

Abstract
Lacrimal glands function to produce an aqueous layer, or tear film, that helps to nourish and protect the ocular surface. Lacrimal glands secrete proteins, electrolytes and water, and loss of gland function can result in tear film disorders such as dry eye syndrome, a widely encountered and debilitating disease in ageing populations. To combat these disorders, understanding the underlying molecular signalling processes that control lacrimal gland function will give insight into corrective therapeutic approaches. Previously, in single lacrimal cells isolated from lacrimal glands, we demonstrated that muscarinic receptor activation stimulates a phospholipase C-coupled signalling cascade involving the inositol trisphosphate-dependent mobilization of intracellular calcium and the subsequent activation of store-operated calcium entry (SOCE). Since intracellular calcium stores are finite and readily exhausted, the SOCE pathway is a critical process for sustaining and maintaining receptor-activated signalling. Recent studies have identified the Orai family proteins as critical components of the SOCE channel activity in a wide variety of cell types. In this study we characterize the role of Orai1 in the function of lacrimal glands using a mouse model in which the gene for the calcium entry channel protein, Orai1, has been deleted. Our data demonstrate that lacrimal acinar cells lacking Orai1 do not exhibit SOCE following activation of the muscarinic receptor. In comparison with wild-type and heterozygous littermates, Orai1 knockout mice showed a significant reduction in the stimulated tear production following injection of pilocarpine, a muscarinic receptor agonist. In addition, calcium-dependent, but not calcium-independent exocytotic secretion of peroxidase was eliminated in glands from knockout mice. These studies indicate a critical role for Orai1-mediated SOCE in lacrimal gland signalling and function.
Introduction
The exocrine lacrimal glands maintain and protect the integrity of the ocular surface by providing a tear film composed of water, electrolytes, surfactants and proteins (Dartt, 2009). Generation and composition of the tear film are under tight neural control so that it can respond rapidly to environmental changes that may occur at the ocular surface. Dysfunction of lacrimal secretion underlies a number of debilitating diseases of the eye (Dartt, 2004). Lacrimal glands are responsive to both muscarinic and α-adrenergic stimulation, and the secretion of both fluid and protein appear to be dependent on the phospholipase C-coupled calcium signalling system (Dartt, 1989; Putney & Bird, 1998).
In addition to their importance to ocular health, the study of signalling pathways in lacrimal cells has been instrumental in defining critical aspects of the underlying phospholipase C-coupled calcium signalling cascade. Activation of muscarinic cholinergic receptors leads to robust formation of inositol 1,4,5-trisphosphate (IP3) (Godfrey & Putney, 1984) and sustained elevations in cytoplasmic Ca2+ (Kwan et al. 1990). Lower, more physiological levels of activation induce cytoplasmic Ca2+ oscillations (Bird et al. 1993). Receptor activation can be bypassed either by uncaging of an agonist for IP3 receptors (Bird et al. 1992b), by use of Ca2+ ionophores (Bird et al. 1992a) or by inhibition of sarcoplasmic–endoplasmic reticulum calcium ATPase pumps with thapsigargin (Kwan et al. 1990; Bird et al. 1992a). In the last case, depletion of endoplasmic reticulum Ca2+ stores by thapsigargin leads to the activation of plasma membrane store-operated channels. Indeed, experiments with lacrimal cells published over 30 years ago (Parod & Putney, 1978) revealed for the first time that the refilling of intracellular Ca2+ stores occurred independently of receptor activation, which ultimately led to the concept of store-operated calcium entry (SOCE; Putney, 1986).
We now understand that SOCE most commonly involves highly Ca2+-selective calcium release-activated calcium (CRAC) channels (Hoth & Penner, 1992). The signal for activation of these channels comes from endoplasmic reticulum-resident STIM proteins that activate one or more of the family of Orai (1–3) channel proteins in the plasma membrane (Cahalan et al. 2007; Hogan et al. 2010). However, it is not known whether SOCE in lacrimal cells involves CRAC channels, or whether the channels consist of Orai subunits. And despite the well-documented role of store-operated entry in Ca2+ signalling in lacrimal cells in vitro, the role of this pathway in in vivo secretion is not known.
In recent years, the development of mouse models deficient in these proteins has been key to understanding the regulation of SOCE, and its physiological context in the intact animal. In this study, we employ a mouse model deficient in the Orai1 channel protein (Orai1-knockout (KO), generated by gene trap (Vig et al. 2008)) to investigate the role of Orai1 in lacrimal gland function and calcium signalling. We find that sustained calcium signalling, as well as activated lacrimal secretion, is substantially compromised in the absence of Orai1.
Methods
Ethical approval and animal procedures
All animal procedures were reviewed and approved by the National Institute of Environmental Health Sciences Animal Care and Use Committee (NIEHS ACUC). All animals were housed, cared for and used in compliance with the Guide for the Care and Use of Laboratory Animals (http://grants.nih.gov/grants/olaw/Guide-for-the-care-and-use-of-laboratory-animals.pdf).
When mice were euthanized for tissue collection, they were placed in a clear, air-filled chamber (10 litre) and then exposed to CO2 at a rate of 2 l min−1. After the animals became unconscious and stopped breathing, the CO2 flow was continued for at least 5 min to avoid recovery when removed from the chamber. A thoracotomy was performed before tissue collection commenced.
For experiments involving measurement of fluid secretion in anaesthetized mice, at the end of the experiment some mice were euthanized for collection of tissue for histological and electron microscopic analysis, and others were allowed to recover. Following anaesthesia, mice were kept on a warming pad and monitored for any adverse effects until fully recovered.
The total number of mice used in this study was 77 (wild-type (WT) 38; KO 39).
Orai1-KO mice
Orai1-KO mice (C57/DBA/129 background; Vig et al. 2008) generally died perinatally, but surviving pups (male and female) were obtained from mice crossed with out-bred ICR mice (Harlan Laboratories Inc. (Indianapolis, IN, USA) strain Hsd: ICR (CD-1)) for two generations. Litters of more than six pups were split and fostered by Swiss Webster mice. Litters with potential KO pups, as suggested by small pup size (Vig et al. 2008), were kept on heating pads until weaning. Mice were typically weaned and genotyped by tail biopsy at 21 days. Potential KO mice were kept with the mother/foster mouse until they were able to eat on their own, usually ∼4 weeks. Due to the infrequency of obtaining surviving KO mice, mice were used at varying ages, but always paired to WT littermates. The average age of mice for the study was 17 weeks.
Isolation of mouse lacrimal acinar cells
Mice were killed and exorbital lacrimal glands excised, finely minced and subjected to enzymatic digestion as previously described (Bird et al. 1992a). Isolated cells were washed and suspended in DMEM containing 10% fetal bovine serum, 5 mm glutamine, 100 units ml−1 penicillin and 100 units ml−1 streptomycin. Cells were allowed to attach to glass coverslips (no. 1.5) coated with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA).
