observations suggest that increased fluctuations of the SF caused by single protonation in PS1 (Movie S2) may expedite transitions between states for the whole PD. In PS1, the transition from state I to state II (t ∼ 0.25 µs) corresponds to an initial bending of S6, in association with a tilt of P2 (SI Appendix, Fig. S13) that pulls on the SF from above. Transition II–III (t ∼ 0.75 µs) sees helix S6 bend further, causing I202 and V204 to press on the base of the SF (via I202–V173 and V204–M174, SI Appendix, Fig. S13), contributing to SF distortion, involving subunit dislocation (Fig. 5A, Right), although maintaining hydration and capacity for three ions (SI Appendix, Fig. S1). Helix deformation reaches a maximum in state IV, then reduces in state V, whereas the SF remains distorted and asymmetrical.
Analysis of S6 backbone dihedral fluctuations (SI Appendix, Fig. S11A) suggests instability between V196 and P200, and at I202, to differing degrees in all simulations (SI Appendix, Fig. S11B). The structure of S6 at the level of F203 is strongly associated with the breaking of backbone I202–T206 H bonding as well as the interfering I202–T206 (backbone to side chain) H bond. Rupture of these H bonds is consistently observed when S6 bends (notably in PS1, PS2, and PS4; SI Appendix, Fig. S11C). Thr’s mechanism of interfering with secondary structure is like that of Ser (a known helix breaker) (39, 40), also known to modulate bending due to Pro (41). Here, the Thr methyl group (that distinguishes it from Ser) is well solvated by intersubunit nonpolar side chains that help direct the hydroxyl group into S6 (SI Appendix, Fig. S12A), leading to weakening of the α-helix forming H bond (SI Appendix, Fig. S12 B and C). In S6, P200 induces deformation that breaks F198–I202 and subsequently the weakened I202–T206 H bond, leading to bending.
These conformational changes have led to protein structures that resemble the proposed inactivated structures of Payandeh et al. (34) (compare Fig. 5C to Fig. 5D). These X-ray structures present asymmetrically collapsed S6 activation gates (asymmetry 5 Å at the level of residues 215–218) and associated narrower SF and reshaped cavity. We have observed similar gate asymmetry, reaching as high as 12 Å after ∼1.5 μs in PS1 (SI Appendix, Fig. S14). We postulate that S6 bending is a natural property of the protein on this microsecond timescale. Although slow inactivation occurs on longer timescales (millisecond to second) (42), the transition from a conducting to an inactivated state may be associated with rapid conformational fluctuations, as seen in these simulations, but require stabilization on longer timescales.
Importantly, the conformational changes can be traced to key residues associated with slow inactivation. Residue I202 is well conserved within the Nav family (2), as is T206 in the NavBac family (6). Mutation of the helix-weakening T206 (T220 in NaChBac) prevents slow inactivation (43), whereas the mutation of the equivalent of residue I202 in Nav1.4 domain IV dramatically affects slow inactivation (44). In addition, mutation of Phe at that same level (F203 in NavAb) strongly inhibits slow inactivation in mammalian Nav (28, 31), reinforcing the significance of the S6 bend around this residue. It is also of much interest that changes in gate-forming helices are correlated with changes in the SF, as suggested by NaChBac studies (10), and akin to C-type (slow) inactivation in K+ channels (27), although apparently via a different mechanism.
Fenestrations and Pore Accessibility.
At the level of the bending between residues 202 and 206, the S6 helices are known to form openings between the membrane and the central cavity (6). These openings have been described as potential pathways to a binding site somewhere within the PD for lipophilic drugs, such as local anesthetics or antiepileptics, to inhibit Na+ current (8). Lipid tails were seen to enter these openings and to exchange with different lipids frequently (Fig. 6A; on average ∼9 times in 2.5 μs). Fig. 6B shows the distribution of fenestration radii for PS0 and PS1, with time series for individual fenestrations (SI Appendix, Fig. S15) revealing rapid fluctuations between 0 and 4 Å. Fig. 6C shows how the size of the openings is controlled by the orientation of the F203 side chain (χ2; red curves), whose phenyl group acts as a lid closing the pathway (indicated in Fig. 6C, Left), but which is strongly modulated by helix bending (blue curves), acting to favor the rotamer that closes the fenestration.
