

Abstract
Systemic treatment of advanced prostate cancer is initiated with androgen deprivation therapy by gonadal testosterone depletion. Response durations are variable and tumors nearly always become resistant as castration-resistant prostate cancer (CRPC), which is driven, at least in part, by a continued dependence on the androgen receptor (AR). The proposed mechanisms that underlie AR function in this clinical setting are quite varied. These include intratumoral synthesis of androgens from inactive precursors, increased AR expression, AR activation through tyrosine kinase-dependent signaling, alterations in steroid receptor coactivators, and expression of a truncated AR with constitutive activity. Various pharmacologic interventions have clinically validated some of these mechanisms, such as those that require the AR ligand-binding domain. Clinical studies have failed to validate other mechanisms, and additional mechanisms have yet to be tested in patients with CRPC. Here, we review the mechanisms that elicit AR activity in CRPC, with a particular focus on recent developments.
A major stimulus for the growth of prostate cancer is the gonadal-testosterone-androgen receptor (AR) axis. AR activation commonly stimulates growth through TMPRSS2-ETS family translocations, which occur in local prostate cancer and permit androgen-driven oncogene expression (1, 2). Ablation of this axis via androgen deprivation therapy has long been the standard initial systemic treatment for advanced prostate cancer (3, 4). Responses to androgen deprivation therapy eventually give way to progression with castration-resistant prostate cancer (CRPC) and the necessity for a change in disease treatment or management (5). The development of CRPC is usually heralded by an increase in serum prostate-specific antigen (PSA), which is indicative of an increase in AR stimulation, despite low serum testosterone (6). An understanding of the mechanisms that underlie AR stimulation in this setting is critical for the development of treatments that reverse AR signaling and tumor progression.
Initial Hormonal Therapy
Gonadal testosterone ablation has long been the upfront treatment modality for metastatic prostate cancer. This is usually accomplished through one of several methods, including surgical orchiectomy and pituitary gonadotropin suppression using GnRH agonists or antagonists (7). Testosterone is physiologically converted by steroid-5α-reductase (SRD5A)-isoenzyme-2 in the prostate to 5α-dihydrotestosterone (DHT), which more potently activates AR (8, 9). Blocking gonadal testosterone works by suppressing intratumoral DHT and depriving AR of its major endogenous agonist. Although initially usually clinically effective, these methods still permit the existence of residual testosterone and DHT in benign prostate and localized prostate cancer tissue (10–12).
Androgen Receptor
AR plays a vital role in development of the normal prostate as well as prostate cancer disease progression. Its role in masculinization and normal prostate development is evidenced by the phenotype conferred by loss-of-function germline mutations that lead to “androgen insensitivity” and the absence of a normal prostate (13). With this condition, the extent of AR loss of transcription function appears to correlate with the magnitude of the defect in masculinization (14).
In contrast to the AR loss of function that occurs in androgen insensitivity, AR-dependent transcription is up-regulated in CRPC. This is probably most clearly clinically illustrated by monitoring serum PSA protein, which is encoded by an androgen-responsive gene, and which often rises prior to radiographic or clinical progression in the setting of low serum testosterone concentrations. Increased AR expression occurs in CRPC and is frequently attributable to AR gene amplification (15–17). Higher levels of AR expression may stimulate androgen-responsive genes because of mass action and may also convert the activity of AR antagonists to AR agonist activity (18). Mutations in the AR ligand-binding domain also subvert the response to AR antagonists, such that these agents instead stimulate agonist activity (19). For example, hydroxyflutamide stimulates the T877A mutation in LNCaP (19, 20). Cessation of AR antagonist therapy in this setting leads to a paradoxical PSA decline with the antiandrogen withdrawal response (21, 22).
