

Abstract
Carbonic anhydrase enzymes (CAs) catalyse the reversible hydration of CO2 to H+ and HCO3− ions. This catalysis is proposed to be harnessed by acid/base transporters, to facilitate their transmembrane flux activity, either through direct protein–protein binding (a ‘transport metabolon’) or local functional interaction. Flux facilitation has previously been investigated by heterologous co-expression of relevant proteins in host cell lines/oocytes. Here, we examine the influence of intrinsic CA activity on membrane HCO3− or H+ transport via the native acid-extruding proteins, Na+–HCO3− cotransport (NBC) and Na+/H+ exchange (NHE), expressed in enzymically isolated mammalian ventricular myocytes. Effects of intracellular and extracellular (exofacial) CA (CAi and CAe) are distinguished using membrane-permeant and –impermeant pharmacological CA inhibitors, while measuring transporter activity in the intact cell using pH and Na+ fluorophores. We find that NBC, but not NHE flux is enhanced by catalytic CA activity, with facilitation being confined to CAi activity alone. Results are quantitatively consistent with a model where CAi catalyses local H+ ion delivery to the NBC protein, assisting the subsequent (uncatalysed) protonation and removal of imported HCO3− ions. In well-superfused myocytes, exofacial CA activity is superfluous, most likely because extracellular CO2/HCO3− buffer is clamped at equilibrium. The CAi insensitivity of NHE flux suggests that, in the native cell, intrinsic mobile buffer-shuttles supply sufficient intracellular H+ ions to this transporter, while intrinsic buffer access to NBC proteins is restricted. Our results demonstrate a selective CA facilitation of acid/base transporters in the ventricular myocyte, implying a specific role for the intracellular enzyme in HCO3− transport, and hence pHi regulation in the heart.
Introduction
Carbonic anhydrase (CA) proteins are expression products of a gene-family that codes for at least 16 isoforms (Sly & Hu, 1995; Supuran, 2008). All except CAVII, CAX and CAXI are functionally active enzymes, widely expressed, which catalyse the reversible hydration of CO2 to H+ and HCO3− ions. Catalytically active CAs fulfil a number of functions (Maren, 1967). They can accelerate the cellular venting of CO2 (Geers & Gros, 2000; Swietach et al. 2008, 2009), the spatial diffusion of intracellular and extracellular H+ ions (Stewart et al. 1999; Spitzer et al. 2002; Swietach et al. 2009; Hulikova et al. 2011), and the speed of CO2/HCO3− (carbonic) buffering (Leem & Vaughan-Jones, 1998). A major role for CAs is proposed to be the catalytic enhancement of acid/base membrane transport. Some reports suggest they achieve this by binding directly to pH regulatory transporters, forming a multimeric protein complex called a transport metabolon (Sterling et al. 2001, 2002; Morgan et al. 2007; Casey et al. 2009; Svastova et al. 2012; Vargas & Alvarez, 2012). The proposal remains controversial, as observations of direct CA binding and flux facilitation have recently been challenged (Boron, 2010; Al-Samir et al. 2013; Parker & Boron, 2013), while the molecular mechanisms involved are far from clear. In some cases, for example, CA protein may stimulate acid transport allosterically, even in the absence of enzymic activity (Becker et al. 2011).
Various CA isoforms are expressed in mammalian heart. In ventricular myocytes, CAII is a cytosolic soluble intracellular isoform, CAIV and XIV are sarcolemmally tethered with exofacially exposed catalytic sites, while CAXIV and IX have also been detected at sarcoplasmic reticular and possibly transverse tubule membranes (Scheibe et al. 2006; Schroeder et al. 2013). CA activity in these cells has been shown to accelerate regulatory pHi recovery following intracellular acid or base loads (Lagadic-Gossmann et al. 1992; Orlowski et al. 2011; Vargas & Alvarez, 2012). Moreover, the principal acid-extruding transporters in these cells, Na+/H+ exchange (NHE1 isoform) and Na+–HCO3− cotransport (NBCe1 and NBCn1) (Aiello et al. 1998; Yamamoto et al. 2005; De Giusti et al. 2011; Garciarena et al. 2013; for review see Vaughan-Jones et al. 2009), when co-expressed heterologously with intracellular CAII or exofacial CAIV in oocytes or cultured cell lines, exhibit enhanced activity that is slowed in the presence of pharmacological CA inhibitors such as acetazolamide (ATZ) (Gross et al. 2002; Li et al. 2002; Alvarez et al. 2003; Loiselle et al. 2004; Li et al. 2006; Becker & Deitmer, 2007; Schueler et al. 2011). It has yet to be shown, however, that NHE and NBC activities are specifically regulated by either intracellular or exofacial CA in native ventricular cells.
In the present work we have examined the influence of the catalytic activity of intracellular and exofacial CA (CAi and CAe, respectively) on NHE-and NBC-mediated acid efflux from rat ventricular myocytes. Intrinsic transporter and CA activity in these cells has been assessed from measurements of intracellular pH, intracellular [Na+], and extracellular surface pH (pHe) using ion-selective fluorophores (Leem et al. 1999; Yamamoto et al. 2005; Schroeder et al. 2013), while applying membrane-permeant and –impermeant CA inhibitors to gauge the relative importance of CAi and CAe. Results suggest a novel and selective role for intracellular catalytic CA activity in controlling membrane acid extrusion, while questioning any obligatory role for exofacial CA. The results have important implications for the cytoplasmic organization of spatial H+ ion movement in the native ventricular myocyte.
Methods
Solutions
Lysis buffer contained: 15 mm NaCl, 105 mm potassium gluconate, 35 mm KCl, 20 mm Mes, 20 mm Hepes, 1 mm MgCl2, 0.5 mm EGTA; pH adjusted to 8.1 at 4°C with 5N HCl and 4M KOH. Hepes-buffered Tyrode solution contained: 135 mm NaCl, 20 mm Hepes, 4.5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 11 mm glucose; pH adjusted to 7.4 at 37°C with 4 m NaOH. CO2/HCO3−-buffered Tyrode solution contained: 125 mm NaCl, 4.5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 11 mm glucose, 22 mm NaHCO3 (or 12 mm NaHCO3 for surface pH measurements); bubbled with 5% CO2–95% O2 to adjust pH to 7.4 at 37°C. High-K+ CO2/HCO3−-buffered Tyrode solution contained: 85 mm NaCl, 45 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 11 mm glucose, 22 mm NaHCO3; bubbled with 5% CO2–95% O2 to adjust pH to 7.4 at 37°C. Ammonium-containing solutions contained NaCl isosmotically replaced with NH4Cl. Superfusion solutions were delivered to the superfusion chamber at 2 ml min–1, 37°C.
Drugs
NHE and NBC inhibitors
Cariporide (NHE inhibitor) and S0859 (NBC inhibitor) were kindly provided by Sanofi Aventis (Germany). Dimethylamiloride (DMA; NHE inhibitor) was purchased from Sigma (Poole, UK).
CA inhibitors
Membrane-permeable acetazolamide (ATZ) and ethoxyzolamide (ETZ) were obtained from Sigma. ATZ and ETZ block both intracellular and exofacial CA isoforms. Membrane-impermeant compound 14v (1-[5-sulfamoyl-1,3,4-thiazol-2-yl-(aminosulfonyl-4-phenyl)]-2,6-dimethyl-4-phenyl-pyridinium perchlorate; Alvarez et al. 2003; Supuran et al. 2004) is a positively charged heterocyclic sulphonamide that inhibits extracellular CA isoforms with nanomolar efficacy (Scozzafava et al. 2000; Supuran et al. 2004).
Isolated rat ventricular myocytes
Male Sprague–Dawley rats (300–400 g) were killed by cervical dislocation in accordance with Home Office regulations for Schedule 1 methods of killing, and guidelines issued by the University of Oxford. Ventricular myocytes were enzymically dispersed from Langendorff-perfused hearts in vitro as described previously (Garciarena et al. 2013), using a mixture of protease and collagenase type II enzymes (Worthington), or Liberase (Roche Pharmaceuticals). Isolated cells were superfused with Hepes or CO2/HCO3−-buffered Tyrode solutions at 37°C. Quiescent, Ca2+-tolerant, rod-shaped cells were selected for experiments.
In vitro carbonic anhydrase activity assay
Whole rat ventricles were dissected from Langendorff-perfused hearts in vitro. They were were homogenized in lysis buffer on ice for 3 min using a Polytron tip. Membranes were disrupted by a repeated freeze–thaw cycle to release all intracellular CA isoforms (Schroeder et al. 2013). For the best results, samples were diluted up to a total protein concentration of between 1 and 10 mg ml−1 (measured by the Bradford assay). Concentrations within this range give adequate enzyme concentration for a reliable CA measurement, without exceeding the resolving power of the measurement system. A 0.67 ml volume of lysate was added to the 2 ml capacity stirred chamber to which a pH electrode (Hamilton Biotrode Fisher Scientific, UK) was inserted to measure medium pH continuously at 1 Hz. The chamber was cooled to 4°C. To initiate the CA-catalysed reaction, 0.33 ml of CO2-saturated water (at 4°C) was added rapidly. The rate of acidification, measured by the electrode, was fitted using a least-squares algorithm to derive a hydration rate constant. The obtained rate constants were normalized to spontaneous rates measured in the presence of 100 μm ATZ, measured for the same dilution of lysate. As a result of this normalization procedure, enzyme concentration is eliminated as a variable.
Fluorescence measurements
Intracellular pH, intracellular [Na+] and surface extracellular pH were measured using fluorescent indicators cSNARF1, SBFI (Yamamoto et al. 2005) and wheat germ agglutinin (WGA)-conjugated fluorescein (Stock et al. 2007; Schroeder et al. 2013) (Invitrogen, UK). Myocytes were loaded with fluorescein dyes (10 min acetoxymethyl ester (AM)-loading, at room temperature, with 10 μm cSNARF1; 60 min AM-loading with 100 μm SBFI; 3 min loading with 10 μm WGA-fluorescein) and allowed to settle at the base of a Perspex superfusion chamber mounted on an epifluorescence setup (Nikon Diaphot 200 microscope (Nikon, UK) with a Cairn monochromator light source (Cairn, UK) and Electron Tubes photomultipliers (Electron Tubes, UK) connected via an in-house designed side-arm) for measurements with cSNARF1 or SBFI, or a confocal fluorescence setup (LSM700, Zeiss, UK) for measurements with WGA-fluorescein. cSNARF1 was excited at 540 ± 5 nm and fluorescence was measured simultaneously at 640 ± 10 nm and 580 ± 10 nm by separating emitted wavelengths using a 605 nm dichroic mirror. SBFI excitation was alternated every 0.25 s between 350 ± 5 nm and 380 ± 5 nm, and the fluorescence emission was measured above 510 nm using a long-pass filter. WGA-fluorescein was excited by a 488 nm laser-line and fluorescence emission was measured above 510 nm using a long-pass filter.