Real time RT-PCR
Total RNA was isolated and purified using an RNeasy Plus Mini Kit (Qiagen, Valencia, CA, USA) from lacrimal acinar cells prepared from WT and Orai1-KO mice. Reverse transcription of RNA was performed using the Omniscript RT Kit (Qiagen), and resulting cDNA was amplified using TaqMan Fast Universal PCR Master Mix and TaqMan Gene Expression Assays (Life Technologies, Carlsbad, CA, USA) (Davis et al. 2013). The following mouse TaqMan Gene Expression Assays were used in this study: Orai1 (Mm00774349_m1), Orai2 (Mm04214089_s1), Orai3 (Mm01612888_m1), Stim1 (Mm01158413_m1), Stim2 (Mm01223103_m1) and Trpc1 (Mm00441975_m1). Reactions were cycled using an Applied Biosystems 7500 Fast Real-Time PCR System (Life Technologies) with universal cycling conditions. Relative quantification was determined with reference to 18S rRNA and was analysed using the comparative CT method (Suchanek et al. 2002).
Single-cell calcium measurement
Lacrimal cells, plated on 30 mm round coverslips and mounted in a Teflon chamber, were incubated in DMEM with 0.5 μm fura-5F/AM (Genolite Biotek, Portland, OR, USA) at 37°C for 10 min. For [Ca2+]i measurements, cells were bathed in Hepes-buffered salt solution (HBSS) at room temperature (21°C) containing (mm): NaCl 120, KCl 5.4, MgCl2 0.8, Hepes 20, CaCl2 1.8 and glucose 10; pH was adjusted to 7.4 with NaOH. Nominally Ca2+-free solutions were HBSS with no added CaCl2. Fluorescence images of the cells were recorded and analysed with a digital fluorescence imaging system (InCyt Im2, Intracellular Imaging Inc., Cincinnati, OH, USA). Changes in intracellular calcium are expressed as the ratio of fura-5F fluorescence (F340/F380). Ratio values were corrected for contributions by autofluorescence. Rmax, determined in situ, averaged 6.5. Peak ratio values for methacholine (MeCh) and thapsigargin were less than 3.5, indicating that the ratio values were less than saturation and thus essentially proportional to Ca2+ concentration.
Measurement of store-operated (ICRAC) and calcium-activated potassium (IK) currents
Lacrimal cells were attached to glass coverslips (no. 1.5) and placed on the stage of a Zeiss Axiovert 25 microscope equipped with a 40× (0.9 N.A) objective. Membrane currents were recorded at room temperature in the whole-cell configuration using an Axopatch 200B amplifier and Digidata 1322A data acquisition system, with Voltage clamp protocols and data acquisition controlled by pClamp 10 (Axon Instruments, Union City, CA, USA). Pipettes were pulled from 7740 thin-walled borosilicate capillary tubing (Sutter Instrument Co., Novato, CA, USA) and were fire polished to a tip diameter of 2∼3 μm with a resistance in bath solution of 2–4 MΩ. Seal resistances of 2–10 GΩ typically were obtained. Membrane potentials were corrected for the measured liquid junction potential.
For measurement of store-operated currents (ICRAC), lacrimal cells were bathed in HBSS containing (in mm): 140 NaCl, 3 KCl, 10 CsCl2, 1.2 MgCl2, 10 CaCl2, 10 glucose, and 10 Hepes (pH adjusted to 7.4 with NaOH). Divalent cation free (DVF) solution was made by removing Ca2+ and Mg2+ from the general external solution containing (in mm): 140 NaCl, 3 KCl, 10 CsCl, 10 Hepes, 10 glucose, and 0.5 EDTA. Fire-polished glass pipettes were filled with pipette solution containing (in mm): 145 cesium methanesulfonate, 10 BAPTA, 10 Hepes, 8 MgCl2 and 25 μm IP3 (pH adjusted to 7.2 with CsOH). Voltage ramps (–120 to +120 mV) of 250 ms from a holding potential of 0 mV were recorded every 2 s immediately after gaining access to the cell, and the currents were normalized based on cell capacitance.
For measurement of calcium-activated K currents (IK), lacrimal cells were bathed in an external solution containing (in mm): 140 NaCl, 5 KCl, 1.2 CaCl2, 2 MgCl2, 10 Hepes, 10 glucose (pH adjusted to 7.4 with NaOH). The internal pipette solution contained (in mm): 140 KCl, 2 MgCl2, 2 MgATP, 10 Hepes (pH adjusted to 7.4 with KOH). IP3, at a concentration of 10 μm, was included in the pipette solution to maximally mobilize calcium from intracellular calcium stores. The calcium buffering capacity of the internal pipette solution was adjusted with Na-BAPTA. Using 0.5 mm Na-BAPTA/0 Ca2+ provides a weak calcium buffer, allowing IP3 to mobilize calcium ions to a high level in the cytoplasm (‘High Ca2+i’ condition) (Trautmann & Marty, 1984). In contrast, 10 mm Na-BAPTA/0 Ca2+ provides a strong calcium buffer, maintaining calcium ions at a low level in the presence of IP3 (‘Low Ca2+i’ condition). Cells were dialysed with pipette solution for ∼1 min prior to the start of recording. Successive 100 ms voltage steps were implemented from a holding potential of −40 mV to test potentials ranging from −100 to +60 mV in +10 mV increments.
Aqueous tear production: phenol red thread
WT and Orai1-KO littermates were anaesthetized (2.5% isoflurane) and then injected i.p. with pilocarpine hydrochloride (2.5 μg g−1 in saline). Three, 8, 15 and 21 min after mouse injection (t = 0), a cotton thread impregnated with phenol red was applied to the ocular surface in the lateral canthus for 60 s (Dursun et al. 2002). Tear production was indicated by measuring (in mm) the ‘wetting’ of the cotton thread. The rate of tear production was normalized against individual mouse body weight.
Histology and electron microscopy
Excised lacrimal glands were harvested from WT and KO mice, both unstimulated and stimulated with pilocarpine, and sectioned into smaller pieces (<1 μm cubes), fixed in modified Karnovsky's fixative (2% paraformaldehyde + 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.4). The samples were bisected with one half processed for electron microscopic studies (see below) and the other half for histological examination.
For histology, the samples were dehydrated through graded alcohol, 80% × 2 for 30 min each, 95% × 2 for 45 min each, 100% × 3 for 45 min each followed by xylene × 3 30 min each. Samples were then infiltrated with four changes of paraffin, two at 30 min each and two at 45 min each. The samples were then embedded in paraffin, sectioned at 5 μm and stained with haematoxylin and eosin (H&E). The tissue sections were placed on glass slides, mounted in Permount™, and examined with an Olympus BX51 light microscope.
For transmission electron microscopy, tissue samples were processed on a Leica EM TP automatic tissue processor. Samples were rinsed in 0.1 m sodium cacodylate buffer (four changes, 10 min each), post-fixed in 2% osmium tetroxide for 1 h, and dehydrated through graded alcohol and propylene oxide (2×, 15 min each), propylene oxide/Poly Bed (1×, 1 h), infiltrated with Poly Bed (2×, 2 h each) and then embedded in Beem capsules with Poly Bed (capsules cured at 60°C for 72 h). The blocks were then trimmed, thick sectioned (700 nm) and stained with 1% toludine blue for examination by light microscopy. Blocks were then selected to be thin sectioned (a section ∼70–90 nm or ‘gold’), placed on Formvar-coated copper grids then stained with uranyl acetate and lead citrate. The grids were examined on an FEI Tecnai 110KV transmission electron microscope.