Fig. 6.

(A) Top view of the PD showing different colored lipids seen within lateral openings during the PS1 simulation. (B) Normalized distributions of fenestration radius for PS0 (green) and PS1 (blue), with red line marking the radius of benzene (∼2.47 Å). (C) Dihedral χ2 of F203 (red curve) and bending angle θS6 (blue curve) for segments D and B in PS0 and PS1, respectively, with corresponding fenestration size as a function of time.
Therefore, fenestration access is regulated by the behavior of residues associated with helix bending (F203, I202, T206, as well as P200), which are also linked to slow inactivation in Na+ channels (8, 28, 44). At their widest (∼4 Å), the fenestrations are large enough to fit typical Nav inhibitor drugs, such as phenytoin (illustrated in SI Appendix, Fig. S15; see also Fig. 6B, which shows openings are large enough to fit an aromatic ring ∼10% of the time and thus easily adapted with strain energies of less than kBT; SI Appendix, SI Text). This demonstrates that these fenestrations are possible pathways for lipophilic drugs.
The key role of the Phe (203 in NavAb) in drug activity has been observed in numerous channels, such as F1764 in rNav1.2 (45), F1710 in rNav1.3 (46), F1579 in hNav1.4 (47), and F1760 in hNav1.5 (28). It has been suggested that drugs can enter the cavity (48) and physically block the channel (49), or bind at the level of the opening via π–π stacking of aromatics (47) and inhibit the current by favoring a nonconductive inactivated state (46). Moreover, it is known that slow inactivation is affected by local anesthetics or antiepileptics drugs (30–32) and by the mutation of the same Phe involved in drug binding (28). Evidence suggests that drug binding affinity varies depending on the conducting state of the channels (46), corroborating our own observations that accessibility of both the Phe and cavity openings are codependent and correlated with changes in the S6 helix and gate collapse, suggesting a transition to the inactivated state.
Conclusion
Multimicrosecond simulations of the Na+ channel NavAb have been used to investigate Nav channel function on timescales that are physiologically relevant for conduction and sufficient to begin to observe the conformational flexibility of the protein related to channel activity. We have reported flexibility of protein conformation, influencing both conduction and key structural features associated with inactivation and lipid accessibility, with implications for understanding Nav drug activity.
The Na+ conduction process in NavAb has been elucidated from unbiased MD simulations on long timescales. Despite the channel having a closed intracellular gate, these simulations mapped out the multiple ion movements that govern Na+ conduction across the channel’s SF. The process involves the movements of three ions via a knock-on mechanism, reminiscent of conduction in K+ channels, or via a pass-by mechanism, made possible by the larger and more flexible NavAb SF. We predict conduction in channels with fully charged or singly protonated EEEE rings, but hypothesize that reduced affinity for a third ion due to protonation may lead to lower conductance, suggesting proton block (17). Interestingly, the dependence of ion occupation on those Glu charges conversely suggests that protonation/deprotonation could occur during conduction events, where SF occupation dynamically switches between two and three ions.
Our simulations with ionic mixtures have suggested a slightly lower affinity for K+ ions and tighter binding of Ca2+ ions, consistent with the nominal relative permeabilities measured experimentally (6, 10). However, dedicated free energy simulations would be needed to capture the differing multiple-ion mechanisms of permeation for each ion type, as well as anomalous mole fraction effects in ionic mixtures. The challenge remains to incorporate the extensive fluctuations of the channel while elucidating those mechanisms.
Bending of helix S6 at the level of P200–T206 appears as a long-timescale fluctuation, leading to an asymmetric arrangement that resembles the inactivated state proposed by Payandeh et al. (34). This is somewhat surprising given that inactivation in bacterial channels occurs on millisecond timescales, but suggests that microsecond conformational fluctuations may be related to a change in state on longer timescales. In addition, we have observed a connection between pairs of residues I202–V173 and V204–M174, as well as P2 helix tilting, communicating structural changes between the gate and the SF, reminiscent of C-type inactivation in K+ channels (27).
