

Abstract
The role of the small GTP-binding protein Rac1 in smooth muscle contraction was examined using small molecule inhibitors (EHT1864, NSC23766) and a novel smooth muscle-specific, conditional, Rac1 knockout mouse strain. EHT1864, which affects nucleotide binding and inhibits Rac1 activity, concentration-dependently inhibited the contractile responses induced by several different modes of activation (high-K+, phenylephrine, carbachol and protein kinase C activation by phorbol-12,13-dibutyrate) in several different visceral (urinary bladder, ileum) and vascular (mesenteric artery, saphenous artery, aorta) smooth muscle tissues. This contractile inhibition was associated with inhibition of the Ca2+ transient. Knockout of Rac1 (with a 50% loss of Rac1 protein) lowered active stress in the urinary bladder and the saphenous artery consistent with a role of Rac1 in facilitating smooth muscle contraction. NSC23766, which blocks interaction between Rac1 and some guanine nucleotide exchange factors, specifically inhibited the α1 receptor responses (phenylephrine) in vascular tissues and potentiated prostaglandin F2α and thromboxane (U46619) receptor responses. The latter potentiating effect occurred at lowered intracellular [Ca2+]. These results show that Rac1 activity is required for active contraction in smooth muscle, probably via enabling an adequate Ca2+ transient. At the same time, specific agonists recruit Rac1 signalling via upstream modulators, resulting in either a potentiation of contraction via Ca2+ mobilization (α1 receptor stimulation) or an attenuated contraction via inhibition of Ca2+ sensitization (prostaglandin and thromboxane receptors).
Introduction
The Rho GTPases constitute a family with 22 described members within the Ras superfamily of small G-proteins (Jaffe & Hall, 2005). They cycle between active GTP-bound and inactive GDP-bound forms, under the control of regulatory proteins, guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and the guanine dissociation inhibitors (RhoGDIs) which influence the cellular translocation of Rho (Cherfils & Zeghouf, 2013). Rho GTPases, in particular Cdc42, Rac1 and RhoA, have been linked to a large number of cellular functions including reorganization of the cytoskeleton, gene expression and enzymatic activities (Jaffe & Hall, 2005).
It was recognized early that small G-proteins have a key role in smooth muscle contraction (Nishimura et al. 1988; Kitazawa et al. 1989). It has been subsequently shown that RhoA signalling inhibits myosin light chain phosphatase activity and thereby increases the Ca2+ sensitivity of the contractile process (Somlyo & Somlyo, 2003; Puetz et al. 2009). This signalling pathway has been implicated in several pathological conditions of smooth muscle and proposed as a possible therapeutic target (Halayko & Solway, 2001; Loirand et al. 2006; Loirand & Pacaud, 2010; Zhang & DiSanto, 2011). Much less is known regarding the function of the other Rho GTPases in smooth muscle. Rac1 has been shown to influence the actin cytoskeleton and modulate smooth muscle cell migration and angiogenesis (Doanes et al. 1998; Sawada et al. 2010). It is involved in the regulation of NADPH oxidase-mediated superoxide production and possibly proliferative/hypertrophic responses (Kim et al. 1998; Lyle & Griendling, 2006). The downstream targets of Rac1 might also include RhoA (Sander et al. 1999; Burridge & Wennerberg, 2004; Rosenfeldt et al. 2006), although this mechanism has not been demonstrated in smooth muscle. Pak1, which is one downstream target of Rac1, has been shown to inhibit the myosin light chain kinase and relax permeabilized intestinal smooth muscle (Wirth et al. 2003). In contrast, evidence has also been presented that Pak1 is part of an activating pathway in some smooth muscles. Knockout or pharmacological inhibition of Pak reduces tone of airway smooth muscle (Hoover et al. 2012). Introduction of Pak3 in permeabilized smooth muscle induces a Ca2+-independent contraction, possibly via phosphorylation of caldesmon (Van Eyk et al. 1998; McFawn et al. 2003). Furthermore, Pak phosphorylates the phosphatase inhibitor CPI17 in vitro (Takizawa et al. 2002).
The upstream control of Rac1 activity is not fully understood. Several GEFs acting on Rac1 have been identified and proposed, and of which the Dbl-homology domain-related GEFs constitute a large group (Rossman et al. 2005; Cherfils & Zeghouf, 2013), which includes Tiam and Vav (Habets et al. 1995; Bustelo, 2000). Very little is known regarding the role of these GEFs in smooth muscle signal transduction and of the physiological events that might lead to their activation. Knockout of the GEFs Vav2 or Vav3 results in severe cardiovascular phenotypes in mice (Sauzeau et al. 2006, 2007), although the mechanisms are not clear. It has been suggested that Vav2, Rac1 and Pak1 are involved in endothelial NO-mediated relaxation in vascular smooth muscle (Sauzeau et al. 2010).
The aim of this study was to investigate the role of Rac1 in smooth muscle contraction. We examined a range of smooth muscle tissues and used different modes of activation. We applied two different small molecule pharmacological inhibitors of Rac1. NSC23766 (Gao et al. 2004) interferes with the interaction between Rac1 and some GEFs (Tiam1 and TrioN) without influencing Cdc42 or RhoA, the interaction between Rac1 and the Bcr GTPase-activating protein, or Pak1 activity. EHT1864 (Desire et al. 2005; Shutes et al. 2007) influences nucleotide binding and the Rac1/Pak1 interaction and provides a more general inhibition of Rac1 when compared to NSC23766. In addition, we developed a new transgenic mouse strain by breeding mice expressing tamoxifen-activated Cre recombinase under the control of the smooth muscle-specific SM22 promoter (Kuhbandner et al. 2000) with mice carrying floxed Rac1 genes (Chrostek et al. 2006) enabling a conditional, smooth muscle-specific knockout of Rac1. We report that Rac1 is differentially activated by different receptor pathways and contributes to the regulation of contraction in smooth muscle.
Methods
Animals and tissue preparations
C57Bl/6 mice (10–15 weeks, female) and conditional smooth muscle-specific Rac1 knockout animals (see below, 10–20 weeks, either sex) were used. The experiments were performed according to European guidelines for animal research, complied with national regulations for the care of experimental animals and were approved by the local animal ethics committee.
The mice were killed by cervical dislocation. The abdominal aorta, saphenous artery (SA), small mesenteric artery (SMA), urinary bladder (UB) and ileum were excised and placed in cold Krebs–Ringer solution (composition in mm: NaCl 123, KCl 4.7, KH2PO4 1.2, MgCl2 1.2, NaHCO3 20, CaCl2 2.5, glucose 5.5). Arteries (endothelium intact) were cut into rings. The UB (urothelium removed) and the outer longitudinal layer of the ileum smooth muscles were cut into strips. The tissues were analysed with isometric force recordings, intracellular Ca2+ measurements or frozen in liquid nitrogen for Western blot analysis.