Digital photomicrographs were taken of each grid/animal and examined for ultrastructural landmarks of lacrimal acinar cells. A blind morphometric analysis was performed on electron micrographs of lacrimal gland tissue prepared from WT or Orai1-KO mice that were either untreated (control) or treated (stimulated) with pilocarpine. In total, 20 electron micrographs were analysed from each mouse group: six WT-control, six KO-control, four WT-stimulated and four KO-stimulated. Identified lumens were assigned a score based on three criteria that represented the extent to which secretion had occurred. The three criteria used were: lumen dilatation, presence of membrane fragments in the lumen and empty secretory granules. A score of 1–3 was assigned based on the extent to which each of these three criteria was met. A score of 0 was assigned when a lumen was present but with none of the criteria for secretion. Based on the extent with which these criteria were met and in combination, the scores were weighted and used to calculate a ‘secretion index’ for each animal preparation. Data were analysed by analysis of variance (ANOVA) using PROC GLM in SAS 9.0, and with post hoc analysis performed using LSMEANS in PROC GLM using a standard t test.
Peroxidase secretion measurement
A pair of lacrimal glands was removed from each mouse and cut into small pieces. The tissue was distributed into three cell strainers (15 mm Netwell Inserts, Corning, New York, USA) contained in a 12-well plate, and incubated in 2 ml of complete DMEM (10% fetal bovine serum, 5 mm glutamine) for 1 h at 37°C. The cell strainers containing lobules were then transferred and bathed in HBSS at 37°C containing (mm): NaCl 120, KCl 5.4, MgCl2 0.8, Hepes 20, CaCl2 1.8 and glucose 10; pH was adjusted to 7.4 with NaOH. HBSS was also supplemented with 0.3% (w/v) albumin. After 15 min, the samples were transferred into wells containing fresh HBSS (2 ml per well) for 30 min to establish a ‘basal’ secretion level. The samples were then transferred into wells containing ‘test’ solutions (2 ml) for an additional 30 min. The test solutions included: (i) control (HBSS + 1.8 mm Ca2+o), (ii) 200 μm MeCh + 1.8 mm Ca2+o and (iii) 200 μm MeCh-Ca2+o (+200 μm BAPTA).
On completion, the tissue and inserts were removed, and the bathing media (basal and test solutions) were assayed for peroxidase activity, an index of protein secretion. Peroxidase activity was measured using Amplex Red (Invitrogen, Carlsbad, CA, USA) (Rios et al. 2005) which, in the presence of hydrogen peroxide, is oxidized by the secreted peroxidase to produce a highly fluorescent molecule. The bathing solutions were incubated with 10 μm Amplex Red and 10 μm hydrogen peroxide and aliquoted in triplicate (150 μl) into the wells of a 96-well plate. The amount of fluorescence was then quantified simultaneously in each well on a FLIPRTETRA fluorescence imaging plate reader (Molecular Devices, Sunnyvale, CA, USA). Fluorescence was detected with a 470–495 nm excitation LED wavelength and a 565–625 nm emission filter. The effect of the test solutions on peroxidase secretion from the lacrimal gland pieces was expressed as a fold increase in fluorescence over that observed in the initial 30 min basal period (set as 1).
Results and discussion
We first confirmed by real-time RT-PCR knockdown of message for Orai1 in Orai1-KO mice (Fig. 1). We also determined message levels for other members of the Orai family (Orai2 and Orai3), TRPC1 – which has previously been shown to contribute to Ca2+ signalling in salivary glands (Liu et al. 2007) – and the two known Orai channel activators, STIM1 and STIM2. Orai1 message was essentially undetectable. Messages for Orai3, STIM1 and STIM2 were not affected by genotype. TRPC1 message increased about 2-fold but this was not statistically significant. Orai2 message increased substantially, as has been observed previously for other cell types from this mouse model (Vig et al. 2008). Interestingly, this increase in Orai2 message did not result in any detectable functional compensation with respect to SOCE, as shown in Fig. 2. Sustained Ca2+ entry due to thapsigargin was completely lost in lacrimal cells from KO mice as compared to their WT littermates (Fig. 2A–D). To determine whether loss of SOCE impacted the ability of acinar cells to maintain intracellular stores, we examined ionomycin-releasable Ca2+ as a measure of the intracellular stores. Treatment of mouse lacrimal cells with ionomycin (10 μm) in the absence of extracellular calcium produces a rapid release of Ca2+ such that the amplitude of the release transient is minimally affected by parallel Ca2+ buffering mechanisms. In unstimulated lacrimal cells, this ionomycin-sensitive store is essentially coincident with the agonist-and thapsigargin-sensitive stores (Bird et al. 1992a). The size of this ionomycin-releasable store was unchanged in cells from KO mice, indicating that intracellular Ca2+ stores were unchanged (Fig. 2E and F). Similar experiments utilizing the muscarinic-cholinergic agonist MeCh showed that the agonist-induced Ca2+ entry was also completely lost in the acinar cells from KO animals (Fig. 3). We previously showed that high concentrations of muscarinic agonists produce sustained Ca2+ signals, while lower concentrations produce sinusoidal oscillations superimposed on an elevated basal Ca2+ level (Bird et al. 1993). The pattern of these oscillations varies somewhat from cell to cell, and in Fig. 3C we show the responses to low MeCh in three different WT and KO acinar cells. In all cases, whether saturating (Fig. 3A) or subsaturating (Fig. 3C) concentrations of MeCh were used, the sustained Ca2+ signal ultimately failed in the KO acinar cells (summarized in Fig. 3B).
Figure 1.
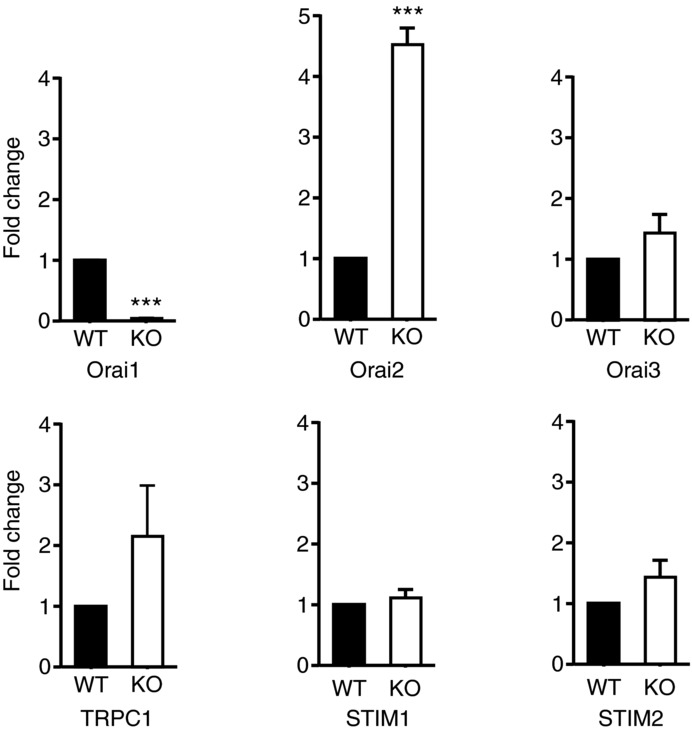
mRNA levels of Orai1, Orai2, Orai3, Stim1, Stim2 and TRPC1 were assessed by real-time RT-PCR as described in Methods. The data are mean ± SEM of three independent experiments (t test, ***P < 0.001).
