

Abstract
Background
Chagas disease is caused by the parasite Trypanosoma cruzi, is endemic in Latin America and leads to an estimated 14,000 deaths per year and around 100 million people at risk of infection. Drugs currently used in the treatment of Chagas are old, partially effective and have numerous side effects.
Methodology
We have previously reported that 3-nitro-1H-1,2,4-triazole-based compounds demonstrate significant and selective activity against T. cruzi amastigotes in infected L6 cells via activation of a type I nitroreductase, specific to trypanosomatids. In the present work we evaluated in vivo 13 of these compounds based on their high in vitro potency against T. cruzi (IC50 < 1 µM) and selectivity (SI: toxicity to L6 cells/toxicity against T. cruzi amastigotes > 200). Representative compounds of different chemical classes were included. A fast luminescence assay with transgenic parasites that express luciferase, and live imaging techniques were used. A total of 11 out of 13 compounds demonstrated significant antichagasic activity when administered intraperitoneally for 5–10 days at relatively small doses. The best in vivo activity was demonstrated by amides and sulfonamide derivatives. ADMET studies were performed for specific compounds.
Conclusion
At least three compounds were identified as effective, non-toxic antichagasic agents suitable for further development.
American trypanosomiasis or Chagas disease is caused by the protozoan parasite Trypanosoma cruzi, which is transmitted by blood-sucking insects and remains a major health problem in Latin America. It is estimated that around 100 million people are at risk of infection with T. cruzi in endemic areas in Latin America [1]. Despite the fact that in the past two decades the number of incidences has significantly declined, primarily due to vector control initiatives, the epidemiology of the disease has changed due to population migration, illegal drug usage and medical practices. Thus, the number of cases in non-endemic regions such as the USA, Australia, Europe and Japan is on the rise [2,3]. In the absence of successful vaccines, chemotherapy remains the only viable option to fight the parasite in the patient.
Currently, two nitro heterocyclic prodrugs are used to treat Chagas disease: nifurtimox (a nitrofuran; Nfx) and benznidazole (a 2-nitroimidazole; Bnz). Both were introduced over 50 years ago [4], have limited efficacy, can cause various side effects, and some strains are refractory to treatment [5]. Recently, inhibitors of the sterol 14α-demethylase enzyme (CYP51), which is part of the ergosterol biosynthesis, are under development as effective antichagasic agents [6]. Unfortunately, the high cost of these inhibitors prohibits their use in poor countries where the disease is most prevalent [7]. Therefore, we urgently need new, affordable and safer drugs to treat Chagas disease.
Most nitroheterocyclic compounds function as prodrugs and must undergo activation before mediating their cytotoxic effects. It was previously demonstrated that an oxygen-insensitive, type I nitroreductase (NTR), absent from most eukaryotes with trypanosomes being a major exception, is responsible for nitrofuran and Bnz trypanocidal activity [8–10]. This enzyme mediates a series of two-electron reduction reactions that result in the fragmentation of the heterocyclic ring and production of toxic metabolites [11]. The fact that the activation of nitroheterocyclic prodrugs can be catalyzed by a type I NTR specific to trypanosomatids has led to a renewed interest in the use of such compounds as antiparasitic agents [12–17].
We have recently reported that 3-nitro-1H-1,2,4-triazole-based amines, amides and sulfonamides demonstrate excellent activity against T. cruzi amastigotes in infected L6 cells with no toxicity towards the host cells [18,19]. The IC50 values of these compounds against the intracellular parasite ranged from low nanomolar to less than 4 µM and have selectivity indices ranging from 66 to 2682. In addition, several of these compounds were up to 56-fold more active than the reference drug Bnz, tested in parallel [18,19]. We have also demonstrated that nitrotriazole-based compounds are activated by the type I NTR and that when this enzyme is overexpressed in the related T. brucei, the recombinant cells displayed hypersensitivity to these compounds [18,19]. However, since there are concerns about the toxicity and potential mutagenicity of nitro-compounds, the ultimate test for any nitro-triazole is their in vivo evaluation for efficacy and adverse effects. Interestingly, in preliminary in vivo studies, we found out that treatment of T. cruzi -infected mice with one nitrotriazole-based aromatic amine, NTLA-1 [20], given at just 2 mg/kg/day × 50 days, resulted in a rapid and persistent drop in peripheral parasite levels and in a fraction of cures [21,22]. More importantly, there was an absolute correlation between treatment efficacy as determined parasitologically and the increase in the fraction of T. cruzi-specific CD8+ T cells with a T-central memory phenotype in the peripheral blood of treated mice [21,22].
In the present study we have evaluated in vivo 13 nitrotriazole-based compounds based on their high in vitro potency against T. cruzi (IC50 <1 µM) and selectivity index ([SI]: toxicity to L6 cells/toxicity against T. cruzi amastigotes >200). Representative compounds of different chemical classes were included. A fast luminescence assay, in which mice are infected with transgenic parasites that express luciferase, and live imaging techniques were used. ADME studies were also performed for specific compounds, to explain discrepancies between in vitro and in vivo activity. Finally, studies were performed to assess potential toxicity/mutagenicity associated with these compounds.
Results & discussion
As was mentioned earlier, the criteria used for the selection of compounds in the present study were their in vitro high potency, selectivity >200 and variability in structure. Thus, compounds 1 and 10 were selected as the most potent in vitro aromatic amines, 2 and 3 as potent aliphatic amines, 4, 7 and 13 as representative potent amides, and 5, 6, 8, 11 and 12 as representative potent sulfonamides (Table 1). Compound 1 is a chloroquinoline-based and 13 a chlorobenzothiazole-based aromatic amine. In the class of aliphatic amines, 2 was selected as a benzyl amine whereas 3 as a piperazine derivative. The phenethylamine 9 was added for in vivo evaluation later on, although its SI is <200, to test the hypothesis that it may demonstrate better in vivo stability than compound 2. In the class of amides we included the benzylamide 4, the benzothiazole amide 13 and the amide 7 in which the nitrotriazole ring is connected through the carbonyl rather than the amino functionality. Finally, in the case of sulfonamides, phenyl- (5), biphenyl- (6) and thiophene sulfonamides (8, 11 & 12) were included. The in vitro evaluation of compounds 1–13 against T. cruzi intracellular amastigotes in L6 host cells was performed by the Drugs for Neglected Diseases initiative in Switzerland and the data are presented in Table 1. The corresponding data for compounds 1–8 have been presented before [18,19], but are included here for comparison purposes with regard to their in vivo activity. A structure–activity relationship discussion, based on the in vitro data of compounds 1–13, is not appropriate since the compounds cover a range of chemical classes with limited number of members in each class. However, comparing compounds in the same chemical class, we can conclude that by increasing lipophilicity (logP) we increase antichagasic potency and toxicity in the host cells (decreasing IC50 values in L6 cells); (compare 5 to 6; 8 to 12). It is worth mentioning that all compounds apart from 9 demonstrated superior activity against T. cruzi amastigotes with IC50 values at nM concentrations and selectivity indices (SI = IC50 in L6 host cells/IC50 in T. cruzi) ≥200, namely they fulfilled the criteria set by us for further in vivo evaluation. In addition, all compounds in Table 1 were from 2- to 56-fold more potent than the reference compound Bnz, tested in parallel. In particular, the sulfonamide 6 and the 2-aminobenzothiazole derivative 10 demonstrated exceptional antichagasic activity with IC50 values of 28 and 59 nM, respectively, exhibiting excellent selectivity of >1700. Interestingly, compounds with the best antichagasic activity (at low nM concentrations) and selectivity (1, 2, 4, 6 & 10) had a clogP value between 3 and 3.5 (with the exception of 6). No correlation seems to exist between PSA value and antichagasic activity or host cell toxicity (Table 1).