Intracellular ratiometric fluorescence signals were calibrated in situ as previously described (Leem et al. 1999; Yamamoto et al. 2005; Schroeder et al. 2013), using high-K+, nigericin-containing superfusates of different pH (for intracellular cSNARF1 signal), superfusates of different pH (for extracellular surface WGA-fluorescein signal), and superfusates of different Na+ concentration containing the cation ionophores gramicidin and monensin (for intracellular SBFI signal). The intracellular SBFI signal recorded during an experiment shows a modest pHi sensitivity (Minta & Tsien, 1989). This was corrected for by recording a pHi time course (cSNARF1; not shown) in parallel experiments, and then applying the equation: [Na+]corr = [Na+]uncorr – α(pHi – 7.2), to the [Na+]i time course, where [Na+]uncorr is the uncorrected calibrated signal, [Na+]corr is the corrected signal, and the best-fitted value for the coefficient, α, was 3.0 (Yamamoto et al. 2005).
Calculating acid flux
Acid efflux on NHE or NBC (or both) was calculated as the product of the rate of pHi recovery from an acid load (dpHi/dt; piecewise fitted to a straight line over a 3 s interval) and buffering capacity (β). For experiments performed in the absence of CO2/HCO3−, β was equal to the intrinsic buffering capacity measured previously (Zaniboni et al. 2003). For experiments performed in the presence of CO2/HCO3−, the additional CO2-dependent component to β was calculated using the equilibrium equation 2.303 × [HCO3−]o × 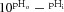 (Leem et al. 1999). The same calculation was performed to derive flux from mathematically simulated pHi time courses.
(Leem et al. 1999). The same calculation was performed to derive flux from mathematically simulated pHi time courses.
Statistics
Data are reported as mean ± standard error of the mean (SEM). Statistical significance between data was assessed by means of Student's unpaired t test.
Results
Catalytic activity of cardiac carbonic anhydrase
Intracellular and extracellular CA
Cardiac homogenization, by lysing cell membranes, exposes both intracellular and extracellular CA activity. Figure 1A shows that addition of 0.33 ml CO2-saturated water to 0.67 ml of buffer solution containing homogenized ventricular tissue reduced its pH over a period of 300 s, while addition of CA inhibitors ATZ (100 μm) or 14v (100 nm) slowed the reaction to a rate seen in the absence of homogenate. The rate constant for CO2 hydration (kf) in cardiac homogenate was reduced 10-fold by the inhibitors, while the residual rate constant equalled that for the spontaneous, uncatalysed reaction at 4°C (Fig. 1B). Ventricular tissue therefore expresses significant CA catalytic activity, as reported previously. In further experiments (not shown), the IC50 values for cardiac CA inhibition by ATZ and 14v were 7.3 ± 1.0 nm and 2.4 ± 0.6 nm, respectively (n = 10).
Figure 1.
A, time course of medium pH measured at 4°C. Addition of CO2-saturated water to cardiac homogenate (prepared in lysis buffer) reduces medium pH at a rate proportional to CA activity. ATZ (100 μm) or 14v (100 nm) reduced the rate of acidification to spontaneous levels (i.e. measured in lysis buffer only). B, CA activity, expressed as the CO2 hydration constant kf, was significantly reduced in the presence 100 μm ATZ or 100 nm 14v to the level measured in ‘blank’ lysis buffer (n = 10 each). ***P < 0.001.
Intracellular CA
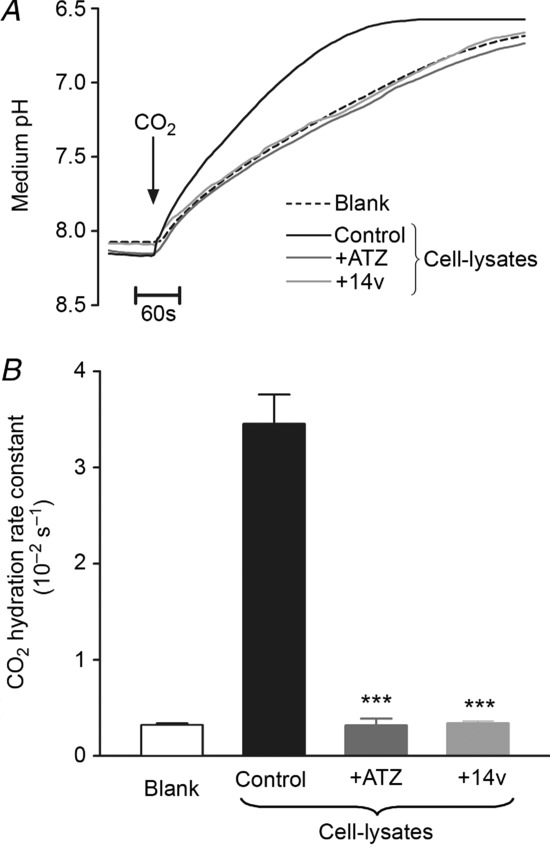
In intact cells, intracellular CA activity was assessed from intracellular cSNARF1 measurements of intracellular pH (pHi). Figure 2A shows that pHi of an isolated myocyte rapidly acidified during superfusion of a 5% CO2/HCO3−-buffered Tyrode solution (CO2 is membrane permeant and hydrates within the cytoplasmic compartment). This acidification was slowed by adding 100 μm ATZ (Fig. 2A and Bi). The suprathreshold dose, similar to that used previously (Schroeder et al. 2013), ensured adequate membrane permeation, thus leading to inhibition of both CAe and CAi. The drug also slowed the subsequent rapid alkalinization of pHi upon CO2 removal (Fig. 2A and Bii). In contrast, the membrane-impermeant CA inhibitor, 14v, had no effect on the pHi changes (Fig. 2B), consistent with its inability to enter the cell. Figure 2C pools data from several experiments, and shows that the intracellular CO2 hydration constant (kf) was reduced about 3–fold by ATZ, as reported previously (Leem & Vaughan-Jones, 1998; Schroeder et al. 2013), but was unaffected by 14v.
Figure 2.
A, time course of intracellular pH (pHi; AM-loaded cSNARF1 fluorescence) during rapid solution switching between Hepes-buffered solution (CO2 free) and 5% CO2/22 mm HCO3−-buffered solution at 37°C (solutions included 30 μm cariporide and 15 μm S0859 to block acid extrusion on Na+/H+ exchange and Na+–HCO3− co-transport, respectively). The initial rate of pHi change is proportional to the intracellular CA activity. Paired experiment in the presence of ATZ to block CA activity. B, details of pHi time courses measured upon switching from Hepes-buffered to CO2/HCO3−-buffered solution (i) or the reverse solution change (ii) under control conditions (black), in the presence of 100 μm ATZ (dark grey) or 100 nm 14v (light grey). C, CO2 hydration constant kf derived from the initial rate of pHi change upon CO2/HCO3− addition (filled bars) or removal (open bars) under control conditions (n = 15) or in the presence of ATZ (n = 15) or 14v (n = 15). *P < 0.05.
Extracellular CA
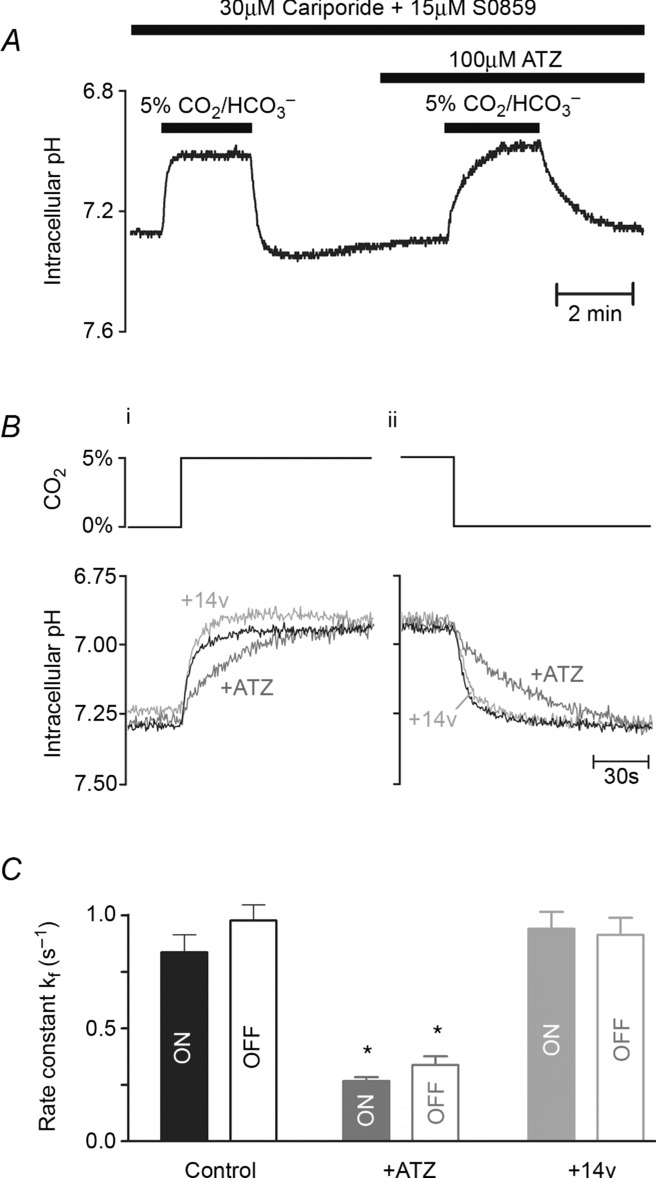
In isolated myocytes, an exofacial, membrane-tethered CA enzyme is also catalytically active. This is illustrated in Fig. 3A, which shows extracellular surface pH (pHe) recorded in the isolated ventricular myocyte, obtained with the membrane-tagging pH fluorophore, wheat germ agglutinin-conjugated fluorescein (WGA-fluorescein). Superfusion of 30 mm NH4Cl induced a large transient acidification of pHe. This is caused by rapid, passive permeation of NH3 molecules into the cell, thereby promoting dissociation of local extracellular NH4+ into NH3 and H+, which reduces pHe. In CO2/HCO3−-buffered superfusates, the H+ ions liberated at the extracellular surface are partly absorbed by extracellular carbonic buffer, as illustrated schematically in the inset diagram. Inhibiting CAe with ATZ would be expected to slow this buffering reaction thus rendering it less effective, and it is notable that in the presence of the drug, the transient fall of pHe became considerably larger. Subsequent removal of extracellular NH4Cl produced transient changes of pHe, this time in the alkaline direction, as NH3 molecules fluxing out of the cell combined locally with extracellular H+. The pHe transient was again enhanced in the presence of ATZ. In seven experiments, the amplitude of the pHe transients was increased in the presence of ATZ by 68 ± 9% and 77 ± 20% on addition and removal, respectively, of NH4Cl (Fig. 3B). Results are thus consistent with inhibition by ATZ of CA activity expressed at the extracellular surface of isolated myocytes.
