

Abstract
Does accelerated cortical atrophy in aging, especially in areas vulnerable to early Alzheimer's disease (AD), unequivocally signify neurodegenerative disease or can it be part of normal aging? We addressed this in 3 ways. First, age trajectories of cortical thickness were delineated cross-sectionally (n = 1100) and longitudinally (n = 207). Second, effects of undetected AD on the age trajectories were simulated by mixing the sample with a sample of patients with very mild to moderate AD. Third, atrophy in AD-vulnerable regions was examined in older adults with very low probability of incipient AD based on 2-year neuropsychological stability, CSF Aβ1-42 levels, and apolipoprotein ɛ4 negativity. Steady decline was seen in most regions, but accelerated cortical thinning in entorhinal cortex was observed across groups. Very low-risk older adults had longitudinal entorhinal atrophy rates similar to other healthy older adults, and this atrophy was predictive of memory change. While steady decline in cortical thickness is the norm in aging, acceleration in AD-prone regions does not uniquely signify neurodegenerative illness but can be part of healthy aging. The relationship between the entorhinal changes and changes in memory performance suggests that non-AD mechanisms in AD-prone areas may still be causative for cognitive reductions.
Keywords: aging, Alzheimer's disease, atrophy, cortical thickness, magnetic resonance imaging
Introduction
Most cognitive functions are affected by age (Reuter-Lorenz and Park 2010). This is likely partly caused by macrostructural brain changes (Raz and Rodrigue 2006; Persson et al. 2012), and neuroanatomical evidence has frequently been used in the debate on whether cognitive decline is continuous throughout the adult lifespan (Salthouse 2009, 2011) or primarily seen in older age (Schaie 2009). Although this debate has a significant bearing on understanding of human aging, it bears on an important practical question of when and how “normal” aging turns into clinically identifiable dementia. Aging-related dementia typically has a slow and gradual onset, with atrophy manifest years in advance of clinical symptoms (Davatzikos et al. 2009; Jack et al. 2010). Some have even suggested that cortical decline in aging is caused by an undetected disease and is not a feature of normal aging (Burgmans et al. 2009). An important implication of the apparent temporal gap between cerebral and cognitive expressions of dementia is that cases with undetected disease in presumably normal samples bias inferences about normal brain aging (Sliwinski and Buschke 1999). The proportion of older adults with undetected neurodegenerative disease is expected to increase with the age of the population studied, which could cause invalid conclusions of accelerated decline in cortical areas vulnerable to such pathology, especially the entorhinal cortex (Braak and Braak 1985; Jack et al. 1997; McDonald et al. 2009). Thus, accurate descriptions of the expected age trajectories of critical brain areas will contribute to a better understanding of normal aging as well as age-related dementia.
Estimations of adult lifespan trajectories in cortical thickness would be the first step to address this important issue. However, examinations of cortical changes across adult life face a multitude of challenges: First, approximations of lifespan trajectories from cross-sectional observations require very large samples to ensure statistical power to detect subtle differences in slopes between age groups. Second, age spans of more than a few years are practically impossible to cover in longitudinal MR studies, and the problems of cohort effects and sampling bias can therefore not easily be resolved. Finally, as discussed above, age-related neurodegenerative conditions may bias the results toward conclusions of accelerated reductions. Thus, it is important to compare the results of cross-sectional and longitudinal approaches.
Previous studies have not yielded consistent results. Cross-sectional findings suggest both linear (Courchesne et al. 2000; Raz, Gunning-Dixon et al. 2004; Fjell et al. 2009a) and nonlinear decline (Sowell et al. 2003; Raz, Gunning-Dixon et al. 2004; Allen et al. 2005; Curiati et al. 2009; Terribilli et al. 2011; Ziegler et al. 2012). Some longitudinal studies have found modest acceleration of regional decline (Raz, Gunning-Dixon et al. 2004, Raz, Rodrigue et al. 2004, Raz et al. 2005; Du et al. 2006; Driscoll et al. 2009; Fjell, Walhovd et al. 2009; Raz et al. 2010; Schuff et al. 2012; Thambisetty et al. 2010), whereas others have shown linear reductions (Resnick et al. 2003; Crivello et al. 2010; Raz et al. 2010). Thus, the question of whether age-related cortical reductions are comparable in young and healthy older adults, only accelerating with neurodegenerative disease, is far from settled.
We addressed this main question by using a novel three-stage approach: First, we delineated lifespan trajectories by combining large cross-sectional (n = 1100) and longitudinal (n = 207) samples. A linear trajectory would indicate that the estimated rate of change is comparable at different ages, and that undetected neurodegenerative disease is unlikely to have influenced the results. Based on previous research (Fjell et al. 2009a), we would expect this to be the case in a majority of cortical areas. However, several previous cross-sectional aging studies have found preservation or even thickening of the anterior cingulate and surrounding areas in healthy older adults (Salat et al. 2002, 2004; Fjell et al. 2009a), and so we expected that cortical decline would level off with higher age in these areas in the cross-sectional analyses.
Second, we explicitly modeled the effects of undetected Alzheimer's disease (AD) by adding AD patients to the sample. We expected inclusion of patients to increase the estimated decline in the areas most vulnerable to early AD, especially in the entorhinal cortex and the lateral temporal cortex. Hippocampus and entorhinal cortex are the areas best distinguishing AD patients from healthy older adults (Jack et al. 1997; Fennema-Notestine et al. 2009; McDonald et al. 2009; Fjell, Walhovd, Fennema-Notestine, McEvoy, Hagler, Holland, Brewer et al. 2010). Thus, it is interesting to detail the adult lifespan trajectory of the entorhinal cortex in presumably healthy and to assess possible influences of undetected age-related neurodegenerative disease. Entorhinal cortex is the most upstream subregion in the hippocampal circuit and thus serves as the gateway into the hippocampal formation (Small et al. 2011), which explains its important role for memory function. One speculation is that areas characterized by high degree of life-long plasticity, such as the entorhinal cortex (Chapman et al. 2008), are vulnerable to detrimental effects of both normal and pathological aging. Thus, accelerated decline of the entorhinal cortex could be expected in healthy aging and further accelerated in preclinical age-related neurodegenerative disorders. We were also interested in testing whether areas around the precuneus and retrosplenial cortex would show increased estimated decline when AD patients were added to the sample. These areas are parts of the so-called default mode network (DMN) (Snyder and Raichle 2012), which undergoes substantial structural and functional changes in aging and AD, with disruptions in presumably healthy individuals being linked to preclinical AD (Andrews-Hanna et al. 2007; Sperling et al. 2009; Walhovd et al. 2010; Addis et al. 2011; Jones et al. 2011).
Finally, we selected subsamples of older adults with very low probability of incipient AD, determined by 2-year neuropsychological stability, CSF Aβ1-42 levels (Blennow et al. 2010) and apolipoprotein (APOE) ɛ3 homozygocity (Corder et al. 1993). We examined the cortical thinning in these low-risk older adults in the regions where AD affected the estimated lifespan trajectories. For regions where contamination of the sample with AD patients caused apparent acceleration of estimated atrophy, it is difficult to decide whether decline in older age can be ascribed to the normal aging process or is a result of accumulation of undetected neurodegenerative disease. By assessing atrophy longitudinally in subgroups of healthy older adults with very low probability of undetected AD, it is possible to better disentangle the normal aging process from effects of incipient degenerative disease. Our hypothesis was that decline would be seen even in older adults at very low AD risk, and that this would be related to cognitive changes.
Materials and Methods
Sample
Cross-Sectional Healthy Lifespan Sample
One thousand one hundred healthy participants (424 men/676 women), spanning an age range of 76 years (18–94 years, mean = 48, SD = 20) were included, pooled from 5 independent studies (see Table 1). The details of each of the subsamples are described in Supplemental Information, as well as in a previous publication with an overlapping data pool (Fjell et al. 2009b). All the healthy samples were screened for diseases and history of neurological conditions, and for cognitive deficits/dementia by questionnaires and standardized tests.
Table 1.
Sample characteristics for the cross-sectional samples
| Sample | Country | Women participants, n (%) | Age mean (range), years | Education mean (range) | Key publications | Main screening instruments/inclusion criteria |
|---|---|---|---|---|---|---|
| Healthy subsamples | ||||||
| 1 | Norway | 69 (57) | 51.3 (20–88) | 15 (7–20) | Walhovd et al. (2005) | Health interview, MMSE > 26, BDI < 16, IQ > 85, RH only |
| 2 | Norway | 208 (71) | 46.8 (19–75) | 14 (9–22) | Espeseth et al. (2008) | Health interview, IQ > 85 |
| 3 | USA | 309 (63) | 44.5 (18–94) | 3.5 (1–5)c | Marcus et al. (2007) | Health interview, CDR = 0b, MMSE > 25b, RH only |
| 4 | USA | 191 (60) | 47.3 (18–81) | 15.7 (12–21) | Raz, Rodrigue et al. (2004) | Health interview, BIMCT > 30, GDQ < 15, RH only, neuroradiology, |
| 5 | Norway | 323 (57) | 50.8 (20–85) | 15.6e (4–26) | Fjell et al. (2008), Westlye et al. (2010b) | Health interview, Neuropsychological evaluation, BDI < 16, IQ > 85, RH only, neuroradiology |
| Mild Alzheimer's disease subsample | ||||||
| 6 | USA | 96 (59) | 76.6 (62–96) | 2.8 (1–5)c | Similar to Sample 4 | Health interview, CDR ≥ 0.5, RH only |
Note: Longitudinal sample 2 overlaps with cross-sectional sample 4, and longitudinal sample 3 overlaps with cross-sectional sample 2.
MMSE, Mini–Mental Status Exam (Folstein et al. 1975); BDI, Beck Depression Inventory (Beck 1987); BIMCT, Blessed Information-Memory-Concentration Test (Blessed et al. 1968); CDR, Clinical Dementia Rating (Berg 1984, 1988; Morris 1993); GDQ, Geriatric Depression Questionnaire (Auer and Reisberg 1997); RH, Right handed; WASI, Wechsler Abbreviated Scale of Intelligence (Wechsler 1999).
aAvailable for 70 participants.
bAvailable for participants ≥ 60 years only.
cAvailable for all participants ≥ 60 years, and sporadically for the rest. 1: less than high school graduation, 2: high school graduation, 3: some college, 4: college graduation, 5: beyond college.
dAlzheimer patients.
eMissing for 4 participants.
Cross-Sectional AD Sample
Ninety-six patients with mild or incipient (clinical dementia rating [CDR] = 0.5), mild (CDR = 1) or moderate (CDR = 2) AD (59 women/37 men) were included from the Open Access Series of Imaging Studies (OASIS) database (www.oasis-brains.org), with an age range of 34 years (62–96 years, mean = 76.6, SD = 7.1). Details of recruitment and diagnostic procedures for the AD group are provided by Marcus et al. (2007). AD participants underwent the Washington University Alzheimer's Disease Research Center's full clinical assessment, yielding CDR (Berg 1984, 1988; Morris 1993; Morris et al. 2001). CDR of 0.5 or higher was taken to indicate mild to moderate AD.
