

Abstract
Although it has long been known that mitochondria take up Ca2+, the molecular identities of the channels and transporters involved in this process were revealed only recently. Here, we discuss the recent work that has led to the characterization of the mitochondrial calcium uniporter complex, which includes the channel-forming subunit MCU (mitochondrial calcium uniporter) and its regulators MICU1, MICU2, MCUb, EMRE, MCUR1 and miR-25. We review not only the biochemical identities and structures of the proteins required for mitochondrial Ca2+ uptake but also their implications in different physiopathological contexts.
Introduction
The publication of back-to-back papers reporting the molecular identification of the mitochondrial calcium uniporter (MCU) complex in the June 19, 2011, issue of Nature (Baughman et al. 2011; De Stefani et al. 2011) represented a definitive culmination of 50 years of intensive research in this field.
Mitochondria rapidly transport Ca2+ across their membranes and accumulate it in the mitochondrial matrix, where several Ca2+ effectors are located (Rizzuto et al. 2012). The driving force of Ca2+ uptake is the mitochondrial membrane potential (ΔΨ), which is present throughout the inner mitochondrial membrane (IMM) and is generated by the respiratory chain. The pumping of protons by the respiratory complexes toward the intermembrane space (IMS) generates an electrochemical gradient (−180 mV) inside the matrix. In response to this gradient, two strong uncouplers of oxidative phosphorylation, dinitrophenol and carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), are typically involved in preventing Ca2+ entry. Thus, mitochondria take up Ca2+ electrophoretically via the Ca2+ uniporter. To export Ca2+ from the mitochondrial matrix, mitochondria release Ca2+ via an antiporter by exchanging Ca2+ with Na+ (in excitable tissues, such as the brain and heart) or H+ (in the liver and many other tissues) (Nicholls & Crompton, 1980), with what is considered an electroneutral stoichiometry for Ca2+ efflux (Ca2+–2H+ antiport) (Brand, 1985). However, the identification of leucine zipper-EF-hand containing transmembrane protein 1 (Letm1) as the Ca2+–H+ antiporter suggests a different stoichiometry (Jiang et al. 2009). Letm1 seems to act as a Ca2+ extrusion mechanism when the [Ca2+] is elevated in the matrix, whereas it may contribute to non-linear Ca2+ uptake at low mitochondrial Ca2+ levels (Jiang et al. 2009). Letm1 has also been proposed to act as a mitochondrial K+–H+ exchanger (Dimmer et al. 2008). Consequently, the role of Letm1 awaits further confirmation.
The recent discovery of NCLX as the mitochondrial Na+–Ca2+ exchanger (Palty et al. 2010) confirms the general understanding that the mitochondrial Na+–Ca2+ antiport is electrogenic (exchanging 3 or 4 Na+ per Ca2+). Nevertheless, the mitochondrial permeability transition pore (mPTP), a key effector of cell death, has been indicated as a putative component of Ca2+ efflux machinery (Altschuld et al. 1992). However, this topic is still controversial because other observations suggest a minimal contribution of mPTP to Ca2+ release (Wei et al. 2011), and further studies are needed to reach a definitive conclusion.
The activity of these Ca2+ efflux pathways shows that mitochondrial Ca2+ accumulation by the MCU complex does not proceed to electrochemical equilibrium, a biological scenario that is incompatible with every cell physiology concept (a ΔΨ of 180 mV implies a [Ca2+] of ∼1 m, based on the Nernst equation).
One of the main properties of the MCU complex is its very low affinity for Ca2+ (KD of 20–30 μm under physiological conditions). Thus, the intracellular (cytosolic) Ca2+ concentration should be approximately 5–10 μm for considerable mitochondrial Ca2+ influx, but such values have never been observed in live, healthy cells. This riddle was solved through the demonstration that mitochondria are juxtaposed with the endoplasmic reticulum (ER) membrane (Rizzuto et al. 1998). The ER is the major intracellular Ca2+ store (Somlyo, 1984; de la Fuente et al. 2013), and the release of the Ca2+ content from the ER into the cytosol is due to the presence of inositol 1,4,5-trisphosphate (IP3), which is generated upon the stimulation of receptors coupled to phospholipase C (Streb et al. 1983). Therefore, microdomains with high Ca2+ concentrations ([Ca2+]>10 μm) can form transiently in regions of close apposition between the mitochondria and the Ca2+ channels of the ER (Patergnani et al. 2011), ensuring a prompt accumulation of Ca2+ inside the mitochondria (see schematization in Fig. 1). However, higher affinity mitochondrial Ca2+ uptake has been observed in many studies (Sparagna et al. 1995; Santo-Domingo & Demaurex, 2010), and patch-clamp experiments have suggested that the uniporter pore has high Ca2+ affinity (dissociation constant of <2 nm; Kirichok et al. 2004).
Figure 1.

Ion fluxes are indicated by arrows. The lower magnification represents the different components of the uniporter complex. MCU Oligomerization at IMM forms a tetramer (shadowed subunits). MCUb is represented as the black shadowed subunit. SERCA, sarcoendoplasmic reticulum calcium transport ATPase; VDAC, voltage-dependent anion channel; TMD, transmembrane domain; RuR, Ruthenium Red. See text for further details.
The development of the Ca2+-sensitive photoprotein aequorin, which targets the mitochondrial matrix (Bonora et al. 2013), together with other GFP-based fluorescent probes (Rudolf et al. 2003), has enabled the direct visualization and measurement of [Ca2+] variations in imaging experiments. Using these tools, we and other independent groups have shown that mitochondria undergo large increases in their Ca2+ levels, reaching >100 μm in some cell types. In summary, during resting conditions, the mitochondrial Ca2+ content is low (Somlyo et al. 1985), but when a cell is stimulated with a cytosolic [Ca2+]-increasing agonist (e.g. histamine or ATP), the mitochondria accumulate high amounts of Ca2+ via the MCU complex (Fig. 1).