Figure 3.
A, time course of extracellular surface pH (pHo; membrane-loaded wheat germ agglutinin-tagged fluorescein) during rapid and transient exposure to 30 mm NH4+/NH3 in 5% CO2/12 mm HCO3−-buffered solutions at 37°C (average of 9 cells). Exposure to NH4+/NH3 triggers surface acidification due to NH3 entry; subsequent withdrawal of NH4+/NH3 drives surface alkalinization. The amplitude of pHe changes depends on CO2/HCO3− buffering kinetics, catalysed by exofacial CA (CAe) activity. Paired experiment in ATZ to inhibit CA activity. B, CAe activity quantified as a ratio of pHe transient amplitude measured in ATZ and in control conditions for the addition (ON) and removal (OFF) of NH4+/NH3 (n = 9).
We conclude that CA is functionally active in the intracellular cytoplasmic and the extracellular exofacial domains of the myocyte.
NHE activity unaffected by CAi or CAe
Various protocols were used to assess NHE activity from pHi measurements in isolated myocytes. Acid extrusion was stimulated by prepulsing a cell with 20 mm NH4Cl to reduce pHi. When using Hepes-buffered superfusates, NHE activity exclusively mediates the subsequent pHi recovery to more alkaline levels (Fig. 4A), while a simultaneous acid efflux on NBC contributes when CO2/HCO3− superfusates are employed (Fig. 4B). Under these latter conditions, the component due to NBC can be revealed by selectively inhibiting NHE, e.g. with 30 μm DMA (Fig. 4C). The relative contributions of NHE and NBC to acid efflux in the presence of CO2/HCO3− buffer have been computed in Fig. 4D. At low pHi, acid extrusion rises steeply, with the larger efflux contributed by NHE. At less acidic levels of pHi, both NHE and NBC contribute about equally, as reported previously in ventricular myocytes (Leem et al. 1999). Estimates of acid extrusion into Hepes media, and DMA-sensitive extrusion into CO2/HCO3−-buffered media, were then used to compare NHE activity in the absence and presence of CO2/HCO3− buffer, as plotted in Fig. 4E. At any given pHi, the two fluxes are virtually identical suggesting that CA, which is likely to be functional only in the presence of CO2/HCO3−, does not directly catalyse the activity of NHE. This result conflicts with previous reports of NHE/CA interaction in heterologous expression systems (Li et al. 2002, 2006), where CO2/HCO3− buffering in combination with intracellular CA catalytic activity appeared to enhance NHE flux.
Figure 4.
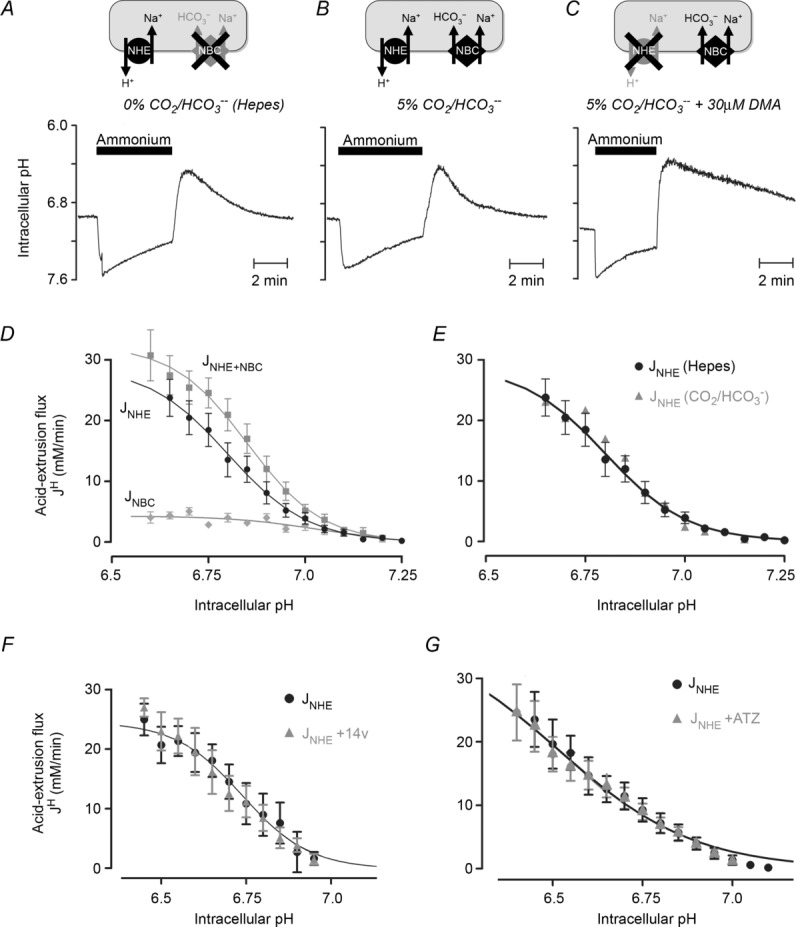
A, time course of intracellular pH during 20 mm ammonium prepulse solution manoeuvre. Recovery of pHi from acid load in the absence of CO2/HCO3− is mediated by Na+/H+ exchange (NHE). B, recovery of pHi in the presence of CO2/HCO3− is mediated by Na+/H+ exchange (NHE) and Na+–HCO3− co-transport (NBC). C, recovery of pHi in the presence of CO2/HCO3− and NHE inhibitor dimethylamiloride (DMA) is mediated by NBC only. D, pHi dependence of NHE-mediated acid efflux (JNHE; measured in Hepes buffer), NBC-mediated acid efflux (JNBC; measured in CO2/HCO3− buffer and DMA) and the sum of NHE and NBC fluxes (JNHE+NBC; measured in CO2/HCO3− buffer). Each data-point is the mean of 10–20 cells. E, flux analysis for NHE measured in Hepes, or calculated from the difference between JNHE+NBC and JNBC measured in the presence of 5% CO2/HCO3− buffer. F, flux analysis for NHE (calculated from difference between JNHE+NBC and JNBC) in the presence or absence of 100 nm 14v (10–15 cells). G, flux analysis for NHE, derived from pHi recovery in CO2/HCO3−-buffered Tyrode solution containing 15 μm S0859 (selective NBC inhibitor) in the presence or absence of 100 μm ATZ (10–15 cells).
The apparent lack of effect of CA on NHE in the native myocyte was investigated further by superfusing 14v, thus selectively removing CAe activity. In CO2/HCO3−-buffered media, the DMA-sensitive component of flux was unaffected by CAe inhibition (Fig. 4F), indicating that NHE activity was not significantly influenced by exofacial enzyme activity. The possible influence of CAi on NHE was also explored by first inhibiting NBC activity in the presence of CO2/HCO3− using the selective NBC antagonist S0859 (Ch'en et al. 2008), thus exposing NHE activity alone. Subsequent addition of membrane-permeant ATZ, which inhibits both CAe and CAi, again had no effect on NHE activity (Fig. 4G). It thus appears that NHE activity in an intact ventricular myocyte is not enhanced by the presence of CAi or CAe, even though these molecules can accelerate the hydration/dehydration kinetics of CO2/HCO3− buffer.
NBC activity not enhanced by CAe
The effect of CAe on NBC activity was investigated by testing the effect of 14v on NBC activity after NHE had been pharmacologically inhibited with DMA or cariporide. In these experiments the superfusates contained 45 mm K+ to depolarize the membrane potential and enhance NBC activity (the dominant NBC isoform in heart is electrogenic (1Na+–2HCO3−) and thus voltage sensitive; Aiello et al. 1998; Yamamoto et al. 2005). As shown in the specimen records of Fig. 5A, 14v had no effect on the time course of NBC-mediated pHi recovery from an acid load (induced by ammonium prepulse). As a result, the computed pHi dependence of acid extrusion via NBC was unaffected (Fig. 5B). The result confirms that NBC activity in superfused myocytes is not catalysed by exofacial CA activity.
Figure 5.
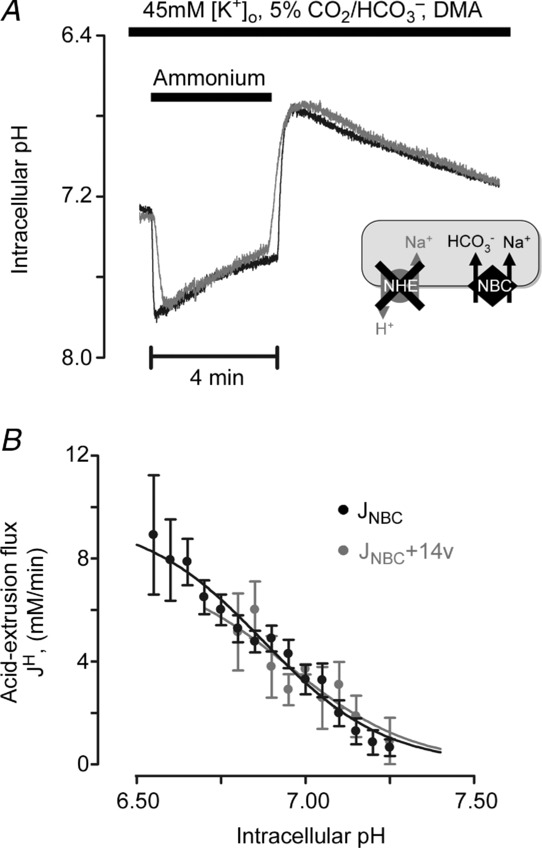
A, 20 mm ammonium prepulse solution manoeuvre in the presence of CO2/HCO3−, 30 μm DMA (NHE inhibitor) and 45 mm K+ (to increase electrochemical gradient for outward NBC current). Superimposed traces of NBC-mediated recovery of pHi in the presence (grey) and absence (black) of 100 nm 14v (membrane-impermeant inhibitor). B, pHi dependence of NBC flux (10–15 cells per data point).
NBC activity enhanced catalytically by CAi
In contrast to the lack of effect of CAe inhibition, there was a significant slowing of NBC activity when CAi was inhibited. Figure 6A shows specimen records where addition of ATZ (or its analogue, ETZ) slowed NBC-mediated pHi recovery from an intracellular acid load. In Fig. 6B, results have been averaged for cells superfused with Tyrode solution containing 4.5 mm K+ (Fig. 6B inset) or 45 mm K+ (to enhance NBC activity; Fig. 6B main panel). With both [K+]o levels, generic NBC flux was halved in the presence of ATZ, although statistical significance was more evident over the pHi range tested when NBC fluxes had initially been enhanced in high [K+]o.
Figure 6.