Longitudinal Sample
The longitudinal sample consisted of 207 participants (60–93 years, mean age = 75.5 years, 56% women). They were drawn from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.ucla.edu) (n = 138) and the Open Access Series of Imaging Studies (OASIS) database (http://www.oasis-brains.org/) (n = 69). For details about the ADNI database, see Supplementary Experimental Procedures. The ADNI was launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies and nonprofit organizations, and the principal investigator is Michael W. Weiner, MD, VA Medical Center and University of California, San Francisco. In the present study, ADNI participants were 55–90 years of age, had an informant able to provide an independent evaluation of functioning, and spoke either English or Spanish. General eligibility criteria were as follows for normal subjects: Mini–Mental State Examination (MMSE) (Folstein et al. 1975) scores between 24 and 30 (inclusive), CDR (Morris 1993) of 0, nondepressed, non-MCI, and nondemented. In addition, to minimize the possibility of including individuals with early, preclinical AD, only participants who had the same or better score on CDR sum of boxes (CDR-sb) at the time of follow-up were included. The subject pool was further restricted to those subjects for whom adequate processed and quality checked MR data were available.
For the OASIS sample, principal investigator is Randy Buckner, and details are available elsewhere (Marcus et al. 2010). Participants were between 60 and 93 years of age (mean 75.3 years, 71% women), and were selected from a larger database of individuals who had participated in MRI studies at Washington University on the basis of the availability of at least 2 separate visits in which clinical and MRI data were obtained, at least 3 acquired T1-weighted images per imaging session, and right-hand dominance. The participants were recruited primarily through media appeals and word of mouth. All subjects were screened for dementia by the ADRC (Washington University Alzheimer's Research Unit) full clinical assessment. Dementia status was established and staged using the CDR scale. The determination of AD or nondemented control status was based solely on clinical methods, derived primarily from a collateral source. To receive a CDR score of ≥0.5, the participant had to experience gradual onset and progression of decline in memory and other cognitive and functional domains. A CDR of 0 at both visits analyzed in the present study was used as inclusion criterion.
In addition, follow-up analyses of entorhinal cortex atrophy were performed on different subsamples from the ADNI where neuropsychological follow-up data, baseline CSF Aβ1-42, and APOE profile were available. First, based on 2-year neuropsychological follow-up results, a group of cognitively superstable participants was identified according to a procedure described previously (Fjell, Westlye et al. 2010). To be regarded as superstable, CDR sum of boxes and MMSE needed to remain identical or improve over 2 years. In addition, raw scores on all of the following 7 neuropsychological tests had to remain at 90% of initial score or higher: Auditory Verbal Learning Test (AVLT) learning (hits: false alarms), AVLT 30-min delayed recall (hits: false alarms) (Rey 1964), digit span (sum of forward and backward), clock copying (Goodglass and Kaplan 1983), digit symbol substitution test (Wechsler 1981), and logical memory test immediate recall and logical memory test delayed recall (Wechsler 1987). Eighteen of 114 participants with 2-year data available fulfilled these extremely strict criteria. Second, participants with available CSF Aβ1-42 samples from baseline (n = 51) were assigned to a low (n = 28) risk group for developing AD based on published cutoff levels for the ADNI sample (CSF Aβ1-42 > 192 pg/mL) (Shaw et al. 2009). Finally, participants with a low risk of developing AD based on both Aβ1-42 and the APOE genotype (ɛ2/ɛ3, or ɛ3/ɛ3) were identified (n = 22).
MR Acquisition and Analysis
Cross-Sectional Cortical Reconstruction
All scans were obtained from 1.5 T magnets from 2 different manufacturers (Siemens, Erlangen, Germany; General Electric CO, Milwaukee, WI), and from 5 different models (Siemens Avanto, Symphony, Sonata and Vision; GE Signa). All participants within each sample were scanned on the same scanner. T1-weighted sequences were acquired (3D magnetization prepared gradient echo for Siemens/3D spoiled gradient recalled pulse sequences for GE). In 6 of the cross-sectional (healthy samples 1, 2, 3, 4, and 6, AD sample) and both the longitudinal samples, multiple scans were acquired within the same scanning session and averaged to increase the signal-to-noise ratio. The details of the sequences are presented in Supplemental Experimental Procedures.
Cross-sectional data were processed and analyzed with FreeSurfer 4.01 (http://surfer.nmr.mgh.harvard.edu/) (Dale and Sereno 1993; Dale et al. 1999) at the Neuroimaging Analysis Lab, Center for the Study of Human Cognition, University of Oslo, with the additional use of computing resources from the titan grid operated by the Research Computing Services Group at USIT, University of Oslo (https://wiki.uio.no/usit/suf/vd/hpc/index.php/TITAN). This procedure yields a measure of cortical thickness for each person at each point on the reconstructed surface and is capable of detecting submillimeter differences between groups (Fischl and Dale 2000; Rosas et al. 2002; Kuperberg et al. 2003). Maps were smoothed using a circularly symmetric Gaussian kernel with a full-width at half-maximum of 30 mm (Fischl et al. 1999). The cortical surface was then parcellated according to procedures described previously (Fischl et al. 2004; Desikan et al. 2006).
Detection of Longitudinal Change
All scans used for the longitudinal analyses were from 1.5 T scanners. For ADNI, DICOMs (including 2 T1-weighted volumes per case) were downloaded from the public ADNI site (http://www.loni.ucla.edu/ADNI/Data/index.shtml). The ADNI sample is largely overlapping with that included in a previous publication (Fjell, Walhovd et al. 2009), but are reanalyzed with a different procedure to measure thickness change to allow direct comparisons with the cross-sectional results, in contrast to the volumetric analyses used in the previous publication (Holland and Dale 2011; Holland et al. 2012). For OASIS, DICOMs were downloaded from the public OASIS site (http://www.oasis-brains.org), at least 3 repeated T1-weighted series of scans per session. Processing of the longitudinal data was performed with the FreeSurfer 5.1 longitudinal stream (Reuter et al. 2010; Reuter and Fischl 2011). The cortical models are built in the same way as for the cross-sectional analyses (see above), but with 1 important exceptions. First, a template volume is created and processed instead of initializing the analyses with a specific time point. By initializing the processing of longitudinal data using the processed results from such an unbiased template, the random variation in the processing procedure is reduced, and the robustness and sensitivity of the longitudinal analysis increased (Reuter et al. 2010). This also ensures inverse consistency, meaning that the inverse transform is obtained when registering time point 2 to time point 1 as opposed to time point 1 to time point 2 (Reuter and Fischl 2011), which is extremely important in longitudinal analyses (Thompson and Holland 2011). Second, new probabilistic methods (temporal fusion) were applied to further reduce the variability across time points. Thus, this longitudinal analysis scheme is designed to be unbiased regarding any of the time points, and is able to detect small changes in cortical thickness between examinations.
Additional Cognitive Testing
To assess cognitive stability over time, clinical and neuropsychological test scores at baseline and after 1 year for the participants from the ADNI sample were included in select analyses. CDR (Morris 1993) sum of boxes (CDR-sb) was calculated as a measure of clinical functioning, and the following neuropsychological tests were included: AVLT learning (hits: false alarms), AVLT 30-min delayed recall (hits: false alarms) (Rey 1964), digit span (sum of forward and backward), clock copying (Goodglass and Kaplan 1983), digit symbol substitution test (Wechsler 1981), and logical memory test immediate recall and logical memory test delayed recall (Wechsler 1987).
Statistical Analyses
To reduce the number of comparisons, mean values for left and right hemisphere regions-of-interest (ROIs) were used in the ROI analyses. A further advantage of this approach is that the reliability of the thickness estimations will increase. A possible disadvantage is that possible interactions between hemisphere and age-thickness effects could confound the results. In our experience, however, age effects tend to be symmetrically distributed across hemispheres so that this is unlikely to represent a concern (Fjell et al. 2009a). Cross-sectional analyses were performed on residuals after the effects of sample and sex were removed by linear regressions. If not specified, an α-level of P < 0.05 was used as threshold for statistical significance in the ROI analyses.
First, to test the strength of the linear effect of age and to show the stability of effects across subsamples, age was correlated with cortical thickness pointwise across the cortical surface in each sample separately and in the pooled sample. To estimate age trajectories from the cross-sectional data without any assumption about the form of the curve, a nonparametric local smoothing model, the smoothing spline, implemented in Matlab, was fit to the data. We have previously shown that this approach yields less biased solutions than the more commonly employed higher order polynomial functions (Fjell, Walhovd, Westlye et al. 2010). We used an algorithm that optimizes smoothing level based on a version of Bayesian Information Criterion (BIC), which provides a way of obviating the need for arbitrarily chosen smoothing levels. The difference between BIC for the model and the lowest BIC (in this case, the difference between the smoothing spline and the ordinary least square [OLS] model) (ΔI) was used to accept or reject the linear model. As a rule of thumb, ΔI < 2 would indicate that the 2 models are essentially indistinguishable with regard to goodness of fit, ΔI > 4 would indicate considerable differences between the models, and ΔI ≥ 10 would indicate that the model with the larger BIC has essentially no support. In addition, surface maps of estimated annual change in cortical thickness was calculated per year, smoothed across time, and displayed per decade. The estimates were adjusted for effects of sample differences and sex. Also, estimated change values for the entorhinal cortex were extracted and plotted as a function of age.
The cross-sectional analyses were first run in the pooled sample (n = 1100). For the longitudinal analyses, annual percent thickness change was calculated for each ROI and averaged across hemispheres. One-sample t-tests were used to determine whether longitudinal change was different from zero. By additional paired-samples t-tests, longitudinal thinning in the ROIs where the cross-sectional analyses indicated accelerated decline in the oldest part of the age range was contrasted with longitudinal atrophy in the ROIs where the cross-sectional models indicated that atrophy would level off. This was done to explore degree of consistency between the smoothing spline models based on the cross-sectional data and the longitudinally measured atrophy. Further comparisons were made between estimated rates of change from the longitudinal and the cross-sectional data. First, the rate of change was estimated cross-sectionally from the same participants that were included in the longitudinal analyses, smoothed across age. Differences in change estimates would then be due to methodological issues of longitudinal versus cross-sectional analysis. Second, the same calculations were done for the full cross-sectional sample of the same age range as the longitudinal sample. Differences between these results and the longitudinal results would represent a mixture of methodological and sampling issues.
The cross-sectional analyses were then repeated in the OASIS subsample only (n = 309), with the inclusion of 96 additional participants with very mild to moderate AD from the same site and scanner. The smoothing spline curves were compared between the full OASIS sample, including both healthy older adults and AD patients, and the healthy part of the OASIS sample only. This was done to mimic the effect of insufficient screening for degenerative conditions on the estimated trajectories.
The area that showed accelerated estimated decline when AD patients were included in the sample (entorhinal cortex) was tested in subsamples of participants with very low probability of AD. The selection of participants with very low probability of AD was based on 2-year neuropsychological stability, baseline levels of Aβ1-42 or a combination of Aβ1-42 and APOE status. The longitudinal rates of change for these subgroups in the AD-prone areas were compared with the larger longitudinal sample by independent samples t-tests. If comparable rates of atrophy were seen in participants with very low probability of AD, then it is unlikely that the atrophy seen is caused by undetected AD-related mechanisms. Finally, to test whether the rate of change in entorhinal cortex was related to cognitive changes, entorhinal change was correlated with change in total learning score and 30-min delayed recall score on the Rey auditory verbal learning test (RAVLT), quantified both in terms of a difference score (1-year follow-up score minus baseline) as well as a “saving score” (ratio of 1 year score to baseline score).
