

Abstract
Herein, we review mechanisms regulating cerebral blood flow (CBF), with specific focus on humans. We revisit important concepts from the older literature and describe the interaction of various mechanisms of cerebrovascular control. We amalgamate this broad scope of information into a brief review, rather than detailing any one mechanism or area of research. The relationship between regulatory mechanisms is emphasized, but the following three broad categories of control are explicated: (1) the effect of blood gases and neuronal metabolism on CBF; (2) buffering of CBF with changes in blood pressure, termed cerebral autoregulation; and (3) the role of the autonomic nervous system in CBF regulation. With respect to these control mechanisms, we provide evidence against several canonized paradigms of CBF control. Specifically, we corroborate the following four key theses: (1) that cerebral autoregulation does not maintain constant perfusion through a mean arterial pressure range of 60–150 mmHg; (2) that there is important stimulatory synergism and regulatory interdependence of arterial blood gases and blood pressure on CBF regulation; (3) that cerebral autoregulation and cerebrovascular sensitivity to changes in arterial blood gases are not modulated solely at the pial arterioles; and (4) that neurogenic control of the cerebral vasculature is an important player in autoregulatory function and, crucially, acts to buffer surges in perfusion pressure. Finally, we summarize the state of our knowledge with respect to these areas, outline important gaps in the literature and suggest avenues for future research.
Introduction
Due to limited capacity for substrate storage (Brown & Ransom, 2007) and the high metabolic rate of brain tissue, the precise regulation of cerebral blood flow (CBF) is critical for maintenance of constant nutrient and oxygen supply to the brain. Substantial reductions in CBF quickly lead to unconsciousness (Van Lieshout et al. 2003) and, if maintained, brain damage and death ensues (Smith et al. 2011). Fastidious control of CBF involves a wide spectrum of overlapping regulatory mechanisms that together work to ensure optimal oxygen and nutrient delivery (Fig. 1). The fact that these mechanisms are present and largely unique to the cerebrovasculature does not obviate the importance of maintained systemic cardiovascular control. Yet, as will be explicated in this review, CBF regulation is often assumed to be so efficacious that it is treated separately, instead of as an integral component of the cardiovascular system.
Figure 1.

The central figure depicts the cerebrovasculature, comprised of two pairs of large arteries that branch from the sublavian arteries, i.e. the internal carotid arteries (ICAs) that carry ∼70% of total cerebral blood flow (CBF) and the vertebral arteries (VAs) that distribute ∼30% of total CBF to the brainstem, cerebellum and occipital cortex. Both the ICAs and VAs anastomose to form the circle of Willis before branching out into the main intracerebral arteries that ramify extensively en route to the brain surface. At the surface, the vessels form a dense network of highly vasoactive arterioles within the pia mater before they penetrate into the cortex (inlay II). The driving pressure in this system is the cerebral perfusion pressure (CPP) that is determined by the difference between mean arterial pressure (MAP) and intracranial pressure (ICP), in conditions where central venous pressure (CVP) is lower than ICP. In these conditions, MAP approximates CPP. Thus, it is important to note that the figure shows a schematic diagram of the cerebrovascular state in a resting supine human, and does not consider the myriad complex adjustments that take place with orthostatic stress (Gisolf et al. 2004; Hicks & Munis, 2005). As a result of the enclosed nature of the skull, ICP acts as a Starling resistor for cerebral venous outflow, a mechanism that is likely to be of greater importance with marked elevations in ICP or CVP, or both. The cerebral arteries (including the ICAs and VAs) are sensitive to changes in blood gases (Heistad et al. 1978a; Faraci et al. 1987a; Willie et al. 2012) and to changes in perfusion pressure, thus serving as a first-line defense in maintaining brain perfusion (Faraci et al. 1987a, b1987b; inlay I). These arteries are also densely innervated with branches of the cranial nerves, the carotid sinus nerve and branches from the superior cervical ganglion. The role of these nerves is contentious, but evidence favours cerebral constriction in response to increased sympathetic outflow and/or increased MAP, particularly at the tortuous segments where the ICA and VA vessels enter the skull. Turbulent blood flow through these segments increases resistance for a given luminal diameter according to Poiseuille's law; constriction of the vessel in these sections in the face of increased MAP attenuates pressure increases distal to the tortuous segment (inlay I). Inlay II shows a neurovascular unit. The pial vessels respond to changes in CPP, arterial partial pressures of O2 (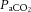 ) and CO2 (
) and CO2 (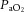 ), oxygen content, and proton concentration (Wolff & Lennox, 1930; Kontos et al. 1978; inlay III). The pial arteriole penetrates the pia mater through the Virchow–Robin space, where it becomes encapsulated by glial processes termed end-feet and pericytes that release vasoactive substances and respond mechanically by constricting or dilating with changes in metabolic demand of the surrounding neural matrix. Gap junctions between the endothelial and vascular smooth muscle cells allow for retrograde conductance of intramural vascular signals such that vasodilatory or constrictive signals pass to the pial arterioles. Thus, the neurovascular unit titrates blood flow to the metabolism of discrete cortical areas. Inlay III shows a qualitative schematic diagram of pial cross-sections against a hypothetical metabolic milieu spectrum. Note the vessels are not only exposed to arterial conditions but also to that of the cerebrospinal fluid that completely surrounds the pial vessel, tethered on all sides by thin processes to the pia mater. The vessels dilate with decreases in perfusion pressure, PaO2 and/or arterial O2 content (
), oxygen content, and proton concentration (Wolff & Lennox, 1930; Kontos et al. 1978; inlay III). The pial arteriole penetrates the pia mater through the Virchow–Robin space, where it becomes encapsulated by glial processes termed end-feet and pericytes that release vasoactive substances and respond mechanically by constricting or dilating with changes in metabolic demand of the surrounding neural matrix. Gap junctions between the endothelial and vascular smooth muscle cells allow for retrograde conductance of intramural vascular signals such that vasodilatory or constrictive signals pass to the pial arterioles. Thus, the neurovascular unit titrates blood flow to the metabolism of discrete cortical areas. Inlay III shows a qualitative schematic diagram of pial cross-sections against a hypothetical metabolic milieu spectrum. Note the vessels are not only exposed to arterial conditions but also to that of the cerebrospinal fluid that completely surrounds the pial vessel, tethered on all sides by thin processes to the pia mater. The vessels dilate with decreases in perfusion pressure, PaO2 and/or arterial O2 content (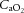 ) and increases in
) and increases in 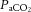 and/or [H+].
and/or [H+].
The partial pressure of arterial carbon dioxide (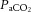 ), mean arterial pressure (MAP), cerebral metabolism and the autonomic nervous system are the principal regulators of CBF. The regulation of CBF should not, therefore, be viewed as being limited to mechanisms within the cranium. Rather, the regulation of CBF should be noted as an integrative process that involves the marked influence of pulmonary gas exchange and cardiovascular function in addition to intracranial mediators of cerebral vessel resistance (and therefore flow). Despite over a century of study (Mosso, 1880; Roy & Sherrington, 1890), establishing an integrative understanding of these mechanisms in humans has been difficult (if not impossible) to achieve for a number of reasons. Over the past 50 years, a reductionist approach to cerebrovascular physiology has dominated the field, largely because of the difficulty of in vivo brain vascular assessment. The prevailing use of transcranial Doppler ultrasound in human CBF research since the late 1980s introduced assumptions of intracerebral vessel characteristics that are increasingly being shown to be incorrect. Finally, there has been limited study of CBF regulatory mechanisms in humans in the literature of the past decade.
), mean arterial pressure (MAP), cerebral metabolism and the autonomic nervous system are the principal regulators of CBF. The regulation of CBF should not, therefore, be viewed as being limited to mechanisms within the cranium. Rather, the regulation of CBF should be noted as an integrative process that involves the marked influence of pulmonary gas exchange and cardiovascular function in addition to intracranial mediators of cerebral vessel resistance (and therefore flow). Despite over a century of study (Mosso, 1880; Roy & Sherrington, 1890), establishing an integrative understanding of these mechanisms in humans has been difficult (if not impossible) to achieve for a number of reasons. Over the past 50 years, a reductionist approach to cerebrovascular physiology has dominated the field, largely because of the difficulty of in vivo brain vascular assessment. The prevailing use of transcranial Doppler ultrasound in human CBF research since the late 1980s introduced assumptions of intracerebral vessel characteristics that are increasingly being shown to be incorrect. Finally, there has been limited study of CBF regulatory mechanisms in humans in the literature of the past decade.
This review aims to concentrate on human cerebrovascular control from an integrative standpoint in the hope of stimulating resurgent interest in a field of study that lags behind those of other systems of the body. We endeavour to synthesize an expansive scope of information into a brief review rather than detail specific areas or mechanisms of cerebrovascular control; there are specific reviews on many of the topics we cover herein, to which the reader will be referred throughout.
Four broad sections, each focusing on an important facet of cerebrovascular control, are delineated below. The first section discusses the metabolic control of CBF, from systemic metabolism and the consequent coupling of arterial blood gases and CBF to the effect of local neuronal metabolism on local CBF. The second section provides an overview of how the cerebrovasculature buffers changes in blood pressure (BP), termed cerebral autoregulation (CA), and how systemic and local metabolism might, in turn, affect CA. The third section covers the role of the autonomic nervous system in CBF regulation and, finally, the fourth section summarizes the state of our knowledge on cerebrovascular regulation, reiterating key gaps in our understanding and suggesting avenues for future research.
Metabolic regulation of cerebral blood flow
Regulation by arterial blood gases
Brain perfusion is highly sensitive to changes in changes in 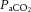 . Studies using transcranial Doppler ultrasound of the middle (Ide et al. 2003; Battisti-Charbonney et al. 2011) and posterior cerebral arteries and the basilar artery (Skow et al. 2013), and Duplex ultrasound of the internal carotid artery (ICA) and vertebral artery (VA; Sato et al. 2012; Willie et al. 2012) all show an approximate 3–6% increase and/or 1–3% decrease in flow per millimetre of mercury change in CO2 above and below eupnoeic
. Studies using transcranial Doppler ultrasound of the middle (Ide et al. 2003; Battisti-Charbonney et al. 2011) and posterior cerebral arteries and the basilar artery (Skow et al. 2013), and Duplex ultrasound of the internal carotid artery (ICA) and vertebral artery (VA; Sato et al. 2012; Willie et al. 2012) all show an approximate 3–6% increase and/or 1–3% decrease in flow per millimetre of mercury change in CO2 above and below eupnoeic 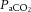 , respectively (see Figure 2, left). Imaging studies show broadly comparable results (Kemna et al. 2001; Mandell et al. 2008; Piechnik et al. 2008). It is important to recognize that methodological differences make comparison between studies difficult. These differences include the following factors: steady-state versus rebreathing methods of CO2 manipulation; linear versus non-linear analyses of the cerebrovascular response to hypocapnia, hypercapnia or the entire manipulated range of CO2; whether arterial partial pressure of O2 (
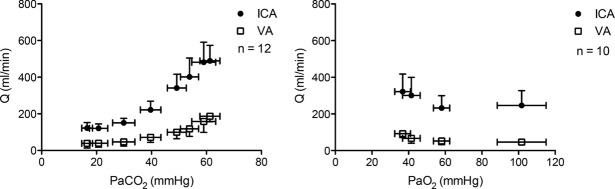
, respectively (see Figure 2, left). Imaging studies show broadly comparable results (Kemna et al. 2001; Mandell et al. 2008; Piechnik et al. 2008). It is important to recognize that methodological differences make comparison between studies difficult. These differences include the following factors: steady-state versus rebreathing methods of CO2 manipulation; linear versus non-linear analyses of the cerebrovascular response to hypocapnia, hypercapnia or the entire manipulated range of CO2; whether arterial partial pressure of O2 (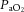 ) is maintained during CO2 manipulation; and the method of CBF measurement. All these factors influence the values of CBF reactivity to changes in
) is maintained during CO2 manipulation; and the method of CBF measurement. All these factors influence the values of CBF reactivity to changes in 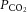 (extensively reviewed by Ainslie & Duffin, 2009; Fierstra et al. 2013). Regardless, this high vascular sensitivity to CO2 is unique to the cerebrovasculature (Ainslie et al. 2005) and is manifest throughout, from the large arteries of the neck (Willie et al. 2012) through the large intracranial arteries (Giller et al. 1993; Wilson et al. 2011; Willie et al. 2013b) to the smallest pial arterioles (Wolff & Lennox, 1930) and parenchymal vessels (Binks et al. 2008; Mandell et al. 2008; Nöth et al. 2008; Piechnik et al. 2008). This sensitivity appears to be similar between brain regions in the hypercapnic range, but dissimilar with hypocapnia (Sato et al. 2012; Willie et al. 2012), as assessed by flow through the arteries of the neck. Based on magnetic resonance imaging (MRI) data, CO2 reactivity of the microvasculature in grey matter is greater than that of white matter, probably because of relatively less vascularization (Mandell et al. 2008; Nöth et al. 2008).
(extensively reviewed by Ainslie & Duffin, 2009; Fierstra et al. 2013). Regardless, this high vascular sensitivity to CO2 is unique to the cerebrovasculature (Ainslie et al. 2005) and is manifest throughout, from the large arteries of the neck (Willie et al. 2012) through the large intracranial arteries (Giller et al. 1993; Wilson et al. 2011; Willie et al. 2013b) to the smallest pial arterioles (Wolff & Lennox, 1930) and parenchymal vessels (Binks et al. 2008; Mandell et al. 2008; Nöth et al. 2008; Piechnik et al. 2008). This sensitivity appears to be similar between brain regions in the hypercapnic range, but dissimilar with hypocapnia (Sato et al. 2012; Willie et al. 2012), as assessed by flow through the arteries of the neck. Based on magnetic resonance imaging (MRI) data, CO2 reactivity of the microvasculature in grey matter is greater than that of white matter, probably because of relatively less vascularization (Mandell et al. 2008; Nöth et al. 2008).
Figure 2.
Cerebrovascular reactivity to changes in CO2 and to hypoxia (%ΔCBF / mmHg CO2) and %ΔCBF / %SaO2) was found to be similar between vessels in the hypercapnic range, ∼10% greater for the VA than the ICA in the hypocapnic range, and 50% greater for the VA with extreme hypoxia.