Truncated Androgen Receptor
AR has an N-terminal domain, a zinc finger DNA-binding domain, and a C-terminal ligand-binding domain (23, 24). Rather than functioning as a positive regulator of AR transcription, the ligand-binding domain instead is a negative regulator, and agonist binding serves to derepress AR function. This is evidenced by the transcriptional activity of AR constructs in which the ligand-binding domain is artificially deleted (25). Several different forms of a truncated “AR variant” have been described in CRPC (26–28). The implication is that such variants lacking the ligand-binding domain would be constitutively active and indifferent to the presence of AR antagonists and androgen synthesis inhibitors. Some evidence suggests that AR variants originate from intragenic deletions and rearrangements that occur in CRPC (29, 30). Other evidence points to alternative splicing in the absence of genomic arrangements that occur through recruitment of RNA splicing factors to the 3′-splicing site (31). Laboratory models suggest that AR variants may play a role in resistance to potent AR antagonists, including enzalutamide (32). However, the dependence of AR variants on full-length AR in preclinical models of enzalutamide resistance may vary and be tied to the specific prostate cancer model employed (32, 33).
Tyrosine Kinase Signaling
A wide variety of growth factors, cytokines, receptor, and nonreceptor tyrosine kinases have been implicated in stimulating AR in the absence of androgens or sensitizing AR to subphysiologic androgen concentrations (34). The cell surface receptors include epidermal growth factor receptor, IL-6 receptor, IGF-1 receptor, and Her2/neu (35–38). The Src nonreceptor tyrosine kinase and other members in this family have also been implicated in AR activation and prostate cancer progression (39, 40). Some of these preclinical mechanisms have been interrogated clinically with pharmacologic interventions, thus far with disappointing therapeutic results in CRPC. Clinical studies of trastuzumab, which targets Her2/neu, and pertuzumab, which is a dimerization inhibitor of the same target, have failed to show any clinically significant activity in men with CRPC (41, 42). Despite the extensive preclinical data developed for the role of Src in prostate cancer, a phase III clinical trial of docetaxel plus the Src inhibitor dasatinib, vs docetaxel alone, failed to demonstrate any survival benefit (43). Clinical evaluation of IGF-1 inhibitors for prostate cancer is underway, and these agents appear to have clinical activity (44, 45). However, the benefit of these agents in CRPC has yet to be determined.
Coregulators
Nuclear receptor coregulators have a variety of roles in disease as well as normal physiology (46). A large number of coregulators has been implicated in stimulating AR-dependent transcription as a mechanism of resistance to castration in various studies, essentially either by loss of repressive function or gain-of-activation function (47, 48). Coregulator expression is transcriptionally regulated by AR, among other factors (49). Genomic profiling suggests that steroid receptor coactivator 2 (SRC-2) is amplified and appears to function as an oncogene in a subset of prostate cancers (50). Protein turnover of another coregulator, SRC-3, is regulated by the E3 ubiquitin ligase adaptor, speckle-type POZ protein (SPOP), such that SRC-3 protein suppression by SPOP suppresses AR-dependent transcription. Missense mutations of SPOP occur in approximately 6%–15% of prostate cancers (51), which may be selected for, in part, by blocking SRC-3 degradation and effectively derepressing AR (52).
Metabolism
Yet another mechanism of AR stimulation in the setting of castration is for the tumor to use the same stimulus used in the eugonadal state in normal physiology, ie, an endogenous AR agonist bound to the ligand-binding domain. Of course, given the state of medical or surgical castration and the depletion of gonadal testosterone, this may at first appear to be a paradox. However, the human adrenal reticularis produces abundant dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS), that can be enzymatically converted to DHT in a limited number of steps (53–55). Several groups have shown that, in the setting of gonadal testosterone depletion, prostatic tissues continue to retain physiologically significant concentrations of DHT (10, 11), which positively correlate with serum DHEA concentrations (10) and may therefore indicate the origins of the initial substrate utilized by prostatic tissues.
Modern techniques have refined the analysis of the presence of intratumoral androgens in prostatic tissues and have brought greater certainty to findings that were initially highly unanticipated. The observation that DHT is present at physiologically significant concentrations in local CRPC was first made over 3 decades ago using an immunoassay (56). Nearly 20 years later, similar studies were done in the same disease state, this time using mass spectrometry, which confirmed elevated DHT concentrations in local CRPC tissues (57). Mass spectrometry analysis of metastatic CRPC tissues followed in a rapid autopsy study and demonstrated that DHT concentrations sufficient to stimulate AR activity are also present in distant disease sites (58).