Figure 2.
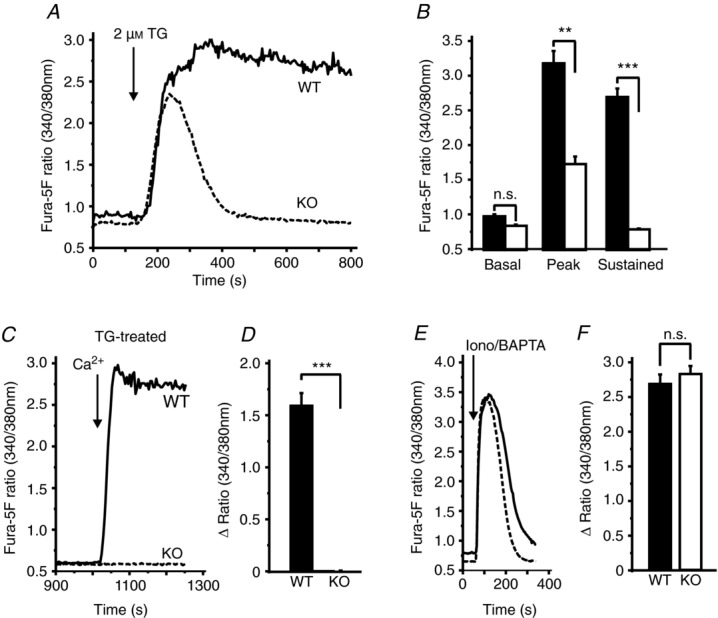
Isolated lacrimal acinar cells were loaded with the fluorescent calcium indicator, fura-5F. A, responses to 2 μm thapsigargin (TG) in the presence of 1.8 mm extracellular Ca2+ in cells from WT (continuous line) and Orai1-KO (broken line) mice. The key characteristics of the data in A are summarized in B, including basal Ca2+ ratio values, peak Ca2+ ratio responses to thapsigargin and sustained Ca2+ ratio values measured at 800 s. Data are mean ± SEM for 14 cells (WT) and 18 cells (KO) captured in five independent experiments (ANOVA; ***P < 0.0001; **P < 0.05; n.s., not significant). C, lacrimal cells were bathed in HBSS nominally free of extracellular Ca2+ and treated with 2 μm thapsigargin for 15 min, then 1.8 mm extracellular Ca2+ was restored to the bathing medium at 900 s. The effect of extracellular Ca2+ was measured as the difference in Ca2+ ratio values (Δ Ratio) measured before (at 850 s) and after (1200 s) the addition at 900 s. The data for WT (continuous line) and Orai1-KO (broken line) cells are summarized in D, as means ± SEM for 17 cells (WT) and 16 cells (KO) captured in three independent experiments (t test; ***P < 0.0001). E, response of lacrimal cells to the addition of 10 μm ionomycin in the presence of 3 mm BAPTA to assess the content of mobilizable intracellular Ca2+ stores in WT (continuous line) and Orai1-KO (broken line) cells, as reflected in the peak response to ionomycin/BAPTA. F, the effect of the extracellular Ca2+ was measured as the difference in Ca2+ ratio values (Δ Ratio) measured before (at 50 s) and after (peak response) the addition of ionomycin/BAPTA. The data are mean ± SEM for 20 cells (WT) and 19 cells (KO) captured in three independent experiments (t test; n.s., not significant).
Figure 3.
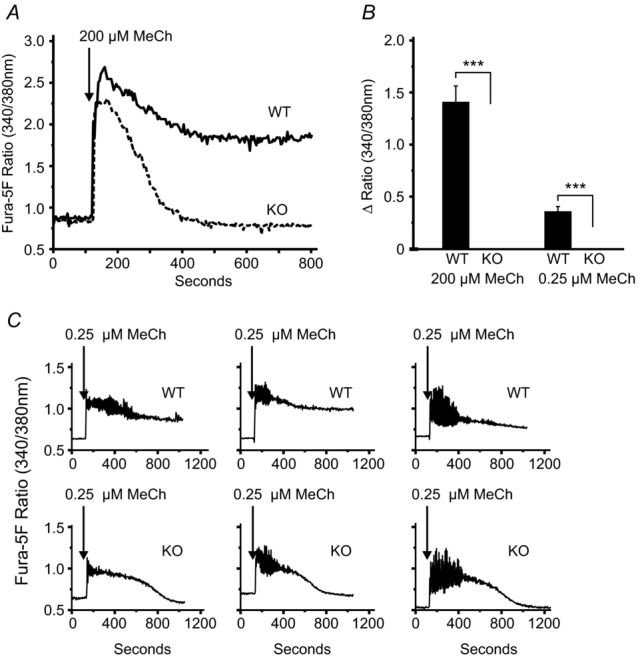
Isolated lacrimal acinar cells were loaded with the fluorescent calcium indicator fura-5F. In the presence of 1.8 mm extracellular Ca2+, the responses to the phospholipase C-coupled agonist MeCh were compared in cells from WT and Orai1-KO mice. A and C, responses to maximal (200 μm) and sub-maximal (0.25 μm) concentrations MeCh, respectively. B, summary of the sustained calcium signal above baseline (Δ Ratio), measured at 800 s for each MeCh concentration tested. For 200 μm MeCh the data are mean ± SEM for 11 cells (WT) and 14 cells (KO) and, for 0.25 μm MeCh, are mean ± SEM for 33 cells (WT) and 47 cells (KO), and captured in three independent experiments (t test; ***P < 0.0001).
To ensure that the diminished Ca2+ signals did in fact result from a defect in SOCE, we also undertook measurement of the classical store-operated Ca2+ current, ICRAC (Hoth & Penner, 1993; Parekh & Penner, 1997). However, as is often the case, Ca2+ currents clearly identifiable as ICRAC could not be detected, perhaps owing to the small size of the current and to the substantial constitutive currents present in these cells. The leak currents were variable in magnitude, slightly outwardly rectifying, but were not significantly different in WT and KO cells (data not shown). In a previous study, a similar problem with small ICRAC in the kidney cell line HEK293 was largely overcome by taking advantage of a property of ICRAC whereby currents are rapidly and transiently increased by removal of extracellular divalent cations (Hoth & Penner, 1993; DeHaven et al. 2007). Figure 4A shows inward (at −120 mV) and outward (at +120 mV) whole cell currents in a lacrimal acinar cell patched with a pipet containing 25 μm IP3 and 10 mm BAPTA to deplete intracellular Ca2+ stores. Inward Ca2+ current is not readily discernible, but a clear inwardly rectifying current is seen immediately upon switching the external medium to one that is free of divalent cations (Fig. 4A and C). This current is transient, owing to the property of de-potentiation, a hallmark of ICRAC (Zweifach & Lewis, 1996). Fig. 4B shows a similar measurement on an acinar cell from an Orai1-KO mouse. There is only a small inward current upon switching to a divalent cation medium, and this small current is mirrored in outward current, indicating that it is not inwardly rectifying and thus is not ICRAC. Figure 4D summarizes leak-subtracted inward current measurements from multiple patch-clamp experiments with cells from WT and KO mice, demonstrating almost complete loss of divalent-free inward current in the KO animals.