Generation of the conditional smooth muscle-specific Rac1 knockout strain
SM22CreER(T2), which is a transgenic mouse strain expressing a tamoxifen-activated cre recombinase under control of the smooth muscle-specific promoter SM22 (Kuhbandner et al. 2000), was crossed with a floxed Rac1 strain (Chrostek et al. 2006) to create mice enabling inducible, smooth muscle-specific Rac1 knockout (SM22Cre/Rac1). The knockout animals had floxed Rac1 genes on both alleles. Controls were carrying the Cre gene only (SM22Cre/Wt). Genotyping was performed with PCR of tail DNA. The SM22CreER allele was detected using CRE1-primer: 5′-GCCTGCATTACCGGTCGATGCAACGA-3′ and CRE2-primer: 5′-GTGGCAGATGGCGCGGCAACACCATT-3′.
Rac1 was genotyped using MG1-primer: 5′-GTCTTGAGTTACATCTCTGG-3′ and MG2-primer: 5′-CTGACGCCAACAACTATGC-3′.
Tamoxifen administration
SM22Cre/Rac1 (conditional knockout animals) and SM22Cre/Wt (controls) were injected intraperitoneally with 100 μl of 30 mg ml−1 4-hydroxy-tamoxifen (Sigma-Aldrich, St Louis, MO, USA) in peanut oil solution once per day for 10–14 consecutive days. Tamoxifen stock solution was prepared and stored as previously described (Feil et al. 2009). The animals were killed and the tissues excised 8–10 weeks after the last tamoxifen treatment. In the experiments, tissues from Rac1 knockout animals were analysed in parallel with controls.
Western blotting
Tissues were frozen in N2, kept at −80°C and homogenized in an SDS lysis buffer. Aliquots were taken for protein determination using the Bradford method and samples (15–20 μg of protein) were separated on 12% precast polyacrylamide gels (BioRad Laboratories, Hercules, CA, USA). The protein bands were stained with rabbit polyclonal or mouse monoclonal primary antibodies (Rac1: C-14, sc-217, Santa Cruz, CA, USA; clone 23A8, EMD Millipore, Darmstadt, Germany; GAPDH: FL-335, Santa Cruz), and detected using chemiluminescence (Pierce's SuperSignal, Thermo Scientific, Waltham, MA, USA; Amersham ECL Detection Reagent, GE Healthcare, Little Chalfont, UK) on a GelDoc XR system (Bio-Rad, Hercules, CA, USA) or on photographic film. Intensities were evaluated using the Quantity One program (Bio-Rad).
Isometric force recordings
Smooth muscle preparations were mounted for isometric force recording in a myograph setup (610M, DMT, Aarhus, Denmark). The Krebs–Ringer solution was constantly gassed with 95% O2/5% CO2 to give a pH of 7.4 at 37°C. The segment length of the vessel preparations was about 2 mm and the weight of the UB and ileum strips was about 0.75 mg. The vessel diameters were adjusted to the optimal for active force generation in each preparation (aorta: ∼800 μm, SA: ∼230 μm, SMA: ∼250 μm). The UB preparations were examined at a passive stress of about 5.9 mN mm–2 (length ∼4 mm), which gives the approximate optimal length in this tissue (Sjuve et al. 1998). The corresponding passive stress for the ileum preparations was 1.9 mN mm–2 (length: ∼4.2 mm). After mounting, the preparations were kept at optimal length for 30 min and then activated (5 min) with high-K+ (80 mm KCl) and relaxed (5 min) 2–3 times to ensure stability of the contractile responses. The maximal force during the last high-K+ contraction was used to normalize subsequent force responses in each experiment.
The Rac1 inhibitors (EHT1864 or NSC23766) and the solvent control were added to the Krebs–Ringer solution at different concentrations for 60 min, after the initial recording of the high-K+ responses. Four to eight preparations were analysed in parallel, each treated with one concentration of the inhibitor compound. After the treatment period the tissues were activated, still in the presence of the respective inhibitors or control, with different agonists, high-K+, prostaglandin F2α (PGF2α), phorbol-12,13-dibutyrate (PDBu), phenylephrine (aorta, SMA, SA) and carbachol (ileum and UB). The maximal force in response to the different agonists was normalized to the initial high-K+ force, recorded in the absence of the inhibitors. In separate series of experiments the effects of the RhoA-kinase inhibitor Y27632 and the protein kinase C (PKC) inhibitor GF103209x were examined. These blockers were introduced 15 min prior to activation with the contractile agonists.
The effects of EHT1864 were investigated in permeabilized UB tissue. Intact preparations were mounted at 22°C, precontracted with 80 mm KCl in Krebs–Ringer solution with composition adjusted for 22°C, to record an initial response, exposed to 0 or 10 μm EHT1864 and again contracted. Thereafter they were permeabilized for 2 h in relaxing solution with 1% Triton X-100, rinsed in relaxing solution and activated with contraction solution, all in the presence of the respective EHT1864 concentration (0 or 10 μm). The relaxation solution (–log (free [Ca2+]) = pCa 9) contained (in mm): EGTA 6, Mg2+ 0.5, MgATP 5, phosphocreatine 15, ionic strength (potassium propionate) 200, DTE 2, Mops 20 and 0.64 mg ml−1 creatine kinase and had a pH 7.0. The contraction solution (pCa 4.7) was made by replacing EGTA with CaEGTA.
The preparations (SA, SMA and UB) from the tamoxifen-treated transgenic mice were mounted as described above. To examine if the passive and active mechanical properties of the vessels were altered following knockout of Rac1, we determined the length–tension relationships of SMA and SA preparations using high-K+ activations at different degrees of stretch. The preparations were shortened to near slack length and then stretched in steps. Five minutes after each length change the preparations were activated, relaxed and then stretched to a new length. At each length the passive and maximal active force were recorded. The maximal active tension (force per segment length, mN mm−1) of the artery preparations was calculated by correcting for segment length. The active stress (force per cross-sectional area, mN mm–2) was calculated in the bladder preparations using the cross-sectional area calculated from preparation length and weight using a density of 1.05 mg mm–3. For the vascular SA and SMA preparations the tissues were fixed at optimal length (overnight in 2.5% glutaraldehyde in 125 mm sodium cacodylate, pH 7.4), embedded in araldite, sectioned and analysed using light microscopy to determine the thickness of the smooth muscle layer in the vessel wall.
Active stress per smooth muscle cross-sectional area was calculated by dividing the wall tension with the thickness of the smooth muscle layer.
Recording of intracellular calcium
Intracellular calcium ([Ca2+]i) oscillations were recorded in the SA preparations using confocal imaging and Fluo-4, as described previously (Rahman et al. 2013). Intracellular calcium was also determined in parallel with isometric force as described previously (Arner et al. 1998; Lucius et al. 1998). In brief, the saphenous artery was cut into segments (∼2 mm) and loaded with 10 μm Fura-2/AM, with 1% DMSO and 0.02% pleuronic F-127 for 2.5 h at 22°C, preceded by a 30 min incubation at 4°C. Prior to recording, the vessel segments were treated with NSC23766 (100 μm) or solvent control for 1 h. The vessel segments were then mounted at optimal length in Krebs–Ringer solution at 37°C (still in the presence of NSC23766 or control, and gassed with 95% O2/5% CO2 to keep pH at 7.4) in a dual wire myograph with glass windows (DMT) on an inverted microscope.