Enzalutamide and abiraterone are both potent hormonal therapies that increase overall survival and have been recently approved for the treatment of metastatic CRPC (59–61). Enzalutamide is a competitive AR antagonist that binds with higher affinity compared with bicalutamide (62). Abiraterone is a potent inhibitor of 17α-hydroxylase/17,20-lyase (CYP17A1), which is an enzyme required for the synthesis of all androgens (63). The clinical successes of both of these agents demonstrate the importance and requirement for the intratumoral genesis of DHT in driving CRPC progression, as well as its ultimate lethality. Although it could be argued that by directly binding AR, enzalutamide may counter mechanisms of AR reactivation that are steroidogenesis independent, the clinical benefit of abiraterone could only be explained by mechanisms that rely on androgen synthesis.
Substrate Sources
In the setting of castration, there are at least 2 possibilities for the initial substrate source for the genesis of intratumoral DHT (64). The first is de novo synthesis of DHT starting from cholesterol, entirely within prostatic tissues (65). This requires 8 enzymatic steps and intratumoral CYP17A1 expression and enzymatic activity. Preferred substrates for human CYP17A1 are Δ5-steroids (ie, pregnenolone and 17OH-pregnenolone) and 5α,3α-steroids (ie, 5α-pregnane-3α-ol-20-one and 5α-pregnane-3α,17α-diol-20-one) (66–68). Therefore, de novo synthesis of androgens may occur through the Δ5-pathway, because it occurs predominantly in the adrenal and testes, or through the “backdoor pathway,” which circumvents testosterone and requires 3-keto-reduction to a 3α-hydroxyl group (eg, conversion of 5α-dihydroprogesterone to 5α-pregnane-3α-ol-20-one) and 3α-hydroxyl-oxidation to a 3-keto-steroid (5α-androstane-3α,17β-diol), which are not otherwise necessary for DHT synthesis (69). When the backdoor pathway occurs, it does so, in part, because progesterone is a very good substrate for SRD5A, in tissues (like prostate) where it is expressed (70).
The second pathway is with the use of precursor steroids that originate from the adrenal reticularis, which possesses robust CYP17A1 enzymatic activity, and produces 19-carbon androgens as its major products. DHEAS and DHEA are the dominant products of the adrenal reticularis that are secreted into circulation. The use of adrenal precursors for DHT synthesis in CRPC would only require 3 enzymatic steps from DHEA. Notably, DHEAS is usually found in concentrations above 1 μM in serum (68), which is greater than 1000-fold the concentration of DHT typically found in CRPC tissues (∼1 nM) (57, 58). Experiments directly comparing DHT synthesis from adrenal precursors vs de novo intermediates suggest that the adrenal pathway is more robust (71).
Steroidogenic Enzymes
Although additional enzymes are required for de novo synthesis of DHT, a common set of enzymatic reactions must take place in CRPC tissues for both the adrenal and the de novo pathways. These include the steps mediated by 3β-hydroxysteroid dehydrogenase/Δ5→4 isomerase (3βHSD), SRD5A, and 17β-hydroxysteroid dehydrogenase-3 (17βHSD3)/aldo-keto reductase 1C3 (AKR1C3). The sites of chemical modification with these enzymatic steps are on carbons 3, 5, and 17 on the steroid backbone (Figure). These modifications result in oxidation of 3β-hydroxyl to 3-keto, Δ5 (carbon-carbon double bond between C5 and C6)-isomerization to Δ4 (carbon-carbon double bond between C4 and C5; as in conversion of DHEA to Δ4-androstenedione [AD]), 5α-reduction (as in conversion of testosterone to DHT), and 17-keto reduction to 17β-hydroxyl (as in conversion of AD to testosterone), all of which are required for the synthesis of DHT.
Figure 1.