Figure 4.
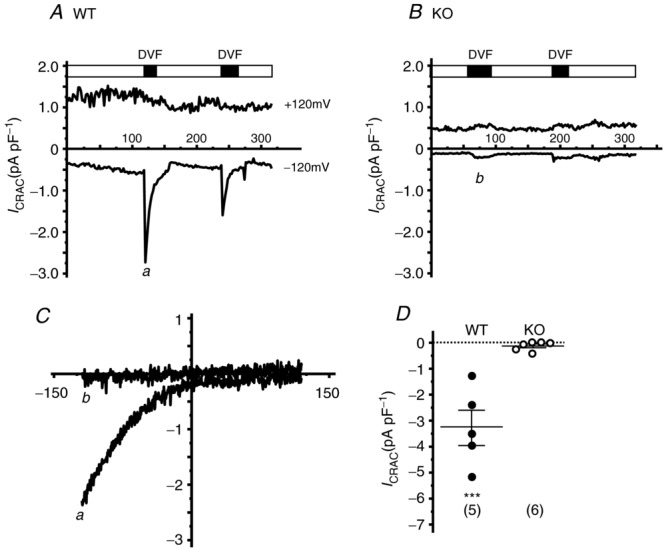
A, patch-clamp recording of whole-cell currents in WT mouse lacrimal cells. Cells were bathed in HBSS containing 10 mm CaCl2 and, on establishing a seal with the patch pipette, intracellular stores were depleted by inclusion of 25 μm IP3 and 10 mm BAPTA in the patch pipette. Under these conditions Ca2+ currents clearly identifiable as ICRAC could not be reliably detected. However, by switching the bathing solution to one free of all extracellular divalent ions (DVF), large transient inwardly rectifying currents appeared, a hallmark of ICRAC. B, little or no inward current appeared in store-depleted Orai-KO cells under whole-cell or DVF conditions. C, leak-subtracted I–V current relationships measured in WT (a from the experiment in A) and Orai1-KO (b from the experiment in B) cells. D, summary of experiments showing large peak inward DVF currents (leak-subtracted) in WT (n = 5 cells) compared to little or no DVF current in Orai1-KO acinar cells (n = 6 cells). Horizontal bars in D represent the mean ± SEM (t test; ***P < 0.005).
To ensure that the loss of ICRAC was not the result of a general loss of plasma membrane channels, and to determine whether other physiological components of the acinar ionic signalling pathway remain intact, we examined Ca2+-activated K+ currents, as described in Methods. Figure 5A shows the I–V relationship for outwardly rectifying K+ currents in cells dialysed with IP3 and 0.5 mm BAPTA, and that these currents are suppressed in the presence of a high (10 mm) concentration of BAPTA. Data from multiple experiments are summarized in Fig. 5B, showing that neither the constitutive nor the Ca2+-activated currents are changed in acinar cells from KO mice.
Figure 5.
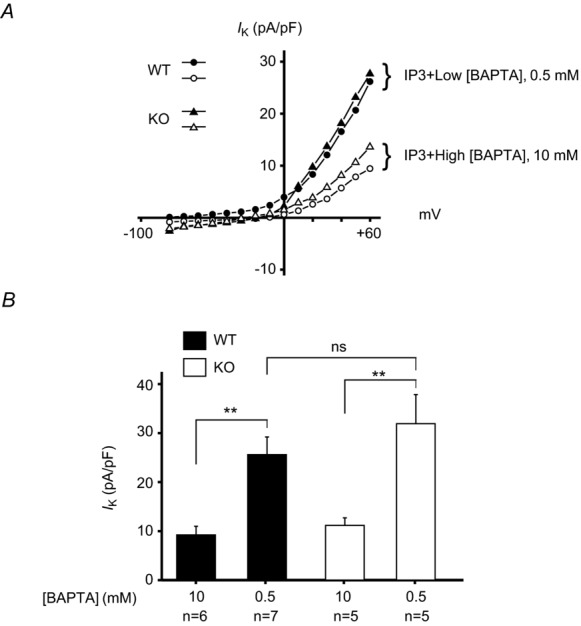
Patch-clamp recording of whole-cell currents was performed on single mouse lacrimal cells isolated from wild-type (WT) and Orai-KO (KO) mice. Cells were bathed in HBSS containing 1.2 mm CaCl2. A whole-cell patch was established with an internal patch pipette solution containing 10 μm IP3 with either 0.5 mm BAPTA (Low [BAPTA]) or 10 mm BAPTA (High [BAPTA]). Under low [BAPTA] conditions, IP3 is able to mobilize and elevate intracellular Ca2+ and activate a K+ current (IK) in both WT and KO lacrimal cells. This effect is suppressed under high Ca2+ buffering conditions (High [BAPTA]). A, I–V relationships for IK under both [BAPTA] conditions were indistinguishable between WT and KO lacrimal cells. B, current densities of IK measured at +60 mV. Data are mean ± SEM (ANOVA; **P < 0.05; n.s., not significant).
We next determined the consequences of loss of SOCE in lacrimal cells to neurogenic secretion of tears in anaesthetized WT and KO mice by examining the flow of tears in response to the muscarinic–cholinergic agonist pilocarpine. As shown in Fig. 6, overflow tear production in WT mice continued at a substantial rate for over 20 min. In the KO mice, the rate of secretion was significantly less and all but ceased within a few minutes.
Figure 6.
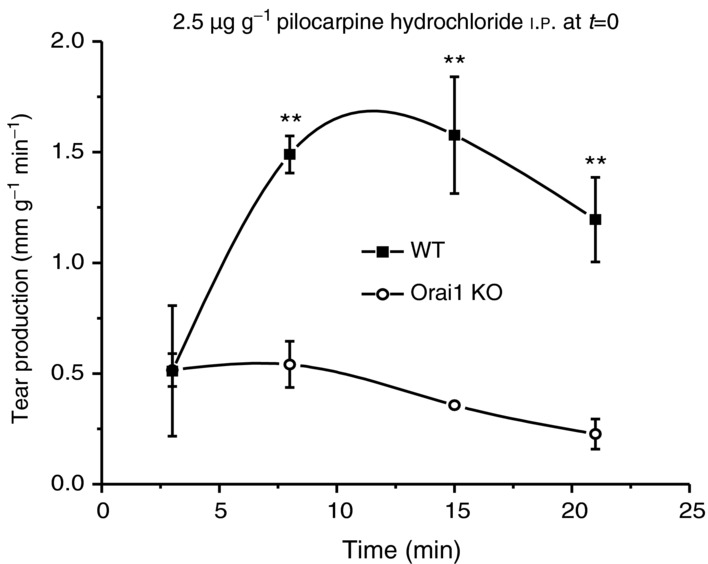
Massive tear production was stimulated in anaesthetized mice by i.p. injection (at t = 0) of the muscarinic agonist pilocarpine (2.5 μg g−1 body weight in saline). Following injection, tear production was measured at the indicated time points using the phenol red string method, and adjusted for animal body weight (mm g−1 min−1; see Methods). With comparisons made with paired littermates, tear production was significantly reduced in the Orai1-KO mice. Data are mean ± SEM from three sets of paired littermates (t test, **P < 0.05).