Research examining the molecular nature of the MCU complex began in the 1970s, when a soluble Ca2+-binding glycoprotein was isolated from liver mitochondria; this protein was found to be capable of inducing a Ca2+ current after reconstitution in lipid bilayers (Sottocasa et al. 1972). Interestingly, Ca2+ import is blocked by both Ruthenium Red, a polycationic stain used to visualize glycoproteins and a well-known inhibitor of MCU, and antibodies directed against this Ca2+-binding glycoprotein (Panfili et al. 1976). Several years later, the idea that a glycoprotein could serve as the mitochondrial Ca2+ uniporter was revived through the isolation of a 40-kDa protein that forms Ca2+-conducting channels in black-lipid membranes from beef heart mitochondria (Saris et al. 1993) and the description of Ruthenium Red-sensitive Ca2+ uptake by proteoliposomes containing mitochondrial proteins with a similar molecular weight (>35 kDa; Zazueta et al. 1991). However, the lack of strong experimental evidence, as well as doubts about the purity of the preparations, gradually dismissed these observations. More recently, some candidates were pinpointed as important components for the mitochondrial Ca2+ uptake machinery (Trenker et al. 2007), but only 3 years ago, the identification of CCDC109A as the channel-forming subunit of the MCU complex and the identification of different MCU regulators provided the key players that fulfil all of the properties predicted for the mitochondrial Ca2+ uptake machinery (Table 1).
Table 1.
Molecular nature of the components of the mitochondrial Ca2+ uniporter complex
| Name | Also known as: | MW (kDa) | Topology | Functions | References |
|---|---|---|---|---|---|
| MCU (Mitochondrial Calcium Uniporter) | C10orf42 CCDC109A | 40→35 | Two transmembrane domains. N-and C-termini span into the matrix | Channel-forming subunit of the uniporter. MCU silencing abolishes Ca2+ entry; MCU overexpression strongly enhances Ca2+ entry. | (Baughman et al., 2011) (De Stefani et al., 2011) |
| MICU1 (Mitochondrial Calcium Uptake 1) | CALC CBARA1 EFHA3 | 54→50 | Single-pass membrane protein. Two EF-Hands span into the IMS | MICU1 interacts with MCU. Thresholding function on MCU. MICU1 silencing enhances the [Ca2+]m under resting conditions or during small cytosolic [Ca2+] elevations. | (Perocchi et al., 2010) (Mallilankaraman et al., 2012b) (Csordas et al., 2013) |
| MICU2 (Mitochondrial Calcium Uptake 2) | EFHA1 | 50→45 | Two conserved EF-Hands, probably faced on IMS | MICU2 associates with the MICU1/MCU complex. MICU2 silencing lowers the [Ca2+]m. | (Plovanich et al., 2013) |
| MCUb (Mitochondrial Calcium Uniporter b) | CCDC109B | 40→37 | Two transmembrane domains. N-and C-termini span into the matrix | Paralog of MCU. Lower expression than MCU. No channel activity. MCUb overexpression reduces the [Ca2+]m. | (Raffaello et al., 2013) |
| MCUR1 (Mitochondrial Calcium Uniporter Regulator 1) | C6ORF79 CCDC90A | 40→37 | Two transmembrane domains. N-and C-termini span into the IMS. Large portion of the protein faced on the matrix. | MCUR1 interacts with MCU but not with MICU1. MCUR1 silencing abrogates Ca2+ uptake. MCUR1 overexpression enhances the [Ca2+]m. | (Mallilankaraman et al., 2012a) |
| EMRE (Essential MCU REgulator) | C22ORF32 SMDT1 | 12→10 | Single-pass membrane protein, with highly conserved aspartate-rich tail. | EMRE silencing abolishes Ca2+ uptake. EMRE is required for interaction of MCU with MICU1/MICU2. | (Sancak et al., 2013) |
The lower molecular weights are consistent with predicted cleavable amino-terminal mitochondrial targeting sequences.
MCU
The inventory of the 1098 mouse (1013 human) nuclear and mtDNA genes that encode proteins with mitochondrial localization, the so-called MitoCarta (Pagliarini et al. 2008), is the foundation for the identification of the MCU components. Among these mitochondrial proteins, coiled-coil domain-containing protein 109A (CCDC109A), renamed MCU, shows all of the typical characteristics of a Ca2+ uniporter:
(1) Using patch-clamp electrophysiology of mitoplasts (mitochondria without an OMM), CCDC109A/MCU has been demonstrated as a unique, gated Ca2+-selective channel in the IMM (Kirichok et al. 2004). The reconstitution of purified CCDC109A/MCU into a planar lipid bilayer generates a Ca2+ current with similar properties to those previously reported by the Clapham group (De Stefani et al. 2011). This result was very recently corroborated and updated with another patch-clamp experiment, which showed that a reduction in MCU transcripts via knock-down and their enhancement via overexpression produce parallel changes in the mitochondrial Ca2+ current (Chaudhuri et al. 2013). Moreover, a single point mutation (S→A at position 259) abolishes the sensitivity of MCU to Ruthenium Red (Baughman et al. 2011; Chaudhuri et al. 2013). These analyses establish that MCU encodes the pore-forming subunit of the uniporter and that Ruthenium Red acts directly on the channel.
(2) All ion channels require at least two transmembrane domains to exert their activity. CCDC109A/MCU possesses two transmembrane α-helixes, which are highly conserved among different species. Moreover, CCDC10A/MCU is ubiquitously expressed in mammals.