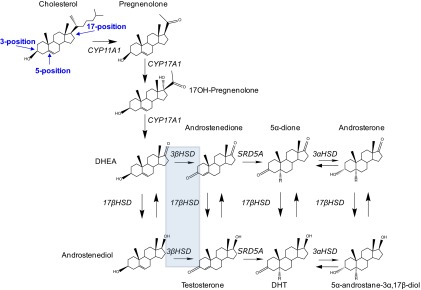
Enzymatic reactions and steroid modifications required for DHT synthesis. The structure of cholesterol, with carbons 3, 5, and 17 labeled, is shown in the upper left. Modifications of carbons 3 and 5 on the steroid A and B rings are catalyzed by 3βHSD and SRD5A in the adrenal pathways and pregnanes in the backdoor pathways (not shown). Conversion from 17-ketosteroids to 17β-hydroxysteroids is catalyzed by the 17βHSD isoenzymes. The conventional pathway occurs through conversion from DHEA → androstenedione → testosterone → DHT. The dominant adrenal pathway (5α-dione pathway) occurs through DHEA → androstenedione → 5α-dione → DHT. A missense mutation in 3βHSD1 (shaded box) increases flux through a metabolic step (DHEA → androstenedione) that is otherwise rate-limiting in the synthesis of DHT, permitting the development of CRPC. Mutant 3βHSD1 may also regulate metabolic flux through the backdoor pathways (not shown).
3βHSD
3βHSD is an enzyme encoded in humans by 2 genes, HSD3B1 and HSD3B2 (72, 73). 3βHSD2 is mainly expressed in steroidogenic organs, including adrenal and gonadal tissues. 3βHSD1 is the isoenzyme dominantly expressed in peripheral tissues, including the prostate. However, the transcripts for both isoenzymes are detectable in CRPC (58, 74). Two steroid modifications are catalyzed by 3βHSD, ie, 3β-hydroxyl oxidation to 3-keto and Δ5 to Δ4-steroid isomerization, which are together essentially irreversible and result in the synthesis of 3-keto, Δ4-steroids. 3βHSD is required for metabolism of 19-carbon androgens in the conversion of DHEA to AD and Δ5-androstenediol to testosterone, as well as metabolism of 21-carbon pregnanes in the conversion of pregnenolone to progesterone and 17OH-progesterone to 17OH-progesterone. The reaction catalyzed by 3βHSD highlights a structural modification necessary for biologically active androgens and is therefore required for all pathways to the synthesis of testosterone and/or DHT. Pharmacologic inhibition of 3βHSD in the dominant adrenal pathway blocks the conversion of DHEA to AD, downstream flux to DHT (as well as conversion of Δ5-androstenediol to testosterone) and AR activation in prostate cancer and may therefore be a viable strategy for the treatment of CRPC (75). Notably, in addition to potent CYP17A1 inhibition, abiraterone weakly inhibits 3βHSD, which is a biochemical feature that may be exploited for more effective therapy (76).
A point mutation in 3βHSD1 has recently emerged as a mechanism of sustaining DHT concentrations in patients with CRPC (77). A germline single-nucleotide polymorphism in HSD3B1 converts A → C at nucleotide position 1245, exchanging asparagine (N) for threonine (T) at amino acid position 367 of 3βHSD1. Three of 25 (12%) human CRPC tumors with homozygous wild-type inheritance have acquired a somatic mutation encoding 3βHSD1(367T), and 3 of 11 (27%) tumors with heterozygous inheritance have lost the wild-type allele to only encode for 3βHSD1(367T). This mutation confers resistance to protein ubiquitination and degradation, leading to an increase in protein half-life, higher steady-state protein levels, increased metabolic flux from DHEA → AD, sustaining DHT concentrations and permitting the development of CRPC (77). This is the first mutation described in the steroidogenic machinery that increases DHT synthesis in CRPC.