Table 1.
In vitro biological data and physical properties of all in vivo tested compounds.
| ID No |
T. cruzi IC-50 (µM) |
Selectivity | Cytotox. L6 IC-50 (µM) |
Bnz/Com | clogP | logD6 | PSA (Å2) | Compound Type |
|---|---|---|---|---|---|---|---|---|
| Bnz | 1.431 µM | Reference | ||||||
| 1 | 0.140 | 976 | 136.6 | 9.6 | 3.0 | 1.9 | 101.5 | 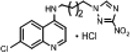 |
| 2 | 0.169 | 816 | 137.9 | 8.0 | 3.5 | 0.48 | 88.6 | 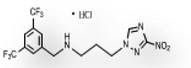 |
| 3 | 0.340 | >562 | >191.1 | 4.0 | 2.9 | 0.56 | 83.0 | 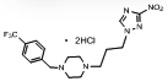 |
| 4 | 0.113 | >2381 | >268 | 13.8 | 3.2 | 3.22 | 114.9 | 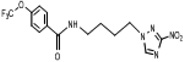 |
| 5 | 0.359 | 656 | 235.3 | 4.4 | 1.9 | 1.90 | 122.7 | 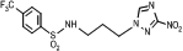 |
| 6 | 0.028 | 1764 | 50 | 55.8 | 2.67 | 2.67 | 122.70 | 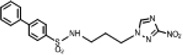 |
| 7 | 0.307 | 468 | 143.77 | 5.1 | 2.2 | 2.17 | 96.8 | |
| 8 | 0.438 | 556 | 243.5 | 3.6 | 1.7 | 1.74 | 122.7 | |
| 9 | 0.775 | 116 | 90.1 | 2.0 | 2.9 | −0.27 | 88.6 | 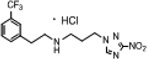 |
| 10 | 0.059 | 1725 | 101.9 | 26.6 | 3.1 | 3.10 | 101.5 | |
| 11 | 0.462 | 277.5 | 128.3 | 4.4 | 2.3 | 2.26 | 122.7 | 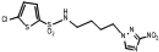 |
| 12 | 0.373 | 261.1 | 97.3 | 6.07 | 2.86 | 2.85 | 122.7 | 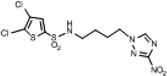 |
| 13 | 0.422 | 364.4 | 153.8 | 5.36 | 2.3 | 2.30 | 118.52 | 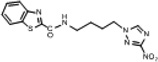 |
T. cruzi, strain Tulahuen C4 amastigotes; IC50 values are means of two independent assays, which varied < ±50%. Selectivity is the ratio: IC50 in L6 cells/IC50 in T. cruzi; Bnz: Benznidazone; Bnz/com: The ratio IC50 of Bnz/IC50 of each compound against T. cruzi; logD6: the logD at pH 6. PSA: polar surface area. All physical properties were predicted using the Marvin Calculator (www.chemaxon.com).
The in vivo antichagasic activity of the compounds in Table 1 was assessed by using a fast luminescence assay [23] in which mice are infected with transgenic parasites that express luciferase [24]. Animals were treated with each candidate compound for 5–10 days and were imaged as described in detail in the Experimental section. The dose used for each compound was selected based on its in vitro activity against T. cruzi and toxicity towards host L6 cells. The results of the in vivo studies are depicted in Figure 1. Mice treatment was continued for up to 10 days and the data were analyzed after 5 (blue bars) and 10 days (red bars) of treatment. For compounds 1 and 5, the data were analyzed only after 10 days of treatment (yellow bars) whereas treatment with compound 6 was continued only for 5 days (purple bar) due to its high in vitro potency (Table 1). Bnz was included in all experiments at an effective low dose of 15 mg/kg/day, since most of the new compounds were tested at this dose. In Figure 1, data for Bnz from two individual experiments with the greatest difference are shown. Compounds 1, 4, 5, 6, 8, 9 & 11 significantly dropped the parasite index more than 80 and up to 100%. In particular, compounds 4 and 11 demonstrated greater activity than Bnz at 15 mg/kg/day, with no detectable parasite signal after 10 days of treatment. Images of mice treated with compounds 4 and 11 are shown in Figure 2. Compound 8, at 15 mg/kg/day, also demonstrated significant antichagasic activity, similar to that of Bnz, dropping the parasite index by 94% after 10-day treatment (Figure 1) Images of mice treated with compound 8 are shown in Figure 3. Although compounds 3, 7, 12 and 13 demonstrated significant in vivo antichagasic activity, they failed to perform better than Bnz after 10-day treatment at 30, 30, 15 and 15 mg/kg/day, respectively (Figure 1). Compounds 2 and 10 failed to demonstrate in vivo antichagasic activity after 5- or 10-day treatment at 40 and 10 mg/kg/day, respectively.
Figure 1. In vivo evaluation of compounds in Table 1.

Parasite index was determined after 5- and 10-day treatment at the indicated doses. For compounds 1 and 5, parasite index was determined only after 10-day treatment, whereas for compound 6 after 5-day treatment only. Errors indicate SD.
*p ≤ 0.05; **p ≤ 0.01.
Figure 2. Images of untreated and treated mice with the indicated compounds.

Groups of five mice were infected with Trypanosoma cruzi trypomastigotes expressing luciferase and imaged before and after 5- and 10-day treatment.
Figure 3. Images of untreated and treated mice with the indicated compounds.

Groups of five mice were infected with Trypanosoma cruzi trypomastigotes expressing luciferase and imaged before and after 5- and 10-day treatment.