(3) CCDC109A/MCU has no orthologue in the yeast S. cerevisiae (De Stefani et al. 2011), which lacks a Ruthenium Red-sensitive mitochondrial Ca2+ uptake system (Carafoli et al. 1970). Nevertheless, CCDC109A/MCU is conserved in trypanosomatidae, a group of parasites possessing a Ca2+ uptake system with properties similar to those described in mammalian mitochondria (Docampo & Lukes, 2012). Interestingly, the essential role of MCU in the regulation of cell bioenergetics in Trypanosoma brucei has been recently reported (Huang et al., 2013).
(4) The overexpression of CCDC109A/MCU almost doubles the mitochondrial Ca2+ content in both intact and permeabilized cells, causing a significant decrease in the cytosolic Ca2+ content due to an enhanced mitochondrial buffer activity (De Stefani et al. 2011). Accordingly, the down-regulation of CCDC109A/MCU strongly inhibits mitochondrial Ca2+ entry (Baughman et al. 2011; De Stefani et al. 2011), and the re-introduction of the wild-type protein in MCU knock-down cells fully rescues Ca2+ uptake (Baughman et al. 2011). Nevertheless, the other classical features of mitochondria, such as ΔΨ, organelle shape, O2 consumption and ATP synthesis, appear unchanged after MCU down-regulation. Thus, MCU is essential for high-capacity Ca2+ transport into the mitochondria but does not alter any other mitochondrial parameters.
The nuclear MCU gene, which is located on chromosome 10, encodes a 40-kDa protein that loses its cleavable target sequence during mitochondrial import, resulting in a 35-kDa mature form (Baughman et al. 2011). Curiously, among the different attempts to identify the mitochondrial Ca2+ uniporter, two studies described MCU-like glycoproteins with similar molecular weights (Zazueta et al. 1991; Saris et al. 1993).
Although the topology of MCU was initially a matter of debate (reviewed in Drago et al. 2011), it is now clear that its N-and C-terminal domains span into the mitochondrial matrix and that its 9-aa linker (the DIME domain) between the two transmembrane domains faces the intermembrane space (Baughman et al. 2011). The orientation of MCU has been definitively solved through the development of APEX, a monomeric 28-kDa peroxidase that is used as an electron microscopy tag, which is active in all cellular compartments and does not require light. The fusion of APEX to the N-and C-termini of MCU clearly stains the mitochondrial matrix but not the intermembrane space (Martell et al. 2012).
The existence of only two putative transmembrane domains strongly suggests that an active and functional uniporter channel could be formed by oligomers of MCU. The predicted quaternary structure is compatible with a tetramer, in which eight helices line the putative pore region, and the clustering of charged residues in proximity of the pore generates a negative electrostatic potential that favours the flux of a cation (Raffaello et al. 2013). In addition, the mutation of two negatively charged residues inside the DIME motif (D261/E264 in human MCU, D260/E263 in the mouse orthologue) abolishes MCU activity (Baughman et al. 2011; De Stefani et al. 2011).
Blue native polyacrylamide gel electrophoresis experiments have confirmed that that MCU oligomerizes in the mitochondrial inner membrane as part of a larger complex, migrating at an apparent molecular weight of ∼480 kDa (Baughman et al. 2011). Thus, the uniporter complex includes different regulatory subunits (Fig. 1), and one of these subunits is represented by the MICU1 protein.
MICU1
The discovery of mitochondrial calcium uptake 1 (MICU1) preceded the identification of MCU by only few months (Perocchi et al. 2010). The Mootha group used MICU1 as a molecular (and computational) bait to identify the core component of the uniporter MCU. Not only do MCU and MICU1 display the same evolutionary pattern of expression and similar RNA expression in a variety of mouse tissues (Bick et al. 2012), but they also physically interact (Baughman et al. 2011).
MICU1 (previously known as CBARA1 and EFHA3) is a 54-kDa single-pass membrane protein that contains two highly conserved EF-hand Ca2+-binding domains. MICU1 was identified through the selective siRNA screening of IMM proteins that are expressed in the majority of mammalian tissues and have homologues in vertebrates and kinetoplastids but not in the yeast S. cerevisiae. The down-regulation of MICU1 drastically reduces the mitochondrial Ca2+ content in an EF-hand-dependent manner without significantly impairing mitochondrial respiration or membrane potential (Perocchi et al. 2010). However, only 2 years after the identification of MICU1, its role as a positive regulator of mitochondrial Ca2+ entry was re-examined. The Foskett and Madesh laboratories proposed an essential role for MICU1 as a gatekeeper for MCU-dependent Ca2+ accumulation (Mallilankaraman et al. 2012b). Contrary to the previous work, MICU1 knock-down cells displayed unchanged histamine-induced mitochondrial Ca2+ uptake but dramatically increased basal Ca2+ content. Thus, MICU1 limits Ca2+ entry via MCU when the intracellular [Ca2+] is low, under resting conditions or during weak agonist stimulation. When MICU1 is lost, the mitochondria become constitutively loaded with Ca2+, suggesting that MICU1 is not required for uniporter-dependent Ca2+ uptake (Mallilankaraman et al. 2012b).
The initially proposed role of MICU1 is radically different from its currently understood role: MICU1 acts as a high-affinity Ca2+ brake on MCU-mediated Ca2+ uptake, and this scenario concurs with the so-called ‘rapid mode of Ca2+ uptake (RaM),’ a phenomenon through which isolated mitochondria seem to sequester Ca2+ very rapidly at the beginning of each Ca2+ pulse in a sequence (Sparagna et al. 1995). Following this model, upon Ca2+ addition, MCU initially takes up Ca2+ very rapidly, but when [Ca2+] inevitably increases inside the matrix, MICU1 binds Ca2+ through its EF-hand domains, exerting an inhibitory role on MCU-dependent Ca2+ entry.