The cerebrovasculature is sensitive to hypoxia, but only below a 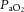 of ∼50 mmHg, (see Figure 2, right). The response is dependent on the prevailing
of ∼50 mmHg, (see Figure 2, right). The response is dependent on the prevailing 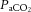 ; hypercapnia increases and hypocapnia decreases cerebrovascular sensitivity to hypoxia (Mardimae et al. 2012). Studies of hypoxic cerebrovascular reactivity are thus confounded by the ventilatory response to hypoxia, which produces hypocapnia and results in cerebrovascular constriction (i.e. poikilocapnia). Studies incorporating a range of techniques that have assessed the CBF response to isocapnic hypoxia have reported CBF reactivities ranging from 0.5 to 2.5% increase in CBF per percentage point reduction in arterial saturation of O2 (Cohen et al. 1967; Shapiro et al. 1970; Jensen et al. 1996; Querido et al. 2008, 2013; Reichmuth et al. 2009; Willie et al. 2012). Variability in the methods of blood gas manipulation, consequent changes in BP and degree of (or lack of)
; hypercapnia increases and hypocapnia decreases cerebrovascular sensitivity to hypoxia (Mardimae et al. 2012). Studies of hypoxic cerebrovascular reactivity are thus confounded by the ventilatory response to hypoxia, which produces hypocapnia and results in cerebrovascular constriction (i.e. poikilocapnia). Studies incorporating a range of techniques that have assessed the CBF response to isocapnic hypoxia have reported CBF reactivities ranging from 0.5 to 2.5% increase in CBF per percentage point reduction in arterial saturation of O2 (Cohen et al. 1967; Shapiro et al. 1970; Jensen et al. 1996; Querido et al. 2008, 2013; Reichmuth et al. 2009; Willie et al. 2012). Variability in the methods of blood gas manipulation, consequent changes in BP and degree of (or lack of) 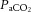 clamping necessitated by the ventilatory response to hypoxia (Shapiro et al. 1970; Kolb et al. 2004; Willie et al. 2012) lead to variation in population norms, and values in the literature vary considerably, perhaps exacerbated by different sensitivities to hypoxia between brain regions. For a given severity of isocapnic hypoxia, blood flow to the brainstem increases more than that to the middle and anterior regions, as assessed by flow through the vertebral and internal carotid arteries, respectfully (Willie et al. 2012; Ogoh et al. 2013). Congruous positron emission tomography (PET) scan data collected during isocapnic hypoxia reveal that cortical blood flow is less responsive to hypoxia than phylogenetically older areas of the brain (Binks et al. 2008). Unlike the response to
clamping necessitated by the ventilatory response to hypoxia (Shapiro et al. 1970; Kolb et al. 2004; Willie et al. 2012) lead to variation in population norms, and values in the literature vary considerably, perhaps exacerbated by different sensitivities to hypoxia between brain regions. For a given severity of isocapnic hypoxia, blood flow to the brainstem increases more than that to the middle and anterior regions, as assessed by flow through the vertebral and internal carotid arteries, respectfully (Willie et al. 2012; Ogoh et al. 2013). Congruous positron emission tomography (PET) scan data collected during isocapnic hypoxia reveal that cortical blood flow is less responsive to hypoxia than phylogenetically older areas of the brain (Binks et al. 2008). Unlike the response to 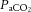 , the CBF response to oxygen appears to be determined by oxygen content rather than
, the CBF response to oxygen appears to be determined by oxygen content rather than 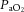 per se, because a reduction in oxygen content resulting from carbon monoxide exposure, acute or chronic anaemia and haemodilution produces increased CBF (Paulson et al. 1973; Brown et al. 1985; Todd et al. 1994; Metry et al. 1999; Hare, 2004; Gottesman et al. 2012). Indeed, the limited data available suggest that the inverse relationship between blood haematocrit (ergo viscosity) and CBF (Muizelaar et al. 1986; Metry et al. 1999) is a function of oxygen delivery rather than viscosity per se (Todd et al. 1994; Tomiyama et al. 1999), but this remains to be studied explicitly in humans.
per se, because a reduction in oxygen content resulting from carbon monoxide exposure, acute or chronic anaemia and haemodilution produces increased CBF (Paulson et al. 1973; Brown et al. 1985; Todd et al. 1994; Metry et al. 1999; Hare, 2004; Gottesman et al. 2012). Indeed, the limited data available suggest that the inverse relationship between blood haematocrit (ergo viscosity) and CBF (Muizelaar et al. 1986; Metry et al. 1999) is a function of oxygen delivery rather than viscosity per se (Todd et al. 1994; Tomiyama et al. 1999), but this remains to be studied explicitly in humans.
Locations of CO2 and O2 sensitivity
Although the entire cerebrovasculature, from the large arteries of the neck to the penetrating cortical arterioles, is sensitive to changes in blood gases, the pial arterioles are generally considered to be the site of resistance modulation. The pial vessel response (dilatation) to asphyxia was observed ∼150 years ago in rabbits (Donders, 1851). Pial arteries dilate up to 40% in response to both increased 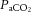 and increased cerebrospinal fluid (CSF)
and increased cerebrospinal fluid (CSF) 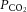 (Wolff & Lennox, 1930; Kontos et al. 1977b). Increased
(Wolff & Lennox, 1930; Kontos et al. 1977b). Increased 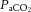 produces smooth muscle relaxation, vessel dilatation and increased flow, whereas hypocapnia increases cerebrovascular resistance and decreases CBF (Kety & Schmidt, 1948; Wasserman & Patterson, 1961). Their anatomical position in the subarachnoid space, tethered to the abutting mater and surrounded by CSF (Fig. 1, inlays II and III), makes them readily exposed to local metabolic conditions. Thus, the tone of the pial vessels is a function of arterial blood gases, arterial pH and local CSF. As described below (see ‘Neurovascular coupling’), pial vessel resistance is also coupled to downstream metabolic activity via retrograde intramural propagation of vascular signals (Segal, 2000; Lagaud et al. 2002; Kawamura et al. 2003; Attwell et al. 2010; Itoh & Suzuki, 2012).
produces smooth muscle relaxation, vessel dilatation and increased flow, whereas hypocapnia increases cerebrovascular resistance and decreases CBF (Kety & Schmidt, 1948; Wasserman & Patterson, 1961). Their anatomical position in the subarachnoid space, tethered to the abutting mater and surrounded by CSF (Fig. 1, inlays II and III), makes them readily exposed to local metabolic conditions. Thus, the tone of the pial vessels is a function of arterial blood gases, arterial pH and local CSF. As described below (see ‘Neurovascular coupling’), pial vessel resistance is also coupled to downstream metabolic activity via retrograde intramural propagation of vascular signals (Segal, 2000; Lagaud et al. 2002; Kawamura et al. 2003; Attwell et al. 2010; Itoh & Suzuki, 2012).
The cerebral arteries (including the internal carotid and vertebral arteries) are also sensitive to changes in blood gases (Heistad et al. 1978a; Faraci et al. 1987a; Willie et al. 2012) and perfusion pressure. While the pial arteriolar bed serves to modulate regional blood flow, the large vessels serve as a ‘first-line’ defense in maintaining brain perfusion (Faraci et al. 1987a,b1987b; Fig. 1, inlay I). It has recently been demonstrated that the ICA and VA of humans are reactive to changes in arterial blood gases, with the ICA showing a ∼20% change in luminal diameter through a 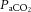 range of 15–65 mmHg (Willie et al. 2012). The fact that the entire cerebrovascular arterial tree is vasoactive is a key feature of this system. Unified vasomotion allows microvascular pressure to remain relatively constant. For example, if constriction of only the large arteries were to occur, decreased pial artery pressure would result, whereas constriction along the entire cerebrovascular tree reduces flow with no changes in pial pressure (Baumbach & Heistad, 1983). Hypocapnia, for example, therefore produces no changes in small arteriole pressure despite attenuated flow, because small and large arteries both constrict in response to reduced
range of 15–65 mmHg (Willie et al. 2012). The fact that the entire cerebrovascular arterial tree is vasoactive is a key feature of this system. Unified vasomotion allows microvascular pressure to remain relatively constant. For example, if constriction of only the large arteries were to occur, decreased pial artery pressure would result, whereas constriction along the entire cerebrovascular tree reduces flow with no changes in pial pressure (Baumbach & Heistad, 1983). Hypocapnia, for example, therefore produces no changes in small arteriole pressure despite attenuated flow, because small and large arteries both constrict in response to reduced 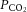 (Faraci et al. 1987a).
(Faraci et al. 1987a).
Mechanisms of cerebrovascular sensitivity to arterial blood gases
The cellular mechanisms responsible for the cerebrovascular response to changes in 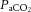 and
and 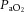 have been subject of countless studies. Yet, the resulting diversity of conclusions serves largely to demonstrate a mechanistic redundancy inherent to the precise cerebrovascular regulation by arterial blood gases. Next, we will focus on human and select in vivo animal studies. Studies employing various in vitro vessel preparations are difficult to compare and apply to in vivo physiology; for detailed reviews of the in vivo literature see Faraci & Heistad, 1998; Yoon et al. 2012.
have been subject of countless studies. Yet, the resulting diversity of conclusions serves largely to demonstrate a mechanistic redundancy inherent to the precise cerebrovascular regulation by arterial blood gases. Next, we will focus on human and select in vivo animal studies. Studies employing various in vitro vessel preparations are difficult to compare and apply to in vivo physiology; for detailed reviews of the in vivo literature see Faraci & Heistad, 1998; Yoon et al. 2012.
An alteration in alveolar gas exchange (either by changes in alveolar ventilation and/or metabolic CO2 production) elicits concomitant changes in both 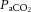 and pH. The latter is thus influenced by both respiratory alkalosis and acidosis in the short term and over longer periods by the degree of renal compensation. However, a long-standing question has been which of these (
and pH. The latter is thus influenced by both respiratory alkalosis and acidosis in the short term and over longer periods by the degree of renal compensation. However, a long-standing question has been which of these (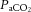 or pH), or both, are responsible for the resultant change in CBF. Direct manipulation of arterial pH does not alter CBF in conditions of maintained
or pH), or both, are responsible for the resultant change in CBF. Direct manipulation of arterial pH does not alter CBF in conditions of maintained 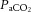 (Lambertsen et al. 1961; Harper & Bell, 1963). In contrast, manipulation of extravascular pH induces changes in arteriolar diameter (Wahl et al. 1970; Kontos et al. 1977a,b1977b). These observations suggest that the CO2 mechanism is independent of arterial pH and is therefore likely to be dependent on the diffusion of non-polar CO2 molecules across the cerebrovascular blood–brain barrier that can induce a change in pH in the extracellular space of the vessel, and thus alter vascular smooth muscle tone (Lambertsen et al. 1961; Lassen, 1968). This model is supported by animal work using in vivo cranial windows and superficial application of solutions with varying
(Lambertsen et al. 1961; Harper & Bell, 1963). In contrast, manipulation of extravascular pH induces changes in arteriolar diameter (Wahl et al. 1970; Kontos et al. 1977a,b1977b). These observations suggest that the CO2 mechanism is independent of arterial pH and is therefore likely to be dependent on the diffusion of non-polar CO2 molecules across the cerebrovascular blood–brain barrier that can induce a change in pH in the extracellular space of the vessel, and thus alter vascular smooth muscle tone (Lambertsen et al. 1961; Lassen, 1968). This model is supported by animal work using in vivo cranial windows and superficial application of solutions with varying 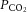 and pH (Wolff & Lennox, 1930; Kontos et al. 1977b). For example, hypercapnic and hypocapnic solutions, respectively, cause pial arteriolar dilatation and constriction, providing the superfusate pH is correspondingly acidic or alkaline. In contrast, application of solutions of neutral pH elicits no vasomotion regardless of superfusate
and pH (Wolff & Lennox, 1930; Kontos et al. 1977b). For example, hypercapnic and hypocapnic solutions, respectively, cause pial arteriolar dilatation and constriction, providing the superfusate pH is correspondingly acidic or alkaline. In contrast, application of solutions of neutral pH elicits no vasomotion regardless of superfusate 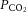 (Wahl et al. 1970; Kontos et al. 1977a,b1977b). Moreover, the effect of arterial hypercapnia is nullified with external (i.e. around the pial vessel) application of alkaline superfusate in rats (Liu et al. 2012), dogs (Koehler & Traystman, 1982) and cats (Kontos et al. 1977b). Likewise, cerebral vascular smooth muscle cells contract with increased pH and relax with decreased pH (Apkon et al. 1997; Peng et al. 1998). Such evidence for the importance of extracellular pH has been corroborated more recently by in vivo vascular preparations as well (e.g. Toda & Okamura, 1998; Dabertrand et al. 2012). The net CBF response is thus a balance between the effects of CO2
per se and endothelial signals in response to changes in flow.
(Wahl et al. 1970; Kontos et al. 1977a,b1977b). Moreover, the effect of arterial hypercapnia is nullified with external (i.e. around the pial vessel) application of alkaline superfusate in rats (Liu et al. 2012), dogs (Koehler & Traystman, 1982) and cats (Kontos et al. 1977b). Likewise, cerebral vascular smooth muscle cells contract with increased pH and relax with decreased pH (Apkon et al. 1997; Peng et al. 1998). Such evidence for the importance of extracellular pH has been corroborated more recently by in vivo vascular preparations as well (e.g. Toda & Okamura, 1998; Dabertrand et al. 2012). The net CBF response is thus a balance between the effects of CO2
per se and endothelial signals in response to changes in flow.
Unlike the cerebrovascular response to changes in 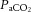 , hypoxaemia causes little change in CBF until a threshold at the steep portion of the oxyhaemoglobin dissociation curve (∼80% arterial saturation of O2). Hypoxia appears to reduce cerebrovascular smooth muscle tone through activation of membrane potassium channels (Bonnet et al. 1991; Gebremedhin et al. 2008) and interference with transmembrane calcium flux (Vinall & Simeone, 1986; Pearce et al. 1992; reviewed by Pearce, 1995). Studies in baboons (James et al. 1969) and dogs (McDowall, 1966; Kogure et al. 1970) showed CBF to begin increasing at a
, hypoxaemia causes little change in CBF until a threshold at the steep portion of the oxyhaemoglobin dissociation curve (∼80% arterial saturation of O2). Hypoxia appears to reduce cerebrovascular smooth muscle tone through activation of membrane potassium channels (Bonnet et al. 1991; Gebremedhin et al. 2008) and interference with transmembrane calcium flux (Vinall & Simeone, 1986; Pearce et al. 1992; reviewed by Pearce, 1995). Studies in baboons (James et al. 1969) and dogs (McDowall, 1966; Kogure et al. 1970) showed CBF to begin increasing at a 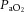 of ∼50 mmHg. Similar data have been published for humans (Ainslie & Poulin, 2004; Willie et al. 2012). The processes involved in the hypoxaemia-induced increase in CBF are multifaceted, probably comprising the following: (1) a retrograde stimulus arising at the neurons/glia of the neurovascular unit, i.e. neurovascular coupling, in response to local decreases in tissue oxygen (Pelligrino et al. 1995; Thompson et al. 2003; Iadecola & Nedergaard, 2007; Gordon et al. 2008; Leithner & Royl, 2014); (2) brain extracellular acidosis due to increased neuronal/glial anaerobic metabolism eliciting vascular dilatation (Kogure et al. 1970; Nolan et al. 1982); and (3) direct vascular mechanisms (see next paragraph). The relative contribution and importance of these processes remain unknown. Particularly with respect to the direct neuronal/glial contribution to hypoxic cerebrovascular dilatation there are few data (Gordon et al. 2008), but a direct CNS effect of hypoxia in ventilatory regulation has been described (reviewed by Powell et al. 2000; Joseph & Pequignot, 2009). In humans, however, there have been surprisingly few studies that have attempted to examine potential mechanisms by which hypoxia leads to cerebral vasodilatation. Those that have been done in humans have focused principally on the role of adenosine and NO and are considered in detail next.
of ∼50 mmHg. Similar data have been published for humans (Ainslie & Poulin, 2004; Willie et al. 2012). The processes involved in the hypoxaemia-induced increase in CBF are multifaceted, probably comprising the following: (1) a retrograde stimulus arising at the neurons/glia of the neurovascular unit, i.e. neurovascular coupling, in response to local decreases in tissue oxygen (Pelligrino et al. 1995; Thompson et al. 2003; Iadecola & Nedergaard, 2007; Gordon et al. 2008; Leithner & Royl, 2014); (2) brain extracellular acidosis due to increased neuronal/glial anaerobic metabolism eliciting vascular dilatation (Kogure et al. 1970; Nolan et al. 1982); and (3) direct vascular mechanisms (see next paragraph). The relative contribution and importance of these processes remain unknown. Particularly with respect to the direct neuronal/glial contribution to hypoxic cerebrovascular dilatation there are few data (Gordon et al. 2008), but a direct CNS effect of hypoxia in ventilatory regulation has been described (reviewed by Powell et al. 2000; Joseph & Pequignot, 2009). In humans, however, there have been surprisingly few studies that have attempted to examine potential mechanisms by which hypoxia leads to cerebral vasodilatation. Those that have been done in humans have focused principally on the role of adenosine and NO and are considered in detail next.
Adenosine is popularly held to mediate hypoxic cerebral vasodilatation. This is based on the following evidence: (1) adenosine is released with hypoxaemia (Winn et al. 1981; Meno et al. 1993); (2) in most animal studies adenosine receptor antagonists attenuate the increase in CBF during hypoxaemia (Haller & Kuschinsky, 1987; Morii et al. 1987; Pinard et al. 1989; Laudignon et al. 1990; Coney & Marshall, 1998; Miekisiak et al. 2008); and (3) in vitro data indicate that adenosine blocks vasoconstrictive signals within the parenchyma (Gordon et al. 2008). The few studies assessing the role of adenosine on the human cerebrovasculature have reported a 20–30% decrease in CBF and cerebral oxygen delivery in normoxia (Wechsler et al. 1950; Gottstein & Paulson, 1972; Magnussen & Hoedt-Rasmussen, 1977), during competitive adenosine receptor antagonism with aminophylline. Nevertheless, following adenosine receptor blockade, the cerebral hypoxic vasodilatory response in fact remains intact; CBF decreases with aminophylline administration in normoxia but increases with hypoxic exposure, albeit to values approximately equal to normoxic control conditions (Bowton et al. 1988; Nishimura et al. 1992, 1993). Thus, while the release of adenosine during hypoxia certainly implicates it in the normal hypoxic cerebral vasodilatory response, it cannot be the sole mediator of the hypoxaemia-induced increase in CBF in humans.