Comparing the in vivo (Figure 1) with the in vitro efficacy (Table 1) of all tested compounds, we observed no direct correlation. This is expected considering the number of additional parameters that determine drug activity in vivo and not modeled in the in vitro assay. Thus, compounds 2 and 10 with IC50 values against T. cruzi at low nM concentrations, failed to show activity in vivo. Moreover, compound 2 with an in vitro selectivity index of 816 also resulted in some deaths at 40 mg/kg/day, presumably due to its bioactivation to reactive intermediates [25]. Furthermore, it has been reported that although both benzyl- and phenethyl-amines substituted with electron-withdrawing groups are substrates of monoamine oxidase B, several such phenethylamines were acting as inhibitors of the enzyme [26]. Therefore, the phenethylamine compound 9 was tested in vivo to compare its activity and toxicity with that of 2. Indeed, compound 9, which was 4.6-fold less active in vitro than 2 (IC50 775 nM vs 169 nM) dropped the parasite index in mice by about 88 and 85% after 5-day and 10-day treatment, respectively, at 30 mg/kg/day without any sign of toxicity. Similarly, compounds 8 and 11 with excellent in vivo activity were not the most potent antichagasic compounds in vitro (Table 1). The lack of a direct correlation between in vitro and in vivo activity confirms that compounds with in vitro IC50 values <2 µM against T. cruzi may be also worthy of in vivo evaluation.
To explain some of the discrepancies observed between in vitro and in vivo activity, we performed some ADME studies for selected compounds (1, 4, 8 & 10). Compounds 4 and 8 were selected because of both their excellent in vitro and in vivo activity, whereas 10 for its lack of in vivo activity despite its excellent in vitro activity. Compound 1 was selected for its excellent in vitro activity and good in vivo activity, despite the fact that it is an aromatic amine similar to the in vivo inactive compound 10. Table 2 shows microsomal stability data for compounds 1, 4, 8 and 10, using verapamil and warfarin as high metabolized and low metabolized controls, respectively. All compounds were stable in the absence of NADPH. However, in the presence of NADPH, mouse microsomal protein highly metabolized the aromatic amines 1 and 10, but left the amide 4 and sulfonamide 8 intact. These data are consistent with the lack of in vivo activity observed for compound 10, especially at the relatively low tested dose of 10 mg/kg/day (Figure 1). However, all compounds were relatively stable in mouse plasma (Table 3), and this perhaps partially explains the good in vivo activity of compound 1. Since amide 4 and sulfonamides 8 and 11 were the best compounds in terms of in vivo activity, their permeability through Caco-2 monolayers was investigated to evaluate whether such compounds can be administered orally at a sufficient blood concentration [27]. Only compounds 4 and 8 were tested in this system: sulfonamides 8 and 11 are closely related analogs, therefore data pertaining to one compound should reflect the situation of the other. Both 4 and 8 (Table 4) demonstrated an excellent permeability since the apparent permeability value (Papp) was more than 5 × 10−6 cm/s [27]. It has also been recently proposed that compounds with a logPapp > −4.96 accurately predict high permeability [28]. In our case the logPapp values of 4 and 8 were −4.61 and −4.55, respectively. Based on the criteria proposed by Chaturvedi et al., it is predicted that compounds 4 and 8 with Papp values >10 × 10−6 cm/s will demonstrate 70–100% oral absorption [29]. Partition coefficients (log D and log P) and molecular surface area (PSA) are also potential predictors of the intestinal permeability of drugs. According to a recent study [30], the logD value at pH 6 (logD6) can more accurately predict intestinal permeability than the other mentioned parameters and a logD6 > −0.42, (the logD6 value of labetalol) is associated with high permeability [30]. As can be seen in Table 1, all compounds 1–13 demonstrate logD6 values > −0.42 and thus they may demonstrate a good intestinal permeability. However, as was shown above, bio-availability is dependent upon a combination of parameters, a crucial one of which is metabolic stability, therefore ADME studies are necessary for reliable predictions. Similarly, in the case of compounds 2 and 9, ADME studies will confirm whether or not extensive metabolism of 2 is responsible for its inactivity in vivo. Such studies are planned in the near future.
Table 2.
Microsomal stability screen data summary.
| Compound | Concentration (µM) |
Mean remaining | Comments | |
|---|---|---|---|---|
| Parent comp (%) | Parent comp (%) | |||
| (+)NADPH | (−)NADPH | |||
| Verapamil | 1 | 5.6 | 113 | High metabolized control |
| Warfarin | 1 | 83.9 | 118 | Low metabolized control |
| 1 | 1 | 5.1 | 112 | |
| 4 | 1 | 90.2 | 115 | |
| 8 | 1 | 91.6 | 120 | |
| 10 | 1 | 1.0 | 108 | |
Table 3.
Plasma stability screen data summary.
| Compound | Concentration (µM) | Mean remaining parent comp (%) |
Comments |
|---|---|---|---|
| Propantheline | 5 | 30.4 | High metabolized control |
| Warfarin | 5 | 97.8 | Low metabolized control |
| 1 | 5 | 96.6 | |
| 4 | 5 | 93.4 | |
| 8 | 5 | 88.6 | |
| 10 | 5 | 87.3 |
Table 4.
Caco-2 permeability summary.
| Compound | Concentration (µM) | Mean A → B | Comments |
|---|---|---|---|
| Papp†10−6 cm s−1 | |||
| Ranitidine | 10 | 0.5 | Low permeability control |
| Warfarin | 10 | 44.2 | High permeability control |
| 4 | 10 | 24.5 | |
| 8 | 10 | 27.9 |
Permeability ranking: Papp × (10−6 cm s−1).
Low: Papp <0.5; Moderate: 0.5 <Papp < 5 ; High: Papp > 5.
Papp: Apparent permeability.
All potent in vivo compounds did not show apparent toxicity at the doses tested. However, since these compounds are nitro-derivatives, there is a concern for potential mutagenicity/genotoxicity. Therefore, representative 3-nitrotriazoles (some of them not yet tested in vivo) were evaluated using the Ames assay [31,32]. Here we present the results for only compound 4, since this compound demonstrated excellent in vivo antichagasic activity. The compound was tested against Salmonella typhimurium TA98 strain (Figure 4) and against a mixed TA7001–7006 strain (MixS; Figure 5), with or without the rat liver metabolic activation system S9, and the mean number of revertants was plotted versus compound concentration [32]. The compound was not mutagenic, with one exception; mutagenicity was seen in mixed strains at the highest tested concentration of 1000 µg/ml and only in the presence of S9 (Figure 5). This concentration was toxic to the L6 cells (Table 1). Since a linear dose response is not observed in the mutagenicity tests (Figures 4 & 5), we can assume that compound 4 is not mutagenic at non0toxic doses, otherwise a safe threshold presumably exists, as has been suggested for certain compounds [33,34].
Figure 4. Mutagenicity study (Ames test) for compound 4 in TA98 strains without/with S9.