These findings imply that the MICU1 EF-hands must face the matrix to enable them to sense the mitochondrial [Ca2+] ([Ca2+]m). Indeed, it has recently been showed as MICU1 might compartmentalize in the mitochondrial matrix side of the IMM, and that MICU1 binding with MCU is defined by a MICU1 N-terminal polybasic domain and two interacting coiled-coil domains of MCU (Hoffman et al., 2013). However, only few months ago, both the topology and functions of MICU1 were reassessed (Fig. 2).
Figure 2.
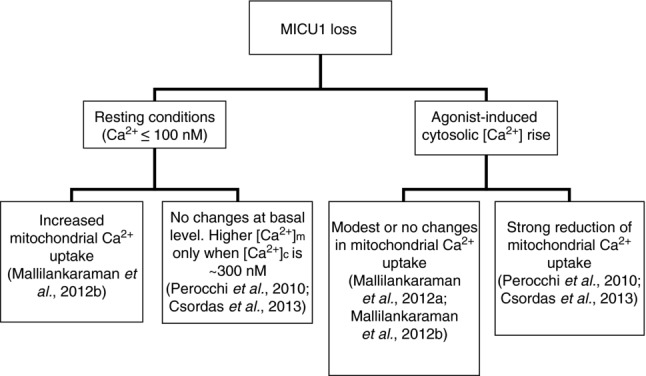
Dynamic diagram showing Ca2+ changes in the different states of cytosolic Ca2+ upon MICU1 knock-down
First, using proteinase K digestion with increasing concentrations of detergent to differently permeabilize the OMM and IMM, MICU1 was shown to be localized to the outer surface of the IMM, facing the intermembrane space rather than the matrix (Csordas et al. 2013). Nevertheless, proteomic mapping of the mitochondrial matrix, based on a combination of mass spectrometry and the employment of matrix-targeted APEX (Rhee et al. 2013; see the ‘MCU’ section), identified a matrix proteome of ∼500 proteins, and MICU1 was absent from this list. Thus, the localization of MICU1 at the IMS suggests a primary response to changes in the cytosolic [Ca2+] rather than the [Ca2+]m. Notably, the existence of a putative intermembrane component that is involved in regulating the activity of the uniporter was described more than 20 years ago (Igbavboa & Pfeiffer, 1991).
Second, Csordas et al. (2013) showed that MICU1 contributes to the cooperative activation of the uniporter at high cytosolic [Ca2+], whereas the same phenomenon was not observed in the previous paper published in Cell. The reason for this difference might be related to the composition of the experimental buffer, as several seminal studies reported that Mg2+ controls the allosteric activation of the uniporter (Bragadin et al. 1979). The effect of MICU1 deletion on MCU cooperativity has been detected exclusively in the presence of Mg2+, and similar measurements by Mallilankaraman et al. were performed in a Mg2+-free bath.
Third, although both of these studies agree that MICU1 is critical in keeping MCU closed (Mallilankaraman et al. 2012b; Csordas et al. 2013), Csordas and co-workers showed that the resting [Ca2+]m is unaltered and that the IP3-dependent Ca2+ uptake is mostly abolished in MICU1 knock-down cells (Csordas et al. 2013), in agreement with previous findings (Perocchi et al. 2010). Importantly, unchanged mitochondrial Ca2+ levels in MICU1-silenced cells have been detected using a non-ratiometric fluorescent dye-based Ca2+ indicator (Rhod-2; Mallilankaraman et al. 2012b), whereas a dramatic decrease in the [Ca2+]m has been measured upon agonist stimulation using genetically encoded Ca2+ sensors in calibrating aequorin-based assays (Perocchi et al. 2010) or ratiometric pericam (Csordas et al. 2013). Thus, the use of different Ca2+ tools is a possible explanation for these discrepancies with regard to the silencing approach. Both papers showed no difference in the MCU protein levels in MICU1-silenced cells (Mallilankaraman et al. 2012b; Csordas et al. 2013). However, the silencing of MICU1 in the mouse liver (based on the same strategy used in Csordas et al. 2013) appeared to have a major impact on the abundance of the MCU protein (but not its mRNA; Plovanich et al. 2013), and impaired mitochondrial Ca2+ handling might reasonably be correlated to the decrease in MCU stabilization.
In summary, on one hand, MICU1 stabilizes the closed state of the MCU complex, limiting mitochondrial Ca2+ entry under resting conditions or during small [Ca2+] elevations through a mechanism that requires its Ca2+-binding EF-hand domains (Mallilankaraman et al. 2012b) or is independent of its EF-hands (Csordas et al. 2013). On the other hand, MICU1 most likely cooperates with MCU to allow Ca2+ accumulation inside the matrix, but the extent of this cooperation is still a matter of debate (Fig. 2). However, the identification of new components of the uniporter complex, such as MICU2, the MCU paralogue MCUb and the regulator MCUR1, introduces additional players in the control of mitochondrial Ca2+ dynamics.
MICU2
MICU1 has two paralogues, namely, the protein products of the human genes EFHA1 and EFHA2, which share 25% sequence identity with MICU1. Both proteins possess N-terminal mitochondrial targeting sequences and are detected in multiple mouse tissues; thus, these proteins have been renamed MICU2 and MICU3, respectively (Plovanich et al. 2013). However, MICU3 does not display a strong or exclusive mitochondrial localization, leading to its exclusion from the MitoCarta list (Pagliarini et al. 2008). In contrast, MICU2 is a mitochondrially localized protein and, similar to MICU1, possesses highly conserved EF-hand domains. MICU2 interacts with MICU1 and MCU (Plovanich et al. 2013) and should reside at the IMS due to its analogy with MICU1 and its absence from the matrix protein list (Rhee et al. 2013). The in vivo silencing of MICU2 does not affect ΔΨ or mitochondrial respiration (Plovanich et al. 2013) but reduces mitochondrial Ca2+ clearance upon the addition of multiple Ca2+ spikes (Plovanich et al. 2013). Moreover, the silencing of both MICU1 and MICU2 shows additive Ca2+ defects, and MICU2 overexpression in MICU1-silenced HeLa cells restores the wild-type phenotype.