SRD5A
3-keto, Δ4-steroids are produced by 3βHSD, which then become substrates for SRD5A to irreversibly produce 5α-reduced steroids (78). There are 2 SRD5A isoenzymes in humans (70). SRD5A1 is the first isoenzyme that was cloned (79) but was found to be unaffected in patients with pseudohermaphroditism and defective 5α-reductase activity. Instead, loss-of-function mutations were found in SRD5A2 in these patients, which is the principal isoenzyme expressed in the prostate and is required for normal male physiology (80). At least 4 independent studies have consistently shown that the SRD5A2 transcript is suppressed and SRD5A1 up-regulated in the transition from untreated prostate cancer to CRPC in patients (58, 81–83). The reason for the isoenzyme switch was unclear but it was generally thought that SRD5A1 up-regulation is probably required to sustain 5α-reduction of testosterone to DHT. However, it turns out that instead of testosterone, an alternative 3-keto, Δ4-steroid, AD, is preferentially 5α reduced by SRD5A1 (84). The substrate preference for SRD5A1 translates to increased flux from AD to 5α-androstanedione (5α-dione) compared with flux from testosterone to DHT, in CRPC cell lines and freshly collected metastatic CRPC tissues from patients (85). Furthermore, this favored 5α-dione pathway also drives CRPC progression to a greater extent than the pathway that requires conversion of testosterone to DHT (85). These findings provide a model that explains increased T:DHT ratios in CRPC (∼3:1) compared with untreated prostate cancer (∼1:10) through a modicum of testosterone synthesis, followed by accumulation because 5α-reduction of testosterone is inefficient compared with 5α-reduction of AD (54).
17β-HSD
Both androgens (DHT and testosterone) that avidly bind AR are 17β-hydroxysteroids and therefore require 17-keto to 17β-hydroxyl reduction by a member of the 17βHSD isoenzyme family, which has at least 14 members (86). Some isoenzymes have a preference for the reductive reaction (17-keto to 17β-hydroxyl), and some have an oxidative preference for the reverse reaction. 17βHSD3 is the reductive isoenzyme required for testosterone synthesis in the Leydig cell of the testes for normal male physiology (87). 17βHSD5, also known as AKR1C3, is another reductive enzyme that is highly up-regulated in CRPC (58, 83, 88). These findings have led to efforts for the development of potent and specific AKR1C3 inhibitors (89, 90), which would block the conversion of AD to testosterone and 5α-dione to DHT (85, 91).
Resistance to Abiraterone and Enzalutamide
The discoveries of increased AR expression, intratumoral androgen synthesis, and the continued dependence on AR have directly led to clinical advances with the development of abiraterone and enzalutamide as newer agents that extend survival in patients with CRPC. However, responses to these agents are usually followed by clinical resistance, which is often accompanied by rises in serum PSA and may again indicate a resurgence of AR activation (59–61). Whether these cases of resistance to abiraterone and enzalutamide occur through steroidogenesis or other mechanisms of AR stimulation remains to be determined and is an area of active investigation (64). The 3βHSD1(367T) somatic mutation occurs in a mouse xenograft model of abiraterone resistance (77); however, the role of this mutation in clinical resistance to abiraterone and enzalutamide remains to be determined.
Conclusions
Although a variety of different mechanisms have been implicated in sustaining AR activity in CRPC, clinical interrogation of these mechanisms of AR stimulation have most clearly validated those that involve androgen biosynthesis and/or the AR ligand-binding domain. Data resulting from these investigations leave no doubt that intratumoral androgen synthesis plays a central role in CRPC progression, and ultimately, the lethality of this disease. Identifying how AR is involved in resistance to abiraterone and enzalutamide should help direct the field to the next set of therapeutic agents.
Acknowledgments
This work was supported by a Howard Hughes Medical Institute Physician-Scientist Early Career Award, the Prostate Cancer Foundation, 1R01CA172382 and 1R01CA168899 from the National Cancer Institute, an American Cancer Society Research Scholar Award, and by Grant PC080193 from the US Army Medical Research and Materiel Command.
Disclosure Summary: N.S. has been a compensated consultant for Janssen and Medivation.
Footnotes
- AD
- Δ4-androstenedione
- AKR1CE
- aldo-keto reductase 1C3
- AR
- androgen receptor
- CRPC
- castration-resistant prostate cancer
- CYP17A1
- 17α-hydroxylase/17,20-lyase
- DHEA
- dehydroepiandrosterone
- DHEAS
- DHEA sulfate
- DHT
- dihydrotestosterone
- HSD
- hydroxysteroid dehydrogenase
- PSA
- prostate-specific antigen
- SPOP
- speckle-type POZ protein
- SRC
- steroid receptor coactivator
- SRD5A
- steroid-5α-reductase.