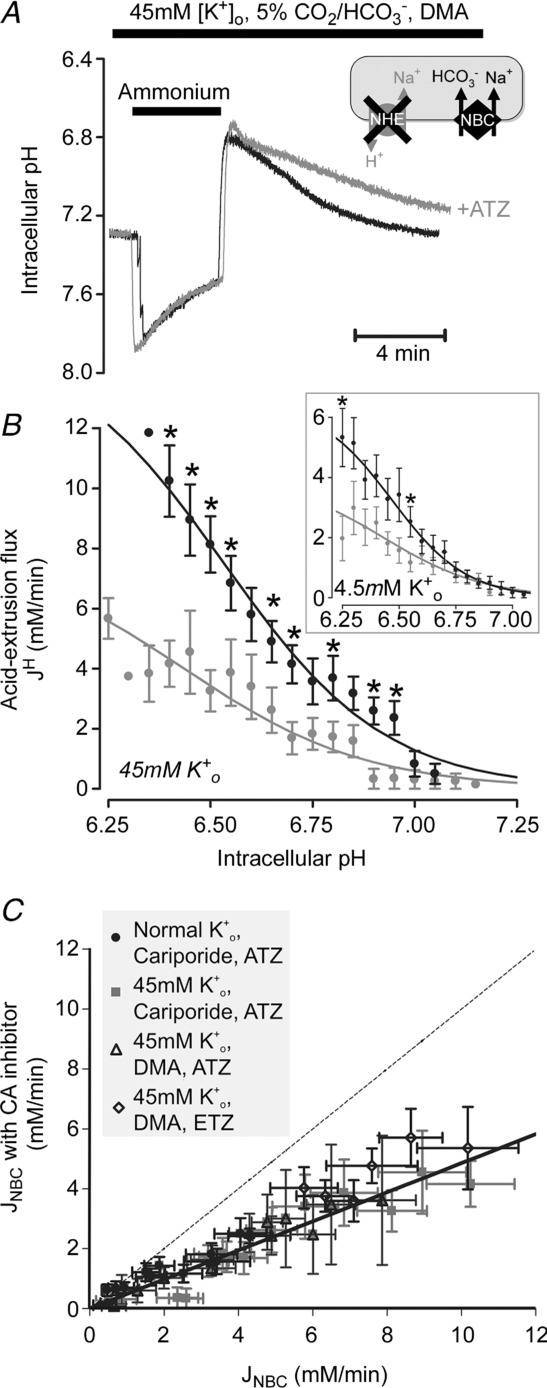
A, 20 mm ammonium prepulse solution manoeuvre in the presence of CO2/HCO3−, 30 μm DMA and 45 mm K+. Superimposed traces of NBC-mediated recovery of pHi in the presence (grey) and absence (black) of 100 μm ATZ. B, pHi dependence of NBC flux. ATZ caused significant reduction (51 ± 2%) of NBC flux (n = 3–12, *P < 0.05). Inset: pHi dependence of NBC flux (n = 8–26; *P < 0.05). C, NBC-mediated acid efflux in the presence versus absence of CA inhibitor (100 μm ATZ or ethoxyzolamide, ETZ) at matching pHi, measured in 4.5 mm or 45 mm K+, in the presence of NHE inhibitors (30 μm DMA or cariporide). Identity shown as dotted line.
Figure 6C plots NBC flux measured at a given pHi in the presence of ATZ or ETZ versus that measured in the absence of CA inhibition. Over almost all of the flux range measured, flux values fall well below the line of identity (dashed line, expected if flux is identical with and without ATZ or ETZ), indicating significant NBC slowing with CA inhibition. Given that selective CAe inhibition exerted no effect, the slowing with membrane-permeant CA inhibitors is likely to be secondary to inhibition of CAi.
NBC slowing by ATZ was confirmed by measuring [Na+]i changes associated with the activity of the transporter, as shown in Fig. 7. In this set of experiments, NBC was stimulated by a pHi reduction induced with superfusion of 80 mm sodium acetate (salt of a membrane-permeant weak acid; cariporide was also added to inhibit NHE). During the activation of NBC, [Na+]i, recorded with intracellular SBFI (Na+ fluorophore, see Methods) increased. This [Na+]i rise did not occur in the absence of CO2/HCO3− buffer (n = 3, Hepes-buffered superfusates containing cariporide, not shown; see also Yamamoto et al. 2005), confirming that it was secondary to NBC activity. Addition of ATZ, which slows NBC-mediated pHi recovery (Fig. 6), reduced the rate of rise of [Na+]i by ∼40%, confirming attenuation of NBC flux.
Figure 7.
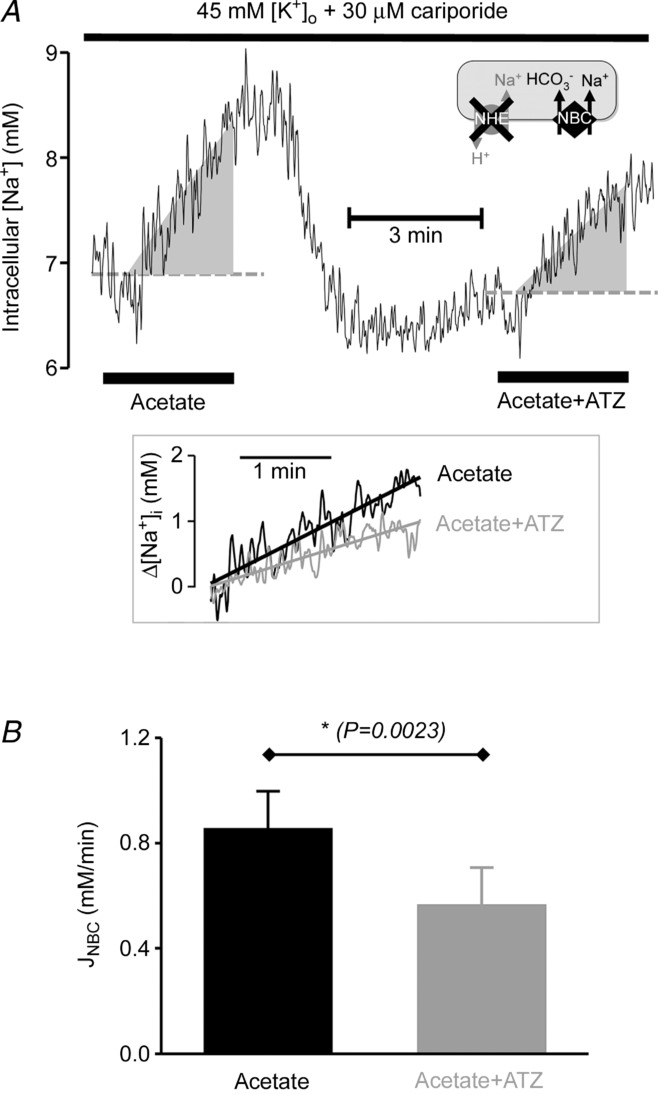
A, time course (average of 9 cells) of intracellular [Na+] ([Na+]i; AM-loaded SBFI fluorescence) calibrated using Na+ ionophores and pHi-corrected using pHi template recorded in separate experiments (not shown; see Methods). Solutions contained 5% CO2/22 mm HCO3− buffer, 45 mm K+ and 30 μm cariporide (NHE inhibitor). Na+ influx on NBC was stimulated by exposure to 80 mm sodium acetate (which rapidly reduces pHi). Paired experiment performed in presence of CA inhibitor ATZ (100 μm). B, rate of intracellular [Na+]i rise, normalized to the starting level, gives a measure of NBC-mediated Na+ influx (JNBC). Initial rate was slower in ATZ (100 μm).
We conclude that NBC activity is enhanced by the catalytic activity of intracellular CA while NHE activity is not.
Discussion
The present work provides the first demonstration in a native cell that CA activity selectively facilitates flux among pH regulatory transporters at the surface membrane. In the ventricular myocyte, selective facilitation relies exclusively upon intracellular CA, which enhances NBC activity, while exerting no effect on NHE1. In contrast, exofacial CA has no catalytic effect on either transporter. These results differ from previous work in heterologous expression systems where the flux activity of NHE1, NBCe1 and NBCn1, expressed in oocytes or various cell lines, has been suggested to be enhanced by the co-expression of either cytoplasmic CAII or exofacial CAIV (Li et al. 2002, 2006; Alvarez et al. 2003; Loiselle et al. 2004; Becker & Deitmer, 2007; Schueler et al. 2011), CA proteins that are endogenously expressed in the myocardium (Scheibe et al. 2006). It is notable that the spatial distribution of NBC (i.e. NBCe1 and NBCn1) and NHE1 protein is not identical at the surface membrane of the ventricular myocyte (Garciarena et al. 2013). NHE1 is expressed at intercalated disks and lateral sarcolemma, while NBC is particularly prominent in the transverse tubules (t-tubules). It is possible, therefore, that the facilitatory influence of CAi activity on NBC flux is dependent on the local geometry at the transporter's expression site (this point is discussed later). Irrespective of the mechanism, however, by facilitating NBC activity in t-tubules, CAi services pHi regulation close to the machinery of excitation–contraction coupling, including L-type Ca2+ channels, ryanodine receptors, local sarcoplasmic reticulum Ca2+-ATPase pumps and Na+/Ca2+ exchangers, all of which are known to be pH sensitive.
No role for exofacial CA in acid extrusion?
Several membrane-tethered CA isoforms with active sites oriented exofacially are expressed in mammalian ventricular myocytes. These include CAIV and CAXIV at the sarcolemma, with the additional possibility of CAIX in transverse tubules (Scheibe et al. 2006). The lack of a detectable influence of exofacial CA activity on H+ or HCO3− transport in the present work is perhaps surprising, given that a stimulatory role has previously been proposed (Sterling et al. 2002; Alvarez et al. 2003), the enzymes collectively acting with pHi regulatory proteins in what has been defined as a ‘transport metabolon’. This effect could represent the activity of a true metabolon, in which one or more exofacial CA molecules bind to the transporter to form a multimeric protein complex that displays enhanced H+-equivalent flux, or it could be a ‘functional metabolon’ that lacks direct protein–protein binding, but where a local sharing of common extracellular chemical substrates (e.g. H+ or HCO3− ions) still enhances overall flux activity. The present work does not address the issue of protein–protein interactions, a phenomenon that is currently disputed (Boron, 2010; Al-Samir et al. 2013). The lack of enhanced NBC or NHE flux in the presence of CAe suggests, however, that even a functional transport metabolon need not automatically result from co-expression of transporters and CA isoforms in the cell membrane. While this may indicate that the protein partners are not sufficiently co-localized in the native cell to result in shared functional activity, an additional possibility, as discussed below, is that experiments on isolated cells under superfusion may not reveal the full functional role of exofacial CA. Whatever the explanation, the present results show that there is no tight, obligatory link between exofacial CA expression and H+ or HCO3− transporter activity in ventricular myocytes.