MICU1, MICU2 and MCU have been shown to reside within a complex (Plovanich et al. 2013). The interrelation of the expression levels of MICU1, MICU2 and MCU have been analysed in three cellular contexts:
(1) In the mouse liver, both MICU1 and MICU2 siRNAs lead to a decrease in MCU protein expression and a shift in the size of the MCU complex (from ∼480 kDa to ∼350 kDa).
(2) In HEK293 cells, MICU1 knock-down leads to a reduction in the level of MICU2 but not vice versa. Moreover, MCU overexpression increases the expression of both MICU1 and MICU2, and MICU1 up-regulation results in a higher level of MICU2.
(3) In HeLa cells, MICU1 down-regulation leads to a reduction in the level of the MICU2 protein (and vice versa). Of note, MCU expression seems to be inversed compared to MICU2 expression.
Further studies are required to clarify whether MICU1–2 and MCU regulate each other with respect to protein expression. Indeed, the silencing approach used might confound the results, changing the indirect activities for specific molecular features.
MCUb
CCDC109B, which was renamed MCUb, is an MCU paralogue/isogene. MCUb is a 33-kDa protein that shares 50% similarity with MCU. Its protein topology and structure is very similar to that of MCU, as it has two transmembrane domains with its N-and C-termini facing the IMS, but MCUb has a lower expression level and a different expression profile than MCU (Raffaello et al. 2013). MCU and MCUb can interact, and the reconstitution of MCUb in the lipid bilayer does not result in channel activity. Accordingly, MCUb overexpression in intact cells reduces mitochondrial Ca2+ uptake (Raffaello et al. 2013). Thus, MCUb might act as an endogenous dominant-negative isoform, and the insertion of one or more MCUb subunits in the multimer might alter Ca2+ permeation. The MCUb primary sequence differs from that of MCU by two residues in the putative pore-forming region. Indeed, the introduction of the same amino acid substitutions in MCU (R251W, D256V) blunted the increase in mitochondrial Ca2+ uptake (Raffaello et al. 2013).
However, the mRNA levels of MCUb are very low compared to those of MCU. MCUb mRNA is highly expressed in the heart and lung and minimally expressed in skeletal muscle. Interestingly, variations in tissue-dependent mitochondrial Ca2+ uptake have been recently reported, with a recorded skeletal muscle Ca2+ influx that is 28-fold greater than that in cardiac mitoplasts (measured in 100 μm [Ca2+] at −160 mV; Fieni et al. 2012).
MCUR1
Mitochondrial Ca2+ uniporter regulator 1 (MCUR1; previously known as CCDC90A) was identified as an essential regulator of Ca2+ uptake in an siRNA screen of 45 mitochondrial proteins that were predicted to be integral to the IMM or that have a well-documented role in mitochondrial Ca2+ homeostasis (Mallilankaraman et al. 2012a). MCUR1 silencing resulted in a dramatic reduction in the [Ca2+]m (approximately −85% in Rhod-2 fluorescence compared to the control) without modifying the cytosolic Ca2+ content. Interestingly, under these experimental conditions, MICU1 knock-down in HEK293 cells caused only minor Ca2+ changes (approximately −15% compared to the control siRNA).
MCUR1 interacts with MCU but not with MICU1, and pull-down assays suggested that these three proteins do not exist in the same complex (Mallilankaraman et al. 2012a). Importantly, MCUR1 overexpression in HeLa cells enhances [Ca2+]m, but this increase is strongly diminished by MCU knock-down. Accordingly, MCU overexpression fails to restore the Ca2+ levels in MCUR1-silenced cells, suggesting that both MCU and MCUR1 are required for efficient Ca2+ uptake through the uniporter. Notably, both MCU mRNA and protein expression are up-regulated in MCUR1-depleted cells, confirming the deep interrelation between these two proteins.
As for MCU and MICU1, MCUR1 itself has a paralogue, CCDC90B, whose function is still not clear. MCUR1 is 40-kDa protein that contains two transmembrane domains and one coiled-coil region, with N-and C-termini facing the same compartment. The topology of MCUR1 has been solved using a proteinase K-based biochemical assay, which showed that its N-and C-terminal residues are projected into the IMS (Mallilankaraman et al. 2012a). Thus, a large portion of the protein (∼250 aa) should span into the matrix, explaining the classification of MCUR1 as a mitomatrix protein (Rhee et al. 2013).
EMRE
Using stable isotope labelling by amino acids in cell culture (SILAC), a mass spectrometry-based proteomic approach, Mootha and co-workers identified EMRE (essential MCU regulator, previously known as C22ORF32) as a component of the uniporter complex (Sancak et al. 2013). EMRE is 10-kDa single-pass membrane protein, located at IMM, with a highly conserved aspartate-rich C-terminal region. EMRE is a member of the MitoCarta list (Pagliarini et al. 2008) and, notably, homologues have not been found in any plants, protozoa and fungi, indicating that EMRE could represent a metazoan innovation (Sancak et al. 2013).
EMRE interacts with MICU1 at IMS and with MCU oligomers in the inner membrane, thus EMRE seems to act as a bridge between the Ca2+-sensing activity of MICU1/MICU2 and the channel properties of MCU. Loss of EMRE induces a reduction of Ca2+ entry to the same extent as MCU depletion, suggesting that MCU requires EMRE for in vivo Ca2+ conductance (Sancak et al. 2013). MCU over-expression in EMRE-silenced cells failed to restore mitochondrial Ca2+ uptake. Interestingly, EMRE protein expression is strictly dependent on MCU levels, a partnership that could be analogous to that of MICU1/MICU2. Indeed, in MCU-depleted cells, EMRE abundance is drastically decreased (but not vice versa), despite no alteration in mRNA levels.