Recordings of the force and fluorescence signals for each group of vessels (control and NSC23766 treated) were made subsequently in the relaxed state (Krebs–Ringer solution, 3 min), following receptor activation (2 μm phenylephrine or 10 μm PGF2α, 5 min) and high-K+ (80 mm added to the solution with agonists and inhibitors still present, 5 min). [Ca2+]i was analysed by averaging the 340/380 nm ratio during the last 30 s of each time period. The force and ratio values are expressed relative to the high-K+ activated levels.
Chemicals
EHT1864, NSC23766 and Y27632 were from Tocris Cookson Inc. (Ellisville, MO, USA), GF109203X was from VWR International (Stockholm, Sweden), PGF2α was from Cayman Chemical Co. (Tallin, Estonia), phenylephrine, carbachol and PDBu were from Sigma Aldrich (Stockholm, Sweden), and Fura-2 AM, Pluronic F-127 and Fluo-4 AM were from Molecular Probes (Carlsbad, CA, USA).
Statistics
Mean values ± SEM are shown with n-values. Student's t test for unpaired data was used to test for differences between groups with P < 0.05 considered significant. All data analysis, curve fitting and statistical analysis was made using SigmaPlot 8 for Windows (Systat Software Inc., Chicago, IL, USA).
Results
Effects of NSC23766 on contractions initiated by membrane depolarization, PKC activation and receptor agonists
We examined the effects of NSC23766 on vascular (aorta, SA, SMA) and visceral (UB, ileum) smooth muscle tissues and applied a range of agonists. A primary focus was on comparing the effects in a visceral and an arterial smooth muscle tissue (UB and SA). Figure 1A shows original recordings of active force in the two tissues before and after incubation for 60 min with or without 30 μm NSC23766. The contractions initiated by depolarization (high-K+) were not affected by NSC23766 in either tissue, as seen by the similar amplitude of the KCl-induced contractions before and after incubation. In contrast, the α1-adrenergic receptor responses (10 μm phenylephrine) were significantly inhibited in the vascular preparations. The contraction of the artery induced by PKC activation (1 μm PDBu) was potentiated by NSC23766. In the UB the muscarinic receptor response (10 μm carbachol) was inhibited to a small extent. Figure 1B and C shows the concentration dependence of the NSC23766 effects in the SA and the UB, respectively. NSC23766 promptly inhibited the α1 adrenergic receptor responses, induced by phenylephrine in the artery to near zero force. By fitting a hyperbolic equation we estimated the EC50 for NSC23766 inhibition (the concentration giving half maximal effect) as 12 ± 1 μm (n = 4) in the vessel. For the muscarinic receptor responses in the UB the highest dose (100 μm) inhibited force by 50–25% consistent with a markedly higher EC50 possibly also including and unspecific effect at higher doses. PDBu only elicited contractions in the vascular tissue. Higher concentrations of NSC23766 potentiated PDBu-induced contractions. These results show that NSC23766 specifically inhibits α1-adrenergic receptor responses with an EC50 of about 10 μm.
Figure 1.
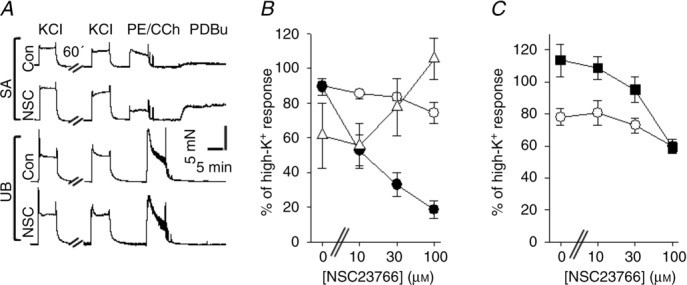
A, original recordings of force responses in mouse saphenous artery (SA) and urinary bladder (UB) preparations, first activated with depolarization (KCl) then incubated without (Con) or with 30 μm NSC23766 (NSC) for 60 min, thereafter again activated with KCl and with agonists (SA: 10 μm phenylephrine (PE) and 1 μm PDBu; UB: 10 μm carbachol (CCh) and 1 μm PDBu). B and C, NSC23766 dose–response relationships for saphenous artery (B) and urinary bladder (C). The force values are normalized to the initial contractile response induced by KCl prior to the 60 min incubation period. Open circles show the responses to high-K+, filled circles phenylephrine (10 μm, B), filled squares carbachol (10 μm, C) and open triangles (PDBu (1 μm, B), n = 4 in each group.
Responses to depolarization were unaffected. At higher doses NSC23766 inhibited muscarinic responses in visceral muscle and potentiated PKC-induced contractions in the vessels.
Corresponding results, with NSC23766-induced inhibition of α1 adrenergic receptor responses and potentiation of PDBu-induced contraction in arterial muscle and minor effects on muscarinic responses in visceral muscle, were observed in the aorta, mesenteric microartery and ileum smooth muscle preparations (supplementary Fig. S1)
Effects of NSC23766 on the dose dependence of phenylephrine, carbachol and PGF2α
We extended the analysis to study the effects of NSC23766 on phenylephrine and carbachol sensitivity in more detail, and the effects on PGF2α responses. The α1 adrenergic (Fig. 2A, artery) and muscarinic (Fig. 2C, bladder) receptor responses were clearly inhibited by NSC23766 (100 μm, cf. Fig. 1). In contrast, NSC23766 significantly potentiated the contractile responses to PGF2α (Fig. 2B, artery; Fig. 2D, bladder) in both tissues. The summary results (Fig. 2E–H) show that NSC23766 has different effects depending on the mode of activation. It shifts the phenylephrine sensitivity towards higher concentrations and inhibits the maximal tension of both phenylephrine-and carbachol-induced contractions with an EC50 of 10–30 μm. By contrast, NSC23766 potentiates the PGF2α responses (Fig. 2F and H) and shifts the sensitivity towards lower concentrations. In the blood vessel, the half-maximal effect for the potentiating action of NSC23766 on PGF2α responses was observed at an NSC23766 concentration of about 10–30 μm. In the UB, NSC23766 inhibited the maximal responses to carbachol (cf. Fig. 1C) and potentiated the PGF2α responses (Fig. 2H). The PGF2α responses were phasic in the bladder and difficult to quantify, but the pattern with NSC23766-induced potentiation of PGF2α responses resembled that observed in the SA. In a separate series, we also examined the effects on contractile responses to U46619 in the SA.
Figure 2.