There is similar confusion concerning the role of nitric oxide in mediating hypoxic cerebrovascular dilatation. Studies in animals have reported substantial effects of nitric oxide synthase inhibition on the CBF response to moderate hypoxia (Hudetz et al. 1998; Santizo et al. 2000; Bauser-Heaton & Bohlen, 2007), whereas others have reported little effect (Pelligrino et al. 1993, 1995). Only two studies in humans have assessed nitric oxide synthase inhibition [both via NG-monomethyl-l-arginine (l-LMMA) infusion] on the cerebrovascular response to 20 min hypoxaemia. Using phase-contrast MRI to measure CBF in healthy young men, Van Mil et al. (2002) reported that CBF returned to near-normoxic values following l-NMMA administration during hypoxia (Oxygen saturation (SpO2) = 80%). Conversely, Ide et al. (2007) found l-NMMA to have no effect on the increased CBF with slightly less severe hypoxia (end-tidal 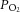 = 50 mmHg; SpO2 not reported) as assessed with transcranial Doppler ultrasound of the middle cerebral artery. Aside from a larger l-NMMA dose used by Ide et al. (2007) and the slightly more severe hypoxia used by Van Mil et al. (2002) there was no apparent difference in either the methodology or the reported physiological response of the subjects in these studies (except for CBF), including end-tidal
= 50 mmHg; SpO2 not reported) as assessed with transcranial Doppler ultrasound of the middle cerebral artery. Aside from a larger l-NMMA dose used by Ide et al. (2007) and the slightly more severe hypoxia used by Van Mil et al. (2002) there was no apparent difference in either the methodology or the reported physiological response of the subjects in these studies (except for CBF), including end-tidal 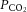 . A speculative explanation for these discrepant findings is a change in middle cerebral artery diameter following hypoxia and/or l-NMMA infusion that would confound the estimations of CBF by Ide et al. (2007); indeed, middle cerebral artery dilatation in hypoxia is now well documented (Wilson et al. 2011; Willie et al. 2012, 2013b), as well as after glycerol trinitrate administration (Hansen et al. 2007).
. A speculative explanation for these discrepant findings is a change in middle cerebral artery diameter following hypoxia and/or l-NMMA infusion that would confound the estimations of CBF by Ide et al. (2007); indeed, middle cerebral artery dilatation in hypoxia is now well documented (Wilson et al. 2011; Willie et al. 2012, 2013b), as well as after glycerol trinitrate administration (Hansen et al. 2007).
Local CBF is tightly coupled to neural metabolism, which is a function of the exquisite physical association between neurons, glia and the microvasculature, together termed the neurovascular unit (see ‘Neurovascular coupling’, below). Excitation of astrocytes by hypoxia in vitro stimulates the direct and indirect production arachidonic acid metabolites associated with local vasodilatation (Liu & Alkayed, 2005; Yamaura et al. 2006). It is difficult to identify in humans the mechanisms of neurovascular coupling involved in the hypoxic vasodilatory response. Given that transient local hypoxia following increased local metabolism is thought to mediate in part the blood oxygen level dependent (BOLD) MRI response during cognitive activation (Vanzetta & Grinvald, 1999; Thompson et al. 2003; Offenhauser et al. 2005), it is likely that the neurovascular unit is only one of the mechanisms involved in cerebral vasodilatation in hypoxia. This finding suggests both mechanistic redundancy of mediators of hypoxic vasodilatation and synergism between the neurovascular unit and the larger arteries and arterioles of the cerebrovasculature due to their similar responses to hypoxia.
Neurovascular coupling
The fact that local cerebral metabolism is tightly coupled to local brain perfusion has been known, although not understood, for over a century (Donders, 1851; Mosso, 1880; Roy & Sherrington, 1890). This coupling is a product of the anatomical and metabolic relationship between neurons, glial cells and cortical penetrating arterioles (Fig. 1, inlay II) that together comprise the neurovascular unit. Excitatory and inhibitory neurons synapse on both astrocytes and GABAergic interneurons, these interneurons being in close association with astrocytes having processes that terminate in end-feet enveloping cortex-penetrating arterioles. By way of gap junctions between adjacent vascular smooth muscle cells, intramural propagation of vascular signals produces remote vasodilatation of upstream pial arterioles (Fig. 1, inlay II; Lagaud et al. 2002; Kawamura et al. 2003; Iadecola, 2004). The result is a robust coupling of neuronal activation to regional CBF that can be observed easily with transcranial Doppler ultrasound, for example, by activation of the occipital cortex by visual stimulation that elicits an immediate ∼20–30% increase in the blood velocity through the posterior cerebral artery feeding the posterior lobe (Rosengarten et al. 2001, 2003; Boms et al. 2010; Willie et al. 2011). This signalling between the local neuronal metabolic state and the vasculature that feeds it is involved in the response to systemic stimuli, such as hypoxaemia (Liu & Alkayed, 2005; Yamaura et al. 2006). There is a rich body of literature based on in vitro experimental data devoted to the mechanisms of communication within the neurovascular unit. Numerous excellent reviews have been written on the topic which, being beyond the purview of this review, will not be detailed here (Iadecola, 2004; Girouard & Iadecola, 2006; Hamel, 2006; Iadecola & Nedergaard, 2007; Jakovcevic & Harder, 2007). Despite this extensive knowledge of the molecular mechanisms based on numerous in vivo studies, it should be noted that, to the best of our knowledge, no studies to date have attempted to delineate the mechanisms of neurovascular coupling in humans.
Cerebral autoregulation
In 1895, Bayliss, Hill and Gulland concluded in The Journal of Physiology that ‘In all physiological conditions a rise in arterial pressure accelerates the flow of blood through the brain, and a fall slackens it’ (Bayliss et al. 1895). This concept prevailed until, in a review paper published in 1959, Lassen constructed a plot of average BP and total brain blood flow from seven studies involving 11 different patient groups having a range of drug-and/or pathology-induced BP levels (Lassen, 1959); see Fig. 3, left panel). The plot revealed a plateau region wherein cerebral blood flow appears to be completely stable across a relatively wide range of blood pressures (∼60–150 mmHg). Such a physiological relationship requires reflex adjustments in cerebrovascular resistance concomitant with changes in BP, and was termed static cerebral autoregulation. Lassen's curve continues to be cited and illustrated in numerous high-impact publications and textbooks (Dagal & Lam, 2009; Barret et al. 2010); the potential consequences of such a parochial view in fields such as anaesthesiology are obvious, as stated previously (Drummond, 1997).
Figure 3.
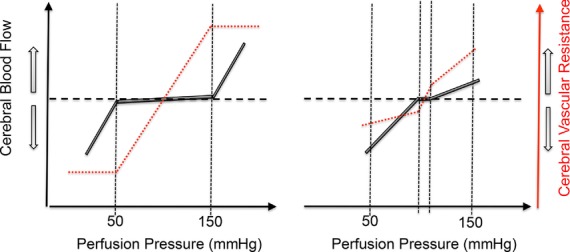
Left panel is a stylized representation of the classical view of the relationships between mean arterial pressure (MAP) and cerebral blood flow (CBF), i.e. autoregulation, put forward by Lassen et al. (1959) based on the between-subject analysis of patients during various pharmacological interventions or pathologies. Right panel is a schematic diagram based on contemporary data indicating a small plateau region (Tan, 2012) and cerebral autoregulation hysteresis. Based on the within-subject reanalysis of 41 studies that reported concomitantly measured CBF and MAP during increases or decreases in blood pressure, the slope of the %ΔCBF versus %ΔMAP relationship was determined to be 0.81 ± 0.77 in the hypotensive range and 0.21 ± 0.47 in the hypertensive range. This indicates a far more pressure-passive CBF than is conventionally believed, and that more efficacious buffering capacity against increases than decreases in perfusion pressure (unpublished observations). See main text for details.
Unfortunately, very few attempts have been made to characterize the normal within-subject relationship between pressure and flow across the brain. The major challenge is that normal baroreflex function limits the effective range of blood pressures, necessitating the use of vasoactive drugs or physical manipulation of central blood volume, both of which confound interpretation of cerebrovascular reflexes per se. Although recent studies have shown a more pressure-passive relationship between MAP and CBF in healthy individuals, this is not a universal finding (e.g. Liu et al. 2013). However, limitations of the majority of these studies are threefold. First, CBF velocity quantified by transcranial Doppler ultrasound represents CBF only if the diameter of the insonated vessel remains constant. This assumption has been evidenced to be violated in conditions of very high  (Willie et al. 2012) or hypoxia (Wilson et al. 2011; Willie et al. 2013a) and may be violated when MAP is changed dramatically (based on animal data showing that the large intracranial vessels react to changes in perfusion pressure; Mchedlishvili, 1964; Mchedlishvili et al. 1973; Faraci & Heistad, 1990). Second, many studies have manipulated CBF pharmacologically with anaesthetics (McCulloch et al. 2005; Ogawa et al. 2008, 2010), angiotensin (Krejcy et al. 1997) and α-adrenergic receptor agonists and/or nitric oxide donors (Zhang et al. 2009; Lucas et al. 2010; Liu et al. 2013; Willie et al. 2013a), the influence of which on cerebrovascular tone is not well understood and is the subject of controversy (e.g. Drummond, 2012; Stewart et al. 2013). Finally, although
(Willie et al. 2012) or hypoxia (Wilson et al. 2011; Willie et al. 2013a) and may be violated when MAP is changed dramatically (based on animal data showing that the large intracranial vessels react to changes in perfusion pressure; Mchedlishvili, 1964; Mchedlishvili et al. 1973; Faraci & Heistad, 1990). Second, many studies have manipulated CBF pharmacologically with anaesthetics (McCulloch et al. 2005; Ogawa et al. 2008, 2010), angiotensin (Krejcy et al. 1997) and α-adrenergic receptor agonists and/or nitric oxide donors (Zhang et al. 2009; Lucas et al. 2010; Liu et al. 2013; Willie et al. 2013a), the influence of which on cerebrovascular tone is not well understood and is the subject of controversy (e.g. Drummond, 2012; Stewart et al. 2013). Finally, although 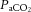 is markedly altered during pharmacological manipulation of BP (Liu et al. 2013), only a few studies have attempted to control for this confounding effect (e.g. Lucas et al. 2010; Chan et al. 2011; Gelinas et al. 2012). Static autoregulation is thus likely to be a function of the experimental conditions in which is it assessed, and its definitive efficacy in healthy humans remains uncertain. Despite this caveat, however, the available within-subject human data indicate that the CBF–MAP relationship is not flat through a broad range of MAP (Fig. 3). We recently reanalysed 41 studies in healthy humans reporting concurrently steady-steady state changes in MAP and CBF for the slope of the %ΔCBF/%ΔMAP relationship above and below resting MAP, which was found to be 0.81 ± 0.77 in the hypotensive range and 0.21 ± 0.47 in the hypertensive range (Unpublished observations: Numan T, Smirl JD, Bain AR, Lewis NC, Hoiland RL & Ainslie PN). Of the 41 studies, 33 estimated CBF by transcranial Doppler ultrasound; however, separate analysis of the studies using other modalities (e.g. MRI, PET or 133Xenon technique) yielded similar CA values. These data indicate that the cerebrovasculature does have autoregulatory capacity, but that its efficacy is not perfect and is dependent on the severity and direction of change in perfusion pressure, as outlined next.
is markedly altered during pharmacological manipulation of BP (Liu et al. 2013), only a few studies have attempted to control for this confounding effect (e.g. Lucas et al. 2010; Chan et al. 2011; Gelinas et al. 2012). Static autoregulation is thus likely to be a function of the experimental conditions in which is it assessed, and its definitive efficacy in healthy humans remains uncertain. Despite this caveat, however, the available within-subject human data indicate that the CBF–MAP relationship is not flat through a broad range of MAP (Fig. 3). We recently reanalysed 41 studies in healthy humans reporting concurrently steady-steady state changes in MAP and CBF for the slope of the %ΔCBF/%ΔMAP relationship above and below resting MAP, which was found to be 0.81 ± 0.77 in the hypotensive range and 0.21 ± 0.47 in the hypertensive range (Unpublished observations: Numan T, Smirl JD, Bain AR, Lewis NC, Hoiland RL & Ainslie PN). Of the 41 studies, 33 estimated CBF by transcranial Doppler ultrasound; however, separate analysis of the studies using other modalities (e.g. MRI, PET or 133Xenon technique) yielded similar CA values. These data indicate that the cerebrovasculature does have autoregulatory capacity, but that its efficacy is not perfect and is dependent on the severity and direction of change in perfusion pressure, as outlined next.
The study of the human cerebrovascular response to rapid changes in MAP has evolved in the last two decades following the introduction of BP and CBF quantification techniques that afford a high temporal resolution. Static versus dynamic CA is an experimental not physiological distinction, where ‘static CA’ refers to the steady-state relationship between MAP and CBF and ‘dynamic CA’ to the cerebral pressure–flow relationship during transient changes in MAP, such as during changes in posture, for example. There are no data, however, that explicitly indicate that short-and long-term regulation of CBF (dynamic and static CA, respectively) are separate mechanistic entities (Tan & Taylor, 2014). Early methods of CBF quantification lacking sufficient temporal resolution to assess the CBF response to rapid changes in MAP were subsequently referred to as static CA, with the inception of the dynamic CA concept arising out of the ability to measure beat-to-beat CBF and MAP concomitantly. Indeed, frequency-domain analysis of CBF was born out of a single technique (transcranial Doppler ultrasound) that allows beat-to-beat analysis of blood velocity in the intracerebral vessels, typically the middle cerebral artery. Two seminal studies in particular have provided a foundation for our understanding of dynamic CA. Aaslid et al. (1989) were the first to study the temporal relationships between middle cerebral artery blood velocity and MAP during the release of inflated thigh-occlusion cuffs that induces rapid transient hypotension. The finding that cerebral blood velocity tracked the drop in MAP but recovered more quickly demonstrated clearly the conceptual inadequacies of the classical view that autoregulation was nearly perfect by showing that CA yielded relative, not absolute, flow buffering. Further characterization of CA as a relative flow buffer followed when Birch et al. (1995) showed that the transfer function characteristics between BP and cerebral blood velocity fluctuations resemble a high-pass filter wherein higher frequency BP fluctuations are more linearly transferred to the cerebral circulation than lower frequency fluctuations.
The above inferences on the nature of dynamic CA are based on the implicit assumption that active and linear changes in cerebrovascular resistance are the primary determinants of dynamic pressure–flow relationships. However, recent studies employing the Windkessel model suggest that the compliant nature of intracranial blood vessels may play an important role in mechanically buffering against dynamic BP fluctuations and that this ability of vessels transiently to ‘store’ blood through a cardiac cycle depends on the speed of MAP change (Zhang et al. 2009; Chan et al. 2011; Tzeng et al. 2011). Transfer function analysis also assumes that the cerebrovascular response to changing MAP is identical whether MAP is rising or falling, but there is clear evidence of hysteresis in CA, i.e. the brain defends more effectively against acute hypertension than hypotension (Aaslid et al. 2007; Schmidt et al. 2009, 2012; Tzeng et al. 2011). At a practical level, the above findings suggest that metrics of CA derived from pressure–flow recordings need to consider not only absolute BP as an input to the cerebral circulation, but also whether BP is accelerating or decelerating and rising or falling (Chan et al. 2011; Tzeng et al. 2011). Such a comprehensive and universal metric does not yet exist, but there is a need to consider important non-stationary components (Panerai, 2013). Recently, Tan (2012), using projection pursuit regression, purported to circumvent some of these linear limitations and reported a small autoregulatory plateau of only ∼10 mmHg when BP oscillations were induced at 0.03 Hz. If the autoregulatory range is in fact so limited, characterization of static CA through incremental changes in MAP may miss the autoregulatory region altogether (e.g. Zhang et al. 2009; Lucas et al. 2010; Liu et al. 2013). It is important to recognize that there remain numerous methods of quantification of CA, each with inherent assumptions and caveats and each specific to some experimental model, and that no one method is considered a ‘gold-standard’ measure. Indeed, the available metrics of CA have been evidenced to yield largely divergent results for the same data (Tzeng et al. 2012) and should thus be scrutinized carefully. There is, however, enough data to support the contention that CA does not maintain constant perfusion through a MAP range of 60–150 mmHg as is so often cited in the literature.