With the identification of EMRE and its ‘bridging activity’, all the members of the uniporter complex should be now defined. Based on SILAC results, the uniplex (uniporter complex) seems to be composed of MCU holomers, MCUb, MICU1, MICU2 and EMRE (Sancak et al. 2013). MCUR1 has not been found using this experimental approach, suggesting a role of this protein in Ca2+ handling outside the uniplex.
Physiopathological implications of the MCU complex
Mitochondrial Ca2+ uptake plays a critical role in the regulation of aerobic metabolism (Bonora et al. 2012) and cell survival (Giorgi et al. 2012). Several oncogenes and tumour suppressors manipulate Ca2+ to exert their anti/pro-apoptotic activities, and mitochondrial Ca2+ overload has been associated with apoptosis or necrosis in many pathological states (Giorgi et al. 2012). Accordingly, upon pro-apoptotic stimuli, MCU-expressing cells display an enhanced sensitivity to apoptosis, confirming that increased Ca2+ loading correlates with a predisposition for cell death (De Stefani et al. 2011). MCU expression and apoptosis are regulated by miRNA (Marchi et al. 2013). The screening of putative MCU-targeting miRNAs showed that miR-25 affects mitochondrial Ca2+ uptake through the specific down-regulation of MCU, conferring reduced mitochondrial Ca2+ content and resistance to Ca2+-dependent apoptotic challenges (Marchi et al. 2013). The alteration of the miRNA expression pattern could lead to a variety of human disorders, including cancer. Thus, miRNAs may function as oncogenes or tumour suppressors. The cancer-related miRNA miR-25 is up-regulated in various human cancers, including prostate and colon carcinomas. Indeed, colon adenocarcinoma samples with high miR-25 levels display low MCU expression (Marchi et al. 2013).
In addition to cancer, fundamental roles for MCU and mitochondrial Ca2+ uptake have been identified in specific cellular processes, which range from the regulation of gastrula morphogenesis in zebrafish (Prudent et al. 2013) to the control of excitotoxicity (Qiu et al. 2013). In cardiomyocytes, MCU silencing amplifies the bulk cytosolic [Ca2+] and is associated with increased contractile responses (Drago et al. 2012). Moreover, Ca2+–calmodulin-dependent protein kinase II (CaMKII), which is highly activated in ischaemia reperfusion and myocardial infarction, promotes myocardial death by increasing the current through the MCU complex (Joiner et al. 2012). CaMKII resides in the matrix, interacts with MCU and promotes mitochondrial Ca2+ entry, most likely by catalysing the phosphorylation of serines 57 and 92 (Joiner et al. 2012).
In pancreatic β-cells, MCU-and MICU1-dependent Ca2+ accumulation regulate the ATP level, glucose metabolism and insulin secretion (Alam et al. 2012; Tarasov et al. 2013). Interestingly, MCU silencing impairs the Ca2+-dependent phase of glucose-induced ATP increase and essentially eliminates secretion stimulated by tolbutamide, a potassium channel blocker used in the management of type II diabetes (Tarasov et al. 2013). Regulation of exocytosis by mitochondrial Ca2+ accumulation could involve both KATP-dependent or -independent hormone secretion. However, the lack of evidence for a role for mitochondrial Ca2+ uptake in the regulation of plasma membrane electrical dynamics might suggest a predominant involvement of the KATP-independent pathway (Tarasov et al. 2012).
The down-regulation of MICU1 dramatically elevates the basal levels of reactive oxygen species (ROS), particularly superoxide anion, and sensitizes the cells to apoptosis (Mallilankaraman et al. 2012b). As the thresholding activity of MICU1 plays a critical role in the regulation of mitochondrial oxidant signalling, the critical roles of MCU and MCUR1 in mitochondrial Ca2+ uptake affect various bioenergetic parameters. The absence of Ca2+ transfer from the ER to the mitochondria results in reduced O2 consumption and ATP levels and the activation of AMP kinase (AMPK), which, in turn, triggers pro-survival autophagy (Cardenas et al. 2010). Furthermore, the knock-down of MCU or MCUR1 induces bioenergetic stress, which is reflected by an increased AMP/ATP ratio and diminished oxidative phosphorylation, and the activation of the autophagic pathway (Mallilankaraman et al. 2012a,b2012b).
However, interesting results have been obtained through the characterization of an MCU-deficient mouse model (Pan et al. 2013). As expected, the drastic reduction in mitochondrial Ca2+ uptake correlates with higher pyruvate dehydrogenase (PDH) phosphorylation and consequent minor PDH activity in knock-out (KO) skeletal muscle mitochondria. MCU-null mice perform less efficiently under situations that require a rapid increase in skeletal muscle work load and a high expenditure of energy (Pan et al. 2013). These findings agree with the widely accepted view that the activation of matrix-located dehydrogenases is crucial for ATP supply under conditions of increased ATP demand. However, the other results of this study were highly unexpected. Surprisingly, no difference in oxygen consumption and autophagy levels were detected in MCU KO cells or tissues, suggesting that basal metabolism is not altered by MCU loss. Nevertheless, the absence of MCU expression does not confer any protection from cell death, although MCU KO mitochondria did not show Ca2+-induced mPTP opening (Pan et al. 2013). Similar effects have also been observed in vivo, using a model of ischaemic-reperfusion injury of the heart. Thus, in the absence of MCU, alternative mPTP-or Ca2+-independent cell death pathways might emerge, acting as predominant mechanisms of death in this scenario. Notably, MCU loss does not reset the matrix [Ca2+] at resting conditions (Pan et al. 2013); therefore, the activity of Ca2+ players that do not reside inside the uniporter complex might be enhanced to compensate for MCU deficiency, ensuring a slow, but continuous, Ca2+ influx inside the matrix.