Results
Cross-Sectional Analyses
The linear effects of age on cortical thickness vertexwise across the cortex, within and across samples, are illustrated in Figure 1. Strong negative correlations were seen across most of the cortex in all samples, reaching −0.80 in the most age-sensitive areas, including medial and lateral sections of the frontal cortex. In the total sample, cortical thickness in all regions except rostral anterior cingulate cortex (ACC) correlated negatively with age (Table 3). The smoothing spline model was used to test the relationship between age and cortical thickness in each region without assuming linearity. The results, presented in Table 2, show that for 7 regions (rostral and caudal anterior cingulate, posterior cingulate, lateral and medial orbitofrontal, lateral occipital cortex and lingual gyrus), BIC for the smoothing spline was >4 compared with the linear fit, indicating considerable differences in fit between the models. For additional 3 regions (entorhinal cortex, pericalcarine cortex, and pars orbitalis), the difference was >2, indicating some improvement of fit from the linear to the smoothing spline models. Lateral occipital, lingual, entorhinal, and pericalcarine cortex showed accelerated decline with increasing age, whereas the other areas leveled off with higher age. Scatter plots for the 9 regions with the highest ΔBIC (i.e., the least linear) are shown in Figure 2, and the scatter plots for 9 representative regions with a linear fit in Figure 3. Figure 4 shows estimated cortical change per decade, color-coded, and projected onto a surface brain.
Figure 1.

Age correlations. Correlations between age and cortical thickness at each vertex in each of the 5 subsamples, as well as in the total sample. The correlations in the total sample are corrected for both offset and slope differences between the subsamples. As can be seen, strong negative correlations are found in all subsamples, being especially strong in medial and lateral frontal cortex. The maps are smoothed with a Gaussian kernel of full-with at half-maximum (FWHM) of 30 mm.
Table 3.
Cross-sectional age relationships and longitudinal change
| Cross-sectional age correlations |
Longitudinal annual atrophy (%) | ||
|---|---|---|---|
| 18–94 years (n = 1100) | 60–94 years (n = 367) | 60–91 years (n = 207) | |
| Cingulate, caudal anterior | −0.10 | .15 | −0.29 |
| Cingulate, rostral anterior | −0.05 | .21 | −0.18 |
| Cingulate, posterior | −0.52 | −0.07 | −0.43 |
| Cingulate, isthmus | −0.47 | −0.12 | −0.59 |
| Frontal, superior | −0.67 | −0.23 | −0.38 |
| Frontal, caudal middle | −0.59 | −0.19 | −0.77 |
| Frontal, rostral middle | −0.55 | −0.14 | −0.52 |
| Frontal, pars opercularis | −0.63 | −0.25 | −0.45 |
| Frontal, pars triangularis | −0.66 | −0.18 | −0.26 |
| Frontal, pars orbitalis | −0.53 | −0.11 | −0.27 |
| Frontal, lateral orbital | −0.51 | −0.07 | −0.38 |
| Frontal, medial orbital | −0.26 | .05 | −0.44 |
| Frontal, pole | −0.34 | −0.06 | −0.09 |
| Parietal, precentral gyrus | −0.60 | −0.27 | −0.75 |
| Parietal, postcentral gyrus | −0.57 | −0.25 | −0.57 |
| Parietal, paracentral gyrus | −0.49 | −0.24 | −0.44 |
| Parietal, superior | −0.52 | −0.29 | −0.46 |
| Parietal, inferior | −0.59 | −0.29 | −0.42 |
| Parietal, supramarginal | −0.62 | −0.25 | −0.43 |
| Parietal, precuneus | −0.60 | −0.30 | −0.11 |
| Temporal, parahippocampal | −0.28 | −0.15 | −0.62 |
| Temporal, entorhinal | −0.22 | −0.06 | −0.90 |
| Temporal, pole | −0.31 | −0.14 | −0.65 |
| Temporal, superior | −0.66 | −0.47 | −0.62 |
| Temporal, middle | −0.52 | −0.37 | −0.53 |
| Temporal, inferior | −0.38 | −0.28 | −0.50 |
| Temporal, transverse | −0.56 | −0.41 | −0.29 |
| Temporal, banks sup temp S | −0.49 | −0.37 | −0.60 |
| Temporal, fusiform | −0.43 | −0.29 | −0.65 |
| Occipital, lateral | −0.50 | −0.25 | −0.64 |
| Occipital, pericalcarine | −0.54 | −0.36 | −0.42 |
| Occipital, lingual | −0.67 | −0.45 | −0.58 |
| Occipital, cuneus | −0.51 | −0.39 | −0.55 |
Note: Bold values indicate P < 0.05. Italic values indicate P < 0.10.
Table 2.
Degree of deviations from linearity
| Cortical ROIs | Linear BIC | Smoothing spline ΔBIC | Direction | Linear OLS standardized βa |
|---|---|---|---|---|
| Rostral anterior cingulate | 7708.95 | 45.66 | Decelerating | −0.05 |
| Lateral orbitofrontal | 7381.81 | 19.7 | Decelerating | −0.51 |
| Medial orbitofrontal | 7632.64 | 12.72 | Decelerating | −0.26 |
| Posteriorcingulate | 7359.24 | 12.18 | Decelerating | −0.52 |
| Caudal anterior cingulate | 7699.99 | 12.02 | Decelerating | −0.10 |
| Lateral occipital | 7394.11 | 10.8 | Accelerating | −0.50 |
| Lingual | 7055.72 | 7.42 | Accelerating | −0.67 |
| Entorhinal | 7656.15 | 3.03 | Accelerating | −0.22 |
| Pericalcarine | 7324.44 | 2.48 | Accelerating | −0.54 |
| Pars orbitalis | 7355.95 | 2.18 | Decelerating | −0.53 |
| Frontal pole | 7572.78 | 1.38 | −0.34 | |
| Pars opercularis | 7147.21 | 0.66 | −0.63 | |
| Pars triangularis | 7093.44 | 0.35 | −0.66 | |
| Precentral | 7217.60 | 0.04 | −0.60 | |
| Inferior temporal | 7541.13 | 0 | −0.38 | |
| Superior frontal | 7057.85 | 0 | −0.67 | |
| Inferior parietal | 7244.26 | −0.01 | −0.59 | |
| Rostral middle frontal | 7318.88 | −0.01 | −0.55 | |
| Superior parietal | 7371.71 | −0.01 | −0.52 | |
| Superior temporal | 7078.20 | −0.01 | −0.66 | |
| Banks Sup Temp Sulc | 7248.30 | −0.02 | −0.49 | |
| Caudal middle frontal | 7248.30 | −0.02 | −0.59 | |
| Middle temporal | 7372.72 | −0.02 | −0.52 | |
| Precuneus | 7230.41 | −0.03 | −0.60 | |
| Temporal pole | 7597.46 | −0.03 | −0.31 | |
| Retrosplenial | 7436.86 | −0.03 | −0.47 | |
| Cuneus | 7381.69 | −0.04 | −0.51 | |
| Paracentral | 7403.67 | −0.04 | −0.49 | |
| Postcentral | 7275.45 | −0.04 | −0.57 | |
| Transverse temporal | 7300.23 | −0.04 | −0.56 | |
| Fusiform | 7486.38 | −0.05 | −0.43 | |
| Parahippocampal | 7622.13 | −0.05 | −0.28 | |
| Supramarginal | 7175.99 | −0.05 | −0.62 |
Note: The column “smoothing spline ΔBIC” denotes the change in BIC between the linear and the smoothing spline model, and the ROIs are sorted according to this value. ΔBIC ≥ 4 indicates that a nonlinear model is considerably better than a linear model, whereas ΔBIC ≤ 2 indicates that the best model is essentially indistinguishable from a linear model.
aCritical P values for the β values: β ≥ ±0.06, P < 0.05; β ≥ ±0.10, P < 0.001.
Figure 2.

Nonlinear lifespan trajectories. The figure show the individual cross-sectional data points and the estimated trajectories for cortical thickness in the 9 cortical regions of interest (ROI) that deviated the most from linearity. Values represent standardized mean thickness across hemispheres, corrected for the influence of sample (Z-scores), and the ROIs are displayed on a semi-inflated template brain surface.
Figure 3.

Linear lifespan trajectories. The figure shows the individual data points and the cross-sectionally estimated trajectories for cortical thickness in 9 representative cortical regions of interest (ROI) where a linear fit was the most appropriate. Values represent mean thickness across hemispheres, corrected for the influence of sample (Z-scores), and the ROIs are displayed on a semi-inflated template brain surface.
Figure 4.
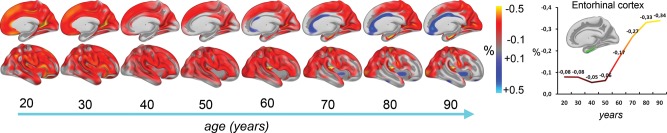
Estimated change per decade from cross-sectional data. Based on cross-sectional data, percentage annual percentage change in cortical thickness was calculated per year, smoothed across time, and displayed per decade. The estimates are adjusted for effects of sample differences and sex. Only right hemisphere is shown. Right panel: Estimated annual change in the right entorhinal cortex, based on cross-sectional data, plotted as a function of age. Note that even for the participants >60 years, these estimates are substantially lower than those obtained from the longitudinal data.
Longitudinal Analyses
The results of the analysis of longitudinal data for 207 participants 60 years or older were compared with the cross-sectional results. Annualized percent change in each of the 32 ROIs measured longitudinally is presented in Table 3. Significant thinning was observed in 24 regions. Pearson correlations between thickness and age in the full cross-sectional sample, and in the subsample with age range identical to that of the longitudinal sample (n = 367), are also shown in Table 3. For the sample above 60 years, significant negative correlations with age were observed in 26 regions and positive in 2 (rostral and caudal anterior cingulate), in accordance with the smoothing spline results. Of the 26 regions showing a negative age-correlation in the cross-sectional analyses in this age span, 20 also showed significant longitudinal thinning (P < 0.05) while 2 showed a trend (P < 0.10). Pars triangularis and pars orbitalis, precuneus and temporal transverse cortices showed significant cross-sectional age correlations without significant longitudinal thinning. This difference may be explained by power and sampling differences between the longitudinal and the cross-sectional samples (see below). Rostral and caudal anterior cingulate and the frontal pole did not show significant thinning and were not significantly related to age in the cross-sectional analyses for the sample >60 years. The P values cannot be directly compared between the cross-sectional and the longitudinal analyses, since they depend on partly different samples and sample sizes, and the statistical tests performed are different. Still, the longitudinal results were to a large extent consistent with the negative age correlations seen in the cross-sectional data.
Further, to test slope differences between ROIs for the last part of the age span, we selected the 10 ROIs for which the cross-sectional smoothing spline function indicated a nonlinear age trajectory. We then tested whether longitudinal thinning in the 6 ROIs for which the cross-sectional analyses indicated deceleration of atrophy in higher age was smaller than the thinning in the 4 ROIs for which the cross-sectional analyses indicated accelerated decline. Averaged annualized longitudinal thinning across the 6 ROIs for which the cross-sectional smoothing spline analyses indicated reduced rate of atrophy was −0.33%, in contrast to −0.64% for the ROIs where accelerated atrophy was indicated from the cross-sectional results. A t-test showed that this difference was significant (t[206] = 2.17, P < 0.05). Post hoc tests revealed that annual thinning was significantly greater than −0.33% in the entorhinal cortex (−0.90%, t[206] = −2.48, P < 0.05), showed a trend in lateral occipital (−0.64%, t[206] = −1.87, P = 0.063) and lingual (−0.58%, t[206] = −1.81, P = 0.066) cortex, while thinning in pericalcarine cortex did not differ significantly from −0.33% (−0.42%, t[206] = −0.48, n.s.). Thus, the main conclusion from the cross-sectional analyses seems to be coherent with the longitudinal data.