Mechanisms of cerebral autoregulation
Changes in cerebrovascular resistance must necessarily underlie CA, whether static or dynamic, yet there remains little consensus, especially in humans, on the mechanisms and location of this cerebral resistance modulation. Most of the attention paid to CA in the last two decades has aimed to characterize CA, to deduce a role of neural regulation of CA and to study the effects of stimuli ranging from exercise to pathology on CA. These studies almost universally ignore the question of where exactly this regulation takes place (e.g. Claassen & Zhang, 2011; Jordan & Powers, 2012). The pial arterioles are dogmatically accepted to serve in this capacity (Fog, 1938; Lassen, 1959). But there are data to the contrary indicating that only the largest of these vessels respond to physiological ranges of MAP and that the large intracranial arteries as well as the even larger vessels of the neck contribute to a substantial portion of cerebrovascular resistance (Heistad et al. 1978a; Kontos et al. 1978; Faraci et al. 1987a). Notwithstanding the exact site(s) where CA is modulated, there seems to be inbuilt mechanistic redundancy. For example, in addition to the apparent roles for the autonomic nervous system (see ‘Autonomic regulation of cerebral blood flow’, below) there is a myogenic role in the regulation of CA. Only two studies in humans have addressed CA following myogenic ‘block’ using Ca2+ channel inhibitors; Tzeng et al. (2011) used the cerebrovascular-specific Ca2+ antagonist (nimodipine), and Tan et al. (2013) used the systemically acting vascular smooth muscle cell Ca2+ channel blocker (nicardipine). Both studies demonstrated altered CA, but only during low-frequency (∼20–30 s) oscillations of MAP as driven by oscillating lower-body negative pressure. Given that buffering of CBF against changes in perfusion pressure must necessarily entail changes in vascular resistance somewhere within the cerebrovasculature, it is perhaps not surprising that compromising the vascular smooth muscle cells alters CA. Equally, however, alterations of CA by Ca2+ channel blockers must not be of elemental importance, because widespread orthostatic intolerance is not necessarily observed in patients prescribed these drugs (Grünig et al. 2013). The relative importance and relationships between myogenic, autonomic and local neuronal mechanisms in CA remains to be understood holistically and will certainly require innovative study design and may necessitate methods of CBF assessment that directly consider cerebral vasomotion (such as Duplex ultrasound or MRI, rather than transcranial Doppler ultrasound alone).
Large conduit arteries and cerebral autoregulation
There is a convincing body of evidence demonstrating that the large arteries of the brain play a much larger and more important role in the regulation of CBF than generally ascribed to them (Mchedlishvili, 1964; Mchedlishvili et al. 1973; Faraci & Heistad, 1990). Indeed, using a canine in situ ICA where the inlet pressure could be controlled, ICA constriction maintained pressure at the circle of Willis nearly constant in the face of increasing perfusion pressure (Mchedlishvili et al. 1973); the feline VA, in contrast, does not buffer increases in MAP (Faraci et al. 1987a). Another group, using a different in vivo technique that allowed calculation of the lumped resistance for all the large arteries in dogs and cats (Heistad et al. 1978a; Faraci et al. 1987a), found similar results, concluding that the large arteries were responsible for a quarter to half of the total cerebrovascular resistance during resting conditions.
Perhaps because these larger arteries of the neck are generally considered to be ‘conduit’ arteries, the idea that they can actively participate in CBF regulation has not been embraced. However, in rabbits and dogs the internal geometry of the ICA and VA was found to change considerably where the vessels bend, at the cavernous sinus for the ICA (the carotid siphon) and at the V3 segment of the VA at the entrance of the foramen magnum (Mchedlishvili, 1964). The turbulent flow resulting from such luminal diameter changes within tortuous segments must dramatically increase resistance compared with non-tortuous segments. That is to say that a smaller decrease in lumen diameter would be required to produce a given increase in resistance within the carotid siphon and V3 segment of the VA (Fig. 1, inlay I).
Recent human MRI data showing complex non-Newtonian flow and attenuated pulsatility along the carotid siphon support this theory (Takeuchi & Karino, 2010; Schubert et al. 2011). In humans, these vessels change diameter in response to changes in 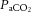 and
and 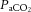 (Wilson et al. 2011; Willie et al. 2012); future studies using such advanced imaging techniques should assess whether they are also involved in the cerebrovascular response to acute changes in MAP.
(Wilson et al. 2011; Willie et al. 2012); future studies using such advanced imaging techniques should assess whether they are also involved in the cerebrovascular response to acute changes in MAP.
Interaction between arterial blood gases and cerebral autoregulation
Traditional tests to assess cerebrovascular CO2 reactivity or CA treat these concepts as separate entities. Clearly they are not; there are persuasive data that both CA and CO2 responses may use the same vascular reserve. In a landmark study, Harper & Glass (1965) examined the brain vascular reactivity to changes in 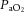 in dogs at various blood pressures (see Fig. 4). Severely hypotensive animals (approximately −60% MAP) showed no change in cerebral vessel diameter in response to increases or decreases in
in dogs at various blood pressures (see Fig. 4). Severely hypotensive animals (approximately −60% MAP) showed no change in cerebral vessel diameter in response to increases or decreases in 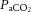 (i.e. CBF reactivity was abolished), because the vessels were already maximally dilated (Harper & Glass, 1965). The reciprocal instance was also demonstrated where, in hypocapnia, CBF was well maintained in the face of controlled haemorrhage-induced hypotension (Iwabuchi et al. 1973). In contrast, with hypercapnia, CBF fell linearly with MAP (Iwabuchi et al. 1973).
(i.e. CBF reactivity was abolished), because the vessels were already maximally dilated (Harper & Glass, 1965). The reciprocal instance was also demonstrated where, in hypocapnia, CBF was well maintained in the face of controlled haemorrhage-induced hypotension (Iwabuchi et al. 1973). In contrast, with hypercapnia, CBF fell linearly with MAP (Iwabuchi et al. 1973).
Figure 4.
Note the abolishment of cerebrovascular reactivity with progressive hypotension. Values are calculated from the data of Harper & Glass (1965).
Broadly comparable findings in both healthy humans (Przybyłowski et al. 2003; Ainslie et al. 2012) and those with pathology (e.g. carotid stenosis; Nishimura et al. 1999) indicate that CBF reductions with transient hypotension lead to a blunting of the CBF response to hypercapnia; hypotension selectively attenuated cerebrovascular CO2 reactivity to hypercapnia but not hypocapnia. Thus, the compromised capacity of the cerebral vessels to dilate in hypotensive conditions when 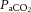 is elevated indicates that the maintenance of cerebral perfusion takes precedence over the maintenance of a normal tissue
is elevated indicates that the maintenance of cerebral perfusion takes precedence over the maintenance of a normal tissue 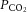 or that there is limited vasodilatory reserve regardless of the combined stimulus. Equally, the latter process may take place regionally. For instance, during times of decreased perfusion or global cerebral vasodilatory stimulus, vascular beds with reduced vasodilatory reserve may have limited ability to dilate sufficiently to compete for limited blood flow to the brain successfully (Mandell et al. 2008). As a result, despite global vasodilatation, regional CBF in these vessels may even decrease, resulting in a cerebrovascular ‘steal’ phenomenon (Faraci & Heistad, 1990; Mandell et al. 2008; Fierstra et al. 2010).
or that there is limited vasodilatory reserve regardless of the combined stimulus. Equally, the latter process may take place regionally. For instance, during times of decreased perfusion or global cerebral vasodilatory stimulus, vascular beds with reduced vasodilatory reserve may have limited ability to dilate sufficiently to compete for limited blood flow to the brain successfully (Mandell et al. 2008). As a result, despite global vasodilatation, regional CBF in these vessels may even decrease, resulting in a cerebrovascular ‘steal’ phenomenon (Faraci & Heistad, 1990; Mandell et al. 2008; Fierstra et al. 2010).
Moreover, given that the brain does not autoregulate perfectly (Fig. 4) and that hypoxia and elevations in 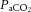 also ‘impair’ the capability of the brain to defend against BP changes (Tzeng et al. 2012), BP is clearly a critical component of CBF. Examples of these integrated changes in and BP occur in a myriad of everyday activities, such as changing posture, laughing, exercise, straining, sexual activity and coughing, to name but a few.
also ‘impair’ the capability of the brain to defend against BP changes (Tzeng et al. 2012), BP is clearly a critical component of CBF. Examples of these integrated changes in and BP occur in a myriad of everyday activities, such as changing posture, laughing, exercise, straining, sexual activity and coughing, to name but a few.
Although there is extensive evidence in support of this proposed interaction between pressure and chemical regulation of the cerebrovasculature (Kety & Schmidt, 1946, 1948; Jordan et al. 2000; Przybyłowski et al. 2003; Ainslie & Tzeng, 2010; Ainslie & Smith, 2011; Tzeng et al. 2012; Willie et al. 2012), integrated consideration of these players is seldom incorporated into contemporary research design. The cerebrovascular conductance index is sometimes used for this reason, because it may provide for more precise estimation of CO2 reactivity that considers the effect of arterial BP on CBF; however, this does not take into account factors that may be manifest during alterations in ventilation, such as large changes in intrathoracic, intracranial and central venous pressures that in turn affect cerebral perfusion pressure. It also assumes that the relationships of 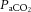 and MAP versus CBF are linear, when this is probably not the case (Battisti-Charbonney et al. 2011). As such, cerebrovascular conductance may oversimplify the issue. In our view, expressing the effects of CO2 and MAP individually (e.g. Willie et al. 2012) gives a better impression of the relative effects of each on CBF.
and MAP versus CBF are linear, when this is probably not the case (Battisti-Charbonney et al. 2011). As such, cerebrovascular conductance may oversimplify the issue. In our view, expressing the effects of CO2 and MAP individually (e.g. Willie et al. 2012) gives a better impression of the relative effects of each on CBF.
Autonomic regulation of cerebral blood flow
The entire cerebrovasculature is extensively innervated by adrenergic and cholinergic fibres of diverse extrinsic (e.g. cervical, sphenopalatine and trigeminal ganglia) and intrinsic origins (e.g. locus coeruleus, fastigial nucleus and dorsal raphe nucleus). Cerebral arteries show three layers of nerve plexi, as follows, with relative distribution varying with the vessel location: (1) the most superficial layer is a paravascular layer of nerve bundles longitudinally arranged superficially to the adventitia; (2) a dense perivascular plexus within the adventia; and (3) a deep perivascular plexus following a transverse course at the adventitial–medial border (Bleys et al. 1996). The extracranial arteries feature a higher proportion of longitudinally arranged paravascular nerve bundles, whereas intracranially there is a greater total density of perivascular nerve fibres, with more following a spiral or annular course, and far more neuron terminals, particularly at bifurcations, communicating arteries and the intracranial curvatures of the ICA and VA (Borodulya & Pletchkova, 1973, 1976; Mchedlishvili, 1986; Bleys et al. 1996). The ICA territory appears to possess denser sympathetic innervation than the vertebrobasilar system (Edvinsson & Hamel, 2002). The necessary anatomy is therefore extant for a neural role in the regulation of CBF.
Sympathetic nervous system (SNS) and CBF
Human studies assessing this role are broadly of two types, namely studies of patients with various diseases treated by ganglionectomy or studies in healthy humans employing local subcutaneous blockade of a ganglion(s) or oral pharmacological blockade of sympathetic ganglia or a systemic receptor group. Four studies have assessed CBF following ganglion excision and each showed increased CBF (Shenkin et al. 1951; Shenkin, 1969; Suzuki et al. 1975; Jeng et al. 1999). Following local cervical ganglion block, five studies reported increased CBF (Linden, 1955; Umeyama et al. 1995; Ide et al. 2000; Treggiari, 2003; Yokoyama et al. 2004), whereas three reported no change in CBF (Harmel et al. 1949; Scheinberg, 1950; Ohta et al. 1990), and in one study a decrease in CBF was found (Kang et al. 2010). Local anaesthesia can result in a partial block of the targeted ganglion, which could explain the lack of effect on CBF in three studies. Although the pathological conditions should be acknowledged, the finding that ganglionectomy universally increased CBF is convincing evidence for a role of sympathetic nerves in CBF regulation, although these data shed little light on the precise nature of such a role.
Invasive animal experiments suggest that the SNS is most important during changes in BP. Most well-controlled animal studies have observed some decrease in CBF at baseline, but especially during hypertension with stimulation of the superior cervical ganglion. This role of the SNS seems especially important in buffering surges in perfusion pressure (Mayhan et al. 1987; Cassaglia et al. 2008a,b2008b, 2009; Ainslie, 2009; Tzeng et al. 2010). Unilateral resection of the superior cervical ganglion during induced hypertension produced ipsilateral disruption of cortical vessel integrity (Ponte & Purves, 1974; Heistad et al. 1978b). This apparent protective function of the SNS in response to increases in perfusion pressure seems to be a function principally of the larger arteries. For example, only the largest pial arterioles respond to sympathetic activation (Wei et al. 1975) and do so to a smaller degree than the large cerebral arteries (Baumbach & Heistad, 1983). The ability of these larger vessels to maintain flow during changes in perfusion pressure is dependent on their innervation, because the vessels become pressure passive once denervated; autoregulation in the large vessels is perhaps modulated, at least in part, neurogenically (Mchedlishvili et al. 1973; Tamaki & Heistad, 1986). Interpretation of studies using animal models to assess the role of the SNS in cerebrovascular function should be done with caution, because profound differences have been reported between species (Heistad et al. 1978b; Busija et al. 1980) and nothing is known of the corresponding relationship to human physiology.
Sympathetic nervous system and CA
Studies employing pharmacological blockade benefit from being completed in healthy humans but are potentially confounded by the systemic effects of the drug. Nonetheless, a number of elegantly studies have reported similar findings, indicating impairment of CA following SNS blockade. Both sympathetic ganglion blockade with trimethaphan (Zhang et al. 2004) and α-adrenoreceptor block with phentolamine (Kimmerly et al. 2003) resulted in a greater rise in CBF for a given increase in MAP produced during a Valsalva manoeuvre or noradrenaline infusion, respectively. Likewise, both these pharmacological interventions increase transfer function gain and decrease phase lead at low frequencies (0.03 Hz), indicating impaired CA (Zhang et al. 2002; Hamner et al. 2010), and α1-adrenoreceptor block with prazosin impairs CBF recovery from transient hypotension induced by thigh-cuff release (Ogoh et al. 2008). It is nonetheless difficult to dissociate the systemic effects of SNS blockade (ganglionic or receptor group) on the peripheral vascular system from direct effects on CA per se. The changes in MAP or the very reflexes responsible should MAP remain constant following SNS block cannot be ruled out from having a direct effect on cerebrovascular regulation.
Sympathetic nervous system and arterial blood gases
The relationship between the SNS and the cerebrovascular response to CO2 and hypoxia also remains vague, because it is difficult to dissociate the peripheral (e.g. MAP) from direct cerebrovascular effects of SNS activity. A convincing study in baboons reported that hypoxaemic stimulation of the carotid bodies increased ipsilateral CBF and that this response was abolished following resection of cranial nerve VII (Ponte & Purves, 1974). Human data are inconsistent. Carbon dioxide reactivity has been reported to be unchanged following augmentation of SNS by hand grip (Ainslie et al. 2005) and lower-body negative pressure (LeMarbre et al. 2003); but conversely, attenuated CO2 reactivity was reported during lower-body negative pressure (Zhang et al. 2011) and following ganglionic blockade with trimethaphan (Jordan et al. 2000).