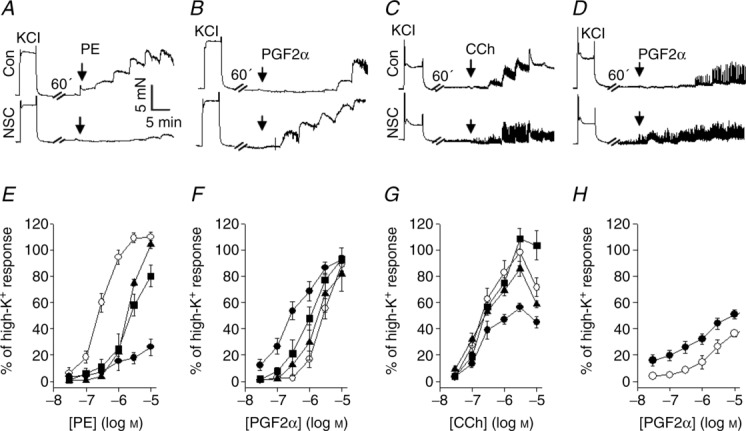
A–D, original recordings of dose–response relationships in mouse saphenous artery (A, phenylephrine (PE); B, PGF2α) and urinary bladder (C, carbachol (CCh); D, PGF2α) in the absence (upper traces) and presence (lower traces) of 100 μm NSC23766. E–H, summary data for the different agonists in saphenous artery (E, F) and urinary bladder (G, H). The force data are expressed as a percentage of an initial high-K+ contraction in the absence of NSC. Open circles, controls; filled triangles, 10 μm NSC23766; filled squares, 30 μm NSC23766; filled circles, 100 μm NSC23766; n = 4–5 in each group.
NSC23766 lowered the EC50 for U46619 in a similar manner as for PGF2α (Control: ∼0.1 μm; NSC23766: ∼0.03 μm; U46619, n = 3). In summary, these results show that the NSC23766 inhibition is associated with a shift in the dose–response relationship of phenylephrine towards higher concentrations. At the same time it increases the PGF2α and U46619 agonist sensitivities. Its inhibitory effect on muscarinic receptor responses in the bladder is associated with a decreased maximal amplitude of the carbachol responses with a minor dose-dependent effect. Note that the inhibitory effect of NSC23766 in the visceral muscle occurred at comparatively high concentrations and that unspecific effects at the higher doses cannot be excluded.
To further explore the interaction between Rac1 signalling, and PKC-and RhoA-mediated effects on contraction, we used GF109203x (Toullec et al. 1991) and Y27632 (Ishizaki et al. 2000) to inhibit the PKC and RhoA/Rho kinase pathways, respectively. We focused on the inhibitory and potentiating effects of NSC23766 on adrenergic and prostaglandin responses in the SA. Exposure to 1 μm GF109203x (which inhibits a contraction induced by 1 μm PDBu in aorta by more then 90%; Davis et al. 2012) inhibited the responses to phenylephrine (Fig. 3A) and to PGF2α (Fig. 3B), by 40 and 60%, respectively. This shows that PKC is recruited following activation of these two receptor pathways. However, when 100 μm NSC23766 was added in the presence of 1 μm GF109203x, it still inhibited the phenylephrine-induced contraction (filled squares, Fig. 3A) and potentiated the PGF2α-induced contraction (filled squares, Fig. 3B). These results show that the inhibitory and potentiating effects of NSC23766 do not involve actions via the PKC pathway.
Figure 3.
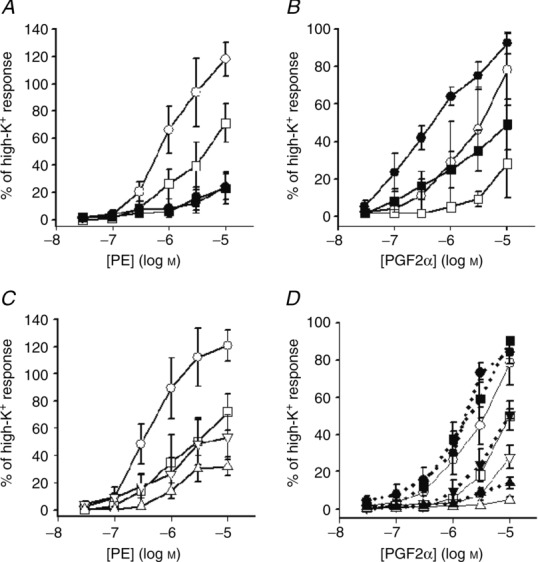
A and B, vessels activated in the absence (circles) and presence (squares) of 1 μm GF109203x. Note that data in the presence of NSC23766 (filled symbols) are superimposed in A. C and D, vessels activated in the absence (circles) and presence of 1 μm (squares), 3 μm (triangles, base up) and 10 μm (triangles, base down) Y27632. Force values are given relative to an initial high-K+-induced contraction in the absence of inhibitors; n = 3–5 in each group.
Rho kinase inhibition with Y27632 significantly attenuated the responses to phenylephrine (Fig. 3C) and to PGF2α (Fig. 3D) with a maximal inhibition of about 75 and 90%, respectively. Rho kinase activation is thus essential for the contractile responses to both these agonists. Since the Y27632 effects (Rho kinase) are so prominent it is difficult to dissect a specific inhibitory action of NSC23766 on this pathway and consequently we did not combine Y27632 and NSC23766 on the phenylephrine-induced contractions. As seen in Fig. 3D, a potentiating effect of NSC23766 was observed at all intermediate Y27632 concentrations following PGFα activation. It has been shown that NSC23766 does not block Rho (Gao et al. 2004). Since Rho kinase inhibition has an inhibitory effect on both phenylephrine and PGF2α responses, it seems unlikely that an effect of NSC23766 on the Rho/Rho kinase pathway is a common mechanism for both the potentiating and the inhibitory effects.
Effects of Rac1 gene ablation on smooth muscle contractions induced by membrane depolarization and agonist stimulation
We examined the expression of Rac1 in a range of smooth muscle tissues and identified the protein with Western blotting in arterial (aorta, mesenteric artery, mesenteric microartery and SA) and visceral smooth muscle (UB and small intestine). We generated smooth muscle-specific conditional knockout mice for Rac1 and examined the effects of Rac1 gene knockout in UB and SA smooth muscle tissues. Two groups, both treated with tamoxifen, were compared: knockout (KO), i.e. animals carrying Cre with conditional expression, and loxP flanked Rac1 genes and controls, i.e. animals with only Cre. No significant differences could be observed in body weight in either sex (Control male: 36.78 ± 1.8 g, n = 8, female: 24.55 ± 0.86 g, n = 3; KO: male: 32.66 ± 0.82 g, n = 6, female: 24.67 ± 0.98 g, n = 5), heart/body weight ratio (Control: 4.3 ± 0.15 mg g−1, n = 11; KO: 4.2 ± 0.1 mg g−1, n = 11) or UB weight (Control: 28.9 ± 2 mg, n = 5; KO: 28.7 ± 2.4 mg, n = 5).
Western blotting analysis (Fig. 4A and D) showed approximately 30 and 50% reduction of Rac1 in the SA and UB in the KO group. Circumference–tension relationships of SA preparations from control and KO animals are shown in supplementary Fig. S2. The active tension at optimal length in response to high-K+ was lower in the KO group. No difference was observed in the optimal circumference for active tension (Control: 847 ± 18 μm; KO: 854 ± 60 μm, n = 6) and in the passive tension at optimal circumference (Control: 1.16 ± 0.09 mN mm−1; KO: 1.00 ± 0.08 mN mm−1, n = 6). Active tension at optimal length in response to activation with high-K+ (Control: 2.96 ± 0.19 mN mm−1; KO: 1.81 ± 0.15 mN mm−1, P < 0.001, n = 11), phenylephrine (Control: 3.73 ± 0.31 mN mm−1; KO: 2.75 ± 0.3 mN mm−1, P < 0.05, n = 11) and PDBu (Control: 1.9 ± 0.22 mN mm−1; KO: 1.3 ± 0.17 mN mm−1, P < 0.05, n = 9) was significantly lower in the SAs of KO animals. To examine whether the lower wall tension in the arterial preparations of the KO animals was due to a difference in wall thickness we determined the thickness of the media layer at optimal length using morphometry (media thickness, control: 17.2 ± 1.2 μm; KO: 15.8 ± 0.7 μm, n = 4) and calculated active stress (i.e. force per unit smooth muscle area). These active stress values were significantly lower in the SA preparations of the KO group, when activated with high-K+ (Fig. 4B).