Finally, to allow a more detailed comparison between the cross-sectional and the longitudinal results, color-coded surface plots of annual percentage change in thickness measured longitudinally are shown in Figure 5. These plots were compared with cross-sectionally estimated annual percentage thickness change for the oldest part of the full sample (≥60 years of age). As this sample only partly overlapped with the longitudinal sample, annual change was estimated cross-sectionally also for the longitudinal sample alone. This enabled comparisons of cross-sectional and longitudinal effects in the same group of participants, which again made it possible to distinguish effects of method (longitudinal vs. cross-sectional) versus effects of sample (the smaller sample of 207 participants for whom longitudinal data were available vs. the larger cross-sectional sample of 367 participants ≥60 years of age). Substantial similarities, but also important differences, were found in the comparison of longitudinally versus cross-sectionally estimated change. First, there was substantial difference in magnitude of estimated change, with mean annual change across the cortical surface of −0.59% versus −0.30% for the longitudinal versus the cross-sectional analyses, respectively. These differences were especially pronounced in the lateral frontal, temporoparietal, superior temporal, and lateral occipital cortices on the lateral side, as well as the isthmus of the cingulate/retrosplenial cortex, the fusiform gyrus, and the right cuneus and left lingual gyrus. Interestingly, while the cross-sectional data indicated preservation or even thickening of the middle and anterior cingulate, as well as partly in the insula, these regions all showed thinning in the longitudinal data. Also, orbitofrontal cortex showed evidence of longitudinal thinning in face of cross-sectional preservation.
Figure 5.
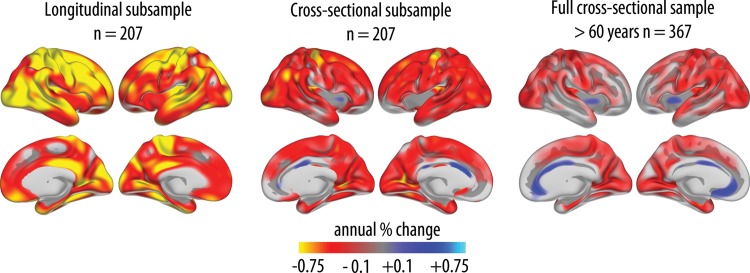
Cortical thinning: longitudinal and cross-sectional comparisons. Left panel: Annualized percentage change in cortical thickness was calculated from the longitudinal data and displayed as a color-coded map on semi-inflated brain models. Middle panel: For comparison, annualized percentage change in thickness was also calculated from the cross-sectional data from the same participants. Right panel: Annualized percentage change in cortical thickness calculated from the cross-sectional data of all participants in the age range 60–90 years (corresponding to the age range of the longitudinal sample).
Second, we wanted to explore whether differences between methods mainly were a matter of magnitude, a scaling effect, or whether bias across methods in terms of topographic distribution of change also could be seen. Thus, we calculated the square root of the squared Z-transformed change maps from both methods to obtain a measure of the relative distribution of change for each method independently of magnitude of change. The difference between these maps was then calculated. Mean difference between the longitudinal and the cross-sectional results across vertices was 0.57 SD. Thresholding the maps by 2 SD revealed differences between methods in the anterior cingulum only. Lowering the threshold to 1.5 SD showed some small scattered differences between methods (Supplementary Material). Generally, however, differences between methods in distribution of effects were modest as long as the differences in magnitude were removed.
Comparing the longitudinal results with the cross-sectionally estimated change rates from the full sample of older adults naturally yielded somewhat lower correspondence, since differences due to method (cross-sectional vs. longitudinal) and differences due to sample (different samples) are added. These factors caused the rate of change to be even lower compared with the longitudinal analyses. Nevertheless, substantial similarities in the topographic distribution of age effects were seen, as described in the section on ROI analyses above (see also Table 3). Of special interest, however, is that the apparent thickening or preservation of the middle and anterior portions of the cingulate and in the insula were seen in both sets of cross-sectional analyses, whereas these results were not replicated in the longitudinal analyses.
Effects of AD on Trajectories
Finally, the analyses were repeated in a subsample of 96 patients with mild to moderate AD and 309 normal participants. The results are presented in Table 4. When patients with mild to moderate AD were included, entorhinal cortex showed accelerating decline with age compared with the non-AD patient group (see Fig. 6 for scatter plots for selected regions). A tendency for stronger age relationships in the oldest age range when AD patients were included was seen for middle temporal cortex also, where a small shift downward from the original curve that leveled off in higher age could be identified.
Table 4.
Effects of inclusion of mild Alzheimer's disease (AD) on estimated lifespan trajectories
| Without Alzheimer patients |
With Alzheimer patients |
||||
|---|---|---|---|---|---|
| Linear BIC | Smoothing spline ΔBIC | Smoothing spline ΔBIC | Linear BIC | Direction of change with age | |
| Lateral orbitofrontal | 524.01 | 21.90 | 15.82 | 788.48 | Decelerating |
| Rostral anterior cingulate | 845.08 | 20.74 | 24.02 | 1212.82 | Decelerating |
| Medial orbitofrontal | 667.35 | 14.47 | 9.68 | 969.18 | Decelerating |
| Pars orbitalis | 676.34 | 8.46 | 7.17 | 1033.09 | Decelerating |
| Lateral occipital | 387.5 | 8.24 | 11.22 | 657.66 | Accelerating |
| Middle temporal | 502.28 | 5.15 | 2.35 | 837.54 | Decelerating |
| Pars triangularis | 443.31 | 3.65 | 4.21 | 739.8 | Decelerating |
| Rostral middle frontal | 646.68 | 1.82 | 1.82 | 646.68 | |
| Superior frontal | 717.9 | 1.63 | 1.63 | 717.9 | |
| Lingual | 574.55 | 1.57 | 1.57 | 574.55 | |
| Pars opercularis | 712.33 | 1.42 | 1.42 | 712.33 | |
| Inferior temporal | 860.68 | 1.33 | 1.33 | 860.68 | |
| Caudal middle frontal | 733.76 | 0.04 | 0.04 | 733.76 | |
| Caudal anterior cingulate | 753.77 | 0.02 | 0.00 | 1139.82 | |
| Entorhinal | 1045.87 | 0.00 | 3.30 | 1559.54 | Accelerating |
| Posterior cingulate | 624.96 | 0.00 | 0.00 | 624.96 | |
| Superior parietal | 753 | 0.00 | 0.00 | 753 | |
| Superior temporal | 826.81 | 0.00 | 0.00 | 826.81 | |
| Frontal pole | 1167.49 | 0.00 | 0.00 | 1167.49 | |
| Banks sup temp sulc | 824.43 | −0.01 | −0.01 | 824.43 | |
| Fusiform | 778.03 | −0.01 | −0.01 | 778.03 | |
| Retrosplenial | 956.25 | −0.01 | −0.01 | 956.25 | |
| Supramarginal | 704.43 | −0.01 | −0.01 | 704.43 | |
| Cuneus | 690.36 | −0.02 | −0.02 | 690.36 | |
| Inferior parietal | 697.65 | −0.02 | −0.02 | 697.65 | |
| Parahippocampal | 1338.99 | −0.02 | −0.02 | 1338.99 | |
| Paracentral | 792.62 | −0.02 | −0.02 | 792.62 | |
| Pericalcarine | 614.01 | −0.02 | −0.02 | 614.01 | |
| Postcentral | 614.01 | −0.02 | −0.02 | 614.01 | |
| Precentral | 726.57 | −0.02 | −0.02 | 726.57 | |
| Precuneus | 728.51 | −0.02 | −0.02 | 728.51 | |
| Temporal pole | 1453.43 | −0.02 | −0.02 | 1453.43 | |
| Transverse temporal | 1074.21 | −0.02 | −0.02 | 1074.21 | |
Note: The column “smoothing spline ΔBIC” denotes the change in BIC between the linear and the smoothing spline model, and the ROIs are sorted according to this value. ΔBIC ≥4 indicates that a nonlinear model is considerably better than a linear model, whereas ΔBIC ≤2 indicates that the best model is essentially indistinguishable from a linear model. In the columns “Without Alzheimer patients,” only the healthy controls are included (n = 309). In the columns “With Alzheimer patients,” a group of mild AD patients (n = 96) was added to the sample, and the analyses rerun. For entorhinal cortex, a low ΔBIC in the group without AD and a higher ΔBIC in the group with AD indicate that inclusion of AD patients had a substantial effect on the estimated lifespan trajectory of this region.
Figure 6.

Nonlinear lifespan trajectories with Alzheimer patients included. Age trajectories for the OASIS subsample (n = 309), with an addition of patients (n = 96) with mild AD in an attempt to mimic the effect of undetected dementing disorder. The healthy controls are illustrated with green dots and the AD patients with pink dots. With the exception of the entorhinal cortex, inclusion of AD patients had relatively minor impacts on the estimated lifespan trajectory for each cortical area. x-Axis values represent the mean thickness across hemispheres, corrected for the influence of sample (Z-scores), and the y-axis represent age in year. ROIs are displayed on a semi-inflated template brain surface.
Entorhinal Cortex Change in Older Adults with Extremely Low Probability of Dementia
Entorhinal cortex is the region typically most vulnerable to very early AD, and inclusion of AD patients in the sample caused accelerated decline in this area. The acceleration of cortical thinning seen both in the cross-sectional and longitudinal analyses could be caused by undetected AD. We performed follow-up analyses to determine whether older adults with very low probability of undetected neurodegenerative disease still evidenced thinning of entorhinal cortex. Annual percentage change in entorhinal thickness in the full ADNI sample of no clinical decline over 1 year was 0.97% (P = 0.002). The rate of thinning in the cognitively superstable part of the sample with no documented cognitive reduction at 2-year follow-up (n = 18) was identical (0.97%), although not significantly different from zero due to the massive reduction in statistical power. These participants showed no clinical decline over 2 years and scored at least 90% of their initial performance on each of 7 different neuropsychological tests. Next, we examined the participants with available CSF Aβ1-42 samples from baseline (n = 51) who formed a low-risk group (n = 28) based on a published criterion (Shaw et al. 2009). This group showed significant 1-year entorhinal thinning (−1.38%, t[27] = 2.08, P < 0.05), that was not significantly different from the thinning observed in the total sample (t[136] = 0.67, n.s.). Finally, a low-risk group based on both CSF Aβ1-42 and APOE status was defined (n = 22). This group showed a trend toward significant entorhinal thinning (1.40%, t[21] = 1.83, P = 0.081), which did not differ from the rate of thinning in the total sample (t[136] = 0.61, n.s.). Thus, entorhinal thinning comparable to that of the total sample was found in participants who remained cognitively superstable over 2 years, participants of low risk of AD based on CSF Aβ1-42, and participants of low AD risk based on both CSF Aβ1-42 and APOE status. These results are illustrated in Figure 7.