Systemic pharmacological manipulation of SNS activity is likely to be confounded, however, by the consequent blunted MAP response to increased 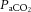 or
or 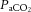 . For example, given that MAP may influence CBF (see ‘Metabolic regulation of cerebral blood flow’, above), lowering of reactivity is likely to be confounded by an attenuated hypercapnia-induced pressure response (Przybyłowski et al. 2003; Ainslie et al. 2012). Moreover, to our knowledge, no study has examined whether sympathetic blockade attenuates the hypoxia-induced CBF increase or, conversely, whether SNS activity might serve to restrain large increases in CBF observed in severe hypercapnia or hypoxia (Wilson et al. 2011; Willie et al. 2012).
. For example, given that MAP may influence CBF (see ‘Metabolic regulation of cerebral blood flow’, above), lowering of reactivity is likely to be confounded by an attenuated hypercapnia-induced pressure response (Przybyłowski et al. 2003; Ainslie et al. 2012). Moreover, to our knowledge, no study has examined whether sympathetic blockade attenuates the hypoxia-induced CBF increase or, conversely, whether SNS activity might serve to restrain large increases in CBF observed in severe hypercapnia or hypoxia (Wilson et al. 2011; Willie et al. 2012).
Parasympathetic nervous system and CA
There are limited data available assessing the cholinergic control of CBF in humans, but again anatomical studies of cerebral vessels indicate rich distribution of cholinergic nerve terminals throughout the intracranial vessels proximal to the Virchow–Robin spaces (Florence & Bevan, 1979; Heistad et al. 1980; Sato et al. 2001; Hamel, 2004). As with the SNS, cholinergic control seems to be species specific, because petrosal nerve resection or stimulation does not affect baseline CBF in cats but does in dogs (D'Alecy & Rose, 1977) and rats (Pinard et al. 1979). Only one study has assessed cerebrovascular control at rest in healthy humans. Hamner et al. (2012) reported increased transfer function coherence between MAP and CBF (as estimated by transcranial Doppler ultrasound) following systemic cholinergic blockade with glycopyrrolate, suggesting impaired CA. As explained in the ‘Cerebral autoregulation’ section above, the precise physiological meaning of transfer function metrics remains ambiguous and, while these data do indicate a cholinergic role in cerebrovasculature function, more work certainly needs be directed to this end.
Conclusions
Our aim in the present review is to call attention to dated concepts in the accepted understanding of cerebrovascular regulation. Despite ample data and past reviews in high-impact journals that have highlighted these erroneous views, they continue to be taught to medical professionals, neuroscientists and physiologists across the globe. Below, we outline four principal points that should be incorporated into contemporary knowledge of cerebrovascular physiology, as well as future directions of research pertaining to each.
(1) Cerebral autoregulation does not maintain constant perfusion through a MAP range of 60–150 mmHg. The cerebral circulation does have more effective autoregulatory ability in the range above baseline MAP, and to a lesser extent below baseline MAP; however, CBF is nonetheless directly affected by changes in perfusion pressure. A definitive, within-individual assessment of global and regional CBF across a range of non-pharmacologically and pharmacologically perturbed blood pressures with maintained
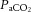 has yet to be completed. How this relationship may alter in pathological conditions is therefore also unknown.
has yet to be completed. How this relationship may alter in pathological conditions is therefore also unknown.(2) There is important stimulatory synergism and regulatory interdependence of arterial blood gases and BP on CBF regulation. The study of cerebrovascular regulation needs to consider this relationship; if BP and
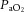 or
or 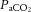 change simultaneously, interpretation of the relative impact of each becomes difficult. The relationship between MAP and cerebrovascular reactivity to
change simultaneously, interpretation of the relative impact of each becomes difficult. The relationship between MAP and cerebrovascular reactivity to 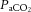 remains unknown in the hypertensive range. Moreover, advanced imaging techniques should facilitate study of the effects of vascular reactivity and resistance heterogeneity in the cerebral vascular bed on the distribution of CBF in response to vasoactive stimuli.
remains unknown in the hypertensive range. Moreover, advanced imaging techniques should facilitate study of the effects of vascular reactivity and resistance heterogeneity in the cerebral vascular bed on the distribution of CBF in response to vasoactive stimuli.(3) Cerebral autoregulation and cerebrovascular sensitivity to changes in arterial blood gases are not modulated solely at the pial arterioles. The large arteries of the neck and cerebrum are critically involved, serving the first-line defense such that the pial and cortical vessels experience minimal changes in pressure and can respond principally to prevailing systemic and local neural metabolism. In order to understand better the role of these vessels in human CBF regulation, high-resolution MRI/magnetic resonance angiography (MRA) should be used to assess their responses to non-pharmacologically driven changes in BP and controlled dynamic and steady-state changes in arterial blood gases
(4) Neurogenic control of the cerebral vasculature is an important player in autoregulatory function, particularly in the large vessels, and acts to buffer surges in perfusion pressure. Although the precise role of cerebral vasomotor nerves is poorly understood after more than 80 years of study, it remains premature to dismiss their role. There are numerous studies yet to be completed in humans to determine precisely the capacity for sympathetic and cholinergic nervous input on the cerebrovasculature. Future studies in healthy humans should not rely solely on transcranial Doppler ultrasound in assessing these questions. Newer imaging modalities, and direct modification of cerebral SNS outflow, rather than global perturbation of SNS receptors, will help to elucidate the role of the autonomic nervous system in cerebrovascular control. Particularly during transient bouts of hypertension, such as those commonly experienced during activity, strain (defaecation, lifting, etc.), pathology (autonomic dysreflexia, subarachnoid haemorrhage, etc.) and during rapid-eye movement sleep, the sympathetically mediated buffering of global CBF and constriction of large cerebral arteries should be assessed. One methodological possibility to this end would be to use a centrally acting α2-adrenoreceptor agonist (e.g. clonidine) to diminish SNS outflow whilst quantifying regional (cerebral) noradrenaline spillover (Mitchell et al. 2009) during non-pharmacologically induced changes in MAP. Another approach would be to assess CBF and large artery vasomotion (by ultrasound or MRI) during transient hypotension and hypertension before and following cervical ganglion block.
Answering these questions in healthy humans will provide new insight into the fundamental mechanisms that regulate CBF. Only by first understanding these mechanisms can we subsequently decipher the role of CBF regulatory impairment in myriad cerebrovascular diseases, such as Alzheimer's disease and dementia (Faraci et al. 1987b; Qiu et al. 2003; Hebert et al. 2004) and even neurogenic hypertension (Waki et al. 2011).
Acknowledgments
We extend our gratitude to Ms Melanie Burger, MScBMC, for her patience and dedicated work through numerous iterations of Fig. 1. Ms Burger not only produced the elegant artwork, but also contributed her scientific expertise to optimize the translation of the concepts described in the text into visual form. The scientific literature benefits from such scientific illustrators (designnuclei.com).
Glossary
- BP
blood pressure
- CA
cerebral autoregulation
- CBF
cerebral blood flow
- CSF
cerebrospinal fluid
- ICA
internal carotid artery
- l-NMMA
NG-monomethyl-l-arginine
- MAP
mean arterial pressure
- MRI
magnetic resonance imaging
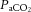
arterial partial pressure of carbon dioxide
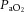
arterial partial pressure of oxygen
- PET
positron emission tomography
- SNS
sympathetic nervous system
- VA
vertebral artery
Biography
Christopher Willie discovered physiology as an undergraduate student in the laboratory of Dr. Richard Wilson while working toward his Bachelors of Health Science degree at the University of Calgary. He met Dr Philip Ainslie at the same time and five years later began his PhD under Dr Ainslie's supervision. Mr. Willie's interests are studying human integrative physiology, with focus on cerebrovascular responses to changes in blood gases and blood pressure. To keep balanced, he enjoys rock, ice and alpine climbing around the world. In the future,MrWillie plans to continue both his scientific and adventurous pursuits. Philip Ainslie is currently a Professor in the School of Health and Exercise Sciences at the University of British Columbia-Okanagan and holds a Canada Research Chair in Cerebrovascular Physiology. His main research focus attempts to examine the fundamental mechanisms that regulate human cerebral blood flow in health and in disease. He has a particular interest in how environmental stress – especially in the context of hypoxia, temperature, pressure and exercise – may impact on cerebral blood flow regulation.
Additional Information
Competing interests
None declared.
Funding
C.K.W. is a Vanier Canada graduate scholar and Killam Doctoral Fellow. P.N.A. is supported by a Canada Research Chair in Cerebrovascular Physiology and NSERC Discovery Grant.
References
- Aaslid R, Blaha M, Sviri G, Douville CM, Newell DW. Asymmetric dynamic cerebral autoregulatory response to cyclic stimuli. Stroke. 2007;38:1465–1469. doi: 10.1161/STROKEAHA.106.473462. [DOI] [PubMed] [Google Scholar]
- Aaslid R, Lindegaard KF, Sorteberg W, Nornes H. Cerebral autoregulation dynamics in humans. Stroke. 1989;20:45–52. doi: 10.1161/01.str.20.1.45. [DOI] [PubMed] [Google Scholar]
- Ainslie PN. Have a safe night: intimate protection against cerebral hyperperfusion during REM sleep. J Appl Physiol. 2009;106:1031–1033. doi: 10.1152/japplphysiol.00091.2009. [DOI] [PubMed] [Google Scholar]
- Ainslie PN, Ashmead JC, Ide K, Morgan BJ, Poulin MJ. Differential responses to CO2 and sympathetic stimulation in the cerebral and femoral circulations in humans. J Physiol. 2005;566:613–624. doi: 10.1113/jphysiol.2005.087320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainslie PN, Duffin J. Integration of cerebrovascular CO2 reactivity and chemoreflex control of breathing: mechanisms of regulation, measurement, and interpretation. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1473–R1495. doi: 10.1152/ajpregu.91008.2008. [DOI] [PubMed] [Google Scholar]
- Ainslie PN, Lucas SJE, Fan J-L, Thomas KN, Cotter JD, Tzeng YC, Burgess KR. Influence of sympathoexcitation at high altitude on cerebrovascular function and ventilatory control in humans. J Appl Physiol. 2012;113:1058–1067. doi: 10.1152/japplphysiol.00463.2012. [DOI] [PubMed] [Google Scholar]
- Ainslie PN, Poulin MJ. Ventilatory, cerebrovascular, and cardiovascular interactions in acute hypoxia: regulation by carbon dioxide. J Appl Physiol. 2004;97:149–159. doi: 10.1152/japplphysiol.01385.2003. [DOI] [PubMed] [Google Scholar]
- Ainslie PN, Smith KJ. Integrated human physiology: breathing, blood pressure and blood flow to the brain. J Physiol. 2011;589:2917. doi: 10.1113/jphysiol.2011.211292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainslie PN, Tzeng YC. On the regulation of the blood supply to the brain: old age concepts and new age ideas. J Appl Physiol (1985) 2010;108:1447–1449. doi: 10.1152/japplphysiol.00257.2010. [DOI] [PubMed] [Google Scholar]
- Apkon M, Weed RA, Boron WF. Motor responses of cultured rat cerebral vascular smooth muscle cells to intra-and extracellular pH changes. Am J Physiol Heart Circ Physiol. 1997;273:H434–H445. doi: 10.1152/ajpheart.1997.273.1.H434. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barret KE, Barman SM, Boitano S, Brooks H. Ganong's Review of Medical Physiology. 23rd edn. New York: McGraw-Hill Medical; 2010. [Google Scholar]
- Battisti-Charbonney A, Fisher J, Duffin J. The cerebrovascular response to carbon dioxide in humans. J Physiol. 2011;589:3039–3048. doi: 10.1113/jphysiol.2011.206052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbach GL, Heistad DD. Effects of sympathetic stimulation and changes in arterial pressure on segmental resistance of cerebral vessels in rabbits and cats. Circ Res. 1983;52:527–533. doi: 10.1161/01.res.52.5.527. [DOI] [PubMed] [Google Scholar]
- Bauser-Heaton HD, Bohlen HG. Cerebral microvascular dilation during hypotension and decreased oxygen tension: a role for nNOS. Am J Physiol Heart Circ Physiol. 2007;293:H2193–H2201. doi: 10.1152/ajpheart.00190.2007. [DOI] [PubMed] [Google Scholar]
- Bayliss WM, Hill L, Gulland GL. On intra-cranial pressure and the cerebral circulation: Part I. Physiological; Part II. Histological. J Physiol. 1895;18:334–362. doi: 10.1113/jphysiol.1895.sp000572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binks AP, Cunningham VJ, Adams L, Banzett RB. Gray matter blood flow change is unevenly distributed during moderate isocapnic hypoxia in humans. J Appl Physiol. 2008;104:212–217. doi: 10.1152/japplphysiol.00069.2007. [DOI] [PubMed] [Google Scholar]
- Birch AA, Dirnhuber MJ, Hartley-Davies R, Iannotti F, Neil-Dwyer G. Assessment of autoregulation by means of periodic changes in blood pressure. Stroke. 1995;26:834–837. doi: 10.1161/01.str.26.5.834. [DOI] [PubMed] [Google Scholar]
- Bleys RL, Cowen T, Groen GJ, Hillen B, Ibrahim NB. Perivascular nerves of the human basal cerebral arteries: I. Topographical distribution. J Cereb Blood Flow Metab. 1996;16:1034–1047. doi: 10.1097/00004647-199609000-00029. [DOI] [PubMed] [Google Scholar]
- Boms N, Yonai Y, Molnar S, Rosengarten B, Bornstein NM, Csiba L, Olah L. Effect of smoking cessation on visually evoked cerebral blood flow response in healthy volunteers. J Vasc Res. 2010;47:214–220. doi: 10.1159/000255964. [DOI] [PubMed] [Google Scholar]
- Bonnet P, Gebremedhin D, Rush NJ, Harder DR. Effects of hypoxia on a potassium channel in cat cerebral arterial muscle cells. Z Kardiol. 1991;80(Suppl 7):25–27. [PubMed] [Google Scholar]