In conclusion, we have summarized the recent findings on the molecular identities of all of the known members of the mitochondrial calcium uniporter complex.
Since the discovery of MICU1, which paved the way for the identification of MCU, the search for the functional roles of these key players has intensified. We are aware that the molecular study of mitochondrial Ca2+ signalling is just getting underway, and we can expect a rapid increase in the body of knowledge on the mitochondrial Ca2+ uptake machinery. In addition, it will be important to consider the link between the Ca2+ uniporter and other players, including Letm1 (Jiang et al. 2009), uncoupling proteins (Trenker et al. 2007), TRPC3 (Feng et al. 2013) and NCLX (Palty et al. 2010), that may influence the properties of the mitochondria with respect to both physiology and physiopathology.
Acknowledgments
None declared.
Glossary
- [Ca2+]m
mitochondrial [Ca2+]
- EMRE
essential MCU regulator
- ER
endoplasmic reticulum
- IMM
inner mitochondrial membrane
- IMS
intermembrane space
- IP3
inositol 1,4,5-trisphosphate
- MCU
mitochondrial calcium uniporter
- mPTP
mitochondrial permeability transition pore
- OMM
outer mitochondrial membrane
Biographies
Saverio Marchi obtained his PhD in ‘Pharmacology and Molecular Oncology’ from University of Ferrara. He studied Ca2+ signalling and mitochondrial dynamics in Dr R. Rizzuto's lab and the mechanisms of autophagy regulation in the lab of Dr G. Kroemer (Paris, France). He is now working in Dr P. Pinton's lab, supported by a FIRC fellowship.
Paolo Pinton is Professor of General Pathology at the University of Ferrara. Dr Pinton graduated from the laboratory of the Drs T. Pozzan and R. Rizzuto. He demonstrated close interactions between the ER and themitochondria. Afterwards, during his PhD, Dr Pinton obtained novel, unexpected information on intracellular Ca2+ homeostasis, including the role of the oncogene Bcl-2 in reducing Ca2+ levels in the ER and the importance of that reduction for its mechanism of action. Currently, Dr Pinton is obtaining interesting results on the signalling occurring at the mitochondria associated membranes in different physio-pathological conditions.
Additional information
Competing interests
None declared.
Author contributions
None declared.
Funding
S.M. is supported by a FIRC fellowship. Dr Pinton's lab is supported by grants from the Italian Association for Cancer Research (AIRC); Telethon (GGP11139B); local funds from the University of Ferrara; the Italian Ministry of Education, University and Research (COFIN, FIRB, and Futuro in Ricerca) and the Italian Ministry of Health to P.P.
References
- Alam MR, Groschner LN, Parichatikanond W, Kuo L, Bondarenko AI, Rost R, Waldeck-Weiermair M, Malli R, Graier WF. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J Biol Chem. 2012;287:34445–34454. doi: 10.1074/jbc.M112.392084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuld RA, Hohl CM, Castillo LC, Garleb AA, Starling RC, Brierley GP. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am J Physiol Heart Circ Physiol. 1992;262:H1699–H1704. doi: 10.1152/ajpheart.1992.262.6.H1699. [DOI] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick AG, Calvo SE, Mootha VK. Evolutionary diversity of the mitochondrial calcium uniporter. Science. 2012;336:886. doi: 10.1126/science.1214977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Giorgi C, Bononi A, Marchi S, Patergnani S, Rimessi A, Rizzuto R, Pinton P. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat Protoc. 2013;8:2105–2118. doi: 10.1038/nprot.2013.127. [DOI] [PubMed] [Google Scholar]
- Bonora M, Patergnani S, Rimessi A, De Marchi E, Suski JM, Bononi A, Giorgi C, Marchi S, Missiroli S, Poletti F, Wieckowski MR, Pinton P. ATP synthesis and storage. Purinergic Signal. 2012;8:343–357. doi: 10.1007/s11302-012-9305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragadin M, Pozzan T, Azzone GF. Kinetics of Ca2+ carrier in rat liver mitochondria. Biochemistry. 1979;18:5972–5978. doi: 10.1021/bi00593a033. [DOI] [PubMed] [Google Scholar]
- Brand MD. Electroneutral efflux of Ca2+ from liver mitochondria. Biochem J. 1985;225:413–419. doi: 10.1042/bj2250413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E, Balcavage WX, Lehninger AL, Mattoon JR. Ca2+ metabolism in yeast cells and mitochondria. Biochim Biophys Acta. 1970;205:18–26. doi: 10.1016/0005-2728(70)90057-5. [DOI] [PubMed] [Google Scholar]
- Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Sancak Y, Mootha VK, Clapham DE. MCU encodes the pore conducting mitochondrial calcium currents. Elife. 2013;2:e00704. doi: 10.7554/eLife.00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnoczky G. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013;17:976–987. doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente S, Fonteriz RI, Montero M, Alvarez J. Ca2+ homeostasis in the endoplasmic reticulum measured with a new low-Ca2+-affinity targeted aequorin. Cell Calcium. 2013;54:37–45. doi: 10.1016/j.ceca.2013.04.001. [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Navoni F, Casarin A, Trevisson E, Endele S, Winterpacht A, Salviati L, Scorrano L. LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum Mol Genet. 2008;17:201–214. doi: 10.1093/hmg/ddm297. [DOI] [PubMed] [Google Scholar]