Figure 7.
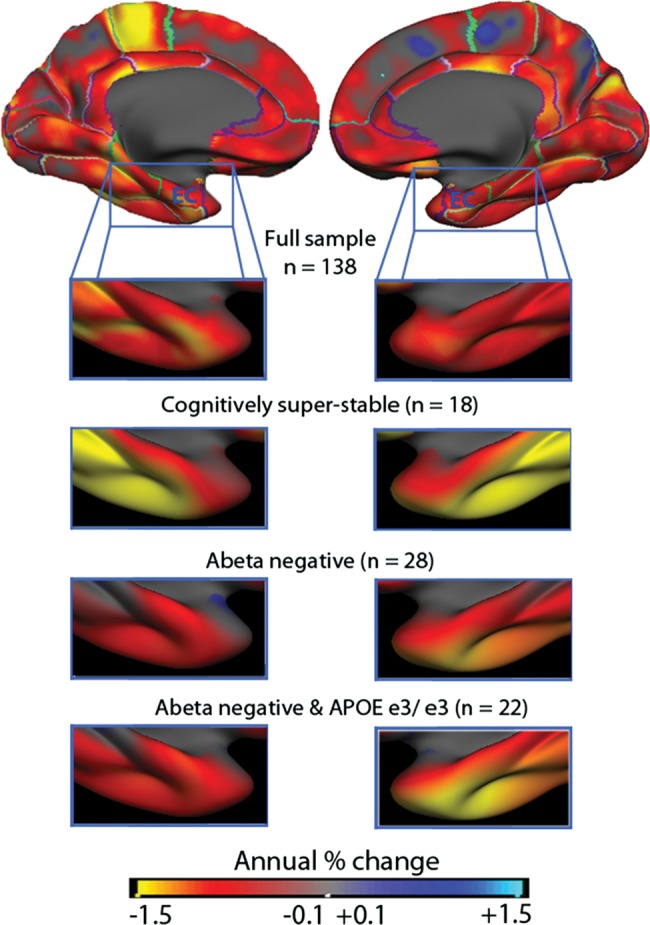
The rate of change in participants with low risk of Alzheimer's disease. From the ADNI sample, subgroups of participants with very low risk of Alzheimer's disease (AD) were selected based on 2-year clinical and neuropsychological stability (n = 18), levels of CSF Aβ1-42 (n = 28) or a combination of CSF Aβ1-42 levels and no APOE ɛ4 alleles (n = 22). The annual rate of change in the entorhinal cortex did not differ statistically between any of the subgroups and the full ADNI sample. Thus, it is very unlikely that thinning in this area in healthy older adults is solely caused by undetected AD processes.
Finally, to test whether the change rate in the entorhinal cortex in the healthy controls had any cognitive correlate, we correlated entorhinal thinning with total learning and 30-min recall scores from the RAVLT in the participants who showed no change in MMSE or CDR-sb score over 2 years (data available for 94 participants). Age was not significantly related to any of the measures. Higher entorhinal change rate was related to lower 30-min recall at follow-up (r = 0.22, P < 0.05) and marginally to lower learning scores (r = 0.20, P = 0.056). Longitudinally, change learning score was significantly related to entorhinal change, both when quantified as the difference between baseline and follow-up (r = 0.29, P < 0.005) and as a saving (ratio) score (r = 0.24, P < 0.05). Change in delayed recall was not significantly related to entorhinal change.
Discussion
The main finding was that even though linear cortical changes seem to be the norm of healthy aging, accelerated decline in select areas does not need to signify neurodegenerative disease. Rather, cortical thinning in advanced age even in areas vulnerable to early AD can be part of a normal aging process. Inclusion of patients with mild to moderate AD had a profound effect on the estimated trajectory of entorhinal cortex only, which is the cortical area most prone to early AD (Jack et al. 1997; Fennema-Notestine et al. 2009; Fjell, Walhovd, Fennema-Notestine, McEvoy, Hagler, Holland, Brewer et al. 2010). Still, similar rates of longitudinal entorhinal thinning were found in participants with very low probability of incipient AD, and the changes were predictive of longitudinal changes in memory scores, thus not being entirely benign. This means that even in AD-prone areas are cortical changes seen in healthy aging. Even though not related to AD, these changes may still impact memory function.
Linearity of Brain Aging: Reduced Change in Higher Age
A linear model of age-related differences in cortical thickness explained the data well in most regions. An exception from linearity was the ACC, where some previous studies have found no or even positive age relationships (Salat et al. 2004; Fjell et al. 2009a). Such a “reversal” of the aging process could reflect neuroplastic adaptation to increased environmental demands (Draganski et al. 2006; Engvig et al. 2010), as ACC plays a role in the allocation of attentional resources (Bush et al. 2000; Posner et al. 2007; Posner 2012), and cortical thickness in this area correlate with executive control and attention (Westlye et al. 2011). However, thickening of caudal and rostral ACC was not seen longitudinally, even in the same sample of participants where preservation/thickening was seen cross-sectionally. This strongly indicates that this is a cross-sectional artifact, possibly caused by covariance between sampling bias and age. Older adults with thinner ACC could be less likely to be considered cognitively healthy, yielding a more select older adults sample. For example, in a recent fMRI study, cross-sectionally observed frontal over-recruitment could be accounted for by a select older adults sample (Nyberg et al. 2010).
Estimated age-related thinning also in the orbitofrontal cortex leveled off with increasing age. In a previous study with an overlapping sample, we found that the medial orbitofrontal cortex was relatively well preserved compared with the rest of the cortex (Fjell et al. 2009a). Sowell et al. (2003) also observed that the age effects in these cortical areas were reduced in higher age. The longitudinal data showed significant thinning of the orbitofrontal cortex, but similar to the ACC, at a slower rate than most ROIs.
Linearity of Brain Aging: Increased Change in Older Age
Minute acceleration of estimated cortical decline with higher age was found in 4 regions, including entorhinal cortex. Entorhinal cortex also showed substantial annual thinning longitudinally (0.9%). This is comparable with the previously reported atrophy rates of between 0.3% and 2.4% (Du et al. 2003, 2006; Ezekiel et al. 2004; Raz et al. 2005; Fjell, Walhovd et al. 2009; Raz et al. 2010; Schuff et al. 2012). A recent large study using voxel-based morphometry found that the medial temporal lobe (hippocampus) was one of few cortical regions showing a significant nonlinear relationship with age (Ziegler et al. 2012), but hippocampal trajectories deviate substantially from other parts of the cortex (Walhovd et al. 2011). Westlye et al., however, showed negative age relationships in entorhinal cortex in a group of older adults compared with young and middle-aged participants, suggesting increased vulnerability in higher age (Westlye et al. 2010a).
Although of modest magnitude, age-related acceleration of decline of the entorhinal cortex is of special importance. AD likely cause atrophy manifest years in advance of clinical symptoms (Davatzikos et al. 2009; Jack et al. 2010), and undetected neurodegenerative disease may therefore bias the results toward accelerated atrophy in AD-prone areas in presumably healthy samples. It has even been argued that insufficient screening for incipient neurodegenerative conditions account for the observed atrophy in studies of healthy aging (Burgmans et al. 2009). Here, by including AD patients in the sample, we demonstrate that undetected neurodegenerative disease is unlikely to produce artifactual observations of nonlinearity in cortical areas outside the entorhinal cortex. Further, the rate of thinning in the entorhinal cortex did not vary between the full sample and the low-risk subsamples. Still, the magnitude of atrophy in entorhinal cortex is several times higher in MCI and AD patients compared with healthy older adults (McDonald et al. 2009), and entorhinal atrophy is a major risk factor for AD. Thus, in AD patients, entorhinal atrophy likely represents a mixture of normal age-related changes and additional neurodegeneration specific to the disease, causing decline at a faster rate.
The relationship between the entorhinal changes and changes in memory performance suggests that non-AD mechanisms in AD-prone areas are still causative for cognitive reductions in healthy older adults. Previous studies have shown relationships between medial temporal structural changes and memory in healthy participants (Rodrigue and Raz 2004; Murphy et al. 2010; Persson et al. 2012). The present results further indicate that entorhinal atrophy may be unrelated to AD pathology, while still being predictive of memory changes. To explain this association, further research needs to focus on neurobiological mechanisms for cognitive reductions that do not include AD pathology. Interestingly, the few studies of healthy controls that have tested the relationship between in vivo biomarkers for amyloid and atrophy generally do not find relationships in the medial temporal lobes (Storandt et al. 2009; Fjell, Walhovd, Fennema-Notestine, McEvoy, Hagler, Holland, Blennow et al. 2010; Tosun et al. 2010; Becker et al. 2011), although the results sometimes are complex (Bourgeat et al. 2010, Chetelat, Villemagne, Bourgeat et al. 2010; Chetelat, Villemagne, Pike et al. 2010). In contrast, relationships between amyloid biomarkers and atrophy in MCI and AD are found mostly in the temporal lobes (Fjell, Walhovd, Fennema-Notestine, McEvoy, Hagler, Holland, Brewer et al. 2010). Thus, it is possible that atrophy in the entorhinal cortex in healthy older adults can occur without being related to the same mechanisms that cause atrophy in AD. In principle, a threshold of cortical thickness or the rate of atrophy could represent dementia, with the entorhinal atrophy of the low-risk participants in the present study being well below such a threshold. However, even though the rate of entorhinal decline is a continuous measure that varies across a clinical spectrum from normal to MCI to AD, this does not imply that the underlying mechanisms causing the atrophy are similar. Entorhinal cortex is vulnerable both to AD and healthy aging, with detrimental impact on memory function in both, but the etiology may at least be partly different.
More puzzling than the increased rate of estimated entorhinal atrophy in older age was the finding of increased estimated change in lingual, pericalcarine and lateral occipital cortices, and areas involved in visual perception. These regions maturated relatively early in development (Shaw et al. 2008; Tamnes et al. 2010; Westlye et al. 2010a), and thus could be expected to be more resilient to the influences of normal aging. The deviations from linearity in these areas, however, were modest, and a linear model explained the data reasonably well. More research is needed to understand these effects.
Linearity of Brain Aging: Longitudinal Changes in Advanced Age
In contrast to cross-sectional analyses, which are based on age “differences,” longitudinal designs can be used to assess “changes” in cortical thickness (Pfefferbaum et al. 1998; Resnick et al. 2003; Raz et al. 2005; Driscoll et al. 2009; Fjell, Walhovd et al. 2009; Raz et al. 2010; Schuff et al. 2012). Drawing inferences about age changes from cross-sectional data alone rests on the assumptions that cohort effects do not exist in the sample, that sampling bias is not correlated with age and that individual development can be captured by population analyses (Lindenberger et al. 2011, see also Maxwell and Cole 2007). Several authors have warned about the fallacies involved in drawing inferences about change from cross-sectional data alone (Nyberg et al. 2010; Raz and Lindenberger 2010, 2011; Lindenberger et al. 2011). When it comes to lifespan studies of brain structures, however, no fully satisfactory solution to this problem exists, and most agree that mean age trends and age-independent individual differences in general can be delineated from cross-sectional data (Raz and Lindenberger 2011). Longitudinal examinations over extended time intervals are not feasible with MRI, and the best compromise may be to combine cross-sectional and longitudinal analyses.