Figure 4.
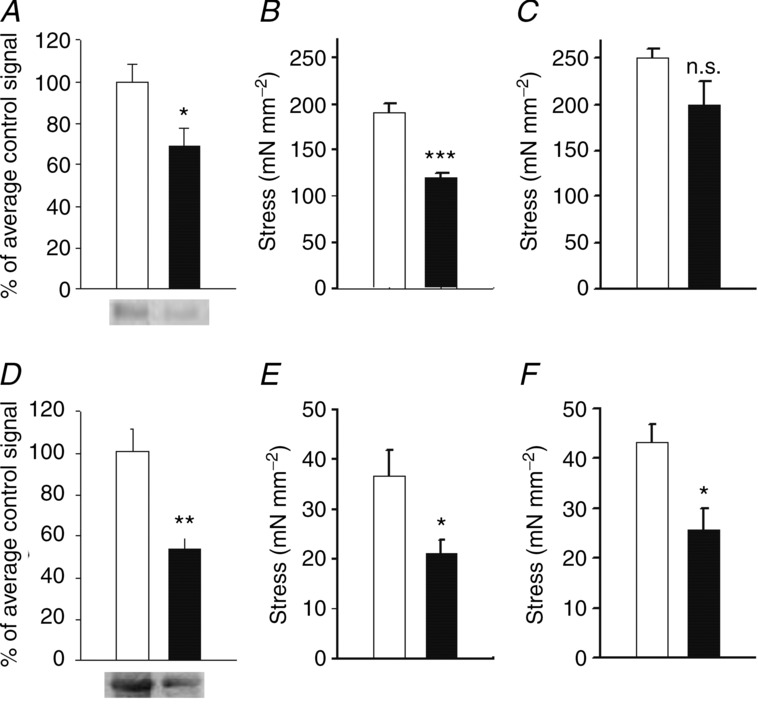
A–C, data from saphenous artery; D–F, data from urinary bladder. Open bars show data from controls and filled bars data from Rac1 knockout animals. A and D, Western blot analysis of Rac1 expression in the saphenous artery (n = 4) and urinary bladder (n = 11–12), respectively. Equal amounts of protein from the two groups were run in parallel on the gels, and the Western blot signals were expressed relative to the signal of the control samples. Samples of Western blots for Rac1 are shown below each diagram. The other panels show active stress of saphenous artery (B, KCl; C, phenylephrine at 10 μm, n = 4) and urinary bladder (E, KCl; F, carbachol at 10 μm, n = 3) of control and knockout mice. *P < 0.05, **P < 0.01, ***P < 0.001.
When activated with phenylephrine the mean active stress was lower but not significantly so (Fig. 4C). In the bladder preparations, active stress generated by both high-K+ and carbachol responses was significantly lower (Fig. 4E and F). In general the knockout of Rac1 in smooth muscle resulted in a lower active force affecting both high-K+ and receptor agonist activation.
Effects of EHT1864 on contractions induced by membrane depolarization, and by α1-adrenergic and muscarinic agonists
Figure 5A shows original recordings of force induced by various modes of activation in the presence and absence of 10 μm EHT1864 in SA (upper traces) and UB (lower traces) preparations. EHT1864 inhibited the contraction induced by all agonists in both tissues. The summary data show that EHT1864 (1–100 μm) concentration-dependently inhibited the agonist-and high-K+-induced contractions in the two tissues (Fig. 5B and C). Similar results were observed in the aorta, SMA and ileum preparations (supplementary Fig. S3) The half-maximal responses were observed between 1 and 10 μm. These results show that the Rac1 inhibitor EHT1864 has a broader inhibitory effect on different modes of activation and tissues compared with NSC23766. EHT1864 (10 μm) inhibited the contractions induced by (10 μm) PGF2α in the SA (n = 3), by about 90%.
Figure 5.
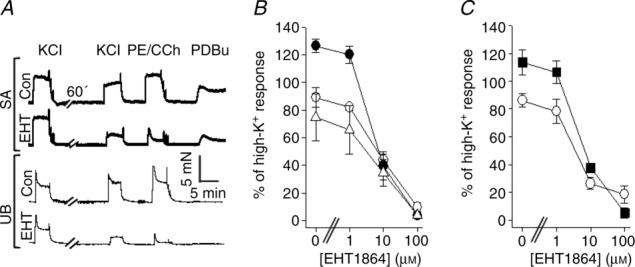
A, original recordings of contractile responses in mouse saphenous artery (SA) and urinary bladder (UB) in the presence of EHT1864 (EHT) and solvent control (Con). B and C, EHT1864 dose–response relationships for saphenous artery (B) and urinary bladder (C). The force values are normalized to the initial contractile response induced by KCl prior to the 60 min incubation period. The open circles show the responses to high-K+, filled circles phenylephrine (10 μm, B), filled squares carbachol (10 μm, C) and open triangles PDBu (1 μm, B); n = 4 in each group.
To examine possible direct effects of EHT1864 on the contractile machinery we examined the effects in permeabilized UB tissue. The preparations were first examined in the intact state and we recorded high-K+ responses before and after a 60 min incubation with 0 or 10 μm EHT1864. The high-K+ responses were significantly (P < 0.001) inhibited in the presence of EHT1864 (Control: 95 ± 2%, n = 8; EHT1864: 51 ± 2%, n = 6; relative to initial high-K+ response). The tissues were then permeabilized and activated at maximal Ca2+ (pCa 4.7), in the presence or absence of EHT1864. The contractile responses of the permeabilized preparations were similar in the two groups (Control: 38 ± 5%, n = 8; EHT1864: 37 ± 6%, n = 6%; relative to initial high-K+ response). These results show that EHT1864 does not cause non-specific contractile failure, but rather acts on upstream pathways lost during the Triton X-100 skinning process. A significant effect of EHT1864 on the myosin light chain phosphorylation/dephosphorylation would have affected the response of skinned fibres, and in view of the significant effects on intracellular Ca2+ levels in the Ca2+ measurements we did not, within this study, further examine potential effects on the Ca2+ sensitivity modulation using other permeabilization techniques and varied Ca2+ levels. It is thus still possible that the effects of Rac1 inhibition also include a component of Ca2+ sensitization/desensitization in addition to the effects on the Ca2+ transient.