One explanation for the absence of CAe interaction with ventricular NHE/NBC function relates to myocyte superfusion, an experimental procedure used in most cellular epifluorescence and imaging experiments. While superfusion provides controlled manipulation of the extracellular compartment, it also maintains the solution at the cell membrane close to CO2/HCO3− equilibrium by continuously replacing it with freshly equilibrated Tyrode solution. Under these conditions, net CAe activity, which accelerates carbonic buffer equilibration, will be close to zero, and thus unable to play a catalytic role in H+-equivalent transport (a similar phenomenon has been reported in cultured tumour cell lines; Swietach et al. 2008, 2009). The extracellular unstirred layer in superfused myocytes may therefore be sufficiently small and well coupled diffusively to the bulk solution such that local changes in extracellular pH, CO2 partial pressure and [HCO3−] are minimized during membrane acid/base transport. Although local pHe changes do occur during high transmembrane H+-equivalent fluxes, and are regulated by exofacial CA activity (as shown in Fig. 3), the fact that CAe inhibition does not influence overall NBC or NHE activity in these cells suggests that the changes are sufficiently small as to exert no significant feedback effect on transporter kinetics. This result does not exclude the possibility that important CAe modulation of H+-equivalent transport might occur in multicellular myocardium, or even experimentally in clusters of NBC-expressing cells subjected to less efficient superfusion, where diffusive coupling in local extracellular spaces could be weaker. The key point is that potential coupling between CAe expression and H+-equivalent flux in a given cell is not obligatory or invariant; any functional link will depend on the geometry of the local extracellular space and its diffusive linkage to the bulk phase.
Catalytic role for intracellular CA in acid extrusion by NBC
While CAe inhibition had no effect on acid extrusion through NBC, the membrane-permeant CA inhibitors ATZ and ETZ reduced NBC flux by up to 50% (Fig. 6), indicating a clear stimulatory role for the catalytic activity of intracellular CA (CAi). The native NBC isoform stimulated by CAi cannot be identified in the present work. Both electrogenic (NBCe1) and electroneutral (NBCn1) isoforms are expressed in rat ventricular myocytes (De Giusti et al. 2011; Garciarena et al. 2013; NBCe2 transcripts have yet to be confirmed in rat myocytes), with electrogenic acid extrusion being a large flux component (Yamamoto et al. 2005). It is notable, therefore, that most previous work has found that the activity of both isoforms, when expressed heterologously in oocytes or cell lines appears enhanced by intracellular CA activity (Gross et al. 2002; Loiselle et al. 2004; Becker & Deitmer, 2007), although one report indicates no effect (Lu et al. 2006). The possibility that membrane-permeant sulphonamides interfere directly with NBC transport, rather than via inhibition of CA activity, is unlikely as ETZ had no effect on NBCe1 when expressed in oocytes in the absence of CA (Lu et al. 2006; Becker & Deitmer, 2007). The specific intracellular CA isoform involved in facilitating NBC flux is also not identified in the present work, but contributions are likely from cytosolic CAII as well as the membrane-tethered intracellular CAs identified in these cells, such as CAIV, XIV and IX, provided their active sites are oriented towards the cytoplasmic compartment (Scheibe et al. 2006; Schroeder et al. 2013).
A striking observation is that, while ATZ and ETZ slow NBC flux in the ventricular myocyte, they have no effect on NHE1, even in the presence of CO2/HCO3− buffer (Fig. 4). This result rules out the possibility that the slowing of NBC-mediated pHi recovery was caused by a general slowing of cytoplasmic CO2/HCO3− buffering (subsequent to the inhibition of CAi), as this would have affected pHi changes mediated by both types of transporter. Indeed mathematical modelling predicts no significant effect of slowing bulk cytoplasmic CO2/HCO3− buffering with ATZ on the time course of NHE-mediated pHi recovery (see Supplemental Fig. S1 available online). The slowing of NBC-mediated recovery must therefore represent a genuine reduction in membrane HCO3− transport rate. The parallel reduction in the rate of rise of [Na+]i associated with NBC activity (Fig. 7) provides independent corroboration of NBC inhibition, as do the results, presented below, of computational NBC modelling. The differential effect of CAi on our estimates of NBC and NHE flux is therefore likely to represent a selective targeting by the enzyme of NBC's transport reactions. In the ventricular myocyte, CAi and NBC appear to be partners in a ‘functional’ transport unit.
Testing models of NBC facilitation by intracellular CA
Standard bicarbonate metabolon model does not predict flux facilitation
The classical explanation of CAi facilitation of acid extrusion on NBC (largely by cytoplasmic CAII) is that the enzyme accelerates the reaction between H+ ions from bulk cytoplasm and HCO3− ions unbinding from NBC, resulting in the formation of CO2 that escapes across the sarcolemma. This chemical reaction can be modelled using a simple mathematical function for pHi-sensitive NBC transport (where activity rises with a fall in pHi), in conjunction with the kinetics of HCO3− protonation in the presence and absence of CA catalysis (measured experimentally, as shown in Figs 1 and 2). The model is presented schematically at the top of Fig. 8A, and full details are given in the Appendix. It predicts that even in the absence of catalytic CAi activity, H+ titration of bulk intracellular HCO3− proceeds sufficiently fast for it not to be rate-limiting for the overall NBC-driven acid-extrusion process, and hence will not significantly affect the overall rate of pHi recovery. With spontaneous kinetics (rate constant kr = 0.22 s−1 μm−1; Fig. 2C), constant 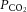 (5%), and a plentiful delivery of H+ ions from intrinsic buffers, the uncatalysed rate of HCO3− protonation in cytoplasm (kr × [H+] × [HCO3−]) will be ∼13 mm min−1, assuming the CO2/HCO3− buffer remains close to equilibrium. This rate would be higher during the stimulation of NBC flux if intracellular [HCO3−] were to rise near the transporter, locally driving cytoplasmic CO2/HCO3− buffer out of equilibrium. These spontaneous rates are in excess of the maximum measured flux on NBC (<12 mm min−1 in Fig. 6), indicating that the catalysis of bulk CO2/HCO3− buffering by CAi is not necessary to support transporter flux. Results of the computational modelling are shown in the main panel of Fig. 8A, which plots predicted NBC flux as a function of pHi, with and without CAi activity (see also Supplemental Fig. S1). Both predictions are virtually coincident, indicating that this type of model fails to simulate the experimentally observed acceleration by CAi of NBC flux. The classical explanation for a functional transport metabolon is thus not sufficient to account for CA facilitation of NBC.
(5%), and a plentiful delivery of H+ ions from intrinsic buffers, the uncatalysed rate of HCO3− protonation in cytoplasm (kr × [H+] × [HCO3−]) will be ∼13 mm min−1, assuming the CO2/HCO3− buffer remains close to equilibrium. This rate would be higher during the stimulation of NBC flux if intracellular [HCO3−] were to rise near the transporter, locally driving cytoplasmic CO2/HCO3− buffer out of equilibrium. These spontaneous rates are in excess of the maximum measured flux on NBC (<12 mm min−1 in Fig. 6), indicating that the catalysis of bulk CO2/HCO3− buffering by CAi is not necessary to support transporter flux. Results of the computational modelling are shown in the main panel of Fig. 8A, which plots predicted NBC flux as a function of pHi, with and without CAi activity (see also Supplemental Fig. S1). Both predictions are virtually coincident, indicating that this type of model fails to simulate the experimentally observed acceleration by CAi of NBC flux. The classical explanation for a functional transport metabolon is thus not sufficient to account for CA facilitation of NBC.
Figure 8.

A, standard metabolon model. NBC imports highly mobile HCO3− anion into the cytoplasm, where it reacts with H+ ions (supplied by intracellular buffers) to form CO2. This reaction can be catalysed by intracellular CA activity. NBC flux simulated by the model is unaffected by removing CA catalysis from the model (to simulate measured effect of ATZ). B, restricted H+ ion delivery model. HCO3− anion, imported by NBC, reacts with H+ ion in a diffusionally restricted space (reaction N). Additionally, NBC activity is regulated allosterically by the pH in this restricted domain (rather than by the pH of bulk cytoplasm). Delivery of H+ ions directly from cytoplasm is rate limiting (e.g. because of binding to immobile titratable sites on proteins; quantified in terms of exchange rate constant kslow) but can be increased by CA activity (reaction C) providing H+ ions from catalytic CO2 hydration near the diffusionally restricted space. Best fit to data from Fig. 6B (45 mm K+ and 4.5 mm K+ in inset) for kslow = 285 s−1. C, simulated time course of pHi recovery (dotted lines) following an ammonium prepulse in the presence (grey) and absence (black) of ATZ, superimposed on experimental time courses (continuous traces). D, plot of NBC flux in the absence of CA inhibitors vs. NBC flux in the presence of ATZ (data points extracted from Fig. 6C), superimposed with model prediction (black line) and identity line (grey dashed line).
Restricted H+ ion delivery model predicts NBC facilitation by CAi
If, because of diffusional restrictions, local H+ ion delivery to the cytoplasmic face of NBC were rate-limiting, then the local HCO3− protonation rate would be slowed (i.e. when kr × [H+] × [HCO3−] < 13 mm min−1), which would reduce HCO3− ion unbinding from the transporter and hence its kinetic turnover rate. If, in addition, the activity of NBC were regulated allosterically by a sensor that probed pH in such a diffusionally restricted domain, then NBC flux would be reduced further, because the protein would sense a pH that is locally more alkaline than in the bulk cytoplasm. Under these conditions, CAi activity could facilitate NBC flux, by catalysing the local provision of H+ ions. The slowing of NBC observed with ATZ or ETZ is consistent with local H+ delivery arising from CAi's catalytic activity (i.e. from CO2 hydration), as illustrated schematically at the top of Fig. 8B. In this model, H+ ions are delivered by CAi, but not by H+-carrying intrinsic mobile buffers, such as histidyl-dipeptides (Vaughan-Jones et al. 2002, 2009). These latter buffers are required, however, to supply the CAi protein with H+ ions to regenerate CO2. Thus, the final H+ donation of the cytoplasmic H+ shuttle to NBC is made catalytically from a CA molecule. The donated H+ ion then combines spontaneously with HCO3− within the NBC protein, resulting in the production of CO2 that escapes across the sarcolemma. On such a model, the H+ donation step from CAi can become rate-limiting for NBC activity, as illustrated in the computational results shown in Fig. 8B. If CA catalysis is set to zero in the model (thus leaving only the slow rate of H+ generation from spontaneous CO2 hydration), then the pHi dependence of HCO3− influx on NBC (plotted in Fig. 8B as H+-equivalent extrusion) is shifted to lower flux levels (as also observed experimentally).
The kinetic model shown in Fig. 8B successfully predicts the time course of NBC-mediated pHi recovery, and its slowing upon CA inhibition with ATZ or ETZ (Fig. 8C). The predicted reduction of NBC flux also matches experimental values over the whole flux range tested (Fig. 8D). The facilitation of NBC activity by intracellular CA is thus consistent with a functional transport unit, in which the enzyme catalytically donates H+ ions to the transport molecule rather than catalysing the subsequent HCO3− titration reaction.