In the present study, the longitudinal results in general supported the main findings from the cross-sectional analyses. However, there were also important differences between the longitudinal and cross-sectional results. First, longitudinal change estimates were substantially higher than the cross-sectional estimates, which corresponds to previous reports (Raz et al. 2005). Given identical thickness estimation techniques, no differences in sample and no covariance between sampling bias and age, the change estimates should be close to identical. When profound differences are found, these may be due to one or more of these 3 factors. First, longitudinal change estimates are based on intrasubject surface registration procedures that yield superior accuracy compared with the intersubject cross-sectional procedures, reducing noise and error estimates (Reuter et al. 2012). As noise tends to reduce effect sizes, this could potentially explain some of the differences in estimates between methods. One approach to estimate the importance of this factor would be to reprocess longitudinal data with both methods and compare the results. Second, sample differences, including differences in scanning protocols, contribute to explain why the deviations between the longitudinal and the cross-sectional estimates were larger for the full sample >60 years compared with the cross-sectional estimates from longitudinal sample itself. Finally, covariance between sampling bias and age likely accounts for the remaining differences between the longitudinal and the cross-sectional change estimates. If the older adults with the highest age are less typical for their age compared with the older adults with the lowest age, then this may bias the cross-sectional analyses, even when the same participants are included in the longitudinal and the cross-sectional analyses. We suspect that this may account for the apparent preservation or thickening of ACC observed cross-sectionally but not longitudinally, raising serious questions about the validity of this observation. The present results, in accordance with volumetric findings (Raz et al. 2005), indicate that cross-sectional designs may underestimate the real change in cortical thickness in aging. With the exception of ACC, this seems mostly to be a scaling issue and does to a lesser degree seem to bias the topographical distribution of effects. This may vary between samples and studies, and represent an important venue for future research. In any case, most trust should be put on effects that are replicated across both longitudinal and cross-sectional analyses. For instance, thinning of entorhinal cortex was observed across samples and methods, supporting the importance of this finding. Unfortunately, longitudinal data were available for the oldest part of the age range only, and longitudinal samples with wider age ranges will be an important improvement to seek in future studies.
Conclusion
The cross-sectional estimates suggest that cortical thickness in most areas declines linearly with age, and longitudinal data confirmed the main pattern of effects for the oldest part of the sample. There are several ways to envision the mechanisms behind the mostly linear course of estimated cortical thinning. One view is that a universal, programmed linear process drives the thinning. More likely, however, brain aging is a process characterized by dynamic equilibrium of multiple linear and nonlinear degenerative and restorative processes causing an apparent linear decline at the macroscopic level. Alternatively, individual participants may follow different nonlinear trajectories that sums up to seemingly linear curves at the group level. In all cases, nonlinear changes may be affected by the added accumulated impact of various negative events, for example, vascular factors (Raz et al. 2005). Notably, the present results indicate that the brain changes, even in the regions most prone to AD pathology, are not necessarily caused by age-related neurodegenerative conditions such as AD. Rather, atrophy in AD-vulnerable areas is also a part of the normal aging process. Adding to the complexity is the brain's life-long ability to morphological change in response to cognitive stimulation (Draganski et al. 2006; Engvig et al. 2010). An important task for future research is to identify specific environmental and genetic factors that impact the rate of cortical thinning in individual participants. To accomplish that task, combined longitudinal and cross-sectional studies with multiple examinations and large age spans are needed (Raz et al. 2010), which will give us the possibility to model the influence of different medical, genetic, and environmental impacts on individual trajectories over time.
Supplementary Material
Supplementary material can be found at: http://www.cercor.oxfordjournals.org/.
Funding
The present study was funded by the following grants: the Norwegian Research Council (177404/W50 to K.B.W., 175066/D15 to A.M.F., 154313/V50 to I.R., 177458/V50 to T.E., 204966 to L.T.W.), University of Oslo (to K.B.W. and A.M.F.), National Institutes of Health, USA (R37 AG011230 to N.R.). The OASIS database is made available by the Washington University Alzheimer's Disease Research Center, Dr. Randy Buckner at the Howard Hughes Medical Institute (HHMI) at Harvard University, the Neuroinformatics Research Group (NRG) at Washington University School of Medicine, and the Biomedical Informatics Research Network (BIRN), and supported by NIH grants P50 AG05681, P01 AG03991, R01 AG021910, P50 MH071616, U24 RR021382, and R01 MH56584. The ADNI data collection and sharing was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Amorfix Life Sciences Ltd.; AstraZeneca; Bayer HealthCare; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for theNational Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129, K01 AG030514, and the Dana Foundation.
Supplementary Material
Notes
Conflict of Interest: Anders M. Dale is a founder and holds equity in CorTechs Labs, Inc., and also serves on its Scientific Advisory Board. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict of interest policies.
References
- Addis DR, Roberts RP, Schacter DL. Age-related neural changes in autobiographical remembering and imagining. Neuropsychologia. 2011;49:3656–3669. doi: 10.1016/j.neuropsychologia.2011.09.021. doi:10.1016/j.neuropsychologia.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JS, Bruss J, Brown CK, Damasio H. Normal neuroanatomical variation due to age: the major lobes and a parcellation of the temporal region. Neurobiol Aging. 2005;26:1245–1260. doi: 10.1016/j.neurobiolaging.2005.05.023. discussion 1279–1282 doi:10.1016/j.neurobiolaging.2005.05.023. [DOI] [PubMed] [Google Scholar]
- Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, Buckner RL. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56:924–935. doi: 10.1016/j.neuron.2007.10.038. doi:10.1016/j.neuron.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer S, Reisberg B. The GDS/FAST staging system. Int Psychogeriatr. 1997;9(Suppl. 1):167–171. doi: 10.1017/s1041610297004869. [DOI] [PubMed] [Google Scholar]
- Beck A. New York: The Psychological Corporation: 1987. Beck Depression Inventory scoring manual. [Google Scholar]
- Becker JA, Hedden T, Carmasin J, Maye J, Rentz DM, Putcha D, Fischl B, Greve DN, Marshall GA, Salloway S, et al. Amyloid-beta associated cortical thinning in clinically normal older adults. Ann Neurol. 2011;69:1032–1042. doi: 10.1002/ana.22333. doi:10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg L. Clinical dementia rating. Br J Psychiatry. 1984;145:339. [PubMed] [Google Scholar]
- Berg L. Clinical dementia rating (CDR) Psychopharmacol Bull. 1988;24:637–639. [PubMed] [Google Scholar]
- Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J sychiatry. 1968;114:797–811. doi: 10.1192/bjp.114.512.797. [DOI] [PubMed] [Google Scholar]
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–144. doi: 10.1038/nrneurol.2010.4. doi:10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- Bourgeat P, Chetelat G, Villemagne VL, Fripp J, Raniga P, Pike K, Acosta O, Szoeke C, Ourselin S, Ames D, et al. Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in older adults subjects without dementia. Neurology. 2010;74:121–127. doi: 10.1212/WNL.0b013e3181c918b5. doi:10.1212/WNL.0b013e3181c918b5. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. On areas of transition between entorhinal allocortex and temporal isocortex in the human brain. Normal morphology and lamina-specific pathology in Alzheimer's disease. Acta Neuropathol. 1985;68:325–332. doi: 10.1007/BF00690836. doi:10.1007/BF00690836. [DOI] [PubMed] [Google Scholar]
- Burgmans S, van Boxtel MP, Vuurman EF, Smeets F, Gronenschild EH, Uylings HB, Jolles J. The prevalence of cortical gray matter atrophy may be overestimated in the healthy aging brain. Neuropsychology. 2009;23:541–550. doi: 10.1037/a0016161. doi:10.1037/a0016161. [DOI] [PubMed] [Google Scholar]
- Bush G, Luu P, Posner MI. Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn Sci. 2000;4:215–222. doi: 10.1016/s1364-6613(00)01483-2. doi:10.1016/S1364-6613(00)01483-2. [DOI] [PubMed] [Google Scholar]
- Chapman CA, Jones RS, Jung M. Neuronal plasticity in the entorhinal cortex. Neural Plast. 2008;2008:314785. doi: 10.1155/2008/314785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat G, Villemagne VL, Bourgeat P, Pike KE, Jones G, Ames D, Ellis KA, Szoeke C, Martins RN, O'Keefe GJ, et al. Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Ann Neurol. 2010;67:317–324. doi: 10.1002/ana.21955. [DOI] [PubMed] [Google Scholar]
- Chetelat G, Villemagne VL, Pike KE, Baron JC, Bourgeat P, Jones G, Faux NG, Ellis KA, Salvado O, Szoeke C, et al. Larger temporal volume in older adults with high versus low beta-amyloid deposition. Brain. 2010;133:3349–3358. doi: 10.1093/brain/awq187. doi:10.1093/brain/awq187. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. doi:10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Chisum HJ, Townsend J, Cowles A, Covington J, Egaas B, Harwood M, Hinds S, Press GA. Normal brain development and aging: quantitative analysis at in vivo MR imaging in healthy volunteers. Radiology. 2000;216:672–682. doi: 10.1148/radiology.216.3.r00au37672. [DOI] [PubMed] [Google Scholar]
- Crivello F, Lemaitre H, Dufouil C, Grassiot B, Delcroix N, Tzourio-Mazoyer N, Tzourio C, Mazoyer B. Effects of ApoE-epsilon4 allele load and age on the rates of grey matter and hippocampal volumes loss in a longitudinal cohort of 1186 healthy older adults persons. Neuroimage. 2010;53:1064–1069. doi: 10.1016/j.neuroimage.2009.12.116. doi:10.1016/j.neuroimage.2009.12.116. [DOI] [PubMed] [Google Scholar]
- Curiati PK, Tamashiro JH, Squarzoni P, Duran FL, Santos LC, Wajngarten M, Leite CC, Vallada H, Menezes PR, Scazufca M, et al. Brain structural variability due to aging and gender in cognitively healthy Elders: results from the Sao Paulo Ageing and Health study. AJNR Am J Neuroradiol. 2009;30:1850–1856. doi: 10.3174/ajnr.A1727. doi:10.3174/ajnr.A1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9:179–194. doi: 10.1006/nimg.1998.0395. [DOI] [PubMed] [Google Scholar]