- Borodulya AV, Pletchkova EK. Distribution of cholinergic and adrenergic nerves in the internal carotid artery. A histochemical study. Acta Anat (Basel) 1973;86:410–425. doi: 10.1159/000144132. [DOI] [PubMed] [Google Scholar]
- Borodulya AV, Pletchkova EK. Cholinergic innervation of vessels of the base of the brain. Acta Anat (Basel) 1976;96:135–147. doi: 10.1159/000144667. [DOI] [PubMed] [Google Scholar]
- Bowton DL, Haddon WS, Prough DS, Adair N, Alford PT, Stump DA. Theophylline effect on the cerebral blood flow response to hypoxemia. Chest. 1988;94:371–375. doi: 10.1378/chest.94.2.371. [DOI] [PubMed] [Google Scholar]
- Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia. 2007;55:1263–1271. doi: 10.1002/glia.20557. [DOI] [PubMed] [Google Scholar]
- Brown MM, Wade JP, Marshall J. Fundamental importance of arterial oxygen content in the regulation of cerebral blood flow in man. Brain. 1985;108:81–93. doi: 10.1093/brain/108.1.81. [DOI] [PubMed] [Google Scholar]
- Busija DW, Heistad DD, Marcus ML. Effects of sympathetic nerves on cerebral vessels during acute, moderate increases in arterial pressure in dogs and cats. Circ Res. 1980;46:696–702. doi: 10.1161/01.res.46.5.696. [DOI] [PubMed] [Google Scholar]
- Cassaglia PA, Griffiths RI, Walker AM. Sympathetic withdrawal augments cerebral blood flow during acute hypercapnia in sleeping lambs. Sleep. 2008a;31:1729–1734. doi: 10.1093/sleep/31.12.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassaglia PA, Griffiths RI, Walker AM. Sympathetic nerve activity in the superior cervical ganglia increases in response to imposed increases in arterial pressure. Am J Physiol Regul Integr Comp Physiol. 2008b;294:R1255–R1261. doi: 10.1152/ajpregu.00332.2007. [DOI] [PubMed] [Google Scholar]
- Cassaglia PA, Griffiths RI, Walker AM. Cerebral sympathetic nerve activity has a major regulatory role in the cerebral circulation in REM sleep. J Appl Physiol. 2009;106:1050–1056. doi: 10.1152/japplphysiol.91349.2008. [DOI] [PubMed] [Google Scholar]
- Chan GSH, Ainslie PN, Willie CK, Taylor CE, Atkinson G, Jones H, Lovell NH, Tzeng YC. Contribution of arterial Windkessel in low-frequency cerebral hemodynamics during transient changes in blood pressure. J Appl Physiol. 2011;110:917–925. doi: 10.1152/japplphysiol.01407.2010. [DOI] [PubMed] [Google Scholar]
- Claassen JAHR, Zhang R. Cerebral autoregulation in Alzheimer's disease. J Cereb Blood Flow Metab. 2011;31:1572–1577. doi: 10.1038/jcbfm.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PJ, Alexander SC, Smith TC, Reivich M, Wollman H. Effects of hypoxia and normocarbia on cerebral blood flow and metabolism in conscious man. J Appl Physiol. 1967;23:183–189. doi: 10.1152/jappl.1967.23.2.183. [DOI] [PubMed] [Google Scholar]
- Coney AM, Marshall JM. Role of adenosine and its receptors in the vasodilatation induced in the cerebral cortex of the rat by systemic hypoxia. J Physiol. 1998;509:507–518. doi: 10.1111/j.1469-7793.1998.507bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabertrand F, Nelson MT, Brayden JE. Acidosis dilates brain parenchymal arterioles by conversion of calcium waves to sparks to activate BK channels. Circ Res. 2012;110:285–294. doi: 10.1161/CIRCRESAHA.111.258145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagal A, Lam AM. Cerebral autoregulation and anesthesia. Curr Opin Anaesthesiol. 2009;22:547–552. doi: 10.1097/ACO.0b013e32833020be. [DOI] [PubMed] [Google Scholar]
- D'Alecy LG, Rose CJ. Parasympathetic cholinergic control of cerebral blood flow in dogs. Circ Res. 1977;41:324–331. doi: 10.1161/01.res.41.3.324. [DOI] [PubMed] [Google Scholar]
- Donders FC. Die Bewegungen des Gehirns und die Veränderungen der Gefässfullung der Pia Mater. Schmid's Fahrbucher. 1851;69:16–20. [Google Scholar]
- Drummond JC. The lower limit of autoregulation: time to revise our thinking? Anesthesiology. 1997;86:1431–1433. doi: 10.1097/00000542-199706000-00034. [DOI] [PubMed] [Google Scholar]
- Drummond JC. Cerebral blood flow and the alpha-1 agonist bogeyman. Anesth Analg. 2012;114:478–479; author reply 479. doi: 10.1213/ANE.0b013e318241ccee. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Hamel E. Perivascular nerves in brain vessels. In: Edvinsson L, Krause DN, editors. Cerebral Blood Flow and Metabolism. Philadelphia: Lippincott, Williams and Wilkins; 2002. pp. 43–67. [Google Scholar]
- Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res. 1990;66:8–17. doi: 10.1161/01.res.66.1.8. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Heistad DD, Mayhan WG. Role of large arteries in regulation of blood flow to brain stem in cats. J Physiol. 1987a;387:115–123. doi: 10.1113/jphysiol.1987.sp016566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci FM, Mayhan WG, Heistad DD. Segmental vascular responses to acute hypertension in cerebrum and brain stem. Am J Physiol Heart Circ Physiol. 1987b;252:H738–H742. doi: 10.1152/ajpheart.1987.252.4.H738. [DOI] [PubMed] [Google Scholar]
- Fierstra J, Poublanc J, Han JS, Silver F, Tymianski M, Crawley AP, Fisher JA, Mikulis DJ. Steal physiology is spatially associated with cortical thinning. J Neurol Neurosurg Psychiatry. 2010;81:290–293. doi: 10.1136/jnnp.2009.188078. [DOI] [PubMed] [Google Scholar]
- Fierstra J, Sobczyk O, Battisti-Charbonney A, Mandell DM, Poublanc J, Crawley AP, Mikulis DJ, Duffin J, Fisher JA. Measuring cerebrovascular reactivity: what stimulus to use? J Physiol. 2013;591:5809–5821. doi: 10.1113/jphysiol.2013.259150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florence VM, Bevan JA. Biochemical determinations of cholinergic innervation in cerebral arteries. Circ Res. 1979;45:212–218. doi: 10.1161/01.res.45.2.212. [DOI] [PubMed] [Google Scholar]
- Fog M. The relationship between the blood pressure and the tonic regulation of the pial arteries. J Neurol Psychiatry. 1938;1:187–197. doi: 10.1136/jnnp.1.3.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Yamaura K, Harder DR. Role of 20-HETE in the hypoxia-induced activation of Ca2+-activated K+ channel currents in rat cerebral arterial muscle cells. Am J Physiol Heart Circ Physiol. 2008;294:H107–H120. doi: 10.1152/ajpheart.01416.2006. [DOI] [PubMed] [Google Scholar]
- Gelinas JC, Marsden KR, Tzeng YC, Smirl JD, Smith KJ, Willie CK, Lewis NC, Binsted G, Bailey DM, Bakker A, Day TA, Ainslie PN. Influence of posture on the regulation of cerebral perfusion. Aviat Space Environ Med. 2012;83:751–757. doi: 10.3357/asem.3269.2012. [DOI] [PubMed] [Google Scholar]
- Giller CA, Bowman G, Dyer H, Mootz L, Krippner W. Cerebral arterial diameters during changes in blood pressure and carbon dioxide during craniotomy. Neurosurgery. 1993;32:737–741. discussion 741–742. [PubMed] [Google Scholar]
- Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- Gisolf J, van Lieshout JJ, van Heusden K, Pott F, Stok WJ, Karemaker JM. Human cerebral venous outflow pathway depends on posture and central venous pressure. J Physiol. 2004;560:317–327. doi: 10.1113/jphysiol.2004.070409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GRJ, Choi HB, Rungta RL, Ellis-Davies GCR, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman RF, Sojkova J, Beason-Held LL, An Y, Longo DL, Ferrucci L, Resnick SM. Patterns of regional cerebral blood flow associated with low hemoglobin in the Baltimore Longitudinal Study of Aging. J Gerontol A Biol Sci Med Sci. 2012;67:963–969. doi: 10.1093/gerona/gls121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottstein U, Paulson OB. The effect of intracarotid aminophylline infusion on the cerebral circulation. Stroke. 1972;3:560–565. doi: 10.1161/01.str.3.5.560. [DOI] [PubMed] [Google Scholar]
- Grünig E, Tiede H, Enyimayew EO, Ehlken N, Seyfarth HJ, Bossone E, D'Andrea A, Naeije R, Olschewski H, Ulrich S, Nagel C, Halank M, Fischer C. Assessment and prognostic relevance of right ventricular contractile reserve in patients with severe pulmonary hypertension. Circulation. 2013;128:2005–2015. doi: 10.1161/CIRCULATIONAHA.113.001573. [DOI] [PubMed] [Google Scholar]
- Haller C, Kuschinsky W. Moderate hypoxia: reactivity of pial arteries and local effect of theophylline. J Appl Physiol. 1987;63:2208–2215. doi: 10.1152/jappl.1987.63.6.2208. [DOI] [PubMed] [Google Scholar]
- Hamel E. Cholinergic modulation of the cortical microvascular bed. Prog Brain Res. 2004;145:171–178. doi: 10.1016/S0079-6123(03)45012-7. [DOI] [PubMed] [Google Scholar]
- Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100:1059–1064. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- Hamner JW, Tan CO, Lee K, Cohen MA, Taylor JA. Sympathetic control of the cerebral vasculature in humans. Stroke. 2010;41:102–109. doi: 10.1161/STROKEAHA.109.557132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamner JW, Tan CO, Tzeng YC, Taylor JA. Cholinergic control of the cerebral vasculature in humans. J Physiol. 2012;590:6343–6352. doi: 10.1113/jphysiol.2012.245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, Pedersen D, Larsen VA, Sánchez-del-Rio M, Alvarez Linera JR, Olesen J, Ashina M. Magnetic resonance angiography shows dilatation of the middle cerebral artery after infusion of glyceryl trinitrate in healthy volunteers. Cephalalgia. 2007;27:118–127. doi: 10.1111/j.1468-2982.2006.01257.x. [DOI] [PubMed] [Google Scholar]
- Hare GMT. Anaemia and the brain. Curr Opin Anaesthesiol. 2004;17:363–369. doi: 10.1097/00001503-200410000-00003. [DOI] [PubMed] [Google Scholar]
- Harmel MH, Hafkenschiel JH, Austin GM, Crumpton CW, Kety SS. The effect of bilateral stellate ganglion block on the cerebral circulation in normotensive and hypertensive patients. J Clin Invest. 1949;28:415–418. doi: 10.1172/JCI102085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AM, Bell RA. The effect of metabolic acidosis and alkalosis on the blood flow through the cerebral cortex. J Neurol Neurosurg Psychiatry. 1963;26:341–344. doi: 10.1136/jnnp.26.4.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AM, Glass HI. Effect of alterations in the arterial carbon dioxide tension on the blood flow through the cerebral cortex at normal and low arterial blood pressures. J Neurol Neurosurg Psychiatry. 1965;28:449–452. doi: 10.1136/jnnp.28.5.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert LE, Scherr PA, Bennett DA, Bienias JL, Wilson RS, Morris MC, Evans DA. Blood pressure and late-life cognitive function change: a biracial longitudinal population study. Neurology. 2004;62:2021–2024. doi: 10.1212/01.wnl.0000129258.93137.4b. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Marcus ML, Abboud FM. Role of large arteries in regulation of cerebral blood flow in dogs. J Clin Invest. 1978a;62:761–768. doi: 10.1172/JCI109187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heistad DD, Marcus ML, Gross PM. Effects of sympathetic nerves on cerebral vessels in dog, cat, and monkey. Am J Physiol Heart Circ Physiol. 1978b;235:H544–H552. doi: 10.1152/ajpheart.1978.235.5.H544. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Marcus ML, Said SI, Gross PM. Effect of acetylcholine and vasoactive intestinal peptide on cerebral blood flow. Am J Physiol Heart Circ Physiol. 1980;239:H73–H80. doi: 10.1152/ajpheart.1980.239.1.H73. [DOI] [PubMed] [Google Scholar]
- Hicks JW, Munis JR. The siphon controversy counterpoint: the brain need not be “baffling. Am J Physiol Regul Integr Comp Physiol. 2005;289:R629–R632. doi: 10.1152/ajpregu.00810.2004. [DOI] [PubMed] [Google Scholar]
- Hudetz AG, Shen H, Kampine JP. Nitric oxide from neuronal NOS plays critical role in cerebral capillary flow response to hypoxia. Am J Physiol Heart Circ Physiol. 1998;274:H982–H989. doi: 10.1152/ajpheart.1998.274.3.H982. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Ide K, Boushel R, Sørensen HM, Fernandes A, Cai Y, Pott F, Secher NH. Middle cerebral artery blood velocity during exercise with β-1 adrenergic and unilateral stellate ganglion blockade in humans. Acta Physiol Scand. 2000;170:33–38. doi: 10.1046/j.1365-201x.2000.00757.x. [DOI] [PubMed] [Google Scholar]
- Ide K, Eliasziw M, Poulin MJ. Relationship between middle cerebral artery blood velocity and end-tidal Pco2 in the hypocapnic-hypercapnic range in humans. J Appl Physiol (1985) 2003;95:129–137. doi: 10.1152/japplphysiol.01186.2002. [DOI] [PubMed] [Google Scholar]
- Ide K, Worthley M, Anderson T, Poulin MJ. Effects of the nitric oxide synthase inhibitor l-NMMA on cerebrovascular and cardiovascular responses to hypoxia and hypercapnia in humans. J Physiol. 2007;584:321–332. doi: 10.1113/jphysiol.2007.138206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Suzuki N. Control of brain capillary blood flow. J Cereb Blood Flow Metab. 2012;32:1167–1176. doi: 10.1038/jcbfm.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabuchi T, Kutsuzawa T, Ikeda K, Nakamura T. Effects of blood gases on the pressure-flow relationships in canine cerebral circulation. Stroke. 1973;4:65–72. doi: 10.1161/01.str.4.1.65. [DOI] [PubMed] [Google Scholar]
- Jakovcevic D, Harder DR. Role of astrocytes in matching blood flow to neuronal activity. Curr Top Dev Biol. 2007;79:75–97. doi: 10.1016/S0070-2153(06)79004-4. [DOI] [PubMed] [Google Scholar]
- James IM, Millar RA, Purves MJ. Observations on the extrinsic neural control of cerebral blood flow in the baboon. Circ Res. 1969;25:77–93. doi: 10.1161/01.res.25.1.77. [DOI] [PubMed] [Google Scholar]
- Jeng JS, Yip PK, Huang SJ, Kao MC. Changes in hemodynamics of the carotid and middle cerebral arteries before and after endoscopic sympathectomy in patients with palmar hyperhidrosis: preliminary results. J Neurosurg. 1999;90:463–467. doi: 10.3171/jns.1999.90.3.0463. [DOI] [PubMed] [Google Scholar]
- Jensen JB, Sperling B, Severinghaus JW, Lassen NA. Augmented hypoxic cerebral vasodilation in men during 5 days at 3,810 m altitude. J Appl Physiol (1985) 1996;80:1214–1218. doi: 10.1152/jappl.1996.80.4.1214. [DOI] [PubMed] [Google Scholar]
- Jordan J, Shannon JR, Diedrich A, Black B, Costa F, Robertson D, Biaggioni I. Interaction of carbon dioxide and sympathetic nervous system activity in the regulation of cerebral perfusion in humans. Hypertension. 2000;36:383–388. doi: 10.1161/01.hyp.36.3.383. [DOI] [PubMed] [Google Scholar]
- Jordan JD, Powers WJ. Cerebral autoregulation and acute ischemic stroke. Am J Hypertens. 2012;25:946–950. doi: 10.1038/ajh.2012.53. [DOI] [PubMed] [Google Scholar]
- Joseph V, Pequignot JM. Breathing at high altitude. Cell Mol Life Sci. 2009;66:3565–3573. doi: 10.1007/s00018-009-0143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang CK, Oh ST, Chung RK, Lee H, Park CA, Kim YB, Yoo JH, Kim DY, Cho ZH. Effect of stellate ganglion block on the cerebrovascular system: magnetic resonance angiography study. Anesthesiology. 2010;113:936–944. doi: 10.1097/ALN.0b013e3181ec63f5. [DOI] [PubMed] [Google Scholar]