Free H+ ion diffusion in cytoplasm is negligible, because of high buffering by poorly mobile proteins (Vaughan-Jones et al. 2002; Swietach et al. 2007). Simple diffusion therefore cannot furnish an adequate H+ ion delivery to NBC. Small mobile buffers other than CA-catalysed carbonic buffer could potentially deliver the H+ ions. But, in this case, NBC activity would be unaffected by CA inhibition, which is contrary to experimental observation. The ability of CAi to facilitate NBC activity therefore requires that local H+ ion delivery from intrinsic buffers be restricted. This might occur if the CA molecule acted as a gate to the NBC protein (see schematic diagram in Fig. 8B). For example, cytoskeletal geometry around the NBC and its local CA molecule might hamper intrinsic buffer access. As mentioned previously, NBC expression in the ventricular myocyte is prominent in the transverse tubules, close to L-type Ca2+ channels that communicate with the intracellular dyadic space (Garciarena et al. 2013). This is a crowded nanoscopic domain, ringed by structural as well as other Ca2+-handling proteins involved in excitation–contraction coupling. Alternatively, a physical barrier in the NBC transporter itself might restrict intrinsic buffer access, but permit a juxtaposition with CAi, thereby allowing the enzyme to donate H+ ions, while still allowing influx and efflux of CO2. Whatever the local geometry, following inhibition of CAi activity with ATZ, spontaneous CO2 hydration would still appear to generate a supply of H+ ions that can fuel the lower uncatalysed rate of NBC activity (cf. Fig. 6 with Fig. 8B). The question of whether CA needs to be bound to NBC, as suggested by the proponents of a structural transport metabolon (Sterling et al. 2001, 2002; Morgan et al. 2007; Casey et al. 2009; Svastova et al. 2012) cannot be answered in the present work. A recent analysis, however, of CA-catalysed HCO3− transport in the red blood cell (by the Band-3 Cl−/HCO3− exchanger) suggests that a physical binding is unlikely (Al-Samir et al. 2013).
Restricted H+-ion diffusion model also predicts CAi facilitation of a carbonate transporter
It should be emphasized that other kinetic models also predict CAi facilitation of acid extrusion on NBC but, again, only when the enzyme catalyses H+ donation to the transporter, and contributions from intrinsic mobile buffers are greatly restricted (implemented in terms of a slow diffusive H+ ion exchange constant between bulk cytoplasm and the NBC transporter). For example, one alternative model (Boron, 2010) is that NBC may import carbonate (CO32−) rather than HCO3− anions, when operating in acid-extrusion mode (as argued experimentally for NBCe1; Zhu et al. 2013). In this model, CAi activity could donate a H+ ion for the first protonation step, in order to release HCO3− from the transporter and into the bulk cytoplasmic compartment, or donate H+ ions for both protonation steps, in order to release CO2 from the transporter. Such H+ donation, if catalysed by CAi, could again become rate-limiting for NBC activity. As a result, mathematical models of CO32− influx would predict enhancement of flux by CAi, providing the enzyme exclusively furnished the H+ donation. The predicted inhibitory effect of ATZ would be greater if CAi were responsible for providing both H+ ions per CO32− transport cycle, rather than supplying the first protonation only. Indeed, any combination of H+ and HCO3− membrane ion transport that encodes net acid efflux, can potentially be enhanced by CAi, as long as the enzyme donates H+ ions during transporter activity. Facilitation in all these models, however, still requires (i) ready H+ ion access to the transporter from active CAi, which implies relatively close proximity of the two proteins, and (ii) restricted H+ ion access to the transporter from intrinsic buffer shuttles, which would otherwise render CAi activity redundant.
Intracellular CA does not facilitate NHE1
The lack of facilitation of NHE1 by CO2/HCO3− or CAi in ventricular myocytes suggests that intracellular H+ ions delivered to this transporter are supplied from intrinsic (non-carbonic) buffers. As mentioned above, intrinsic H+ shuttling within cytoplasm is mediated by cytoplasmic mobile buffers, such as the histidyl dipeptides, carnosine, anserine and homocarnosine, which are collectively expressed at high concentration (∼15 mm) in ventricular myocytes (O'Dowd et al. 1988; Vaughan-Jones et al. 2002; Swietach et al. 2013). This H+ shuttle has a high diffusive capacity. Previous work in these cells has demonstrated that the shuttle can support longitudinal H+ fluxes of 50–100 mm min−1, while the CA-catalysed carbonic shuttle starts to saturate at rates above ∼8 mm min−1 (Swietach et al. 2005). These intracellular diffusive H+ fluxes should be compared with membrane transport fluxes stimulated at low pHi on NHE1 (up to 25 mm min−1; Fig. 4D), and NBC (up to ∼12 mm min−1; Fig. 4D). The ATZ insensitivity and high capacity of acid extrusion through NHE1 therefore suggests that its H+ ion transport is supplied principally from the intrinsic dipeptide shuttle, while ATZ/ETZ sensitivity suggests that the terminal end, at least, of the cytoplasmic H+ shuttle to NBC is composed of a CA-catalysed delivery of H+ ions. These differing arrangements are illustrated schematically in Fig. 9. H+ ions from intrinsic mobile buffers thus appear to have ready access to the cytoplasmic face of NHE1 proteins. While this does not exclude delivery of CAi-derived H+ ions, the lack of inhibition of NHE1 flux by ATZ suggests the latter route must be small. The mechanism determining intrinsic buffer access to NHE1 but not NBCs is unknown. It may, for example, be related to the different spatial domains in which the two types of transporter are expressed in the ventricular myocyte.
Figure 9.
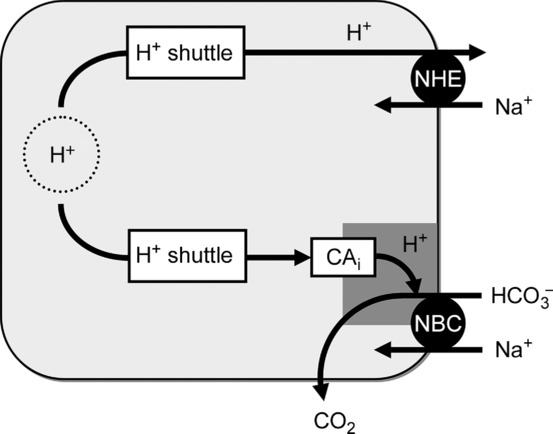
Schematic representation of proposed model. NBC: intracellular CA activity delivers H+ ions to a diffusionally restricted domain at the cytoplasmic surface of NBC protein, the H+ ions being necessary for the uncatalysed titration of its HCO3− anion cargo. Reaction releases CO2, which diffuses freely across the myocyte and its surface membrane. The relevant CAi is itself supplied with H+ ions derived from the bulk cytoplasmic H+ ion shuttle (composed mainly of mobile histidyl dipeptides). NHE: this transporter is not located in a diffusionally restricted space and can thus be supplied directly by H+ ions derived from the bulk cytoplasmic H+ ion shuttle.
Previous reports indicate that acid extrusion by NHE1 proteins, when over-expressed heterologously in Xenopus oocytes, is facilitated by CO2/HCO3− (Li et al. 2002, 2006), provided intracellular CA (largely CAII) is also functionally active. Given the lack of CAi facilitation in wild-type ventricular myocytes, this result is initially puzzling. One possibility is that intrinsic H+ ion mobility in oocytes is much lower than in myocytes, because of lower levels of diffusible intrinsic buffer. In the absence of an efficient intrinsic buffer shuttle, NHE transport will become more reliant on servicing by the intracellular carbonic buffer shuttle, and will thus be expected to display a more prominent facilitation by CAi activity. This reliance of membrane flux on cytoplasmic H+ mobility may be exacerbated by the large size of the oocyte, resulting in unusually long diffusion delays for H+ ions from the cytoplasmic core to NHE transporters at the periphery, delays that may be lengthened further by cytoplasmic CA inhibition. Whatever the cause of CAi facilitation of NHE1 in oocytes, the present work shows clearly that CAi activity is not necessary for physiological NHE1 function in the native ventricular myocyte.
Conclusions
We conclude that, in the native ventricular myocyte, intracellular CA enzymes form a functional transport unit with NBC, thereby enhancing its flux. Our results disagree in important respects with several previous reports of a HCO3− transport metabolon, based on heterologous expression systems. Although catalytic CAi activity appears to enhance NBC flux in expression systems and wild-type cardiac cells, the native cardiac transporter is insensitive to extracellular (exofacial) CA activity. In addition, native cardiac NHE1 activity is entirely CA insensitive. The coupling of CAi to NBC flux in myocytes does not necessarily imply physical binding between the two molecules (a classical transport metabolon), a non-covalent juxtaposition may suffice, with the proteins interacting functionally. A novel finding is that CAi facilitation of NBC is quantitatively consistent with the enzyme catalysing the delivery of H+ ions to the cytoplasmic face of the transporter, rather than the subsequent protonation of the HCO3− substrate. In contrast, the lack of modulation of NHE1 flux by CAi suggests that, in the ventricular cell, this particular transporter is adequately supplied with H+ ions via intrinsic mobile buffers. CAi activity therefore plays a key role in regulating native NBC but not NHE activity in cardiac cells (Fig. 9). By spatially trading H+ ions, the myocyte's intracellular CA enzymes behave as proton-coupling proteins, linking pHi to membrane HCO3− transport, in order to stabilize [H+]i levels and thus physiological function in the heart.
Key points
Carbonic anhydrase enzymes are said to enhance membrane H+ and HCO3− transport, an idea tested almost exclusively in heterologous cell-expression systems.
In ventricular myocytes, inhibiting cytoplasmic enzyme activity slows membrane acid extrusion (HCO3− influx) on Na+–HCO3− co-transporters, with no effect on Na+/H+ exchangers. Inhibiting exofacial enzyme activity has no effect on either transporter.
Mathematical modelling simulates the influence of carbonic anhydrase on Na+–HCO3− co-transport, provided the enzyme catalytically delivers H+ ions to the transporter (across a cytoplasmic nanodomain that is poorly accessible to intrinsic buffers), rather than catalysing protonation of the imported bicarbonate. Intrinsic cytoplasmic mobile buffers appear to deliver H+ to Na+/H+ exchangers, thus obviating the need for carbonic anhydrase.
Conclusion: in native cardiac cells, intracellular carbonic anhydrase molecules partner Na+–HCO3− co-transporters but not Na+/H+ exchangers, to enhance flux activity. The enzyme thus plays a key role in supporting bicarbonate transport and pH control in the heart.
Acknowledgments
None.