PC was 625/1200 ng/ml 4-nitroquinoline-N-oxide/2-nitrofluorene (−S9) and 2-aminoanthracene (+S9).
PC: Positive control.
Figure 5. Mutagenicity study (Ames test) for compound 4 in mixed TA7001–7006/strains without/with S9.

PC: −S9-Mix: 625/1200 ng/ml 4-nitroquinoline-N-oxide/2-nitrofluorene; +S9-Mix:10 µg/ml 2-aminoanthracene.
*p ≤ 0.05; **p ≤ 0.01.
For comparison purposes a 2-nitroimidazole-based compound was also tested in the Ames assay. Mutagenicity was demonstrated against TA98/TA98NR strains in the presence or absence of S9 at doses as low as 20 µg/plate (data not shown). In addition, this compound was highly mutagenic in the TA100/TA100NR strains in the presence or absence of S9 at doses ≥0.8 µg/plate (data not shown). Furthermore, the 2-nitroimidazole-based compound was toxic at doses ≥350 µg/plate to all strains. In general, most 3-nitrotriazole-based compounds that were tested in the Ames test did not show mutagenicity, suggesting that mutagenicity is associated to a greater degree with 2-nitroimidazole rather than 3-nitrotriazole systems, although further compounds should be tested for more accurate conclusions. In addition, the 3-nitrotriazolic compounds that exhibited mutagenicity did so at concentrations significantly higher than their IC50 value in the L6 host cells (Table 1). It should also be mentioned that mutagenicity in S. typhimurium strains is not necessarily translated to mutagenicity in humans. For instance, although Bnz shows significant mutagenicity at relatively low concentrations (<62 µg/ml) in S. typhimurium strains [35], mutagenicity in humans has never been reported. A mutagenicity study of serum and urine from guinea pigs treated with Bnz showed that Bnz is not metabolized by the mammalian host into stable mutagenic derivatives detectable by the Ames test, suggesting that the potential cancer risk in humans is minimal [36].
We also investigated the effect of compound 1 on zebrafish embryos’ development. Drugs were applied to developing zebrafish embryos at 24 h post-fertilization (hpf), at the end of the segmentation stage when the primary stages of organogenesis are complete and the fish have begun to move. Groups of six embryos per dose were examined at three developmental time-intervals (24, 48 and 72 hpf) and each experiment repeated in triplicate. The data are summarized in Table 5. No compound-related toxicity or phenotypic changes were observed at all doses and time intervals. Concentrations up to 300 µM were tested. Similar results were obtained with two other 3-nitrotriazole-based amides, analogs of 4 (data not shown). In contrast, incubation of embryos in nifurtimox resulted in weakened heart beat, pericardial oedema or death (data not shown).
Table 5.
Zebrafish embryos toxicity data for compound 1.
| Compound I (µM) | Time (hours post-fertilization)† | |||
|---|---|---|---|---|
| 24 | 48 | 72 | ||
| 3.7 | Embryos | 18/18 | 18/18 | 17/18 |
| 11.1 | Embryos | 18/18 | 18/18 | 17/18 |
| 33.3 | Embryos | 18/18 | 17/18 | 17/18 |
| 100.0 | Embryos | 18/18 | 17/18 | 17/18 |
| 300.0 | Embryos | 18/18 | 18/18 | 18/18 |
| Control | Embryos | 18/18 | 18/18 | 16/18 |
| DMSO | Embryos | 18/18 | 17/18 | 16/18 |
The data show the ratio of number of surviving zebrafish embryos at different developmental time points (in hours post fertilization) at each compound 1 concentration versus the total number of zebrafish embryos used in the assay.
The in vivo luminescence assay combined with in vitro ADMET data provides a rapid method to identify safe, stable compounds with in vivo activity and potentially good oral bioavailability before any other expensive pharmacokinetic/pharmacodynamic evaluation. Thus, this strategy can lead to an accelerated drug discovery process [23]. Compounds with good in vivo activity seem to also have good metabolic stability. Through this process we have identified at least three compounds, 4, 8 and 11 as candidates for further development. However, the question of whether or not a 10-day treatment resulted in the sterile cure of these animals has not been answered yet, since the animals were not kept long after treatment. Experiments in which mice will be kept for an extended period of time post-treatment and treated with an immunosuppressant will be the next step to provide us with an answer [22]. In addition, studies should be done to determine if these compounds can treat the chronic stage of the disease. However, current studies have clearly demonstrated that 3-nitrotriazole-based amides and sulfonamides have a significant chance to be developed as antichagasic drugs. They can be easily synthesized in high yields and purity with low cost [19], they show very good mouse plasma and metabolic stability (Tables 2 & 3) and, in general, are not mutagenic at nontoxic concentrations. However, not all in vivo active compounds have been tested yet for mutagenicity. Moreover, additional compounds with IC50 values against T. cruzi <2 µM might be good candidates for ADMET and subsequent in vivo evaluation.
Experimental
Chemistry
All starting materials and solvents were purchased from Sigma-Aldrich (WI, USA), were of research-grade quality and were used without further purification. Solvents used were anhydrous and the reactions were carried out under a nitrogen atmosphere and exclusion of moisture. Melting points were determined by using a Mel-Temp II Laboratory Devices apparatus (MA, USA) and are uncorrected. Proton NMR spectra were obtained on a Varian Inova-500 or a Bruker Avance-III-500 spectrometer at 500 MHz and are referenced to Me4Si or to the corresponding protonated solvent, if the solvent was not CDCl3. High-resolution electrospray ionization (HRESIMS) MS were obtained on a Agilent 6210 LC–TOF MS at 11000 resolution. Thin-layer chromatography was carried out on aluminum oxide N/UV254 or polygram silica gel G/UV254-coated plates (0.2 mm, Analtech, DE, USA). Chromatography was carried out on preparative TLC alumina GF (1000 microns) or silica gel GF (1500 microns) plates (Analtech). All compounds were purified by preparative TLC chromatography on silica gel GF plates (≥95% purity). The synthesis of compounds 1–8 has been described before [18,19]. Similar synthetic procedures were followed to obtain compounds 9–13. For compound 9, 3-(trifluoromethyl)phenethyl bromide (1.035 mmol) was added dropwise (15 min) to a solution of 3-nitro-1H-1,2,4-triazolyl-propylamine (1.035 mmol) [37] in the presence of potassium carbonate (9.52 mmol) in dry acetonitrile (15 ml), and the reaction mixture was refluxed under a nitrogen atmosphere for 10 h. The reaction mixture was cooled down, filtered, the solids were washed with acetonitrile, the organic filtrate was evaporated and the residue extracted from water-ethyl acetate. The organic layer was separated and dried over anhydrous Na2SO4. The solvent was evaporated and the residue was purified by preparative TLC on alumina plates with ethyl acetate:MeOH (99:1). A yellowish oil was obtained (Rf = 0.53), which was the desired monoalkylated product. This was dissolved in ethyl acetate and converted to its HCl salt by treating with HCl gas in dry ether (1 M solution).