References
- 1. Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648 [DOI] [PubMed] [Google Scholar]
- 2. Tomlins SA, Mehra R, Rhodes DR, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51 [DOI] [PubMed] [Google Scholar]
- 3. Huggins C, Hodges CV. Studies on prostate cancer, I: the effect of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:293–297 [Google Scholar]
- 4. Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. JAMA. 2005;294:238–244 [DOI] [PubMed] [Google Scholar]
- 5. Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferraldeschi R, Sharifi N, Auchus RJ, Attard G. Molecular pathways: inhibiting steroid biosynthesis in prostate cancer. Clin Cancer Res. 2013;19:3353–3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schroder F, Crawford ED, Axcrona K, Payne H, Keane TE. Androgen deprivation therapy: past, present and future. BJU Int 109 Suppl. 2012;6:1–12 [DOI] [PubMed] [Google Scholar]
- 8. Bruchovsky N, Wilson JD. The conversion of testosterone to 5-α-androstan-17-β-ol-3-one by rat prostate in vivo and in vitro. J Biol Chem. 1968;243:2012–2021 [PubMed] [Google Scholar]
- 9. Bruchovsky N, Wilson JD. The intranuclear binding of testosterone and 5-α-androstan-17-β-ol-3-one by rat prostate. J Biol Chem. 1968;243:5953–5960 [PubMed] [Google Scholar]
- 10. Page ST, Lin DW, Mostaghel EA, et al. Persistent intraprostatic androgen concentrations after medical castration in healthy men. J Clin Endocrinol Metab. 2006;91:3850–3856 [DOI] [PubMed] [Google Scholar]
- 11. Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clin Cancer Res. 2004;10:7121–7126 [DOI] [PubMed] [Google Scholar]
- 12. Labrie F. Blockade of testicular and adrenal androgens in prostate cancer treatment. Nat Rev Urol. 2011;8:73–85 [DOI] [PubMed] [Google Scholar]
- 13. McPhaul MJ. Androgen receptor mutations and androgen insensitivity. Mol Cell Endocrinol. 2002;198:61–67 [DOI] [PubMed] [Google Scholar]
- 14. Gottlieb B, Beitel LK, Wu JH, Trifiro M. The androgen receptor gene mutations database (ARDB): 2004 update. Hum Mutat. 2004;23:527–533 [DOI] [PubMed] [Google Scholar]
- 15. Bubendorf L, Kononen J, Koivisto P, et al. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999;59:803–806 [PubMed] [Google Scholar]
- 16. Attard G, Swennenhuis JF, Olmos D, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69:2912–2918 [DOI] [PubMed] [Google Scholar]
- 17. Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39 [DOI] [PubMed] [Google Scholar]
- 19. Veldscholte J, Ris-Stalpers C, Kuiper GG, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534–540 [DOI] [PubMed] [Google Scholar]
- 20. Wilding G, Chen M, Gelmann EP. Aberrant response in vitro of hormone-responsive prostate cancer cells to antiandrogens. Prostate. 1989;14:103–115 [DOI] [PubMed] [Google Scholar]
- 21. Kelly WK, Scher HI. Prostate specific antigen decline after antiandrogen withdrawal: the flutamide withdrawal syndrome. J Urol. 1993;149:607–609 [DOI] [PubMed] [Google Scholar]
- 22. Figg WD, Sartor O, Cooper MR, et al. Prostate specific antigen decline following the discontinuation of flutamide in patients with stage D2 prostate cancer. Am J Med. 1995;98:412–414 [DOI] [PubMed] [Google Scholar]
- 23. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–3015 [DOI] [PubMed] [Google Scholar]
- 24. Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol. 2011;29:3651–3658 [DOI] [PubMed] [Google Scholar]
- 25. Gao T, Marcelli M, McPhaul MJ. Transcriptional activation and transient expression of the human androgen receptor. J Steroid Biochem Mol Biol. 1996;59:9–20 [DOI] [PubMed] [Google Scholar]