Glossary
- ATZ
acetazolamide
- CA
carbonic anhydrase
- CAi
intracellular CA
- CAe
extracellular exofacial CA
- DMA
dimethylamiloride
- ETZ
ethoxyzolamide
- NBC
Na+–HCO3− cotransport
- NHE
Na+/H+ exchange
- WGA-fluorescein
wheat germ agglutinin-conjugated fluorescein
Appendix
Appendix
A mathematical model was developed to explore the effect of carbonic anhydrase (CA) catalytic activity on pHi regulation by NBC flux (JNBC). The transporter flux was modelled using the Hill equation (Yamamoto et al. 2005):
where [H+]c represents cytoplasmic [H+]. In comparison to JNBC, CO2 fluxes are not rate-limiting because of rapid membrane permeability and diffusivity across all cell compartments. To simplify the equations, CO2 levels were clamped (d[CO2]/dt = 0) at 1.2 mm. Table 1 lists the parameters of the model.
Variables used in the mathematical models (see Appendix)
| Parameter | Symbol | Value | Reference |
|---|---|---|---|
| Intrinsic buffering capacity | βint | (a function of pHi) | Zaniboni et al. (2003) |
| Myocyte length | L | 120 μm | Present work |
| Myocyte radius | R | 10 μm | Present work |
| NBC maximal flux (basal) | Vmax | 6.5 mm min−1 | Fig. 6B |
| NBC maximal flux (stimulated) | Vmax | 22 mm min−1 | Fig. 6B |
| NBC internal H+ affinity constant | Km | 10−6.5 m | Fig. 6B |
| NBC Hill co-operativity for internal H+ ions | n | 2 (unitless) | Fig. 6B |
| Cytoplasm-averaged carbonic anhydrase activity | CA | 3 (unitless) | Fig. 2C |
| Spontaneous CO2 hydration constant | kf | 0.18 s−1 | Fig. 2C |
| Spontaneous HCO3− dehydration constant | kr | 0.22 μm−1 s−1 | Fig. 2C |
| CO2 acid-dissociation constant | K | 10−6.1 m | Leem & Vaughan-Jones (1998) |
| Rapid transfer constant between compartments | krapid | >104 s−1 | Best fit |
| Slow transfer constant between compartments | kslow | 250 s−1 | Best fit |
Standard metabolon model
The first model was based on the scheme shown in Fig. 8A. The equations for cytoplasmic [HCO3−] ([HCO3−]c) and [H+]c were:
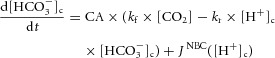 |
where CA is a measure of carbonic anhydrase activity and βint is the intrinsic buffering capacity in units of mm (pH unit)–1 (converted to dimensionless buffer ratio by dividing by 2303 × [H+]).
H+ ion-limited model
In the second model (Fig. 8B), additional equations were included to simulate the behaviour of [HCO3−] and [H+] in a sub-membrane diffusionally restricted (unstirred) domain ([HCO3−]u, [H+]u) and a transporter-associated CA domain ([HCO3−]ca, [H+]ca):
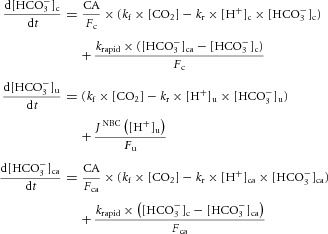 |
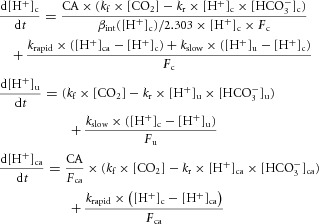 |
In this model, flux JNBC is a function of [H+] in the unstirred layer. The volume fractions of the cytoplasmic, unstirred and carbonic anhydrase domains were given by constants Fc, Fu and Fca. The bulk cytoplasm was assumed to be, by far, the largest (Fc ≈ 1). Diffusive transfer of ions between compartments was modelled with a rapid or slow (rate-limiting) exchange rate constant, krapid or kslow, respectively. H+ ions exchanged rapidly between the CA and unstirred domains, but only slowly between the cytoplasmic and unstirred domains. The experimentally observed CA dependence of NBC activity might be explained by allowing a direct H+ ion delivery route from the CA catalytic site to the transporter protein. The basis for this H+-shuttling mechanism may be the recently proposed H+-collecting antenna in CAII (Becker et al. 2011). To simulate this privileged H+-shuttling pathway, diffusive exchange of H+ ions was allowed to be fast between CA and NBC, but negligible between CA and the bulk cytoplasm. HCO3− ions were permitted to exchange freely between the cytoplasmic and CA domains, but not between the unstirred and cytoplasmic domains (a condition that is expected if HCO3− ions reacted to form CO2 faster than they would unbind from the transporter). To simulate pharmacological CA inhibition, reactions in the CA domain were set to zero and hydration kinetics in the cytoplasmic domain were set to spontaneous levels (NB: reactions in the unstirred domain proceed at spontaneous kinetics irrespective of the presence or absence of ATZ). The inhibitory effect of ATZ or ETZ on NBC flux could be simulated mathematically by setting kslow to a rate-limiting value (relative to krapid). By selecting an adequately large krapid, the transfer of H+ and HCO3− between the cytoplasmic and CA domains becomes non-rate-limiting. The results of the simulation are therefore not sensitive to the values for Fca and Fu, provided these are small. The best fit to the experimental data (i.e. a ∼50% decrease in NBC flux in the presence of ATZ) could be simulated for kslow = 285 s−1 (krapid > 105 s−1; Fu > 10−6, Fca > 10−6). For comparison, the Ca2+ diffusion exchange constant between the cytoplasm and subsarcolemmal fuzzy space is 2500 s−1 (Snyder et al. 2000). The value of kslow is related to H+ ion diffusivity, DH, and diffusion distance δ between the cytoplasmic and unstirred domains:
H+ ion diffusivity in water is 1.2 × 104 μm2 s–1, and this value yields a distance δ = 7 μm, which is unrealistic for the geometry of the myocyte. However, H+ ion diffusivity is reduced due to binding with fixed titratable sites (such as those presented by proteins). If the ratio of bound to free H+ ions is B, diffusivity will be reduced by a factor of (B + 1), assuming that none of the buffers is mobile in the unstirred domain. A further reduction is expected due to volume exclusion by macromolecules. For a volume exclusion of 50% and an intracellular buffering capacity of 30 mm (pH unit)–1 (B = 2 × 105), distance δ is predicted to be 10 nm, i.e. comparable to the width of a lipid bilayer.
The model above assumes that the transported substrate is HCO3−. If, instead, the transporter protein handled CO32− ions, as proposed recently (Boron, 2010; Zhu et al. 2013), then an additional protonation step would be required in the transport cycle. The diffusive barrier to cytoplasmic H+ delivery could apply to the first (H+ + CO32− → HCO3−), second (H+ + HCO3− → CO2) or both protonation steps. If both protonation steps were subject to the diffusive barrier, the best-fitting distance δ would be smaller.
Additional information
Competing interests
None declared.
Author contributions
All the experiments were carried out in the Department of Physiology, Anatomy and Genetics, University of Oxford. All authors approved the final submitted version of the manuscript. Conception and design of the experiments: RDV-J, FCV, PS. Collection, analysis and interpretation of data: FCV, PS, JBY, KF, RC, CTS, PMC, MR, RDVJ. Drafting the article or revising it critically for important intellectual content: RDV-J, PS, FCV.
Funding
This work was funded by a British Heart Foundation Programme grant (to R.D.V.-J.), a research fellowship from the Royal Society (to P.S.) and a Wellcome Trust studentship (to F.C.V.).
References
- Aiello EA, Petroff MG, Mattiazzi AR, Cingolani HE. Evidence for an electrogenic Na+–HCO3– symport in rat cardiac myocytes. J Physiol. 1998;512:137–148. doi: 10.1111/j.1469-7793.1998.137bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Samir S, Papadopoulos S, Scheibe RJ, Meissner JD, Cartron JP, Sly WS, Alper SL, Gros G, Endeward V. Activity and distribution of intracellular carbonic anhydrase II and their effects on the transport activity of anion exchanger AE1/SLC4A1. J Physiol. 2013;591:4963–4982. doi: 10.1113/jphysiol.2013.251181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez BV, Loiselle FB, Supuran CT, Schwartz GJ, Casey JR. Direct extracellular interaction between carbonic anhydrase IV and the human NBC1 sodium/bicarbonate co-transporter. Biochemistry. 2003;42:12321–12329. doi: 10.1021/bi0353124. [DOI] [PubMed] [Google Scholar]
- Becker HM, Deitmer JW. Carbonic anhydrase II increases the activity of the human electrogenic Na+/HCO3– cotransporter. J Biol Chem. 2007;282:13508–13521. doi: 10.1074/jbc.M700066200. [DOI] [PubMed] [Google Scholar]
- Becker HM, Klier M, Schuler C, McKenna R, Deitmer JW. Intramolecular proton shuttle supports not only catalytic but also noncatalytic function of carbonic anhydrase II. Proc Natl Acad Sci U S A. 2011;108:3071–3076. doi: 10.1073/pnas.1014293108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boron WF. Evaluating the role of carbonic anhydrases in the transport of HCO3–-related species. Biochim Biophys Acta. 2010;1804:410–421. doi: 10.1016/j.bbapap.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JR, Sly WS, Shah GN, Alvarez BV. Bicarbonate homeostasis in excitable tissues: role of AE3 Cl–/HCO3– exchanger and carbonic anhydrase XIV interaction. Am J Physiol Cell Physiol. 2009;297:C1091–C1102. doi: 10.1152/ajpcell.00177.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'en FF-T, Villafuerte FC, Swietach P, Cobden PM, Vaughan-Jones RD. S0859, an N–cyanosulphonamide inhibitor of sodium-bicarbonate cotransport in the heart. Br J Pharmacol. 2008;153:972–982. doi: 10.1038/sj.bjp.0707667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Giusti VC, Orlowski A, Villa-Abrille MC, de Cingolani GE, Casey JR, Alvarez BV, Aiello EA. Antibodies against the cardiac sodium/bicarbonate co-transporter (NBCe1) as pharmacological tools. Br J Pharmacol. 2011;164:1976–1989. doi: 10.1111/j.1476-5381.2011.01496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garciarena CD, Ma YL, Swietach P, Huc L, Vaughan-Jones RD. Sarcolemmal localisation of Na+/H+ exchange and Na+-HCO3– co-transport influences the spatial regulation of intracellular pH in rat ventricular myocytes. J Physiol. 2013;591:2287–2306. doi: 10.1113/jphysiol.2012.249664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geers C, Gros G. Carbon dioxide transport and carbonic anhydrase in blood and muscle. Physiol Rev. 2000;80:681–715. doi: 10.1152/physrev.2000.80.2.681. [DOI] [PubMed] [Google Scholar]