Effects of Rac1 inhibitors on intracellular calcium transients induced by phenylephrine and PGF2α
Using confocal microscopy we found that 10 μm EHT1864 or 100 μm NSC23766 reversibly inhibited intracellular Ca2+ oscillations in phenylephrine-and PGF2α-activated SAs (from about 2–3 to about 0.1 waves per minute and cell). We then proceeded with combined force and Ca2+ measurements using Fura-2. Figure 6A–D shows original traces from simultaneous recordings of force and [Ca2+]i with and without 100 μm NSC23677. The arterial segments were loaded with Fura-2 and activated by agonists (2 μm phenylephrine or 10 μm PGF2α). At the plateau of contraction high-K+ was added. NSC23766 inhibited phenylephrine-induced contractions (Fig. 6A, lower trace and Fig. 6E), which was correlated with decreased [Ca2+] (Fig. 6B, lower trace and Fig. 6F). Both force and [Ca2+] increased in high-K+ when introduced at the plateau of the agonist response, in both the presence and the absence of NSC23766. NSC23766 potentiated PGF2α-induced force generation (Fig. 6C, lower trace and Fig. 6G). However, [Ca2+] levels during PGF2α activation were lower in NSC23766 (Fig. 6D, lower trace and Fig. 6H). These results show that the inhibitory action of NSC23766 on α1 receptor responses can be correlated with an attenuated Ca2+ transient. Potentiation of the PGF2α responses occurred at lower [Ca2+], suggesting an increased Ca2+ sensitivity.
Figure 6.
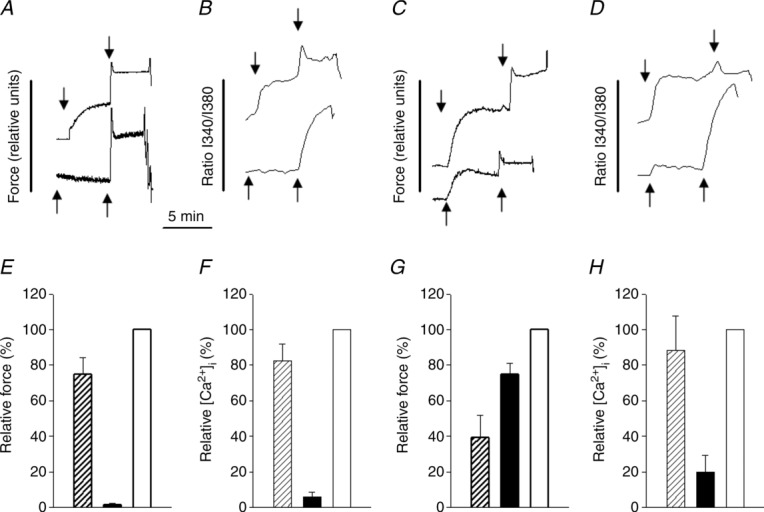
Original recordings of force (A and C) and intracellular calcium levels (B and D) in the mouse saphenous artery. Force is shown in arbitrary units and intracellular Ca2+ as a ratio between emitted intensities at 340 and 380 nm. The ratio data were smoothed using a Gaussian weight function and a quadratic fit as implemented in SigmaPlot 8. Upper and lower traces show recordings in the absence and presence of 100 μm NSC23766, respectively. Contraction was induced with receptor agonists (first arrow in each trace): 2 μm phenylephrine (A and B) or 10 μm PGF2α (C and D). After 5 min exposure to the receptor agonists, 80 mm KCl was added (second arrow). E–H, summary force and [Ca2+] data (E and F, phenylephrine; G and H, PGF2α) normalized to values in high-K+. Open bars show high-K+-induced force and calcium signals; filled and hatched bars show the agonists-induced responses (Phen/PGF2a) in the absence (hatched) and presence (filled) of 100 μm NSC23766); n = 4–5 in each group.
Discussion
We show that Rac1 is expressed in both arterial and visceral smooth muscle and has an essential role in regulating the contraction, probably via a modulation of the level of activator Ca2+. EHT1864 is an inhibitor of Rac1 activity, via action on the nucleotide binding site (Shutes et al. 2007) and thereby affects the interaction between Rac1 and its downstream targets. EHT1864 almost completely blocks active tension generation of all smooth muscles examined, in response to a range of agonists. This inhibition is reversible. Experiments on skinned fibres showed that the effect of EHT1864 cannot simply be fully explained by an irreversible structural reorganization or major effects on myosin light chain phosphorylation/dephosphorylation. We cannot, however, at present fully exclude a contribution via Ca2+ sensitization/desensitization mechanisms. The absence of EHT1864 effects in permeabilized tissue shows that Rac1, or a Rac1-dependent process, affecting tension, is lost during permeabilization or that the high and controlled Ca2+ level in the permeabilized muscle bypasses the action of Rac1. The Ca2+ measurements in the intact tissue are consistent with the latter hypothesis, and suggest that Ca2+ mobilization is an important target for Rac1. Phospholipase C-β2 (PLC-β2) has been shown to be an effector of Rac1 (Jezyk et al. 2006; Harden et al. 2009), and inhibition of PLC could affect the Ca2+ mobilization. However, Rac1 effects on PLC do not seem to be the sole mechanism, since depolarization with high-K+, PKC activation and receptor agonist stimulation (phenylephrine, carbachol and PGF2α) are all affected by EHT1864, suggesting that Rac1 activity affects a common step in their signalling pathways, downstream of receptor activation, depolarization and PKC. Receptor agonists, depolarization and PKC activation lead to influx of Ca2+ via L-type Ca2+ channels (Ekman et al. 2006; Snetkov et al. 2006; Rahman et al. 2007; Mukherjee et al. 2013), in addition to Ca2+ release and sensitization of the contractile activation systems. To our knowledge, the effects of Rac1 on L-type channel activity have not been examined in smooth muscle. However, cardiac L-type channels are proposed to be activated by RhoA (Yatani et al. 2005), and one possibility is that Rac1 is required for function or activation of the smooth muscle L-type Ca2+ channels.
This might involve direct effects or be secondary to other Rac1 targets. A cross-talk with RhoA (Sander et al. 1999; Burridge & Wennerberg, 2004; Rosenfeldt et al. 2006) cannot be excluded. However, Rac1 seems to inhibit RhoA activity, which is not consistent with our results that inhibition of Rac1 inhibits the Ca2+ transient. We cannot, at present, determine if Rac1 is constitutively active or if it is activated in response to the different agonists discussed above. It might be difficult to find a common signalling pathway recruited by PKC, membrane depolarization and the receptors. Calcium/calmodulin-dependent kinase has been shown to affect RacGEFs in some cell types (Penzes et al. 2008). One intriguing possibility is that Rac1 activation in smooth muscle includes a Ca2+-dependent pathway whereby a small mobilization of Ca2+ activates Rac1 to enable a larger increase in activator Ca2+ via subsequent Rac1 activation of membrane influx.
Knockout of Rac1, using conditional smooth muscle-specific Rac1 knockout mice, showed that a 50% loss of the protein in the tissue results in a significant decrease in the contractile responses to different agonists in smooth muscle. This is consistent with the results of experiments with EHT1864, which show that Rac1 activity is required for contractile activation of smooth muscle. Our mechanical data from the microarteries of these knockout animals do not reveal any major changes in vascular diameter or in the passive properties of the vessels, excluding any major remodelling of the vessels during the 8–10 week period after induction of cre and ablation of Rac1. The similar heart/body weight and UB weight suggest that the lowering of Rac1 activity in the smooth muscle tissue does not introduce major changes in the cardiovascular system or UB structure. It is possible that compensatory mechanisms maintain body homeostasis and that further in vivo analysis, beyond the scope of this study, might reveal more subtle changes in animal physiology associated with the lowering of Rac1.