- Docampo R, Lukes J. Trypanosomes and the solution to a 50-year mitochondrial calcium mystery. Trends Parasitol. 2012;28:31–37. doi: 10.1016/j.pt.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci U S A. 2012;109:12986–12991. doi: 10.1073/pnas.1210718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago I, Pizzo P, Pozzan T. After half a century mitochondrial calcium in-and efflux machineries reveal themselves. EMBO J. 2011;30:4119–4125. doi: 10.1038/emboj.2011.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Li H, Tai Y, Huang J, Su Y, Abramowitz J, Zhu MX, Birnbaumer L, Wang Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc Natl Acad Sci U S A. 2013;110:11011–11016. doi: 10.1073/pnas.1309531110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieni F, Lee SB, Jan YN, Kirichok Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun. 2012;3:1317. doi: 10.1038/ncomms2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A, Suski JM, Wieckowski MR, Pinton P. Mitochondrial Ca2+ and apoptosis. Cell Calcium. 2012;52:36–43. doi: 10.1016/j.ceca.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NE, Chandramoorthy HC, Shamugapriya S, Zhang X, Rajan S, Mallilankaraman K, Gandhirajan RK, Vagnozzi RJ, Ferrer LM, Sreekrishnanilayam K, Natarajaseenivasan K, Vallem S, Force T, Choi ET, Cheung JY, Madesh M. MICU1 Motifs Define Mitochondrial Calcium Uniporter Binding and Activity. Cell Rep. 2013 doi: 10.1016/j.celrep.2013.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Vercesi AE, Docampo R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat Commun. 2013;4 doi: 10.1038/ncomms3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igbavboa U, Pfeiffer DR. Regulation of reverse uniport activity in mitochondria by extramitochondrial divalent cations. Dependence on a soluble intermembrane space component. J Biol Chem. 1991;266:4283–4287. [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–147. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–273. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012a;14:1336–1343. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell. 2012b;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, Poletti F, Gafa R, Lanza G, Negrini M, Rizzuto R, Pinton P. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol. 2013;23:58–63. doi: 10.1016/j.cub.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol. 2012;30:1143–1148. doi: 10.1038/nbt.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Crompton M. Mitochondrial calcium transport. FEBS Lett. 1980;111:261–268. doi: 10.1016/0014-5793(80)80806-4. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panfili E, Sandri G, Sottocasa GL, Lunazzi G, Liut G, Graziosi G. Specific inhibition of mitochondrial Ca2+ transport by antibodies directed to the Ca2+-binding glycoprotein. Nature. 1976;264:185–186. doi: 10.1038/264185a0. [DOI] [PubMed] [Google Scholar]
- Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Calcium signalling around Mitochondria Associated Membranes (MAMs) Cell Commun Signal. 2011;9:19. doi: 10.1186/1478-811X-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One. 2013;8:e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent J, Popgeorgiev N, Bonneau B, Thibaut J, Gadet R, Lopez J, Gonzalo P, Rimokh R, Manon S, Houart C, Herbomel P, Aouacheria A, Gillet G. Bcl-wav and the mitochondrial calcium uniporter drive gastrula morphogenesis in zebrafish. Nat Commun. 2013;4:2330. doi: 10.1038/ncomms3330. [DOI] [PubMed] [Google Scholar]
- Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun. 2013;4:2034. doi: 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32:2362–2376. doi: 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rudolf R, Mongillo M, Rizzuto R, Pozzan T. Looking forward to seeing calcium. Nat Rev Mol Cell Biol. 2003;4:579–586. doi: 10.1038/nrm1153. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–1382. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santo-Domingo J, Demaurex N. Calcium uptake mechanisms of mitochondria. Biochim Biophys Acta. 2010;1797:907–912. doi: 10.1016/j.bbabio.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Saris NE, Sirota TV, Virtanen I, Niva K, Penttila T, Dolgachova LP, Mironova GD. Inhibition of the mitochondrial calcium uniporter by antibodies against a 40-kDa glycoproteinT. J Bioenerg Biomembr. 1993;25:307–312. doi: 10.1007/BF00762591. [DOI] [PubMed] [Google Scholar]
- Somlyo AP. Cell physiology: cellular site of calcium regulation. Nature. 1984;309:516–517. doi: 10.1038/309516b0. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Bond M, Somlyo AV. Calcium content of mitochondria and endoplasmic reticulum in liver frozen rapidly in vivo. Nature. 1985;314:622–625. doi: 10.1038/314622a0. [DOI] [PubMed] [Google Scholar]
- Sottocasa G, Sandri G, Panfili E, De Bernard B, Gazzotti P, Vasington FD, Carafoli E. Isolation of a soluble Ca2+ binding glycoprotein from ox liver mitochondria. Biochem Biophys Res Commun. 1972;47:808–813. doi: 10.1016/0006-291x(72)90564-5. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu SS, Gunter TE. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J Biol Chem. 1995;270:27510–27515. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 1983;306:67–69. doi: 10.1038/306067a0. [DOI] [PubMed] [Google Scholar]
- Tarasov AI, Semplici F, Li D, Rizzuto R, Ravier MA, Gilon P, Rutter GA. Frequency-dependent mitochondrial Ca2+ accumulation regulates ATP synthesis in pancreatic beta cells. Pflugers Arch. 2013;465:543–554. doi: 10.1007/s00424-012-1177-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic beta-cells. PLoS One. 2012;7:e39722. doi: 10.1371/journal.pone.0039722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol. 2007;9:445–452. doi: 10.1038/ncb1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei AC, Liu T, Cortassa S, Winslow RL, O'Rourke B. Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: low and high affinity effects of cyclosporine A. Biochim Biophys Acta. 2011;1813:1373–1381. doi: 10.1016/j.bbamcr.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zazueta C, Holguin JA, Ramirez J. Calcium transport sensitive to ruthenium red in cytochrome oxidase vesicles reconstituted with mitochondrial proteins. J Bioenerg Biomembr. 1991;23:889–902. doi: 10.1007/BF00786007. [DOI] [PubMed] [Google Scholar]