- Dale AM, Sereno MI. Improved localization of cortical activity by combining EEG and MEG with MRI cortical surface reconstruction: a linear approach. J Cogn Neurosci. 1993;5:162–176. doi: 10.1162/jocn.1993.5.2.162. doi:10.1162/jocn.1993.5.2.162. [DOI] [PubMed] [Google Scholar]
- Davatzikos C, Xu F, An Y, Fan Y, Resnick SM. Longitudinal progression of Alzheimer's-like patterns of atrophy in normal older adults: the SPARE-AD index. Brain. 2009;132:2026–2035. doi: 10.1093/brain/awp091. doi:10.1093/brain/awp091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. doi:10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Draganski B, Gaser C, Kempermann G, Kuhn HG, Winkler J, Buchel C, May A. Temporal and spatial dynamics of brain structure changes during extensive learning. J Neurosci. 2006;26:6314–6317. doi: 10.1523/JNEUROSCI.4628-05.2006. doi:10.1523/JNEUROSCI.4628-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll I, Davatzikos C, An Y, Wu X, Shen D, Kraut M, Resnick SM. Longitudinal pattern of regional brain volume change differentiates normal aging from MCI. Neurology. 2009;72:1906–1913. doi: 10.1212/WNL.0b013e3181a82634. doi:10.1212/WNL.0b013e3181a82634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du AT, Schuff N, Chao LL, Kornak J, Jagust WJ, Kramer JH, Reed BR, Miller BL, Norman D, Chui HC, et al. Age effects on atrophy rates of entorhinal cortex and hippocampus. Neurobiol Aging. 2006;27:733–740. doi: 10.1016/j.neurobiolaging.2005.03.021. doi:10.1016/j.neurobiolaging.2005.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du AT, Schuff N, Zhu XP, Jagust WJ, Miller BL, Reed BR, Kramer JH, Mungas D, Yaffe K, Chui HC, et al. Atrophy rates of entorhinal cortex in AD and normal aging. Neurology. 2003;60:481–486. doi: 10.1212/01.wnl.0000044400.11317.ec. doi:10.1212/01.WNL.0000044400.11317.EC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engvig A, Fjell AM, Westlye LT, Moberget T, Sundseth O, Larsen VA, Walhovd KB. Effects of memory training on cortical thickness in the older adults. NeuroImage. 2010;52:1667–1676. doi: 10.1016/j.neuroimage.2010.05.041. doi:10.1016/j.neuroimage.2010.05.041. [DOI] [PubMed] [Google Scholar]
- Espeseth T, Westlye LT, Fjell AM, Walhovd KB, Rootwelt H, Reinvang I. Accelerated age-related cortical thinning in healthy carriers of apolipoprotein E epsilon 4. Neurobiol Aging. 2008;29:329–340. doi: 10.1016/j.neurobiolaging.2006.10.030. doi:10.1016/j.neurobiolaging.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Ezekiel F, Chao L, Kornak J, Du AT, Cardenas V, Truran D, Jagust W, Chui H, Miller B, Yaffe K, et al. Comparisons between global and focal brain atrophy rates in normal aging and Alzheimer disease: boundary shift integral versus tracing of the entorhinal cortex and hippocampus. Alzheimer Dis Assoc Disord. 2004;18:196–201. [PMC free article] [PubMed] [Google Scholar]
- Fennema-Notestine C, Hagler DJ, Jr, McEvoy LK, Fleisher AS, Wu EH, Karow DS, Dale AM. Structural MRI biomarkers for preclinical and mild Alzheimer's disease. Hum Brain Mapp. 2009;30:3238–3253. doi: 10.1002/hbm.20744. doi:10.1002/hbm.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA. 2000;97:11050–11055. doi: 10.1073/pnas.200033797. doi:10.1073/pnas.200033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, Tootell RB, Dale AM. High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp. 1999;8:272–284. doi: 10.1002/(SICI)1097-0193(1999)8:4<272::AID-HBM10>3.0.CO;2-4. doi:10.1002/(SICI)1097-0193(1999)8:4<272::AID-HBM10>3.0.CO;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Segonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, et al. Automatically parcellating the human cerebral cortex. Cereb Cortex. 2004;14:11–22. doi: 10.1093/cercor/bhg087. doi:10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Blennow K, Brewer JB, Dale AM. Brain atrophy in healthy aging is related to CSF levels of Abeta1-42. Cereb Cortex. 2010;20:2069–2079. doi: 10.1093/cercor/bhp279. doi:10.1093/cercor/bhp279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Brewer JB, Dale AM. CSF biomarkers in prediction of cerebral and clinical change in mild cognitive impairment and Alzheimer's disease. J Neurosci. 2010;30:2088–2101. doi: 10.1523/JNEUROSCI.3785-09.2010. doi:10.1523/JNEUROSCI.3785-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Brewer JB, Dale AM. One-year brain atrophy evident in healthy aging. J Neurosci. 2009;29:15223–15231. doi: 10.1523/JNEUROSCI.3252-09.2009. doi:10.1523/JNEUROSCI.3252-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Westlye LT, Ostby Y, Tamnes CK, Jernigan TL, Gamst A, Dale AM. When does brain aging accelerate? Dangers of quadratic fits in cross-sectional studies. Neuroimage. 2010;50:1376–1383. doi: 10.1016/j.neuroimage.2010.01.061. doi:10.1016/j.neuroimage.2010.01.061. [DOI] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat DH, Greve DN, Fischl B, et al. High consistency of regional cortical thinning in aging across multiple samples. Cereb Cortex. 2009a;19:2001–2012. doi: 10.1093/cercor/bhn232. doi:10.1093/cercor/bhn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat DH, Greve DN, Fischl B, et al. Minute effects of sex on the aging brain: a multisample magnetic resonance imaging study of healthy aging and Alzheimer's disease. J Neurosci. 2009b;29:8774–8783. doi: 10.1523/JNEUROSCI.0115-09.2009. doi:10.1523/JNEUROSCI.0115-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Espeseth T, Reinvang I, Dale AM, Holland D, Walhovd KB. Cortical gray matter atrophy in healthy aging cannot be explained by undetected incipient cognitive disorders: a comment on Burgmans et al. (2009) Neuropsychology. 2010;24:258–263. doi: 10.1037/a0018827. discussion 264–266 doi:10.1037/a0018827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Greve DN, Fischl B, Benner T, van der Kouwe AJ, Salat D, Bjornerud A, Due-Tonnessen P, Walhovd KB. The relationship between diffusion tensor imaging and volumetry as measures of white matter properties. Neuroimage. 2008;42:1654–1668. doi: 10.1016/j.neuroimage.2008.06.005. doi:10.1016/j.neuroimage.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. doi:10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Goodglass H, Kaplan E. The assessment of aphasia and related disorders. Philadelphia, PA: Lea & Febiger; 1983. [Google Scholar]
- Holland D, Dale AM. Nonlinear registration of longitudinal images and measurement of change in regions of interest. Med Image Anal. 2011;15:489–497. doi: 10.1016/j.media.2011.02.005. doi:10.1016/j.media.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland D, McEvoy LK, Dale AM. Unbiased comparison of sample size estimates from longitudinal structural measures in ADNI. Hum Brain Mapp. 2012;33:2586–2602. doi: 10.1002/hbm.21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. doi:10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Petersen RC, Xu YC, Waring SC, O'Brien PC, Tangalos EG, Smith GE, Ivnik RJ, Kokmen E. Medial temporal atrophy on MRI in normal aging and very mild Alzheimer's disease. Neurology. 1997;49:786–794. doi: 10.1212/wnl.49.3.786. doi:10.1212/WNL.49.3.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Machulda MM, Vemuri P, McDade EM, Zeng G, Senjem ML, Gunter JL, Przybelski SA, Avula RT, Knopman DS, et al. Age-related changes in the default mode network are more advanced in Alzheimer disease. Neurology. 2011;77:1524–1531. doi: 10.1212/WNL.0b013e318233b33d. doi:10.1212/WNL.0b013e318233b33d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperberg GR, Broome MR, McGuire PK, David AS, Eddy M, Ozawa F, Goff D, West WC, Williams SC, van der Kouwe AJ, et al. Regionally localized thinning of the cerebral cortex in schizophrenia. Arch Gen Psychiatry. 2003;60:878–888. doi: 10.1001/archpsyc.60.9.878. doi:10.1001/archpsyc.60.9.878. [DOI] [PubMed] [Google Scholar]
- Lindenberger U, von Oertzen T, Ghisletta P, Hertzog C. Cross-sectional age variance extraction: what's change got to do with it? Psychol Aging. 2011;26:34–47. doi: 10.1037/a0020525. doi:10.1037/a0020525. [DOI] [PubMed] [Google Scholar]
- Marcus DS, Fotenos AF, Csernansky JG, Morris JC, Buckner RL. Open access series of imaging studies: longitudinal MRI data in nondemented and demented older adults. J Cogn Neurosci. 2010;22:2677–2684. doi: 10.1162/jocn.2009.21407. doi:10.1162/jocn.2009.21407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus DS, Wang TH, Parker J, Csernansky JG, Morris JC, Buckner RL. Open Access Series of Imaging Studies (OASIS): cross-sectional MRI data in young, middle aged, nondemented, and demented older adults. J Cogn Neurosci. 2007;19:1498–1507. doi: 10.1162/jocn.2007.19.9.1498. doi:10.1162/jocn.2007.19.9.1498. [DOI] [PubMed] [Google Scholar]
- Maxwell SE, Cole DA. Bias in cross-sectional analyses of longitudinal mediation. Psychol Methods. 2007;12:23–44. doi: 10.1037/1082-989X.12.1.23. doi:10.1037/1082-989X.12.1.23. [DOI] [PubMed] [Google Scholar]
- McDonald CR, McEvoy LK, Gharapetian L, Fennema-Notestine C, Hagler DJ, Jr, Holland D, Koyama A, Brewer JB, Dale AM. Regional rates of neocortical atrophy from normal aging to early Alzheimer disease. Neurology. 2009;73:457–465. doi: 10.1212/WNL.0b013e3181b16431. doi:10.1212/WNL.0b013e3181b16431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. doi:10.1212/WNL.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH, Berg L. Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001;58:397–405. doi: 10.1001/archneur.58.3.397. doi:10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- Murphy EA, Holland D, Donohue M, McEvoy LK, Hagler DJ, Jr, Dale AM, Brewer JB. Six-month atrophy in MTL structures is associated with subsequent memory decline in older adults controls. NeuroImage. 2010;53:1310–1317. doi: 10.1016/j.neuroimage.2010.07.016. doi:10.1016/j.neuroimage.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg L, Salami A, Andersson M, Eriksson J, Kalpouzos G, Kauppi K, Lind J, Pudas S, Persson J, Nilsson LG. Longitudinal evidence for diminished frontal cortex function in aging. Proc Natl Acad Sci USA. 2010;107:22682–22686. doi: 10.1073/pnas.1012651108. doi:10.1073/pnas.1012651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson J, Pudas S, Lind J, Kauppi K, Nilsson LG, Nyberg L. Longitudinal Structure-Function Correlates in Older adults Reveal MTL Dysfunction with Cognitive Decline. Cereb Cortex. 2012;22:2297–2304. doi: 10.1093/cercor/bhr306. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Rosenbloom MJ, Mathalon DH, Lim KO. A controlled study of cortical gray matter and ventricular changes in alcoholic men over a 5-year interval. Arch Gen Psychiatry. 1998;55:905–912. doi: 10.1001/archpsyc.55.10.905. doi:10.1001/archpsyc.55.10.905. [DOI] [PubMed] [Google Scholar]
- Posner MI. Imaging attention networks. NeuroImage. 2011;61:450–456. doi: 10.1016/j.neuroimage.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner MI, Rothbart MK, Sheese BE, Tang Y. The anterior cingulate gyrus and the mechanism of self-regulation. Cogn Affect Behav Neurosci. 2007;7:391–395. doi: 10.3758/cabn.7.4.391. doi:10.3758/CABN.7.4.391. [DOI] [PubMed] [Google Scholar]