[3-(3-nitro-1H-1,2,4-triazol-1-yl)propyl] ({2-[3-(trifluoromethyl)phenyl]ethyl})amine hydrochloride (9)
Fine white powder (40%): mp 103–104°C; 1H NMR (500 MHz, CD3OD) δ: 8.66 (s, 1H), 7.64–7.55 (m, 4H), 4.50 (t, J = 7.0 Hz, 2H), 3.34 (t, J = 7.0 Hz, 2H), 3.17 (t, J = 8.0 Hz, 2H), 3.10 (t, J = 8.0 Hz, 2H), 2.35 (quintet, J = 8.0 Hz, 2H). HRESIMS calculated for C14H17F3N5O2 and C14H16F3N5NaO2 m/z [M+H]+ and [M+Na]+ 344.1329, 345.1356 and 366.1148, 367.1176, found 344.1336, 345.1363 and 366.1152, 367.1181, respectively.
For compound 10, the commercially available 2,6-dichloro-1,3-benzothiazole (1.24 mmol) was coupled with 3-nitro-1H-1,2,4-triazolyl-propylamine (1.24 mmol) [37], by refluxing in absolute propanol (8 ml) for 16 h, and in the presence of fivefold excess of triethyl amine.
6-chloro-N-[3-(3-nitro-1H-1,2,4-triazol-1-yl) propyl]-1,3-benzothiazol-2-amine (10)
Orange powder (65%): mp 194–195°C (dec.); 1H NMR (500 MHz, CD3COCD3) δ: 8.71 (s, 1H), 7.70 (d, J = 2.5 Hz, 1H), 7.39 (d, J = 8.5 Hz, 1H), 7.37 (br s, 1H), 7.26 (dd J1 = 8.5, J2 = 2.5 Hz, 1H), 4.57 (t, J = 7.0 Hz, 2H), 3.62 (m, 2H), 2.39 (quintet, J = 6.5 Hz, 2H). HRESIMS calculated for C12H12ClN6O2S m/z [M+H]+ 339.0425, 341.0398, found 339.0427, 341.0395.
General synthetic procedure of arylamides/sulfonamides 11–13
For compounds 11–13: the appropriate commercially available arylcarbonyl/arylsulfonyl chloride (1.24 mmol) was dissolved in 2–3 ml dry dichloromethane and added dropwise to a solution of 3-nitro-1H-1,2,4-triazolyl-butylamine) (1.24 mmol) [37],and triethylamine (2.48 mmol) in 6–8 ml of dry dichloromethane, at room temperature and under an inert atmosphere. The reaction mixture was stirred for 12 h. Consequently, the inorganic salts were filtered off, the filtrate was evaporated and the residue was chromatographed on silica gel.
5-chloro-N-[4-(3-nitro-1H-1,2,4-triazol-1-yl) butyl]thiophene-2-sulfonamide (11)
White crystallic powder (84%): mp 81–83 C; 1H NMR (500 MHz, CD3COCD3) δ: 8.65 (s, 1H), 7.46 (d, J = 4.0 Hz, 1H), 7.14 (d, J = 4.0 Hz, 1H), 6.84 (br t, 1H), 4.44 (t, J = 7.0 Hz, 2H), 3.10 (t, J = 7.0 Hz, 2H), 2.05 (m, 2H), 1.65 (m, 2H). HRESIMS calculated for C10H13ClN5O4S2 and C10H12ClN5NaO4S2 m/z [M+H]+ and [M+Na]+ 366.0092, 368.0063 and 387.9911, 389.9882, found 366.0091, 368.0063 and 387.9909, 389.9884, respectively.
4,5-dichloro-N-[4-(3-nitro-1H-1,2,4-triazol-1-yl)butyl]thiophene-2-sulfonamide (12)
White crystallic powder (75%): mp 104–105°C; 1H NMR (500 MHz, CDCl3) δ: 8.19 (s, 1H), 7.39 (s, 1H), 4.82 (br t, 1H), 4.34 (t, J = 7.0 Hz, 2H), 3.13 (q, J = 6.5 Hz, 2H), 2.09 (quintet, J = 7.5 Hz, 2H), 1.65 (quintet, J = 7.5 Hz, 2H). HRESIMS calculated for C10H12Cl2N5O4S2 and C10H11Cl2N5NaO4S2 m/z [M+H]+ and [M+Na]+ 399.9702, 401.9673 and 421.9522, 423.9492 found 399.9704, 401.9671 and 421.9521, 423.9490.
N-[4-(3-nitro-1H-1,2,4-triazol-1-yl)butyl]-1,3-benzothiazole-2-carboxamide (13)
Off-white powder (71%): mp 92–94°C; 1H NMR (500 MHz, CDCl3) δ: 8.22 (s, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.57–7.51 (m, 3H), 4.39 (t, J = 7.0 Hz, 2H), 3.59 (t, J = 7.0 Hz, 2H), 2.10 (quintet, J = 7.5 Hz, 2H), 1.75 (quintet, J = 7.5 Hz, 2H). HRESIMS calculated for C14H15N6O3S m/z [M+H]+ 347.0921, found 347.0922.
ADME in vitro studies
ADME in vitro studies were performed by APREDICA (MA, USA) for several compounds. Samples were analyzed by LC/MS/MS using an Agilent 6410 MS coupled with an Agilent 1200 HPLC and a CTC PAL chilled autosampler, all controlled by MassHunter software (Agilent). After separation on a C18 reverse-phase HPLC column (Agilent, Waters or equivalent) using an acetonitrile-water gradient system, peaks were analyzed by MS using ESI ionization in MRM mode.
Microsomal stability screen
Each test compound was dissolved in DMSO and incubated (37°C) at 1 µM final concentration with 0.3 mg/ml of mouse microsomal protein in 100 mM potassium phosphate, 3 mM MgCl2, pH 7.4, in the presence or absence of 2 mM NADPH (to detect NADPH-free degradation) for up to 60 min. At indicated times (0 and 60 min), an aliquot was removed from each experimental and control reaction then mixed with an equal volume of ice-cold Stop Solution (0.3% acetic acid in acetonitrile containing haloperidol, diclofenac, or other internal standard). Stopped reactions were incubated for at least 10 min at −20°C, and an additional volume of water was added. The samples were centrifuged to remove precipitated protein, and the supernatants were analyzed by LC/MS/MS to quantitate the remaining parent compound. Data were reported as % remaining by dividing by the time zero concentration value [38].