Our second pharmacological approach was to apply NSC23766, a blocker of the upstream interaction between Rac1 and GEFs (mainly Tiam1 and Trio). Using this compound we identified a specific pathway from the α1-and FP (prostaglandin) receptors, to Rac1 activation and to the Ca2+ influx. Interference with this GEF/Rac1 interaction is sufficient to block both phenylephrine-and PGF2α−mediated activation of Ca2+ influx. This shows that both the α1 adrenergic and FP receptors use the Rac1 signalling to activate the receptor-operated Ca2+ influx. This can explain the inhibition of phenylephrine-induced contractions in the presence of NSC23766. However, the PGF2α responses were potentiated, which cannot simply be explained by Ca2+ changes. An additional Rac1 effect that inhibits the Ca2+ sensitization, possibly via effects on RhoA or other steps in the Ca2+ sensitization pathway, has to be included. The difference between the NSC23766 effects on phenylephrine and PGF2α responses might reside in the relative recruitment of different sensitizing pathways.
Since Rac1 can affect endothelial signalling (Sauzeau et al. 2010), a contribution from endothelial factors cannot be excluded in the arteries.
In summary, this is the first study showing a key role of Rac1 signalling in the activation of smooth muscle contraction. Specifically, we conclude that Rac1 is required for Ca2+ influx, a pathway recruited for several modes of activation. Details of the link between Rac1 and the Ca2+ channels, as well as the mechanisms by which the Rac1 pathway is activated by different agonists, remain to be elucidated. The latter could include Ca2+-dependent activation of Rac1. We have further identified two receptors (the α1-adrenoceptor and the FP-prostaglandin receptor) that activate Rac1 via a separate mechanism, probably via specific GEFs and shown that inhibition of this pathway reveals differences in Ca2+ sensitization mechanisms between the two receptors.
Key points
The role of the small G-protein Rac1 was investigated in smooth muscle, using a smooth muscle-specific knockout mouse and pharmacological blockers.
Inhibition of the interaction between Rac1 and upstream regulators inhibited the α-receptor contractions and potentiated prostaglandin F2α contractions in vascular tissue.
The inhibition was mediated via an attenuation of the Ca2+ transient.
A global inhibition of Rac1 activity inhibited contractions in response to several agonists in a range of smooth muscle tissues.
The results demonstrate a novel Rac1-associated signalling pathway for regulation of smooth muscle contraction.
Acknowledgments
We are very grateful for the able technical assistance from Mrs Lilian Sundberg.
Glossary
- EC50
concentration giving half maximal effect
- GEFs
guanine nucleotide exchange factors
- PDBu
phorbol-12,13-dibutyrate
- PGF2α
prostaglandin F2α
- PKC
protein kinase C
- SA
saphenous artery
- SMA
small mesenteric artery
- UB
urinary bladder
Additional information
Competing interests
None declared.
Author contributions
A.R., B.D., C.L. and V.T.H. carried out the physiological recordings and biochemical experiments, participated in analysis of the mouse properties and contributed to the writing of the manuscript. R.F and C.B. generated the original transgenic mouse strains and contributed to the writing of the manuscript. A.A. conceived and supervised the project, wrote the manuscript and is responsible for the work. All the authors have read and approved the final version.
Funding
This study was supported by grants from the Swedish Research Council and the Swedish Heart Lung Foundation and an FP7 EU grant (INComb).
References
- Arner A, Malmqvist U, Rigler R. Calcium transients and the effect of a photolytically released calcium chelator during electrically induced contractions in rabbit rectococcygeus smooth muscle. Biophys J. 1998;75:1895–1903. doi: 10.1016/S0006-3495(98)77630-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac take centre stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Bustelo XR. Regulatory and signalling properties of the Vav family. Mol Cell Biol. 2000;20:1461–1477. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- Chrostek A, Wu X, Quondamatteo F, Hu R, Sanecka A, Niemann C, Langbein L, Haase I, Brakebusch C. Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol Cell Biol. 2006;26:6957–6970. doi: 10.1128/MCB.00075-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis B, Rahman A, Arner A. AMP-activated kinase relaxes agonist induced contractions in the mouse aorta via effects on PKC signalling and inhibits NO-induced relaxation. Eur J Pharmacol. 2012;695:88–95. doi: 10.1016/j.ejphar.2012.07.025. [DOI] [PubMed] [Google Scholar]
- Desire L, Bourdin J, Loiseau N, Peillon H, Picard V, De Oliveira C, Bachelot F, Leblond B, Taverne T, Beausoleil E, Lacombe S, Drouin D, Schweighoffer F. RAC1 inhibition targets amyloid precursor protein processing by γ-secretase and decreases Aβ production in vitro and in vivo. J Biol Chem. 2005;280:37516–37525. doi: 10.1074/jbc.M507913200. [DOI] [PubMed] [Google Scholar]
- Doanes AM, Irani K, Goldschmidt-Clermont PJ, Finkel T. A requirement for rac1 in the PDGF-stimulated migration of fibroblasts and vascular smooth cells. Biochem Mol Biol Int. 1998;45:279–287. doi: 10.1080/15216549800202652. [DOI] [PubMed] [Google Scholar]
- Ekman M, Andersson KE, Arner A. Developmental regulation of nerve and receptor mediated contractions of mammalian urinary bladder smooth muscle. Eur J Pharmacol. 2006;532:99–106. doi: 10.1016/j.ejphar.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Feil S, Valtcheva N, Feil R. Inducible Cre mice. Methods Mol Biol. 2009;530:343–363. doi: 10.1007/978-1-59745-471-1_18. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habets GG, van Der Kammen RA, Stam JC, Michiels F, Collard JG. Sequence of the human invasion-inducing TIAM1 gene, its conservation in evolution and its expression in tumor cell lines of different tissue origin. Oncogene. 1995;10:1371–1376. [PubMed] [Google Scholar]
- Halayko AJ, Solway J. Molecular mechanisms of phenotypic plasticity in smooth muscle cells. J Appl Physiol. 2001;90:358–368. doi: 10.1152/jappl.2001.90.1.358. [DOI] [PubMed] [Google Scholar]
- Harden TK, Hicks SN, Sondek J. Phospholipase C isozymes as effectors of Ras superfamily GTPases. J. Lipid Res. 2009;50(Suppl):S243–S248. doi: 10.1194/jlr.R800045-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover WC, Zhang W, Xue Z, Gao H, Chernoff J, Clapp DW, Gunst SJ, Tepper RS. Inhibition of p21 activated kinase (PAK) reduces airway responsiveness in vivo and in vitro in murine and human airways. PloS One. 2012;7:e42601. doi: 10.1371/journal.pone.0042601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol. 2000;57:976–983. [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jezyk MR, Snyder JT, Gershberg S, Worthylake DK, Harden TK, Sondek J. Crystal structure of Rac1 bound to its effector phospholipase C-β2. Nat Struct Mol Biol. 2006;13:1135–1140. doi: 10.1038/nsmb1175. [DOI] [PubMed] [Google Scholar]