The behavior of S6 affects the openings to the membrane interior, thought to be involved in the binding of Nav drugs (3, 8). The size of these fenestrations is controlled by rotational isomerization of F203, a residue that is key to inactivation and drug binding (28, 45–47). Fenestration size is modulated by S6 bending, which we have proposed may be associated with a collapse into an inactivated state. These observations suggest that the binding of drugs at this crucial site could modulate conversion to a slow-inactivated state, as suggested experimentally (28, 31, 32, 46). These simulations have therefore proven to be important for exploring the roles of protein flexibility in ion conduction mechanisms and have given us clues as to the molecular-level processes that underscore inactivation and drug inhibition of Nav channels.
Methods
The NavAb protein [Protein Data Bank (PDB) 3rvy (6)] was embedded in a dipalmitoylphosphatidylcholine (DPPC) membrane with explicit three-point transferable intermolecular potential (TIP3P) water molecules. All six possible protonation states—PS0, PS1, PS2A (neighboring), PS2B (opposing), PS3, and PS4—with 0–4 glutamates (E177) protonated were considered for the EEEE SF ring. Systems were simulated in 150 mM NaCl solution, and protonation states PS0 and PS1 were additionally simulated in mixtures of NaCl/KCl (300/300 mM) and NaCl/CaCl2 (150/300 mM). Systems were built and relaxed initially with Chemistry at Harvard Macromolecular Mechanics (CHARMM) (50), then further equilibrated with NAMD (51) before long production runs on Anton supercomputer (52), summarized in SI Appendix, Table S1. PMFs were calculated from unbiased simulation as 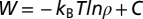 , where
, where 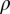 is the unbiased probability distribution as a function of axial (z) and/or radial
is the unbiased probability distribution as a function of axial (z) and/or radial 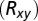 positions of one or more ions and C is a constant. Permeation free energy maps are a function of two or three ionic coordinates—z1, z2, or z3—being the z components of the distance to the SF COM (residues 175–178). See SI Appendix, SI Text for full details.
positions of one or more ions and C is a constant. Permeation free energy maps are a function of two or three ionic coordinates—z1, z2, or z3—being the z components of the distance to the SF COM (residues 175–178). See SI Appendix, SI Text for full details.
Supplementary Material
Acknowledgments
We thank Chris French for helpful discussions. This work was supported by Australian Research Council DP120103548, National Science Foundation MCB1052477, DE Shaw Anton (PSCA00061P; National Resource for Biomedical Supercomputing, through National Institutes of Health RC2GM093307), Victorian Life Science Computation Initiative (VR0200), National Computational Infrastructure (dd7), and Teragrid (MCB050005N).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320907111/-/DCSupplemental.
References
- 1.Hille B. Ionic Channels of Excitable Membranes. 3rd Ed. Sunderland, MA: Sinauer; 2001. [Google Scholar]
- 2.England S, de Groot MJ. Subtype-selective targeting of voltage-gated sodium channels. Br J Pharmacol. 2009;158(6):1413–1425. doi: 10.1111/j.1476-5381.2009.00437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mike A, Lukacs P. The enigmatic drug binding site for sodium channel inhibitors. Curr Mol Pharmacol. 2010;3(3):129–144. doi: 10.2174/1874467211003030129. [DOI] [PubMed] [Google Scholar]
- 4.Ren D, et al. A prokaryotic voltage-gated sodium channel. Science. 2001;294(5550):2372–2375. doi: 10.1126/science.1065635. [DOI] [PubMed] [Google Scholar]
- 5.Koishi R, et al. A superfamily of voltage-gated sodium channels in bacteria. J Biol Chem. 2004;279(10):9532–9538. doi: 10.1074/jbc.M313100200. [DOI] [PubMed] [Google Scholar]