We noted that some but not all KO mice showed some signs of inflammation in their eyes. In an earlier report, using another global Orai1 knockout mouse, some degree of eyelid irritation was reported (Gwack et al. 2008). However, in a global knockout, it is not possible to determine if this results from diminished secretion or the compromised immunity expected in these mice. Furthermore, in a previous report utilizing mice with STIM1 and STIM2 deleted from T cells, disruption of salivary gland function was observed, and this was attributed to an increase in autoimmunity and pathological lymphocytic infiltration into the glands, leading ultimately to progressive salivary gland destruction. Thus, we next examined the structure of WT and KO lacrimal acinar cells by both light and electron microscopy. The general morphology of the glands and acinar cells was not notably different in WT and KO animals. As shown in Fig. 7, in both cases glands were composed of closely packed acini (asterisks), intralobular ducts (long arrows) and interlobular ducts (short arrows). At the electron microscopic level, Fig. 8 shows similar populations of immature and mature secretory granules (Fig. 8A and C, respectively), and following stimulation with the muscarinic–cholinergic agonist pilocarpine, both WT and KO animals showed signs of exocytotic secretion (Fig. 8B and D). No ductal abnormalities were noted in either the WT or KO glands. No consistent or widespread infiltrates of lymphocytes were observed in the glands of either the WT or the KO animals, nor was there any evidence of destructive glandular changes, similar to those described by Cheng et al. (2012). Electron microscopy revealed evidence of a few inflammatory cells (lymphocytes); however, these cells were few in number and were not found in acinar lumens but most often within the surrounding ducts or connective tissue. There was no gland destruction at all, especially destruction due to lymphocyte infiltrates noted by electron microscopy.
Figure 7.
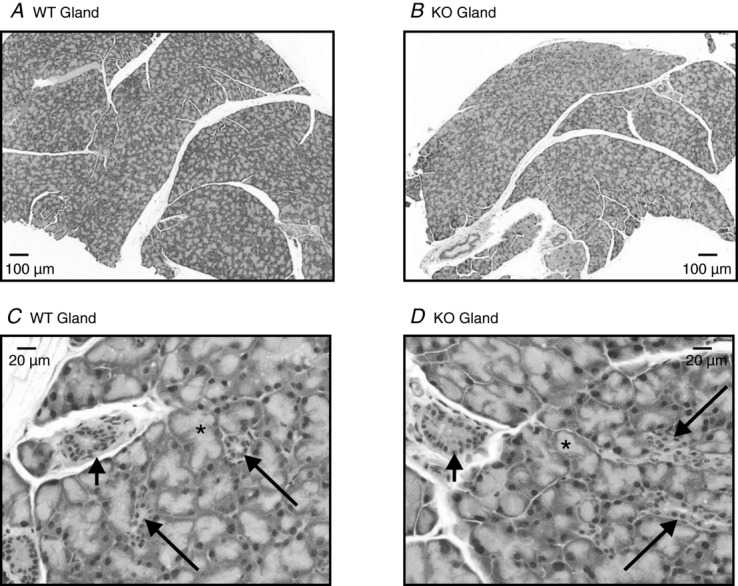
Excised lacrimal glands were harvested from wild-type (WT) and Orai1-KO (KO) mice, fixed in modified Karnovsky's fixative, embedded in paraffin and stained with H&E for histological examination as described in Methods. A and C, low magnification (4×) and a higher magnification (20×) of a representative lacrimal gland isolated from a WT mouse. B and D, low magnification (4×) and a higher magnification (20×) of a representative lacrimal gland isolated from an Orai1-KO mouse. WT and KO lacrimal glands were indistinguishable, composed of closely packed acini (asterisks) with interspersed intralobular ducts (long arrows) that convey the acinar secretions into the interlobular ducts (short arrows). Observations based on pairs of lacrimal glands isolated from five WT and KO mice (two male and three female mice used in each case).
Figure 8.
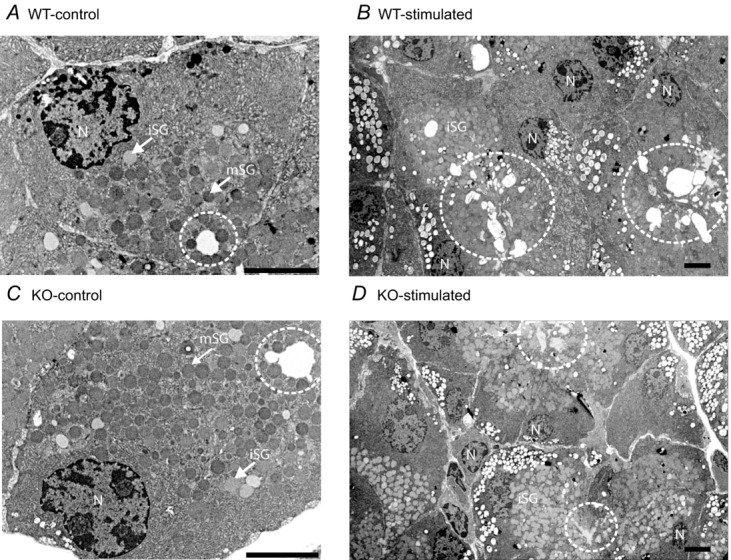
A and C, representative electron micrographs of lacrimal tissue from control wild-type (WT-control) and Orai-KO (KO-control) mice, respectively. B and D, representative electron micrographs of lacrimal tissue from pilocarpine-stimulated (20 min), wild-type (WT-stimulated) and Orai-KO (KO-stimulated) mice respectively. Indicated on the micrographs are nuclei (N), immature secretory granules (iSG), mature secretory granules (mSG) and acinar lumens (dashed circle). Scale bars = 5 μm.