- Gross E, Pushkin A, Abuladze N, Fedotoff O, Kurtz I. Regulation of the sodium bicarbonate cotransporter kNBC1 function: role of Asp986, Asp988 and kNBC1-carbonic anhydrase II binding. J Physiol. 2002;544:679–685. doi: 10.1113/jphysiol.2002.029777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulikova A, Vaughan-Jones RD, Swietach P. Dual role of CO2/HCO3– buffer in the regulation of intracellular pH of three-dimensional tumor growths. J Biol Chem. 2011;286:13815–13826. doi: 10.1074/jbc.M111.219899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadic-Gossmann D, Buckler KJ, Vaughan-Jones RD. Role of bicarbonate in pH recovery from intracellular acidosis in the guinea-pig ventricular myocyte. J Physiol. 1992;458:361–384. doi: 10.1113/jphysiol.1992.sp019422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem CH, Lagadic-Gossmann D, Vaughan-Jones RD. Characterization of intracellular pH regulation in the guinea-pig ventricular myocyte. J Physiol. 1999;517:159–180. doi: 10.1111/j.1469-7793.1999.0159z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem CH, Vaughan-Jones RD. Out-of-equilibrium pH transients in the guinea-pig ventricular myocyte. J Physiol. 1998;509:471–485. doi: 10.1111/j.1469-7793.1998.471bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Alvarez B, Casey JR, Reithmeier RA, Fliegel L. Carbonic anhydrase II binds to and enhances activity of the Na+/H+ exchanger. J Biol Chem. 2002;277:36085–36091. doi: 10.1074/jbc.M111952200. [DOI] [PubMed] [Google Scholar]
- Li X, Liu Y, Alvarez BV, Casey JR, Fliegel L. A novel carbonic anhydrase II binding site regulates NHE1 activity. Biochemistry. 2006;45:2414–2424. doi: 10.1021/bi051132d. [DOI] [PubMed] [Google Scholar]
- Loiselle FB, Morgan PE, Alvarez BV, Casey JR. Regulation of the human NBC3 Na+/HCO3– cotransporter by carbonic anhydrase II and PKA. Am J Physiol Cell Physiol. 2004;286:C1423–C1433. doi: 10.1152/ajpcell.00382.2003. [DOI] [PubMed] [Google Scholar]
- Lu J, Daly CM, Parker MD, Gill HS, Piermarini PM, Pelletier MF, Boron WF. Effect of human carbonic anhydrase II on the activity of the human electrogenic Na/HCO3 cotransporter NBCe1-A in Xenopus oocytes. J Biol Chem. 2006;281:19241–19250. doi: 10.1074/jbc.M602181200. [DOI] [PubMed] [Google Scholar]
- Maren TH. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev. 1967;47:595–781. doi: 10.1152/physrev.1967.47.4.595. [DOI] [PubMed] [Google Scholar]
- Minta A, Tsien RY. Fluorescent indicators for cytosolic sodium. J Biol Chem. 1989;264:19449–19457. [PubMed] [Google Scholar]
- Morgan PE, Pastorekova S, Stuart-Tilley AK, Alper SL, Casey JR. Interactions of transmembrane carbonic anhydrase, CAIX, with bicarbonate transporters. Am J Physiol Cell Physiol. 2007;293:C738–C748. doi: 10.1152/ajpcell.00157.2007. [DOI] [PubMed] [Google Scholar]
- O'Dowd JJ, Robins DJ, Miller DJ. Detection, characterisation, and quantification of carnosine and other histidyl derivatives in cardiac and skeletal muscle. Biochim Biophys Acta. 1988;967:241–249. doi: 10.1016/0304-4165(88)90015-3. [DOI] [PubMed] [Google Scholar]
- Orlowski A, De Giusti VC, Morgan PE, Aiello EA, Alvarez BV. Binding of carbonic anhydrase IX to extracellular loop 4 of the NBCe1 Na+/HCO3– cotransporter enhances NBCe1-mediated HCO3– influx in the rat heart. Am J Physiol Cell Physiol. 2011;303:C69–C80. doi: 10.1152/ajpcell.00431.2011. [DOI] [PubMed] [Google Scholar]
- Parker MD, Boron WF. The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol Rev. 2013;93:803–959. doi: 10.1152/physrev.00023.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibe RJ, Gros G, Parkkila S, Waheed A, Grubb JH, Shah GN, Sly WS, Wetzel P. Expression of membrane-bound carbonic anhydrases IV, IX, and XIV in the mouse heart. J Histochem Cytochem. 2006;54:1379–1391. doi: 10.1369/jhc.6A7003.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder MA, Ali MA, Hulikova A, Supuran CT, Clarke K, Vaughan-Jones RD, Tyler DJ, Swietach P. Extramitochondrial domain rich in carbonic anhydrase activity improves myocardial energetics. Proc Natl Acad Sci U S A. 2013;110:E958–E967. doi: 10.1073/pnas.1213471110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler C, Becker HM, McKenna R, Deitmer JW. Transport activity of the sodium bicarbonate cotransporter NBCe1 is enhanced by different isoforms of carbonic anhydrase. PLoS One. 2011;6:e27167. doi: 10.1371/journal.pone.0027167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scozzafava A, Briganti F, Ilies MA, Supuran CT. Carbonic anhydrase inhibitors: synthesis of membrane-impermeant low molecular weight sulfonamides possessing in vivo selectivity for the membrane-bound versus cytosolic isozymes. J Med Chem. 2000;43:292–300. doi: 10.1021/jm990479+. [DOI] [PubMed] [Google Scholar]
- Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem. 1995;64:375–401. doi: 10.1146/annurev.bi.64.070195.002111. [DOI] [PubMed] [Google Scholar]
- Snyder SM, Palmer BM, Moore RL. A mathematical model of cardiocyte Ca2+ dynamics with a novel representation of sarcoplasmic reticular Ca2+ control. Biophys J. 2000;79:94–115. doi: 10.1016/S0006-3495(00)76276-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer KW, Skolnick RL, Peercy BE, Keener JP, Vaughan-Jones RD. Facilitation of intracellular H+ ion mobility by CO2/HCO3– in rabbit ventricular myocytes is regulated by carbonic anhydrase. J Physiol. 2002;541:159–167. doi: 10.1113/jphysiol.2001.013268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling D, Alvarez BV, Casey JR. The extracellular component of a transport metabolon. Extracellular loop 4 of the human AE1 Cl–/HCO3– exchanger binds carbonic anhydrase IV. J Biol Chem. 2002;277:25239–25246. doi: 10.1074/jbc.M202562200. [DOI] [PubMed] [Google Scholar]
- Sterling D, Reithmeier RA, Casey JR. A transport metabolon. Functional interaction of carbonic anhydrase II and chloride/bicarbonate exchangers. J Biol Chem. 2001;276:47886–47894. doi: 10.1074/jbc.M105959200. [DOI] [PubMed] [Google Scholar]
- Stewart AK, Boyd CA, Vaughan-Jones RD. A novel role for carbonic anhydrase: cytoplasmic pH gradient dissipation in mouse small intestinal enterocytes. J Physiol. 1999;516:209–217. doi: 10.1111/j.1469-7793.1999.209aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock C, Mueller M, Kraehling H, Mally S, Noel J, Eder C, Schwab A. pH nanoenvironment at the surface of single melanoma cells. Cell Physiol Biochem. 2007;20:679–686. doi: 10.1159/000107550. [DOI] [PubMed] [Google Scholar]
- Supuran C, Casini A, Scozzafava A. Development of sulfonamide carbonic anhydrase inhibitors. In: Supuran C, Scozzafava A, Conway J, editors. Carbonic Anhydrase: Its Activators and Inhibitors. Boca Raton, FL, USA: CRC Press; 2004. [Google Scholar]
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Svastova E, Witarski W, Csaderova L, Kosik I, Skvarkova L, Hulikova A, Zatovicova M, Barathova M, Kopacek J, Pastorek J, Pastorekova S. Carbonic anhydrase IX interacts with bicarbonate transporters in lamellipodia and increases cell migration via its catalytic domain. J Biol Chem. 2012;287:3392–3402. doi: 10.1074/jbc.M111.286062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swietach P, Leem CH, Spitzer KW, Vaughan-Jones RD. Experimental generation and computational modelling of intracellular pH gradients in cardiac myocytes. Biophys J. 2005;88:3018–3037. doi: 10.1529/biophysj.104.051391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swietach P, Patiar S, Supuran CT, Harris AL, Vaughan-Jones RD. The role of carbonic anhydrase 9 in regulating extracellular and intracellular pH in three-dimensional tumor cell growths. J Biol Chem. 2009;284:20299–20310. doi: 10.1074/jbc.M109.006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swietach P, Spitzer KW, Vaughan-Jones RD. pH-dependence of extrinsic and intrinsic H+-ion mobility in the rat ventricular myocyte, investigated using flash photolysis of a caged-H+ compound. Biophys J. 2007;92:641–653. doi: 10.1529/biophysj.106.096560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swietach P, Wigfield S, Cobden P, Supuran CT, Harris AL, Vaughan-Jones RD. Tumor-associated carbonic anhydrase 9 spatially coordinates intracellular pH in three-dimensional multicellular growths. J Biol Chem. 2008;283:20473–20483. doi: 10.1074/jbc.M801330200. [DOI] [PubMed] [Google Scholar]
- Swietach P, Youm JB, Saegusa N, Leem CH, Spitzer KW, Vaughan-Jones RD. Coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling. Proc Natl Acad Sci U S A. 2013;110:E2064–2073. doi: 10.1073/pnas.1222433110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas LA, Alvarez BV. Carbonic anhydrase XIV in the normal and hypertrophic myocardium. J Mol Cell Cardiol. 2012;52:741–752. doi: 10.1016/j.yjmcc.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Vaughan-Jones RD, Peercy BE, Keener JP, Spitzer KW. Intrinsic H+ ion mobility in the rabbit ventricular myocyte. J Physiol. 2002;541:139–158. doi: 10.1113/jphysiol.2001.013267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol. 2009;46:318–331. doi: 10.1016/j.yjmcc.2008.10.024. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Swietach P, Rossini A, Loh SH, Vaughan-Jones RD, Spitzer KW. Functional diversity of electrogenic Na+-HCO3– cotransport in ventricular myocytes from rat, rabbit and guinea pig. J Physiol. 2005;562:455–475. doi: 10.1113/jphysiol.2004.071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaniboni M, Swietach P, Rossini A, Yamamoto T, Spitzer KW, Vaughan-Jones RD. Intracellular proton mobility and buffering power in cardiac ventricular myocytes from rat, rabbit, and guinea pig. Am J Physiol Heart Circ Physiol. 2003;285:H1236–H1246. doi: 10.1152/ajpheart.00277.2003. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Shao XM, Kao L, Azimov R, Weinstein AM, Newman D, Liu W, Kurtz I. Missense mutation T485S alters NBCe1-A electrogenicity causing proximal renal tubular acidosis. Am J Physiol Cell Physiol. 2013;305:C392–C405. doi: 10.1152/ajpcell.00044.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.