- 6.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475(7356):353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCusker EC, et al. Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat Commun. 2012;3:1102. doi: 10.1038/ncomms2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee S, Goodchild SJ, Ahern CA. Local anesthetic inhibition of a bacterial sodium channel. J Gen Physiol. 2012;139(6):507–516. doi: 10.1085/jgp.201210779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charalambous K, Wallace BA. NaChBac: The long lost sodium channel ancestor. Biochemistry. 2011;50(32):6742–6752. doi: 10.1021/bi200942y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yue L, Navarro B, Ren D, Ramos A, Clapham DE. The cation selectivity filter of the bacterial sodium channel, NaChBac. J Gen Physiol. 2002;120(6):845–853. doi: 10.1085/jgp.20028699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finol-Urdaneta RK, et al. Sodium channel selectivity and conduction: Prokaryotes have devised their own molecular strategy. J Gen Physiol. 2014;143(2):157–171. doi: 10.1085/jgp.201311037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boda D, et al. Ionic selectivity in L-type calcium channels by electrostatics and hard-core repulsion. J Gen Physiol. 2009;133(5):497–509. doi: 10.1085/jgp.200910211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corry B, Allen TW, Kuyucak S, Chung SH. Mechanisms of permeation and selectivity in calcium channels. Biophys J. 2001;80(1):195–214. doi: 10.1016/S0006-3495(01)76007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisenman G. Cation selective glass electrodes and their mode of operation. Biophys J. 1962;2(2 Pt 2):259–323. doi: 10.1016/s0006-3495(62)86959-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker MD, Bostock H. The pH dependence of late sodium current in large sensory neurons. Neuroscience. 1999;92(3):1119–1130. doi: 10.1016/s0306-4522(99)00058-5. [DOI] [PubMed] [Google Scholar]
- 16.Khan A, Kyle JW, Hanck DA, Lipkind GM, Fozzard HA. Isoform-dependent interaction of voltage-gated sodium channels with protons. J Physiol. 2006;576(Pt 2):493–501. doi: 10.1113/jphysiol.2006.115659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X-H, Bezprozvanny I, Tsien RW. Molecular basis of proton block of L-type Ca2+ channels. J Gen Physiol. 1996;108(5):363–374. doi: 10.1085/jgp.108.5.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15(10):396–402. doi: 10.1016/0166-2236(92)90191-a. [DOI] [PubMed] [Google Scholar]
- 19.Katz AM. Physiology of the Heart. Lippincott Williams & Wilkins, Philadelphia; 2010. [Google Scholar]
- 20.Amaral C, Carnevale V, Klein ML, Treptow W. Exploring conformational states of the bacterial voltage-gated sodium channel NavAb via molecular dynamics simulations. Proc Natl Acad Sci USA. 2012;109(52):21336–21341. doi: 10.1073/pnas.1218087109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corry B, Thomas M. Mechanism of ion permeation and selectivity in a voltage gated sodium channel. J Am Chem Soc. 2012;134(3):1840–1846. doi: 10.1021/ja210020h. [DOI] [PubMed] [Google Scholar]
- 22.Furini S, Domene C. On conduction in a bacterial sodium channel. PLOS Comput Biol. 2012;8(4):e1002476. doi: 10.1371/journal.pcbi.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chakrabarti N, et al. Catalysis of Na+ permeation in the bacterial sodium channel NaVAb. Proc Natl Acad Sci USA. 2013;110(28):11331–11336. doi: 10.1073/pnas.1309452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70(5):567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ulbricht W. Sodium channel inactivation: Molecular determinants and modulation. Physiol Rev. 2005;85(4):1271–1301. doi: 10.1152/physrev.00024.2004. [DOI] [PubMed] [Google Scholar]
- 26.Vilin YY, Fujimoto E, Ruben PC. A single residue differentiates between human cardiac and skeletal muscle Na+ channel slow inactivation. Biophys J. 2001;80(5):2221–2230. doi: 10.1016/S0006-3495(01)76195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuello LG, Jogini V, Cortes DM, Perozo E. Structural mechanism of C-type inactivation in K+ channels. Nature. 2010;466(7303):203–208. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carboni M, Zhang ZS, Neplioueva V, Starmer CF, Grant AO. Slow sodium channel inactivation and use-dependent block modulated by the same domain IV S6 residue. J Membr Biol. 2005;207(2):107–117. doi: 10.1007/s00232-005-0805-0. [DOI] [PubMed] [Google Scholar]