We further explored the consequences of loss of SOCE in lacrimal glands to neurogenic stimulation in response to stimulation. The morphometry of lacrimal acinar lumens under control and pilocarpine-stimulated (20 min) conditions was analysed blindly to generate a ‘secretion index’ based on three characteristics: lumen dilatation, presence of membrane fragments and empty secretory granules (Fig. 9A). Cholinergic stimulation resulted in a significant increase in the secretory index for both WT and Orai1-KO lacrimal tissue. However, the secretory index was significantly less in pilocarpine-treated Orai1-KO tissue than in the WT tissue (Fig. 9A). To confirm the decreased protein secretion, we carried out assays of agonist-induced release of peroxidase, a major secretory protein in rodent exorbital lacrimal glands (Dartt, 1989; Rios et al. 2005). As shown in Fig. 9B, the muscarinic cholinergic agonist MeCh induced a significant increase in the release of peroxidase from lacrimal gland fragments. This release of peroxidase was reduced, although not entirely blocked, by removal of extracellular Ca2+. The residual Ca2+-independent secretion could result either from the transient release of intracellular Ca2+ or possibly from a contribution of the protein kinase C pathway (Dartt, 1989). Importantly, when secretion was measured in fragments from KO mice, the release of peroxidase was indistinguishable in the presence or absence of Ca2+, and was similar to that seen in the absence of Ca2+ with fragments from WT animals. These morphological and biochemical data demonstrate, first, that knockdown of Orai1 and loss of SOCE has minimal effect on the development and differentiation of lacrimal glands, and, second, that cholinergic stimulated secretion (Ding et al. 2003) occurs both in WT and, to a lesser extent, in Orai1-KO mouse lacrimal glands. The lack of change in the Ca2+-independent peroxidase secretion is consistent with the morphological analyses indicating that the glands from KO animals lack SOCE, but have no other non-specific alterations in lacrimal gland function
Figure 9.
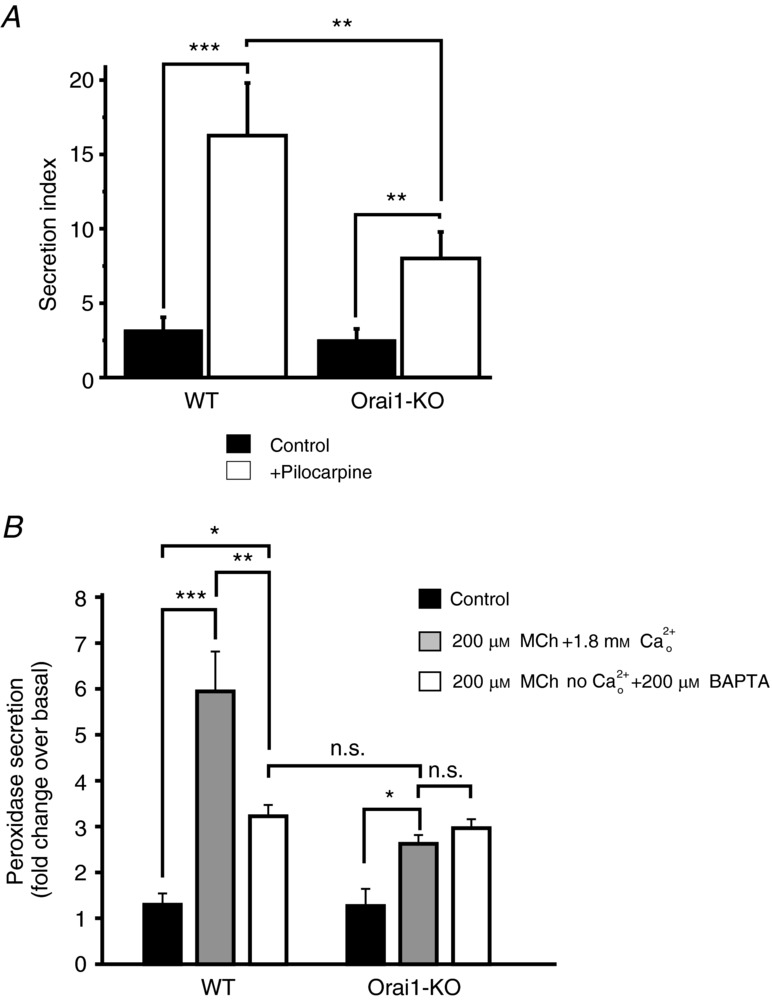
A, comparison of ‘secretion index’ (see Methods) from morphometric analysis of electron micrographs of lacrimal acini (as in Fig. 8). In total, 20 electron micrographs were analysed from each mouse group, which numbered: six WT-control, six KO-control, four WT-stimulated and four KO-stimulated. Data are mean ± SEM as described in Methods (***P < 0.0001; **P < 0.05). B, effects of MeCh on peroxidase secretion from lacrimal gland pieces isolated from wild-type (WT) and Orai1-KO (KO) mice. The effect of MeCh was monitored both in the presence and absence of extracellular calcium (1.8 mm). The data are mean ± SEM from four sets of paired littermates (ANOVA; ***P < 0.0001; **P < 0.01; *P < 0.05; n.s., not significant).
Lacrimal glands from mice or rats have long served as useful models for basic studies of Ca2+ signalling (Putney & Bird, 1994). For example, rat lacrimal gland studies provided early evidence that Ca2+ entry could be activated independently of receptor activation (Parod & Putney, 1978). One of the earliest demonstrations of the ability of thapsigargin to activate SOCE was carried out with lacrimal acinar cells (Kwan & Putney, 1990). In recent years, attention has turned to haematopoietic cells because of the ability to measure the current underlying SOCE, ICRAC (Hoth & Penner, 1992). It has traditionally been difficult to measure ICRAC in non-haematopoetic cells because of its extremely small size; here we utilize a technique whereby ICRAC is substantially amplified by removal of divalent cations (Hoth & Penner, 1993; DeHaven et al. 2007) to provide the first demonstration of an ICRAC-like current in this cell type. In addition, the results of this study demonstrate the necessary role of SOCE, involving channels comprising Orai1 subunits, in Ca2+ signalling and function of mouse lacrimal glands.
As mentioned above, a strategy that compromises SOCE in T cells also leads to a Sjögren's syndrome-like diminished exocrine secretion, through an increase in autoimmunity (Cheng et al. 2012). Thus, SOCE obviously impacts on exocrine gland function at both the local level, as shown here, and the level of the immune system. We can speculate that alleviation of debilitating exocrinopathies, such as dry eye syndrome, may involve modulation of SOCE or other Ca2+ entry pathways.
Key points
Lacrimal acinar cells from mice whose gene for Orai1 has been deleted have no detectable store-operated Ca2+ entry, whether assessed by measurement of cytoplasmic Ca2+ changes or as a store-operated current.
Mice lacking Orai1 have diminished lacrimal fluid secretion in response to muscarinic–cholinergic stimulation.
Mice lacking Orai1 also show diminished exocytosis, both in vivo and in vitro.
The development and morphology of lacrimal glands, as well as responses not dependent on Ca2+ entry were unchanged in the knockout mice.
The results demonstrate the central importance of store-operated Ca2+ entry in lacrimal exocrine function, and suggest possible strategies for combating diseases associated with diminished lacrimal secretion.
Acknowledgments
We gratefully acknowledge the help of Dr Connie Cummings, Dr Gordon Flake and Ms Deloris Sutton with electron microscopy and of Dr Shyamal Peddada with the statistical analysis of the morphological data. We are also grateful to Mr John Brodie and Ms Page Myers of the NIEHS animal facility for their assistance with care and husbandry of the mice. Drs Stephen Shears and Lutz Birnbaumer read the manuscript and provided helpful comments.
Glossary
- CRAC
calcium release-activated calcium
- DVF
divalent cation free
- HBSS
Hepes-buffered saline solution
- IP3
inositol 1,4,5-trisphosphate
- KO
knockout
- MeCh
methacholine
- Orai1-KO
Orai1 knockout
- SOCE
store-operated Ca2+ entry
- WT
wild type.