Plasma stability screen
Each test compound (in a DMSO stock solution) was incubated at 5 µM final concentration with mouse plasma and 2% DMSO at 37°C in duplicate. At indicated times (0 and 60 min), an aliquot was removed from each experimental and control reaction and mixed with three volumes of ice-cold Stop Solution (methanol containing haloperidol, diclofenac or other internal standard). Stopped reactions were incubated at least for 10 min at −20°C. The samples were centrifuged to remove precipitated protein, and the supernatants were analyzed by LC/MS/MS to quantitate the remaining parent compound. Data were converted to % remaining by dividing by the time zero concentration value [39].
Caco-2 monolayer permeability studies
Caco-2 cells grown in tissue culture flasks were trypsinized, suspended in medium, and the suspensions were applied to wells of a collagen-coated BioCoat Cell Environment in 96-well format. The cells were allowed to grow and differentiate for 3 weeks, feeding at 2-day intervals. For apical to basolateral (A→B) permeability, the test agent was added to the apical (A) side at 10 µM final concentration and amount of permeation was determined on the basolateral (B) side. The A-side buffer contained 100 µM Lucifer yellow dye, in Transport Buffer (1.98 g/l glucose in 10 mM HEPES, 1×Hank’s Balanced Salt Solution) pH 6.5, and the B-side buffer was Transport Buffer, pH 7.4. Caco-2 cells were incubated with these buffers for 2 h and the receiver side buffer was removed for analysis by LC/MS/MS. To verify that the Caco-2 cell monolayers were properly formed, aliquots of the cell buffers were analyzed by fluorescence to determine the transport of the impermeable dye Lucifer Yellow. Data were expressed as permeability (Papp): Papp = (dQ/dt)/C0A, where dQ/dt is the rate of permeation, C0 is the initial concentration of test agent, and A is the area of the monolayer [27].
In vitro evaluation against T. cruzi
In vitro activity against T. cruzi intracellular amastigotes and cytotoxicity assessment in the host L6 cells (rat skeletal myoblasts) was determined using a 96-well plate format as previously described [40]. Data were analyzed with the graphic program Softmax Pro (Molecular Devices, CA, USA), which calculated IC50 values by linear regression from the sigmoidal dose inhibition curves.
In vivo anti-T. cruzi activity assessment
Trypomastigote forms from transgenic T. cruzi Y strain expressing firefly luciferase [24] were purified, diluted in PBS and injected intraperitoneally in Balb/c mice (105 trypomastigotes per mouse). 3 days after infection the mice were anesthetized by inhalation of isofluorane (controlled flow of 1.5% isofluorane in air was administered through a nose cone via a gas anesthesia system). Mice were injected with 150 mg/kg of d-luciferin potassium-salt (Goldbio) dissolved in PBS. Mice were imaged 5–10 min after injection of luciferin with an IVIS 100 (Xenogen, CA, USA) and the data acquisition and analysis were performed with the software LivingImage (Xenogen) as described before [23]. 1 day later (4 days after infection) treatment with compounds at a specific concentration (usually 15 mg/kg/day) or vehicle control (2% methylcellulose + 0.5% Tween 80) was started by intraperitoneal injection in groups of five mice and continued daily for 5–10 days. On the days indicated, mice were imaged again after anesthesia and injection of luciferin as described above. Parasite index is calculated as the ratio of parasite levels in treated mice compared with the control group and is multiplied by 100. The ratio of parasite levels is calculated for each animal dividing the luciferase signal after treatment by the luciferase signal on the first imaging (before treatment). Mean values of all animals in each group ± SD were then used to calculate the parasite index [23].
Toxicity studies in zebrafish embryos
Wildtype (WT) zebrafish strains (Tubingen and Tupfel long fin) were bred and raised in-house at the zebrafish facility of Queen Mary College, University of London, UK. Embryos were collected by natural spawning and staged according to Kimmel and colleagues [41] – given in the text as standard developmental time at 28.5°C (hpf). Work on zebrafish embryos (prior to independent feeding) is exempt under the UK Animals (Scientific Procedures) Act 1986 and does not require ethical approval. For each experiment six zebrafish embryos (three embryos per well in a 24-well plate) were treated per compound concentration (concentration was varied from 3.7 to 300 µM) and the viability of the developing embryos assessed with respect to time (hpf) as the ratio:number of live zebrafish at the time indicated/number of zebrafish at time 0 per each concentration. Each experiment was conducted in triplicate using a total of 18 embryos being analyzed per each concentration.
Mutagenicity studies
The Ames mutagenicity test was performed with S. typhimurium TA98, TA100, TA98NR (nitroredutase deficient) and TA100NR (nitroreductase deficient) strains according to a method described before [31]. Concurrently, nitrofurantoin (NFT), 2-nitrofluorene, 4-nitroquinoline-N-oxide and benzo[a]-pyrene were included in the assays with TA98/TA98NR strains or sodium azide, nitrofurantoin, metronidazole and 2-aminoanthracene in the assays with TA100/TA100NR strains. In one case, mixed TA7001– 7006 series of Salmonella, his− mutant strains were used [32]. The assays were performed in the presence (for metabolic activation) or absence of the liver S9 mix [31,32]. All tested compounds were dissolved in DMSO and the same amount of DMSO was delivered to each plate. Prior to starting the assay, the concentrations to be tested were selected in terms of solubility and toxicity results in the test system. Concentrations up to 1000 µg/plate were tested. Triplicate (in one case duplicate) plates were used for each dose and mean values of His+ revertants per plate are indicated as the results.
Statistical analysis
Data were analyzed by using the t-test (Prism vs 4.0c, GraphPad). Statistics were considered significant if p was ≤ 0.05 (*) or p ≤ 0.01 (**).
Future perspective
Recently Chagas disease was characterized as ‘the new AIDS of the Americas’ because its spread resembles the early dissemination of HIV [42]. Although this characterization is an exaggeration, there are similarities in the sense that both HIV and T. cruzi cause life-long infections and, like all blood-borne pathogens, are potentially transmitted by blood transfusion and congenitally from mother to newborn. As a result of reactivation among immigrant populations, an increase in Chagas disease infections has been reported in non-endemic settings and this ‘globalization’ will be of concern [2,3]. Currently, around 8–10 million individuals are infected with T. cruzi in endemic areas, while it has been estimated there are around 325,000 cases in the USA and about 100,000 cases in Europe, 87,000 of which are in Spain [1]. In addition, Chagas disease creates financial and social burdens to individuals, their households and countries. The early mortality and substantial disability caused by this disease, which often occurs in the most productive population, young adults, results in a devastating economic loss in the Americas.