- 29.Irie K, et al. Comparative study of the gating motif and C-type inactivation in prokaryotic voltage-gated sodium channels. J Biol Chem. 2010;285(6):3685–3694. doi: 10.1074/jbc.M109.057455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Errington AC, Stöhr T, Heers C, Lees G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol Pharmacol. 2008;73(1):157–169. doi: 10.1124/mol.107.039867. [DOI] [PubMed] [Google Scholar]
- 31.Chen Z, et al. Lidocaine induces a slow inactivated state in rat skeletal muscle sodium channels. J Physiol. 2000;524(Pt 1):37–49. doi: 10.1111/j.1469-7793.2000.t01-1-00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fozzard HA, Lee PJ, Lipkind GM. Mechanism of local anesthetic drug action on voltage-gated sodium channels. Curr Pharm Des. 2005;11(21):2671–2686. doi: 10.2174/1381612054546833. [DOI] [PubMed] [Google Scholar]
- 33.Kuo CC, Bean BP. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12(4):819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 34.Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012;486(7401):135–139. doi: 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th Ed. New York: Freeman; 2002. [Google Scholar]
- 36.Hodgkin AL, Keynes RD. The potassium permeability of a giant nerve fibre. J Physiol. 1955;128(1):61–88. doi: 10.1113/jphysiol.1955.sp005291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noskov SY, Roux B. Control of ion selectivity in LeuT: Two Na+ binding sites with two different mechanisms. J Mol Biol. 2008;377(3):804–818. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, et al. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature. 2012;486(7401):130–134. doi: 10.1038/nature11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGregor MJ, Islam SA, Sternberg MJ. Analysis of the relationship between side-chain conformation and secondary structure in globular proteins. J Mol Biol. 1987;198(2):295–310. doi: 10.1016/0022-2836(87)90314-7. [DOI] [PubMed] [Google Scholar]
- 40.Ballesteros JA, Deupi X, Olivella M, Haaksma EE, Pardo L. Serine and threonine residues bend alpha-helices in the χ1 = g- conformation. Biophys J. 2000;79(5):2754–2760. doi: 10.1016/S0006-3495(00)76514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deupi X, et al. Ser and Thr residues modulate the conformation of pro-kinked transmembrane alpha-helices. Biophys J. 2004;86(1 Pt 1):105–115. doi: 10.1016/S0006-3495(04)74088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gamal El-Din TM, Martinez GQ, Payandeh J, Scheuer T, Catterall WA. A gating charge interaction required for late slow inactivation of the bacterial sodium channel NavAb. J Gen Physiol. 2013;142(3):181–190. doi: 10.1085/jgp.201311012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Y, Scheuer T, Catterall WA. Reversed voltage-dependent gating of a bacterial sodium channel with proline substitutions in the S6 transmembrane segment. Proc Natl Acad Sci USA. 2004;101(51):17873–17878. doi: 10.1073/pnas.0408270101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zarrabi T, et al. A molecular switch between the outer and the inner vestibules of the voltage-gated Na+ channel. J Biol Chem. 2010;285(50):39458–39470. doi: 10.1074/jbc.M110.132886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265(5179):1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 46.Li HL, Galue A, Meadows L, Ragsdale DS. A molecular basis for the different local anesthetic affinities of resting versus open and inactivated states of the sodium channel. Mol Pharmacol. 1999;55(1):134–141. doi: 10.1124/mol.55.1.134. [DOI] [PubMed] [Google Scholar]
- 47.Ahern CA, Eastwood AL, Dougherty DA, Horn R. Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ Res. 2008;102(1):86–94. doi: 10.1161/CIRCRESAHA.107.160663. [DOI] [PubMed] [Google Scholar]
- 48.O’Reilly AO, et al. Bisphenol A binds to the local anesthetic receptor site to block the human cardiac sodium channel. PLoS ONE. 2012;7(7):e41667. doi: 10.1371/journal.pone.0041667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramos E, O’leary ME. State-dependent trapping of flecainide in the cardiac sodium channel. J Physiol. 2004;560(Pt 1):37–49. doi: 10.1113/jphysiol.2004.065003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brooks BR, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaw DE, et al. Anton, a special-purpose machine for molecular dynamics simulation. Commun ACM. 2008;51(7):91–97. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.