Additional information
Competing interests
All authors ascertain no conflict of interests associated with this work.
Author contributions
J.X.: carried out experiments providing published data; conception and design, or analysis and interpretation of data; drafting the article or revising it critically for important intellectual content. J.G.P.: assisted in experiments providing published data; conception and design, or analysis and interpretation of data; drafting the article or revising it critically for important intellectual content. F.M.D.: carried out experiments providing published data; conception and design, or analysis and interpretation of data; drafting the article or revising it critically for important intellectual content. P.N.D.: carried out experiments providing published data; conception and design, or analysis and interpretation of data; drafting the article or revising it critically for important intellectual content. J.W.P.: conception and design, or analysis and interpretation of data; drafting the article or revising it critically for important intellectual content. G.S.B.: carried out experiments providing published data; conception and design, or analysis and interpretation of data; drafting the article or revising it critically for important intellectual content. All authors gave final approval of the version to be published.
Funding
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
References
- Bird GS, Rossier MF, Obie JF, Putney JW. Sinusoidal oscillations in intracellular calcium requiring negative feedback by protein kinase C. J Biol Chem. 1993;268:8425–8428. [PubMed] [Google Scholar]
- Bird GStJ, Obie JF, Putney JW. Functional homogeneity of the non-mitochondrial Ca2+-pool in intact mouse lacrimal acinar cells. J Biol Chem. 1992a;267:18382–18386. [PubMed] [Google Scholar]
- Bird GStJ, Obie JF, Putney JW. Sustained Ca2+ signalling in mouse lacrimal acinar cells due to photolysis of ‘caged’ glycerophosphoryl-myo-inositol 4,5-bisphosphate. J Biol Chem. 1992b;267:17722–17725. [PubMed] [Google Scholar]
- Cahalan MD, Zhang SL, Yeromin AV, Ohlsen K, Roos J, Stauderman KA. Molecular basis of the CRAC channel. Cell Calcium. 2007;42:133–144. doi: 10.1016/j.ceca.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Alevizos I, Liu X, Swaim WD, Yin H, Feske S, Oh-hora M, Ambudkar IS. STIM1 and STIM2 protein deficiency in T lymphocytes underlies development of the exocrine gland autoimmune disease, Sj+¦gren's syndrome. Proc Natl Acad Sci U S A. 2012;109:14544–14549. doi: 10.1073/pnas.1207354109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartt DA. Signal transduction and control of lacrimal gland protein secretion: a review. Current Eye Res. 1989;8:619–636. doi: 10.3109/02713688908995762. [DOI] [PubMed] [Google Scholar]
- Dartt DA. Dysfunctional neural regulation of lacrimal gland secretion and its role in the pathogenesis of dry eye syndromes. Ocul Surf. 2004;2:76–91. doi: 10.1016/s1542-0124(12)70146-5. [DOI] [PubMed] [Google Scholar]
- Dartt DA. Neural regulation of lacrimal gland secretory processes: relevance in dry eye diseases. Prog Retin Eye Res. 2009;28:155–177. doi: 10.1016/j.preteyeres.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Parsonage MT, Cabot PJ, Parat MO, Thompson EW, Roberts-Thomson SJ, Monteith GR. Assessment of gene expression of intracellular calcium channels, pumps and exchangers with epidermal growth factor-induced epithelial-mesenchymal transition in a breast cancer cell line. Cancer Cell Int. 2013;13:76. doi: 10.1186/1475-2867-13-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Putney JW. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem. 2007;282:17548–17556. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- Ding C, Walcott B, Keyser KT. Sympathetic neural control of the mouse lacrimal gland. Invest Ophthalmol Vis Sci. 2003;44:1513–1520. doi: 10.1167/iovs.02-0406. [DOI] [PubMed] [Google Scholar]
- Dursun D, Wang M, Monroy D, Li DQ, Lokeshwar BL, Stern ME, Pflugfelder SC. A mouse model of keratoconjunctivitis sicca. Invest Ophthalmol Vis Sci. 2002;43:632–638. [PubMed] [Google Scholar]
- Godfrey PP, Putney JW. Receptor-mediated metabolism of the phosphoinositides and phosphatidic acid in rat lacrimal acinar cells. Biochem J. 1984;218:187–195. doi: 10.1042/bj2180187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, Prakriya M, Rajewsky K, Rao A. Hair loss and defective T-and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28:5209–5222. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signalling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–355. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan CY, Putney JW. Uptake and intracellular sequestration of divalent cations in resting and methacholine-stimulated mouse lacrimal acinar cells. Dissociation by Sr2+ and Ba2+ of agonist-stimulated divalent cation entry from the refilling of the agonist-sensitive intracellular pool. J Biol Chem. 1990;265:678–684. [PubMed] [Google Scholar]
- Kwan CY, Takemura H, Obie JF, Thastrup O, Putney JW. Effects of methacholine, thapsigargin and La3+ on plasmalemmal and intracellular Ca2+ transport in lacrimal acinar cells. Am J Physiol. 1990;258:C1006–C1015. doi: 10.1152/ajpcell.1990.258.6.C1006. [DOI] [PubMed] [Google Scholar]
- Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L, Ambudkar IS. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(/) mice. Proc Natl Acad Sci U S A. 2007;104:17542–17547. doi: 10.1073/pnas.0701254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Parod RJ, Putney JW. The role of calcium in the receptor mediated control of potassium permeability in the rat lacrimal gland. J Physiol. 1978;281:371–381. doi: 10.1113/jphysiol.1978.sp012428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW, Bird GS. The inositol phosphate-calcium signalling system in lacrimal gland cells. Adv Exp Med Biol. 1994;350:115–119. doi: 10.1007/978-1-4615-2417-5_20. [DOI] [PubMed] [Google Scholar]
- Putney JW, Bird GStJ. Calcium signalling in lacrimal acinar cells. Adv Exp Med Biol. 1998;438:123–128. doi: 10.1007/978-1-4615-5359-5_16. [DOI] [PubMed] [Google Scholar]
- Rios JD, Horikawa Y, Chen LL, Kublin CL, Hodges RR, Dartt DA, Zoukhri D. Age-dependent alterations in mouse exorbital lacrimal gland structure, innervation and secretory response. Exp Eye Res. 2005;80:477–491. doi: 10.1016/j.exer.2004.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchanek KM, May FJ, Robinson JA, Lee WJ, Holman NA, Monteith GR, Roberts-Thomson SJ. Peroxisome proliferator-activated receptor α in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol Carcinog. 2002;34:165–171. doi: 10.1002/mc.10061. [DOI] [PubMed] [Google Scholar]
- Trautmann A, Marty A. Activation of Ca-dependent K channels by carbamoylcholine in rat lacrimal glands. Proc Natl Acad Sci U S A. 1984;81:611–615. doi: 10.1073/pnas.81.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Calcium-dependent potentiation of store-operated calcium channels in T lymphocytes. J Gen Physiol. 1996;107:597–610. doi: 10.1085/jgp.107.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]