As was mentioned earlier, with no immediate prospect for vaccines, chemotherapy is the only way to fight the parasite in the patient. One way to develop effective drugs is by targeting enzymes specific to the parasite, for example, cruzipain or CYP51. However, such approaches may lead to drug-resistant phenotypes that will create additional searches for new targets. Another approach is to utilize an enzyme specific to the parasite that activates a prodrug. We have followed the latter strategy. We have shown that 3-nitrotriazole-based compounds can be very effective in vitro against T. cruzi amastigotes via NTR-activation, without showing toxicity to the host cells [18,19]. In vitro active compounds demonstrating good metabolic and plasma stability also showed in vivo effectiveness against the parasite. Furthermore, our data have shown that 3-nitrotriazole-based compounds do not cause developmental toxicity, they are not mutagenic at non-toxic doses and are significantly less mutagenic than 2-nitroimidazoles. Therefore, further in vivo evaluation of these compounds is necessary to determine whether or not we can obtain cures without long-term toxicity and whether or not such compounds have an effect against the chronic phase of the disease. In addition, studies in combination with target-specific or even currently used antichagasic drugs may reveal a synergistic interaction, which could result in lowering of doses and shortening of the treatment-period in humans. Therefore, there is a considerable future in drug development research against Chagas disease.
Moreover, the treatment for Chagas disease is currently expensive and effective agents with low cost are desperately needed. Nitrotriazole-based compounds could be a potential future solution. However, additional studies are necessary to determine the efficacy of these compounds in the chronic stage of the disease and under oral administration.
Executive summary.
-
▪
American trypanosomiasis or Chagas disease is a neglected disease that is expanding recently in non-endemic countries in North America, Europe and Asia.
-
▪
Due to the absence of a vaccine and in view of problems associated with current drugs, there is an urgent need for the development of effective, non-toxic and affordable new drugs.
-
▪
We have discovered that 3-nitro-1,2,4-triazole-based amines, amides and sulfonamides demonstrate significant antichagasic activity against Trypanosoma cruzi amastigotes in infected L6 cells with high selectivity for the parasite.
-
▪
Such compounds are prodrugs that exert their antiparasitic activity via a type I nitroreductase activation, specific to the trypanosomatids, as has been previously demonstrated.
-
▪
At least three such compounds have demonstrated excellent in vivo activity against T. cruzi and are superior to benznidazole, at the acute phase of infection, without systemic or developmental toxicity.
-
▪
Limited mutagenicity studies suggest that several of these compounds do not demonstrate mutagenic toxicity, at least at concentrations up to their in vitro toxicity level.
Acknowledgements
The authors thank Y Wu (Northwestern University) for obtaining the NMR spectra of the compounds, M Cal, S Sax and C Stalder (Swiss TPH) for the in vitro parasite assay results, G Courtemanche (Sanofi, Toulouse, France) for initiating the mutagenicity studies of compound 4, B Bourdin (Drugs for Neglected Diseases initiative, Geneva, Switzerland) for initiating all other mutagenicity studies, and J Djumpah and the zebrafish facility at Queen Mary University of London for zebrafish developmental toxicity studies.
This work was partially supported by an NIH Challenge Grant: 1R01AI082542 – 01, Subaward No: RU374-063/4693578.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
Animal studies were approved by the Institutional Animal Care and Use Committee of New York University School of Medicine (protocol #81213). This protocol adheres to the guidelines of the Association For Assessment and Accreditation Of Laboratory Animal Care International (AAALAC).
References
- 1.Schmunis GA, Yadon ZE. Chagas disease: a Latin American health problem becoming a world health problem. Act a Trop. 2010;115:14–21. doi: 10.1016/j.actatropica.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Rassi A, Jr, Rassi A, Marin-Neto JA. Chagas disease (review) Lancet. 2010;375(9723):1388–1402. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]
- 3.Leslie M. Infectious diseases. A tropical disease hits the road. Science. 2011;333:934. doi: 10.1126/science.333.6045.934. [DOI] [PubMed] [Google Scholar]
- 4.Bern C. Antitrypanosomal therapy for chronic Chagas’ disease. N. Engl. J. Med. 2011;364:2527–2534. doi: 10.1056/NEJMct1014204. [DOI] [PubMed] [Google Scholar]
- 5.Urbina JA. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop. 2010;115:55–68. doi: 10.1016/j.actatropica.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 6.Urbina JA. Ergosterol biosynthesis and drug development for Chagas disease. Mem. Inst. Oswaldo Cruz. 2009;104(Suppl. 1):311–318. doi: 10.1590/s0074-02762009000900041. [DOI] [PubMed] [Google Scholar]
- 7.Clayton J. Chagas disease: pushing through the pipeline. Nature. 2010;465:S12–S15. doi: 10.1038/nature09224. [DOI] [PubMed] [Google Scholar]
- 8.Wilkinson SR, Taylor MC, Horn D, et al. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl Acad. Sci. USA. 2008;105(13):5022–5027. doi: 10.1073/pnas.0711014105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alsford S, Eckert S, Baker N, et al. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature. 2010;482:232–236. doi: 10.1038/nature10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker N, Alsford S, Horn D. Genome-wide RNAi screens in African trypanosomes identify the nifurtimox activator NTR and the eflornithine transporter AAT6. Mol. Biochem. Parasitol. 2011;176:55–57. doi: 10.1016/j.molbiopara.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson SR, Bot C, Kelly JM, et al. Trypanocidal activity of nitroaromatic prodrugs: current treatments and future perspectives. Curr. Top. Med. Chem. 2011;11:2072–2084. doi: 10.2174/156802611796575894. [DOI] [PubMed] [Google Scholar]