- Raz N, Ghisletta P, Rodrigue KM, Kennedy KM, Lindenberger U. Trajectories of brain aging in middle-aged and older adults: regional and individual differences. NeuroImage. 2010;51:501–511. doi: 10.1016/j.neuroimage.2010.03.020. doi:10.1016/j.neuroimage.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Gunning-Dixon F, Head D, Rodrigue KM, Williamson A, Acker JD. Aging, sexual dimorphism, and hemispheric asymmetry of the cerebral cortex: replicability of regional differences in volume. Neurobiol Aging. 2004;25:377–396. doi: 10.1016/S0197-4580(03)00118-0. doi:10.1016/S0197-4580(03)00118-0. [DOI] [PubMed] [Google Scholar]
- Raz N, Lindenberger U. News of cognitive cure for age-related brain shrinkage is premature: a comment on Burgmans et al. (2009) Neuropsychology. 2010;24:255–257. doi: 10.1037/a0018828. doi:10.1037/a0018828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Lindenberger U. Only time will tell: cross-sectional studies offer no solution to the age-brain-cognition triangle: comment on Salthouse (2011) Psychol Bull. 2011;137:790–795. doi: 10.1037/a0024503. doi:10.1037/a0024503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Lindenberger U, Rodrigue KM, Kennedy KM, Head D, Williamson A, Dahle C, Gerstorf D, Acker JD. Regional brain changes in aging healthy adults: general trends, individual differences and modifiers. Cereb Cortex. 2005;15:1676–1689. doi: 10.1093/cercor/bhi044. doi:10.1093/cercor/bhi044. [DOI] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM. Differential aging of the brain: patterns, cognitive correlates and modifiers. Neurosci Biobehav Rev. 2006;30:730. doi: 10.1016/j.neubiorev.2006.07.001. doi:10.1016/j.neubiorev.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Head D, Kennedy KM, Acker JD. Differential aging of the medial temporal lobe: a study of a five-year change. Neurology. 2004;62:433–438. doi: 10.1212/01.wnl.0000106466.09835.46. doi:10.1212/01.WNL.0000106466.09835.46. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Pham DL, Kraut MA, Zonderman AB, Davatzikos C. Longitudinal magnetic resonance imaging studies of older adults: a shri6nking brain. J Neurosci. 2003;23:3295–3301. doi: 10.1523/JNEUROSCI.23-08-03295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Fischl B. Avoiding asymmetry-induced bias in longitudinal image processing. Neuroimage. 2011;57:19–21. doi: 10.1016/j.neuroimage.2011.02.076. doi:10.1016/j.neuroimage.2011.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Rosas HD, Fischl B. Highly accurate inverse consistent registration: a robust approach. Neuroimage. 2010;53:1181–1196. doi: 10.1016/j.neuroimage.2010.07.020. doi:10.1016/j.neuroimage.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage. 2012;61:1402–1418. doi: 10.1016/j.neuroimage.2012.02.084. doi:10.1016/j.neuroimage.2012.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter-Lorenz PA, Park DC. Human neuroscience and the aging mind: a new look at old problems. J Gerontol B Psychol Sci Soc Sci. 2010;65:405–415. doi: 10.1093/geronb/gbq035. doi:10.1093/geronb/gbq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey A. L'examen clinique en psychologie. Paris: Presses Univeritaires de France; 1964. [Google Scholar]
- Rodrigue KM, Raz N. Shrinkage of the entorhinal cortex over five years predicts memory performance in healthy adults. J Neurosci. 2004;24:956–963. doi: 10.1523/JNEUROSCI.4166-03.2004. doi:10.1523/JNEUROSCI.4166-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH, van der Kouwe A, Jenkins BG, Dale AM, Fischl B. Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology. 2002;58:695–701. doi: 10.1212/wnl.58.5.695. doi:10.1212/WNL.58.5.695. [DOI] [PubMed] [Google Scholar]
- Salat DH, Buckner RL, Snyder AZ, Greve DN, Desikan RS, Busa E, Morris JC, Dale AM, Fischl B. Thinning of the cerebral cortex in aging. Cereb Cortex. 2004;14:721–730. doi: 10.1093/cercor/bhh032. doi:10.1093/cercor/bhh032. [DOI] [PubMed] [Google Scholar]
- Salat DH, Kaye JA, Janowsky JS. Greater orbital prefrontal volume selectively predicts worse working memory performance in older adults. Cereb Cortex. 2002;12:494–505. doi: 10.1093/cercor/12.5.494. doi:10.1093/cercor/12.5.494. [DOI] [PubMed] [Google Scholar]
- Salthouse TA. Neuroanatomical substrates of age-related cognitive decline. Psychol Bull. 2011;137:753–784. doi: 10.1037/a0023262. doi:10.1037/a0023262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salthouse TA. When does age-related cognitive decline begin? Neurobiol Aging. 2009;30:507–514. doi: 10.1016/j.neurobiolaging.2008.09.023. doi:10.1016/j.neurobiolaging.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaie KW. "When does age-related cognitive decline begin?" Salthouse again reifies the "cross-sectional fallacy". Neurobiol Aging. 2009;30:528–529. doi: 10.1016/j.neurobiolaging.2008.12.012. discussion 530–533 doi:10.1016/j.neurobiolaging.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuff N, Tosun D, Insel PS, Chiang GC, Truran D, Aisen PS, Jack CR, Jr, Weiner MW. Nonlinear time course of brain volume loss in cognitively normal and impaired elders. Neurobiol Aging. 2012;33:845–855. doi: 10.1016/j.neurobiolaging.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. doi:10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Kabani NJ, Lerch JP, Eckstrand K, Lenroot R, Gogtay N, Greenstein D, Clasen L, Evans A, Rapoport JL, et al. Neurodevelopmental trajectories of the human cerebral cortex. J Neurosci. 2008;28:3586–3594. doi: 10.1523/JNEUROSCI.5309-07.2008. doi:10.1523/JNEUROSCI.5309-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwinski M, Buschke H. Cross-sectional and longitudinal relationships among age, cognition, and processing speed. Psychol Aging. 1999;14:18–33. doi: 10.1037//0882-7974.14.1.18. doi:10.1037/0882-7974.14.1.18. [DOI] [PubMed] [Google Scholar]
- Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci. 2011;12:585–601. doi: 10.1038/nrn3085. doi:10.1038/nrn3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder AZ, Raichle ME. A brief history of the resting state: the Washington University perspective. Neuroimage. 2012;62:902–910. doi: 10.1016/j.neuroimage.2012.01.044. doi:10.1016/j.neuroimage.2012.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW. Mapping cortical change across the human life span. Nat Neurosci. 2003;6:309–315. doi: 10.1038/nn1008. doi:10.1038/nn1008. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O'Keefe K, O'Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. doi:10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storandt M, Mintun MA, Head D, Morris JC. Cognitive decline and brain volume loss as signatures of cerebral amyloid-beta peptide deposition identified with Pittsburgh compound B: cognitive decline associated with Abeta deposition. Arch Neurol. 2009;66:1476–1481. doi: 10.1001/archneurol.2009.272. doi:10.1001/archneurol.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamnes CK, Ostby Y, Fjell AM, Westlye LT, Due-Tonnessen P, Walhovd KB. Brain maturation in adolescence and young adulthood: regional age-related changes in cortical thickness and white matter volume and microstructure. Cereb Cortex. 2010;20:534–548. doi: 10.1093/cercor/bhp118. doi:10.1093/cercor/bhp118. [DOI] [PubMed] [Google Scholar]
- Terribilli D, Schaufelberger MS, Duran FL, Zanetti MV, Curiati PK, Menezes PR, Scazufca M, Amaro E, Jr, Leite CC, Busatto GF. Age-related gray matter volume changes in the brain during non-older adults adulthood. Neurobiol Aging. 2011;32:354–368. doi: 10.1016/j.neurobiolaging.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambisetty M, Wan J, Carass A, An Y, Prince JL, Resnick SM. Longitudinal changes in cortical thickness associated with normal aging. Neuroimage. 2010;52:1215–1223. doi: 10.1016/j.neuroimage.2010.04.258. doi:10.1016/j.neuroimage.2010.04.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WK, Holland D. Bias in tensor based morphometry Stat-ROI measures may result in unrealistic power estimates. Neuroimage. 2011;57:1–4. doi: 10.1016/j.neuroimage.2010.11.092. discussion 5–14 doi:10.1016/j.neuroimage.2010.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosun D, Schuff N, Truran-Sacrey D, Shaw LM, Trojanowski JQ, Aisen P, Peterson R, Weiner MW. Relations between brain tissue loss, CSF biomarkers, and the ApoE genetic profile: a longitudinal MRI study. Neurobiol Aging. 2010;31:1340–1354. doi: 10.1016/j.neurobiolaging.2010.04.030. doi:10.1016/j.neurobiolaging.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhovd KB, Fjell AM, Dale AM, McEvoy LK, Brewer J, Karow DS, Salmon DP, Fennema-Notestine C. Multi-modal imaging predicts memory performance in normal aging and cognitive decline. Neurobiol Aging. 2010;31:1107–1121. doi: 10.1016/j.neurobiolaging.2008.08.013. doi:10.1016/j.neurobiolaging.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhovd KB, Fjell AM, Reinvang I, Lundervold A, Dale AM, Eilertsen DE, Quinn BT, Salat D, Makris N, Fischl B. Effects of age on volumes of cortex, white matter and subcortical structures. Neurobiol Aging. 2005;26:1261–1270. doi: 10.1016/j.neurobiolaging.2005.05.020. discussion 1275–1268 doi:10.1016/j.neurobiolaging.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Walhovd KB, Westlye LT, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat DH, Greve DN, Fischl B, et al. Consistent neuroanatomical age-related volume differences across multiple samples. Neurobiol Aging. 2011;32:916–932. doi: 10.1016/j.neurobiolaging.2009.05.013. doi:10.1016/j.neurobiolaging.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D. The Wechsler Adult Intelligence Scale-revised. New York, USA: Psychological Corporation; 1981. [Google Scholar]
- Wechsler D. Wechsler Memory Scale-revised. San Antonia, TX: Psychological Corporation; 1987. [Google Scholar]
- Wechsler D. Wechsler Abbreviated Scale of Intelligence. San Antonio (TX): Psychological Corporation: 1999. [Google Scholar]
- Westlye LT, Grydeland H, Walhovd KB, Fjell AM. Associations between regional cortical thickness and attentional networks as measured by the attention network test. Cereb Cortex. 2011;21:345–356. doi: 10.1093/cercor/bhq101. doi:10.1093/cercor/bhq101. [DOI] [PubMed] [Google Scholar]
- Westlye LT, Walhovd KB, Dale AM, Bjornerud A, Due-Tonnessen P, Engvig A, Grydeland H, Tamnes CK, Ostby Y, Fjell AM. Differentiating maturational and aging-related changes of the cerebral cortex by use of thickness and signal intensity. Neuroimage. 2010a;52:172–185. doi: 10.1016/j.neuroimage.2010.03.056. doi:10.1016/j.neuroimage.2010.03.056. [DOI] [PubMed] [Google Scholar]
- Westlye LT, Walhovd KB, Dale AM, Bjornerud A, Due-Tonnessen P, Engvig A, Grydeland H, Tamnes CK, Ostby Y, Fjell AM. Lifespan changes of the human brain White matter: diffusion tensor imaging (DTI) and volumetry. Cereb Cortex. 2010b;20:2055–2068. doi: 10.1093/cercor/bhp280. doi:10.1093/cercor/bhp280. [DOI] [PubMed] [Google Scholar]
- Ziegler G, Dahnke R, Jancke L, Yotter RA, May A, Gaser C. Brain structural trajectories over the adult lifespan. Hum Brain Mapp. 2012;33:2377–2389. doi: 10.1002/hbm.21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.