- Kawamura H, Sugiyama T, Wu DM, Kobayashi M, Yamanishi S, Katsumura K, Puro DG. ATP: a vasoactive signal in the pericyte-containing microvasculature of the rat retina. J Physiol. 2003;551:787–799. doi: 10.1113/jphysiol.2003.047977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemna LJ, Posse S, Tellmann L, Schmitz T, Herzog H. Interdependence of regional and global cerebral blood flow during visual stimulation: an O-15-butanol positron emission tomography study. J Cereb Blood Flow Metab. 2001;21:664–670. doi: 10.1097/00004647-200106000-00004. [DOI] [PubMed] [Google Scholar]
- Kety SS, Schmidt CF. The effects of active and passive hyperventilation on cerebral blood flow, cerebral oxygen consumption, cardiac output, and blood pressure of normal young men. J Clin Invest. 1946;25:107–119. [PubMed] [Google Scholar]
- Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest. 1948;27:484–492. doi: 10.1172/JCI101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmerly DSDS, Tutungi EE, Wilson TDTD, Serrador JMJM, Gelb AWAW, Hughson RLRL, Shoemaker JKJK. Circulating norepinephrine and cerebrovascular control in conscious humans. Clin Physiol Funct Imaging. 2003;23:314–319. doi: 10.1046/j.1475-0961.2003.00507.x. [DOI] [PubMed] [Google Scholar]
- Koehler RC, Traystman RJ. Bicarbonate ion modulation of cerebral blood flow during hypoxia and hypercapnia. Am J Physiol Heart Circ Physiol. 1982;243:H33–H40. doi: 10.1152/ajpheart.1982.243.1.H33. [DOI] [PubMed] [Google Scholar]
- Kogure K, Scheinberg P, Reinmuth OM, Fujishima M, Busto R. Mechanisms of cerebral vasodilatation in hypoxia. J Appl Physiol. 1970;29:223–229. doi: 10.1152/jappl.1970.29.2.223. [DOI] [PubMed] [Google Scholar]
- Kolb JC, Ainslie PN, Ide K, Poulin MJ. Protocol to measure acute cerebrovascular and ventilatory responses to isocapnic hypoxia in humans. Respir Physiol Neurobiol. 2004;141:191–199. doi: 10.1016/j.resp.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Raper AJ, Patterson JL. Analysis of vasoactivity of local pH, Pco2 and bicarbonate on pial vessels. Stroke. 1977a;8:358–360. doi: 10.1161/01.str.8.3.358. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Wei EP, Navari RM, Levasseur JE, Rosenblum WI, Patterson JL. Responses of cerebral arteries and arterioles to acute hypotension and hypertension. Am J Physiol Heart Circ Physiol. 1978;234:H371–H383. doi: 10.1152/ajpheart.1978.234.4.H371. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Wei EP, Raper AJ, Patterson JL. Local mechanism of CO2 action of cat pial arterioles. Stroke. 1977b;8:226–229. doi: 10.1161/01.str.8.2.226. [DOI] [PubMed] [Google Scholar]
- Krejcy K, Wolzt M, Kreuzer C, Breiteneder H, Schütz W, Eichler HG, Schmetterer L. Characterization of angiotensin-II effects on cerebral and ocular circulation by noninvasive methods. Br J Clin Pharmacol. 1997;43:501–508. doi: 10.1046/j.1365-2125.1997.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagaud G, Karicheti V, Knot HJ, Christ GJ, Laher I. Inhibitors of gap junctions attenuate myogenic tone in cerebral arteries. Am J Physiol Heart Circ Physiol. 2002;283:H2177–H2186. doi: 10.1152/ajpheart.00605.2001. [DOI] [PubMed] [Google Scholar]
- Lambertsen CJ, Semple SJ, Smyth MG, Gelfand R. H+ and pCO2 as chemical factors in respiratory and cerebral circulatory control. J Appl Physiol. 1961;16:473–484. doi: 10.1152/jappl.1961.16.3.473. [DOI] [PubMed] [Google Scholar]
- Lassen NA. Cerebral blood flow and oxygen consumption in man. Physiol Rev. 1959;39:183–238. doi: 10.1152/physrev.1959.39.2.183. [DOI] [PubMed] [Google Scholar]
- Lassen NA. Brain extracellular pH: the main factor controlling cerebral blood flow. Scand J Clin Lab Invest. 1968;22:247–251. doi: 10.3109/00365516809167060. [DOI] [PubMed] [Google Scholar]
- Laudignon N, Farri E, Beharry K, Rex J, Aranda JV. Influence of adenosine on cerebral blood flow during hypoxic hypoxia in the newborn piglet. J Appl Physiol (1985) 1990;68:1534–1541. doi: 10.1152/jappl.1990.68.4.1534. [DOI] [PubMed] [Google Scholar]
- Leithner C, Royl G. The oxygen paradox of neurovascular coupling. J Cereb Blood Flow Metab. 2014;34:19–29. doi: 10.1038/jcbfm.2013.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMarbre G, Stauber S, Khayat RN, Puleo DS, Skatrud JB, Morgan BJ. Baroreflex-induced sympathetic activation does not alter cerebrovascular CO2 responsiveness in humans. J Physiol. 2003;551:609–616. doi: 10.1113/jphysiol.2003.046987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden L. The effect of stellate ganglion block on cerebral circulation in cerebrovascular accidents. Acta Med Scand Suppl. 1955;301:1–110. [PubMed] [Google Scholar]
- Liu J, Zhu YS, Hill C, Armstrong K, Tarumi T, Hodics T, Hynan LS, Zhang R. Cerebral autoregulation of blood velocity and volumetric flow during steady-state changes in arterial pressure. Hypertension. 2013;62:973–979. doi: 10.1161/HYPERTENSIONAHA.113.01867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Alkayed NJ. Hypoxic preconditioning and tolerance via hypoxia inducible factor (HIF) α-linked induction of P450 2C11 epoxygenase in astrocytes. J Cereb Blood Flow Metab. 2005;25:939–948. doi: 10.1038/sj.jcbfm.9600085. [DOI] [PubMed] [Google Scholar]
- Liu X, Li C, Falck JR, Harder DR, Koehler RC. Relative contribution of cyclooxygenases, epoxyeicosatrienoic acids, and pH to the cerebral blood flow response to vibrissal stimulation. Am J Physiol Heart Circ Physiol. 2012;302:H1075–H1085. doi: 10.1152/ajpheart.00794.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas SJ, Tzeng YC, Galvin SD, Thomas KN, Ogoh S, Ainslie PN. Influence of changes in blood pressure on cerebral perfusion and oxygenation. Hypertension. 2010;55:698–705. doi: 10.1161/HYPERTENSIONAHA.109.146290. [DOI] [PubMed] [Google Scholar]
- McCulloch TJ, Boesel TW, Lam AM. The effect of hypocapnia on the autoregulation of cerebral blood flow during administration of isoflurane. Anesth Analg. 2005;100:1463–1467. doi: 10.1213/01.ANE.0000148623.06596.7E. table of contents. [DOI] [PubMed] [Google Scholar]
- McDowall DG. Interrelationships between blood oxygen tensions and cerebral blood flow. Int Anesthesiol Clin. 1966;4:205–219. [PubMed] [Google Scholar]
- Magnussen I, Hoedt-Rasmussen K. The effect of intraarterial administered aminophylline on cerebral hemodynamics in man. Acta Neurol Scand. 1977;55:131–136. doi: 10.1111/j.1600-0404.1977.tb05633.x. [DOI] [PubMed] [Google Scholar]
- Mandell DM, Han JS, Poublanc J, Crawley AP, Kassner A, Fisher JA, Mikulis DJ. Selective reduction of blood flow to white matter during hypercapnia corresponds with leukoaraiosis. Stroke. 2008;39:1993–1998. doi: 10.1161/STROKEAHA.107.501692. [DOI] [PubMed] [Google Scholar]
- Mardimae A, Balaban DY, Machina MA, Battisti-Charbonney A, Han JS, Katznelson R, Minkovich LL, Fedorko L, Murphy PM, Wasowicz M, Naughton F, Meineri M, Fisher JA, Duffin J. The interaction of carbon dioxide and hypoxia in the control of cerebral blood flow. Pflugers Arch. 2012;464:345–351. doi: 10.1007/s00424-012-1148-1. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Werber AH, Heistad DD. Protection of cerebral vessels by sympathetic nerves and vascular hypertrophy. Circulation. 1987;75:I107–I112. [PubMed] [Google Scholar]
- Mchedlishvili GI. In: Arterial Behavior and Blood Circulation in the Brain. Bevan JA, editor. New York: Plenum Press; 1986. [Google Scholar]
- Mchedlishvili GI. Vascular mechanisms pertaining to the intrinsic regulation of the cerebral circulation. Circulation. 1964;30:597–610. doi: 10.1161/01.cir.30.4.597. [DOI] [PubMed] [Google Scholar]
- Mchedlishvili GI, Mitagvaria NP, Ormotsadze LG. Vascular mechanisms controlling a constant blood supply to the brain (“autoregulation”) Stroke. 1973;4:742–750. doi: 10.1161/01.str.4.5.742. [DOI] [PubMed] [Google Scholar]
- Meno JR, Ngai AC, Winn HR. Changes in pial arteriolar diameter and CSF adenosine concentrations during hypoxia. J Cereb Blood Flow Metab. 1993;13:214–220. doi: 10.1038/jcbfm.1993.26. [DOI] [PubMed] [Google Scholar]
- Metry G, Wikström B, Valind S, Sandhagen B, Linde T, Beshara S, Långström B, Danielson BG. Effect of normalization of hematocrit on brain circulation and metabolism in hemodialysis patients. J Am Soc Nephrol. 1999;10:854–863. doi: 10.1681/ASN.V104854. [DOI] [PubMed] [Google Scholar]
- Miekisiak G, Kulik T, Kusano Y, Kung D, Chen J-F, Winn HR. Cerebral blood flow response in adenosine 2a receptor knockout mice during transient hypoxic hypoxia. J Cereb Blood Flow Metab. 2008;28:1656–1664. doi: 10.1038/jcbfm.2008.57. [DOI] [PubMed] [Google Scholar]
- Mitchell D, Lambert G, Secher N, Raven P, Van Lieshout J, Esler M. Jugular venous overflow of noradrenaline from the brain: a neurochemical indicator of cerebrovascular sympathetic nerve activity in humans. J Physiol. 2009;587:2589–2597. doi: 10.1113/jphysiol.2008.167999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morii S, Ngai AC, Ko KR, Winn HR. Role of adenosine in regulation of cerebral blood flow: effects of theophylline during normoxia and hypoxia. Am J Physiol Heart Circ Physiol. 1987;253:H165–H175. doi: 10.1152/ajpheart.1987.253.1.H165. [DOI] [PubMed] [Google Scholar]
- Mosso A. Sulla circolazione del cervello dell'uomo. Att R Accad Lincei. 1880;5:237–358. [Google Scholar]
- Muizelaar JP, Wei EP, Kontos HA, Becker DP. Cerebral blood flow is regulated by changes in blood pressure and in blood viscosity alike. Stroke. 1986;17:44–48. doi: 10.1161/01.str.17.1.44. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Suzuki A, Yoshioka A, Yamamoto M, Akiyama Y, Miyamoto K, Kishi F, Kawakami Y. Effect of aminophylline on brain tissue oxygenation in patients with chronic obstructive lung disease. Thorax. 1992;47:1025–1029. doi: 10.1136/thx.47.12.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Yoshioka A, Yamamoto M, Akiyama Y, Miyamoto K, Kawakami Y. Effect of theophylline on brain tissue oxygenation during normoxia and hypoxia in humans. J Appl Physiol (1985) 1993;74:2724–2728. doi: 10.1152/jappl.1993.74.6.2724. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Suzuki A, Hatazawa J, Nishimura H, Shirane R, Yasui N, Yoshimoto T. Cerebral blood-flow responses to induced hypotension and to CO2 inhalation in patients with major cerebral artery occlusive disease: a positron-emission tomography study. Neuroradiology. 1999;41:73–79. doi: 10.1007/s002340050709. [DOI] [PubMed] [Google Scholar]
- Nolan WF, Houck PC, Thomas JL, Davies DG. Ventral medullary extracellular fluid pH and blood flow during hypoxia. Am J Physiol Regul Integr Comp Physiol. 1982;242:R195–R198. doi: 10.1152/ajpregu.1982.242.3.R195. [DOI] [PubMed] [Google Scholar]
- Nöth U, Kotajima F, Deichmann R, Turner R, Corfield DR. Mapping of the cerebral vascular response to hypoxia and hypercapnia using quantitative perfusion MRI at 3 T. NMR Biomed. 2008;21:464–472. doi: 10.1002/nbm.1210. [DOI] [PubMed] [Google Scholar]
- Offenhauser N, Thomsen K, Caesar K, Lauritzen M. Activity-induced tissue oxygenation changes in rat cerebellar cortex: interplay of postsynaptic activation and blood flow. J Physiol. 2005;565:279–294. doi: 10.1113/jphysiol.2005.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Iwasaki K, Aoki K, Gokan D, Hirose N, Kato J, Ogawa S. The different effects of midazolam and propofol sedation on dynamic cerebral autoregulation. Anesth Analg. 2010;111:1279–1284. doi: 10.1213/ANE.0b013e3181f42fc0. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Iwasaki K, Aoki K, Kojima W, Kato J, Ogawa S. Dexmedetomidine weakens dynamic cerebral autoregulation as assessed by transfer function analysis and the thigh cuff method. Anesthesiology. 2008;109:642–650. doi: 10.1097/ALN.0b013e3181862a33. [DOI] [PubMed] [Google Scholar]
- Ogoh S, Brothers R, Eubank W, Raven P. Autonomic neural control of the cerebral vasculature: acute hypotension. Stroke. 2008;39:1979–1987. doi: 10.1161/STROKEAHA.107.510008. [DOI] [PubMed] [Google Scholar]
- Ogoh S, Nakahara H, Okazaki K, Bailey DM, Miyamoto T. Cerebral hypoperfusion modifies the respiratory chemoreflex during orthostatic stress. Clin Sci (Lond) 2013;125:37–44. doi: 10.1042/CS20120335. [DOI] [PubMed] [Google Scholar]
- Ohta S, Hadeishi H, Suzuki M. Effect of stellate ganglion block on cerebral blood flow in normoxemic and hyperoxemic states. J Neurosurg Anesthesiol. 1990;2:272–279. doi: 10.1097/00008506-199012000-00004. [DOI] [PubMed] [Google Scholar]
- Panerai RB. Nonstationarity of dynamic cerebral autoregulation. Med Eng Phys. 2013;35:111–116. doi: 10.1016/j.medengphy.2013.09.004. [DOI] [PubMed] [Google Scholar]
- Paulson OB, Parving HH, Olesen J, Skinhoj E. Influence of carbon monoxide and of hemodilution on cerebral blood flow and blood gases in man. J Appl Physiol. 1973;35:111–116. doi: 10.1152/jappl.1973.35.1.111. [DOI] [PubMed] [Google Scholar]
- Pearce WJ. Mechanisms of hypoxic cerebral vasodilatation. Pharmacol Ther. 1995;65:75–91. doi: 10.1016/0163-7258(94)00058-b. [DOI] [PubMed] [Google Scholar]
- Pearce WJ, Ashwal S, Long DM, Cuevas J. Hypoxia inhibits calcium influx in rabbit basilar and carotid arteries. Am J Physiol Heart Circ Physiol. 1992;262:H106–H113. doi: 10.1152/ajpheart.1992.262.1.H106. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Koenig HM, Albrecht RF. Nitric oxide synthesis and regional cerebral blood flow responses to hypercapnia and hypoxia in the rat. J Cereb Blood Flow Metab. 1993;13:80–87. doi: 10.1038/jcbfm.1993.10. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Wang Q, Koenig HM, Albrecht RF. Role of nitric oxide, adenosine, N-methyl-d-aspartate receptors, and neuronal activation in hypoxia-induced pial arteriolar dilation in rats. Brain Res. 1995;704:61–70. doi: 10.1016/0006-8993(95)01105-6. [DOI] [PubMed] [Google Scholar]
- Peng HL, Ivarsen A, Nilsson H, Aalkjaer C. On the cellular mechanism for the effect of acidosis on vascular tone. Acta Physiol Scand. 1998;164:517–525. doi: 10.1111/j.1365-201x.1998.tb10701.x. [DOI] [PubMed] [Google Scholar]
- Piechnik SK, Chiarelli PA, Jezzard P. Modelling vascular reactivity to investigate the basis of the relationship between cerebral blood volume and flow under CO2 manipulation. Neuroimage. 2008;39:107–118. doi: 10.1016/j.neuroimage.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Pinard E, Puiroud S, Seylaz J. Role of adenosine in cerebral hypoxic hyperemia in the unanesthetized rabbit. Brain Res. 1989;481:124–130. doi: 10.1016/0006-8993(89)90492-7. [DOI] [PubMed] [Google Scholar]
- Pinard E, Purves MJ, Seylaz J, Vasquez JV. The cholinergic pathway to cerebral blood vessels. II. Physiological studies. Pflugers Arch. 1979;379:165–172. doi: 10.1007/BF00586943. [DOI] [PubMed] [Google Scholar]
- Ponte J, Purves MJ. The role of the carotid body chemoreceptors and carotid sinus baroreceptors in the control of cerebral blood vessels. J Physiol. 1974;237:315–340. doi: 10.1113/jphysiol.1974.sp010484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell FL, Huey KA, Dwinell MR. Central nervous system mechanisms of ventilatory acclimatization to hypoxia. Respir Physiol. 2000;121:223–236. doi: 10.1016/s0034-5687(00)00130-4. [DOI] [PubMed] [Google Scholar]