- Kim KS, Takeda K, Sethi R, Pracyk JB, Tanaka K, Zhou YF, Yu ZX, Ferrans VJ, Bruder JT, Kovesdi I, Irani K, Goldschmidt-Clermont P, Finkel T. Protection from reoxygenation injury by inhibition of rac1. J Clin Invest. 1998;101:1821–1826. doi: 10.1172/JCI1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Kobayashi S, Horiuti K, Somlyo AV, Somlyo AP. Receptor-coupled, permeabilized smooth muscle. Role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+ J Biol Chem. 1989;264:5339–5342. [PubMed] [Google Scholar]
- Kuhbandner S, Brummer S, Metzger D, Chambon P, Hofmann F, Feil R. Temporally controlled somatic mutagenesis in smooth muscle. Genesis. 2000;28:15–22. doi: 10.1002/1526-968x(200009)28:1<15::aid-gene20>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006;98:322–334. doi: 10.1161/01.RES.0000201960.04223.3c. [DOI] [PubMed] [Google Scholar]
- Loirand G, Pacaud P. The role of Rho protein signalling in hypertension. Nat Rev Cardiol. 2010;7:637–647. doi: 10.1038/nrcardio.2010.136. [DOI] [PubMed] [Google Scholar]
- Lucius C, Arner A, Steusloff A, Troschka M, Hofmann F, Aktories K, Pfitzer G. Clostridium difficile toxin B inhibits carbachol-induced force and myosin light chain phosphorylation in guinea-pig smooth muscle: role of Rho proteins. J Physiol. 1998;506:83–93. doi: 10.1111/j.1469-7793.1998.083bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle AN, Griendling KK. Modulation of vascular smooth muscle signalling by reactive oxygen species. Physiology. 2006;21:269–280. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- McFawn PK, Shen L, Vincent SG, Mak A, Van Eyk JE, Fisher JT. Calcium-independent contraction and sensitization of airway smooth muscle by p21-activated protein kinase. Am J Physiol Lung Cell Mol Physiol. 2003;284:L863–L870. doi: 10.1152/ajplung.00068.2002. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Duan F, Kolb MR, Janssen LJ. Platelet derived growth factor-evoked Ca2+ wave and matrix gene expression through phospholipase C in human pulmonary fibroblast. Int J Biochem Cell Biol. 2013;45:1516–1524. doi: 10.1016/j.biocel.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Nishimura J, Kolber M, van Breemen C. Norepinephrine and GTP-γ-S increase myofilament Ca2+ sensitivity in α-toxin permeabilized arterial smooth muscle. Biochem Biophys Res Commun. 1988;157:677–683. doi: 10.1016/s0006-291x(88)80303-6. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, Srivastava DP. Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol. 2008;18:405–413. doi: 10.1016/j.tcb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiology. 2009;24:342–356. doi: 10.1152/physiol.00023.2009. [DOI] [PubMed] [Google Scholar]
- Rahman A, Hughes A, Matchkov V, Nilsson H, Aalkjaer C. Antiphase oscillations of endothelium and smooth muscle [Ca2+]i in vasomotion of rat mesenteric small arteries. Cell Calcium. 2007;42:536–547. doi: 10.1016/j.ceca.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Rahman A, Ekman M, Shakirova Y, Andersson KE, Morgelin M, Erjefalt JS, Brundin P, Li JY, Sward K. Late onset vascular dysfunction in the R6/1 model of Huntington's disease. Eur J Pharmacol. 2013;698:345–353. doi: 10.1016/j.ejphar.2012.10.026. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt H, Castellone MD, Randazzo PA, Gutkind JS. Rac inhibits thrombin-induced Rho activation: evidence of a Pak-dependent GTPase crosstalk. J Mol Signal. 2006;1:8. doi: 10.1186/1750-2187-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behaviour. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauzeau V, Sevilla MA, Rivas-Elena JV, de Alava E, Montero MJ, Lopez-Novoa JM, Bustelo XR. Vav3 proto-oncogene deficiency leads to sympathetic hyperactivity and cardiovascular dysfunction. Nat Med. 2006;12:841–845. doi: 10.1038/nm1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauzeau V, Jerkic M, Lopez-Novoa JM, Bustelo XR. Loss of Vav2 proto-oncogene causes tachycardia and cardiovascular disease in mice. Mol Biol Cell. 2007;18:943–952. doi: 10.1091/mbc.E06-09-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauzeau V, Sevilla MA, Montero MJ, Bustelo XR. The Rho/Rac exchange factor Vav2 controls nitric oxide-dependent responses in mouse vascular smooth muscle cells. J Clin Invest. 2010;120:315–330. doi: 10.1172/JCI38356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada N, Li Y, Liao JK. Novel aspects of the roles of Rac1 GTPase in the cardiovascular system. Curr Opin Pharmacol. 2010;10:116–121. doi: 10.1016/j.coph.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- Sjuve R, Arner A, Li Z, Mies B, Paulin D, Schmittner M, Small JV. Mechanical alterations in smooth muscle from mice lacking desmin. J Muscle Res Cell Motil. 1998;19:415–29. doi: 10.1023/a:1005353805699. [DOI] [PubMed] [Google Scholar]
- Snetkov VA, Knock GA, Baxter L, Thomas GD, Ward JP, Aaronson PI. Mechanisms of the prostaglandin F2α-induced rise in [Ca2+]i in rat intrapulmonary arteries. J Physiol. 2006;571:147–163. doi: 10.1113/jphysiol.2005.101394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Takizawa N, Koga Y, Ikebe M. Phosphorylation of CPI17 and myosin binding subunit of type 1 protein phosphatase by p21-activated kinase. Biochem Biophys Res Commun. 2002;297:773–778. doi: 10.1016/s0006-291x(02)02302-1. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Van Eyk JE, Arrell DK, Foster DB, Strauss JD, Heinonen TY, Furmaniak-Kazmierczak E, Cote GP, Mak AS. Different molecular mechanisms for Rho family GTPase-dependent, Ca2+-independent contraction of smooth muscle. J Biol Chem. 1998;273:23433–23439. doi: 10.1074/jbc.273.36.23433. [DOI] [PubMed] [Google Scholar]
- Wirth A, Schroeter M, Kock-Hauser C, Manser E, Chalovich JM, De Lanerolle P, Pfitzer G. Inhibition of contraction and myosin light chain phosphorylation in guinea-pig smooth muscle by p21-activated kinase 1. J Physiol. 2003;549:489–500. doi: 10.1113/jphysiol.2002.033167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatani A, Irie K, Otani T, Abdellatif M, Wei L. RhoA GTPase regulates L-type Ca2+ currents in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2005;288:H650–659. doi: 10.1152/ajpheart.00268.2004. [DOI] [PubMed] [Google Scholar]
- Zhang X, DiSanto ME. Rho-kinase, a common final path of various contractile bladder and ureter stimuli. Handb Exp Pharmacol. 2011;202:543–568. doi: 10.1007/978-3-642-16499-6_24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.