- 12.Baliani A, Gerpe A, Aran VJ, et al. Design and synthesis of a series of melamine-based nitroheterocycles with activity against trypanosomatid parasites. J. Med. Chem. 2005;48:5570–5579. doi: 10.1021/jm050177+. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez J, Aran VJ, Boiani L, et al. New potent 5-nitroindazole derivatives as inhibitors of Trypanosoma cruzi growth: synthesis, biological evaluation, and mechanism of action studies. Bioorg. Med. Chem. 2009;17:8186–8196. doi: 10.1016/j.bmc.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 14.Boiani L, Gerpe A, Aran VJ, et al. In vitro and in vivo antitrypanosomatid activity of 5-nitroindazoles. Eur. J. Med. Chem. 2009;44:1034–1040. doi: 10.1016/j.ejmech.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 15.Hall BS, Wu X, Hu L, et al. Exploiting the drug-activating properties of a novel trypanosomal nitroreductase. Antimicrob. Agents Chemother. 2010;54:1193–1199. doi: 10.1128/AAC.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bot C, Hall BS, Bashir N, et al. Trypanocidal activity of aziridinyl nitrobenzamide prodrugs. Antimicrob. Agents Chemother. 2010;54(10):4246–4252. doi: 10.1128/AAC.00800-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu L, Wu X, Han J, et al. Synthesis and structure-activity relationships of nitrobenzyl phosphoramide mustards as nitroreductase-activated prodrugs. Bioorg. Med. Chem. Lett. 2011;21(13):3986–3991. doi: 10.1016/j.bmcl.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Papadopoulou MV, Bourdin B, Bloomer W, et al. Novel 3-nitro-1,2,4-triazole-based aliphatic and aromatic amines as anti-Chagasic agents. J. Med. Chem. 2011;54(23):8214–8223. doi: 10.1021/jm201215n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papadopoulou MV, Bloomer W, Rosenzweig HS, et al. Novel 3-Nitro-1H-1,2,4-triazole-based amides and sulfonamides as potential anti-trypanosomal agents. J. Med. Chem. 2012;55(11):5554–5565. doi: 10.1021/jm300508n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenzweig HS, Papadopoulou MV, Bloomer WD. Interaction of strong DNA-intercalating bioreductive compounds with topoisomerases I and II. Oncol. Res. 2005;15:219–231. doi: 10.3727/096504005776382288. [DOI] [PubMed] [Google Scholar]
- 21.Bustamante JM, Evans A, Papadopoulou MV, et al. Use of CD8+ T central memory characteristics as immunologic evidence for treatment efficacy in mice infected with Trypanosoma cruzi; Presented at: 12th Woods Hole Immunoparasitology Meeting; Woods Hole, MA, USA. Apr, 2008. pp. 27–29. [Google Scholar]
- 22.Canavaci AMC, Bustamante JM, Padilla AM, et al. In vitro and in vivo high-throughput assays for the testing of anti-Trypanosoma cruzi compounds. PLoS Negl. Trop. Dis. 2010;4(7):e740. doi: 10.1371/journal.pntd.0000740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andriani G, Chessler A-DC, Courtemanche G, et al. Activity in vivo of anti-trypanosoma cruzi compounds selected from a high throughput screening. PLoS Negl. Trop. Dis. 2011;5(8):e1298. doi: 10.1371/journal.pntd.0001298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vazquez MP, Levin MJ. Functional analysis of the intergenic regions of TcP2beta gene loci allowed the construction of an improved Trypanosoma cruzi expression vector. Gene. 1999;239:217–225. doi: 10.1016/s0378-1119(99)00386-8. [DOI] [PubMed] [Google Scholar]
- 25.Mutlib AE, Dickenson P, Chen S-Y, et al. Bioactivation of benzylamine to reactive intermediates in rodents: formation of glutathione, glutamate, and peptide conjugates. Chem. Res. Toxicol. 2002;15:1190–1207. doi: 10.1021/tx020063q. [DOI] [PubMed] [Google Scholar]
- 26.Silverman RB, Hawe WP. SAR studies of fluorine-substituted benzylamines and substituted 2-penylethylamines as substrates and inactivators of monoamine oxidase B. J. Enzyme Inhib. 1995;9:203–215. doi: 10.3109/14756369509021486. [DOI] [PubMed] [Google Scholar]
- 27.Stewart BH, Chan OH, Lu RH, et al. Comparison of intestinal permeabilities determined in multiple in vitro and in situ models: relationship to absorption in humans. Pharm. Res. 1995;12:693. doi: 10.1023/a:1016207525186. [DOI] [PubMed] [Google Scholar]
- 28.Gozalbes R, Jacewicz M, Annand R, et al. QSAR-based permeability model for drug-like compounds. Bioorg. Med. Chem. 2011;19:2615–2624. doi: 10.1016/j.bmc.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 29.Chaturvedi PR, Decker CJ, Odinecs A. Prediction of pharmacokinetic properties using experimental approaches during early drug discovery. Curr. Opin. Chem. Biol. 2001;5:452–463. doi: 10.1016/s1367-5931(00)00228-3. [DOI] [PubMed] [Google Scholar]
- 30.Shawahna R, Rahman NU. Evaluation of the use of partition coefficients and molecular surface properties as predictors of drug absorption: a provisional biopharmaceutical classification of the list of national essential medicines of Pakistan. DARU. 2011;19(2):83–99. [PMC free article] [PubMed] [Google Scholar]
- 31.Maron DM, Ames BN. Revised methods for the Salmonella mutagenicity test. Mutat. Res. 1983;113:173–215. doi: 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- 32.Gee P, Maron DM, Ames BN. Detection and classification of mutagens: a set of base-specific Salmonella tester strains. Proc. Natl Acad. Sci. USA. 1994;91:11606–11610. doi: 10.1073/pnas.91.24.11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ames BN, Gold LS. Chemical carcinogenesis: too many rodent carcinogens. Proc. Natl Acad. Sci. USA. 1990;87(19):7772–7776. doi: 10.1073/pnas.87.19.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jenkins GJ, Doak SH, Johnson GE, et al. Do dose response thresholds exist for genotoxic alkylating agents? Mutagenesis. 2005;20(6):389–398. doi: 10.1093/mutage/gei054. [DOI] [PubMed] [Google Scholar]
- 35.Cabrera M, Lavaggi ML, Hernandez P, et al. Cytotoxic, mutagenic and genotoxic effects of new anti-T. cruzi 5-phenylethenyl-benzofuroxans. Contribution of Phase I metabolites on the mutagenicity induction. Toxicol. Lett. 2009;190:140–149. doi: 10.1016/j.toxlet.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Ferreira RC, de Melo ME, Moraes Junior MA, et al. Evaluation of genotoxic activity in the blood and urine of guinea pigs treated with nifurtimox and benznidazole. Brazilian J. Med. Biol. Res. 1988;21(5):1069–1077. [PubMed] [Google Scholar]
- 37.Papadopoulou MV, Bloomer WD. Nitroheterocyclic-linked acridines as DNA-targeting bioreductive agents. Drugs Fut. 1993;18:231–238. [Google Scholar]
- 38.Houston JB. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol. 1994;47:1469. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 39.Di L, Kerns EH, Hong Y, et al. Development and application of high throughput plasma stability assay for drug discovery. Int. J. Pharm. 2005;297:110–119. doi: 10.1016/j.ijpharm.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 40.Orhan I, Sener B, Kaiser M, et al. Inhibitory activity of marine sponge-derived natural products against parasitic protozoa. Mar. Drugs. 2010;8:47–58. doi: 10.3390/md8010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimmel CB, Ballard WW, Kimmel SR, et al. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 42.Hotez P, Dumonteil E, Woc-Colburn L, et al. Chagas disease: ‘the new HIV/AIDS of the Americas.’. PLoS Negl. Trop. Dis. 2012;9:e1498. doi: 10.1371/journal.pntd.0001498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Website
- 101.Chemaxon- cheminformatics platforms and desktop applications. www.chemaxon.com.