- Przybyłowski T, Bangash MF, Reichmuth K, Morgan BJ, Skatrud JB, Dempsey JA. Mechanisms of the cerebrovascular response to apnoea in humans. J Physiol. 2003;548:323–332. doi: 10.1113/jphysiol.2002.029678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C, von Strauss E, Fastbom J, Winblad B, Fratiglioni L. Low blood pressure and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Arch Neurol. 2003;60:223–228. doi: 10.1001/archneur.60.2.223. [DOI] [PubMed] [Google Scholar]
- Querido JS, Ainslie PN, Foster GE, Henderson WR, Halliwill JR, Ayas NT, Sheel AW. Dynamic cerebral autoregulation during and following acute hypoxia: role of carbon dioxide. J Appl Physiol (1985) 2013;114:1183–1190. doi: 10.1152/japplphysiol.00024.2013. [DOI] [PubMed] [Google Scholar]
- Querido JS, Godwin JB, Sheel AW. Intermittent hypoxia reduces cerebrovascular sensitivity to isocapnic hypoxia in humans. Respir physiol neurobiol. 2008;161:1–9. doi: 10.1016/j.resp.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Reichmuth K, Dopp JM, Barczi SR, Skatrud JB, Wojdyla P, Hayes D, Jr, Morgan BJ. Impaired vascular regulation in patients with obstructive sleep apnea: effects of CPAP treatment. Am J Respir Crit Care Med. 2009;180:1143–1150. doi: 10.1164/rccm.200903-0393OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengarten B, Aldinger C, Kaufmann A, Kaps M. Comparison of visually evoked peak systolic and end diastolic blood flow velocity using a control system approach. Ultrasound Med Biol. 2001;27:1499–1503. doi: 10.1016/s0301-5629(01)00464-1. [DOI] [PubMed] [Google Scholar]
- Rosengarten B, Aldinger C, Spiller A, Kaps M. Neurovascular coupling remains unaffected during normal aging. J Neuroimaging. 2003;13:43–47. [PubMed] [Google Scholar]
- Roy CS, Sherrington CS. On the regulation of the blood-supply of the brain. J Physiol. 1890;11:85–158.17. doi: 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santizo R, Baughman VL, Pelligrino DA. Relative contributions from neuronal and endothelial nitric oxide synthases to regional cerebral blood flow changes during forebrain ischemia in rats. Neuroreport. 2000;11:1549–1553. [PubMed] [Google Scholar]
- Sato A, Sato Y, Uchida S. Regulation of regional cerebral blood flow by cholinergic fibers originating in the basal forebrain. Int J Dev Neurosci. 2001;19:327–337. doi: 10.1016/s0736-5748(01)00017-x. [DOI] [PubMed] [Google Scholar]
- Sato K, Sadamoto T, Hirasawa A, Oue A, Subudhi AW, Miyazawa T, Ogoh S. Differential blood flow responses to CO2 in human internal and external carotid and vertebral arteries. J Physiol. 2012;590:3277–3290. doi: 10.1113/jphysiol.2012.230425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinberg P. Cerebral blood flow in vascular disease of the brain; with observations on the effects of stellate ganglion block. Am J Med. 1950;8:139–147. doi: 10.1016/0002-9343(50)90354-8. [DOI] [PubMed] [Google Scholar]
- Schmidt B, Czosnyka M, Klingelhöfer J. Asymmetry of cerebral autoregulation does not correspond to asymmetry of cerebrovascular pressure reactivity. Perspectives in Medicine. 2012;1:285–289. [Google Scholar]
- Schmidt B, Klingelhöfer J, Perkes I, Czosnyka M. Cerebral autoregulatory response depends on the direction of change in perfusion pressure. J Neurotrauma. 2009;26:651–656. doi: 10.1089/neu.2008.0784. [DOI] [PubMed] [Google Scholar]
- Schubert T, Santini F, Stalder AF, Bock J, Meckel S, Bonati L, Markl M, Wetzel S. Dampening of blood-flow pulsatility along the carotid siphon: does form follow function. AJNR Am J Neuroradiol. 2011;32:1107–1112. doi: 10.3174/ajnr.A2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal SS. Integration of blood flow control to skeletal muscle: key role of feed arteries. Acta Physiol Scand. 2000;168:511–518. doi: 10.1046/j.1365-201x.2000.00703.x. [DOI] [PubMed] [Google Scholar]
- Shapiro W, Wasserman AJ, Baker JP, Patterson JL. Cerebrovascular response to acute hypocapnic and eucapnic hypoxia in normal man. J Clin Invest. 1970;49:2362–2368. doi: 10.1172/JCI106455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkin HA. Cervical sympathectomy on patients with occlusive cerebrovascular disease. Arch Surg. 1969;98:317–320. doi: 10.1001/archsurg.1969.01340090093015. [DOI] [PubMed] [Google Scholar]
- Shenkin HA, Cabieses F, Van Den Noordt G. The effect of bilateral stellectomy upon the cerebral circulation of man. J Clin Invest. 1951;30:90–93. doi: 10.1172/JCI102421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skow RJ, MacKay CM, Tymko MM, Willie CK, Smith KJ, Ainslie PN, Day TA. Differential cerebrovascular CO2 reactivity in anterior and posterior cerebral circulations. Respir Physiol Neurobiol. 2013;189:76–86. doi: 10.1016/j.resp.2013.05.036. [DOI] [PubMed] [Google Scholar]
- Smith BA, Clayton EW, Robertson D. Experimental arrest of cerebral blood flow in human subjects: the red wing studies revisited. Perspect Biol Med. 2011;54:121–131. doi: 10.1353/pbm.2011.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JM, Medow MS, DelPozzi A, Messer ZR, Terilli C, Schwartz CE. Middle cerebral O2 delivery during the modified Oxford maneuver increases with sodium nitroprusside and decreases during phenylephrine. Am J Physiol Heart Circ Physiol. 2013;304:H1576–H1583. doi: 10.1152/ajpheart.00114.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Iwabuchi T, Hori S. Cervical sympathectomy for cerebral vasospasm after aneurysm rupture. Neurol Med Chir (Tokyo) 1975;15:41–50. doi: 10.2176/nmc.15pt1.41. [DOI] [PubMed] [Google Scholar]
- Takeuchi S, Karino T. Flow patterns and distributions of fluid velocity and wall shear stress in the human internal carotid and middle cerebral arteries. World Neurosurg. 2010;73:174–185. doi: 10.1016/j.surneu.2009.03.030. discussion e27. [DOI] [PubMed] [Google Scholar]
- Tamaki K, Heistad DD. Response of cerebral arteries to sympathetic stimulation during acute hypertension. Hypertension. 1986;8:911–917. doi: 10.1161/01.hyp.8.10.911. [DOI] [PubMed] [Google Scholar]
- Tan CO. Defining the characteristic relationship between arterial pressure and cerebral flow. J Appl Physiol (1985) 2012;113:1194–1200. doi: 10.1152/japplphysiol.00783.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CO, Hamner JW, Taylor JA. The role of myogenic mechanisms in human cerebrovascular regulation. J Physiol. 2013;591:5095–5105. doi: 10.1113/jphysiol.2013.259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CO, Taylor JA. Integrative physiological and computational approaches to understand autonomic control of cerebral autoregulation. Exp Physiol. 2014;99:3–15. doi: 10.1113/expphysiol.2013.072355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JK, Peterson MR, Freeman RD. Single-neuron activity and tissue oxygenation in the cerebral cortex. Science. 2003;299:1070–1072. doi: 10.1126/science.1079220. [DOI] [PubMed] [Google Scholar]
- Toda N, Okamura T. Cerebral vasodilators. Jpn J Pharmacol. 1998;76:349–367. doi: 10.1254/jjp.76.349. [DOI] [PubMed] [Google Scholar]
- Todd MM, Wu B, Maktabi M, Hindman BJ, Warner DS. Cerebral blood flow and oxygen delivery during hypoxemia and hemodilution: role of arterial oxygen content. Am J Physiol Heart Circ Physiol. 1994;267:H2025–H2031. doi: 10.1152/ajpheart.1994.267.5.H2025. [DOI] [PubMed] [Google Scholar]
- Tomiyama Y, Jansen K, Brian JEJ, Todd MM. Hemodilution, cerebral O2 delivery, and cerebral blood flow: a study using hyperbaric oxygenation. Am J Physiol Heart Circ Physiol. 1999;276:H1190–H1196. doi: 10.1152/ajpheart.1999.276.4.H1190. [DOI] [PubMed] [Google Scholar]
- Treggiari MM. Cervical sympathetic block to reverse delayed ischemic neurological deficits after aneurysmal subarachnoid hemorrhage. Stroke. 2003;34:961–967. doi: 10.1161/01.STR.0000060893.72098.80. [DOI] [PubMed] [Google Scholar]
- Tzeng YC, Ainslie PN, Cooke WH, Peebles KC, Willie CK, MacRae BA, Smirl JD, Horsman HM, Rickards CA. Assessment of cerebral autoregulation: the quandary of quantification. Am J Physiol Heart Circ Physiol. 2012;303:H658–H671. doi: 10.1152/ajpheart.00328.2012. [DOI] [PubMed] [Google Scholar]
- Tzeng Y-C, Chan GS, Willie CK, Ainslie PN. Determinants of human cerebral pressure–flow velocity relationships: new insights from vascular modeling and Ca2+ blockade. J Physiol. 2011;589:3263–3274. doi: 10.1113/jphysiol.2011.206953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng Y-C, Willie CK, Atkinson G, Lucas SJE, Wong A, Ainslie PN. Cerebrovascular regulation during transient hypotension and hypertension in humans. Hypertension. 2010;56:268–273. doi: 10.1161/HYPERTENSIONAHA.110.152066. [DOI] [PubMed] [Google Scholar]
- Umeyama T, Kugimiya T, Ogawa T, Kandori Y, Ishizuka A, Hanaoka K. Changes in cerebral blood flow estimated after stellate ganglion block by single photon emission computed tomography. J Auton Nerv Syst. 1995;50:339–346. doi: 10.1016/0165-1838(94)00105-s. [DOI] [PubMed] [Google Scholar]
- Van Lieshout JJ, Wieling W, Karemaker JM, Secher NH. Syncope, cerebral perfusion, and oxygenation. J Appl Physiol (1985) 2003;94:833–848. doi: 10.1152/japplphysiol.00260.2002. [DOI] [PubMed] [Google Scholar]
- Van Mil AH, Spilt A, Van Buchem MA, Bollen EL, Teppema L, Westendorp RG, Blauw GJ. Nitric oxide mediates hypoxia-induced cerebral vasodilation in humans. J Appl Physiol (1985) 2002;92:962–966. doi: 10.1152/japplphysiol.00616.2001. [DOI] [PubMed] [Google Scholar]
- Vanzetta I, Grinvald A. Increased cortical oxidative metabolism due to sensory stimulation: implications for functional brain imaging. Science. 1999;286:1555–1558. doi: 10.1126/science.286.5444.1555. [DOI] [PubMed] [Google Scholar]
- Vinall PE, Simeone FA. Effects of oxygen and glucose deprivation on vasoactivity in isolated bovine middle cerebral arteries. Stroke. 1986;17:970–975. doi: 10.1161/01.str.17.5.970. [DOI] [PubMed] [Google Scholar]
- Wahl M, Deetjen P, Thurau K, Ingvar DH, Lassen NA. Micropuncture evaluation of the importance of perivascular pH for the arteriolar diameter on the brain surface. Pflugers Archiv. 1970;316:152–163. doi: 10.1007/BF00586483. [DOI] [PubMed] [Google Scholar]
- Waki H, Bhuiyan ME, Gouraud SS, Takagishi M, Hatada A, Kohsaka A, Paton JF, Maeda M. Acute reductions in blood flow restricted to the dorsomedial medulla induce a pressor response in rats. J Hypertens. 2011;29:1536–1545. doi: 10.1097/HJH.0b013e3283484106. [DOI] [PubMed] [Google Scholar]
- Wasserman AJ, Patterson JLJ. The cerebral vascular response to reduction in arterial carbon dioxide tension. J Clin Invest. 1961;40:1297–1303. doi: 10.1172/JCI104359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler RL, Kleiss LM, Kety SS. The effects of intravenously administered aminophylline on cerebral circulation and metabolism in man. J Clin Invest. 1950;29:28–30. doi: 10.1172/JCI102230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei EP, Raper AJ, Kontos HA, Patterson JL. Determinants of response of pial arteries to norepinephrine and sympathetic nerve stimulation. Stroke. 1975;6:654–658. doi: 10.1161/01.str.6.6.654. [DOI] [PubMed] [Google Scholar]
- Willie CK, Ainslie PN, Taylor CE, Eves ND, Tzeng YC. Maintained cerebrovascular function during post-exercise hypotension. Eur J Appl Physiol. 2013a;113:1597–1604. doi: 10.1007/s00421-012-2578-3. [DOI] [PubMed] [Google Scholar]
- Willie CK, Cowan EC, Ainslie PN, Taylor CE, Smith KJ, Sin PY, Tzeng YC. Neurovascular coupling and distribution of cerebral blood flow during exercise. J Neurosci Methods. 2011;198:270–273. doi: 10.1016/j.jneumeth.2011.03.017. [DOI] [PubMed] [Google Scholar]
- Willie CK, Macleod DB, Shaw AD, Smith KJ, Tzeng YC, Eves ND, Ikeda K, Graham J, Lewis NC, Day TA, Ainslie PN. Regional brain blood flow in man during acute changes in arterial blood gases. J Physiol. 2012;590:3261–3275. doi: 10.1113/jphysiol.2012.228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willie CK, Smith KJ, Day TA, Ray LA, Lewis NC, Bakker A, Macleod DB, Ainslie PN. Regional cerebral blood flow in humans at high altitude: gradual ascent and two weeks at 5050m. J Appl Physiol. 2013b doi: 10.1152/japplphysiol.00594.2013. doi: 10.1152/japplphysiol.00594.2013. [DOI] [PubMed] [Google Scholar]
- Wilson MH, Edsell MEG, Davagnanam I, Hirani SP, Martin DS, Levett DZH, Thornton JS, Golay X, Strycharczuk L, Newman SP, Montgomery HE, Grocott MPW, Imray CHE Caudwell Xtreme Everest Research Group. Cerebral artery dilatation maintains cerebral oxygenation at extreme altitude and in acute hypoxia-an ultrasound and MRI study. J Cereb Blood Flow Metab. 2011;31:2019–2029. doi: 10.1038/jcbfm.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn HR, Rubio R, Berne RM. Brain adenosine concentration during hypoxia in rats. Am J Physiol Heart Circ Physiol. 1981;241:H235–H242. doi: 10.1152/ajpheart.1981.241.2.H235. [DOI] [PubMed] [Google Scholar]
- Wolff HG, Lennox WG. Cerebral circulation. XII. The effect on pial vessels of variations in the oxygen and carbon dioxide content of the blood. Arch Neurol Psychiatry. 1930;23:1097–1120. [Google Scholar]
- Yamaura K, Gebremedhin D, Zhang C, Narayanan J, Hoefert K, Jacobs ER, Koehler RC, Harder DR. Contribution of epoxyeicosatrienoic acids to the hypoxia-induced activation of Ca2+-activated K+ channel current in cultured rat hippocampal astrocytes. Neuroscience. 2006;143:703–716. doi: 10.1016/j.neuroscience.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Yokoyama K, Kishida T, Sugiyama K. Stellate ganglion block and regional cerebral blood volume and oxygenation. Can J Anaesth. 2004;51:515–516. doi: 10.1007/BF03018319. [DOI] [PubMed] [Google Scholar]
- Yoon S, Zuccarello M, Rapoport RM. pCO2 and pH regulation of cerebral blood flow. Front Physiol. 2012;3:365. doi: 10.3389/fphys.2012.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Huang G, Shi X. Cerebral vasoreactivity during hypercapnia is reset by augmented sympathetic influence. J Appl Physiol (1985) 2011;110:352–358. doi: 10.1152/japplphysiol.00802.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Behbehani K, Levine BD. Dynamic pressure–flow relationship of the cerebral circulation during acute increase in arterial pressure. J Physiol. 2009;587:2567–2577. doi: 10.1113/jphysiol.2008.168302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Crandall CG, Levine BD. Cerebral hemodynamics during the Valsalva maneuver: insights from ganglionic blockade. Stroke. 2004;35:843–847. doi: 10.1161/01.STR.0000120309.84666.AE. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zuckerman JH, Iwasaki K, Wilson TE, Crandall CG, Levine BD. Autonomic neural control of dynamic cerebral autoregulation in humans. Circulation. 2002;106:1814–1820. doi: 10.1161/01.cir.0000031798.07790.fe. [DOI] [PubMed] [Google Scholar]