- 26. Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011;71:2108–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Y, Hwang TH, Oseth LA, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31:4759–4767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells [published online July 15, 2013]. Oncogene. doi:10.1038/onc.2013.284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watson PA, Chen YF, Balbas MD, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci USA. 2010;107:16759–16765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lamont KR, Tindall DJ. Minireview: alternative activation pathways for the androgen receptor in prostate cancer. Mol Endocrinol. 2011;25:897–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ponguta LA, Gregory CW, French FS, Wilson EM. Site specific androgen receptor serine phosphorylation linked to epidermal growth factor dependent growth of castration-recurrent prostate cancer. J Biol Chem. 2008;283:20989–21001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dutt SS, Gao AC. Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncol. 2009;5:1403–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308 [DOI] [PubMed] [Google Scholar]
- 38. Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517–527 [DOI] [PubMed] [Google Scholar]
- 39. Guo Z, Dai B, Jiang T, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309–319 [DOI] [PubMed] [Google Scholar]
- 40. Posadas EM, Al-Ahmadie H, Robinson VL, et al. FYN is overexpressed in human prostate cancer. BJU Int. 2009;103:171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ziada A, Barqawi A, Glode LM, et al. The use of trastuzumab in the treatment of hormone refractory prostate cancer; phase II trial. Prostate. 2004;60:332–337 [DOI] [PubMed] [Google Scholar]
- 42. de Bono JS, Bellmunt J, Attard G, et al. Open-label phase II study evaluating the efficacy and safety of two doses of pertuzumab in castrate chemotherapy-naive patients with hormone-refractory prostate cancer. J Clin Oncol. 2007;25:257–262 [DOI] [PubMed] [Google Scholar]
- 43. Araujo JC, Trudel GC, Saad F, et al. Overall survival (OS) and safety of dasatinib/docetaxel versus docetaxel in patients with metastatic castration-resistant prostate cancer (mCRPC): Results from the randomized phase III READY trial. J Clin Oncol. 2013;31 (suppl 6): abstract LBA8 [Google Scholar]
- 44. Chi KN, Gleave ME, Fazli L, et al. A phase II pharmacodynamic study of preoperative figitumumab in patients with localized prostate cancer. Clin Cancer Res. 2012;18:3407–3413 [DOI] [PubMed] [Google Scholar]
- 45. Dean JP, Sprenger CC, Wan J, et al. Response of the insulin-like growth factor (IGF) system to IGF-IR inhibition and androgen deprivation in a neoadjuvant prostate cancer trial: effects of obesity and androgen deprivation. J Clin Endocrinol Metab. 2013;98:E820–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lonard DM, O'Malley BW. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol. 2012;8:598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Agoulnik IU, Weigel NL. Coactivator selective regulation of androgen receptor activity. Steroids. 2009;74:669–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. Urol Oncol. 2009;27:36–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heemers HV, Regan KM, Schmidt LJ, Anderson SK, Ballman KV, Tindall DJ. Androgen modulation of coregulator expression in prostate cancer cells. Mol Endocrinol. 2009;23:572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Geng C, He B, Xu L, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A. 2013;110:6997–7002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sharifi N, Auchus RJ. Steroid biosynthesis and prostate cancer. Steroids. 2012;77:719–726 [DOI] [PubMed] [Google Scholar]
- 54. Sharifi N. Minireview: androgen metabolism in castration-resistant prostate cancer. Mol Endocrinol. 2013;27:708–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Luu-The V, Bélanger A, Labrie F. Androgen biosynthetic pathways in the human prostate. Best Pract Res Clin Endocrinol Metab. 2008;22:207–221 [DOI] [PubMed] [Google Scholar]
- 56. Geller J, Albert J, Loza D, Geller S, Stoeltzing W, de la Vega D. DHT concentrations in human prostate cancer tissue. J Clin Endocrinol Metab. 1978;46:440–444 [DOI] [PubMed] [Google Scholar]
- 57. Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–4657 [DOI] [PubMed] [Google Scholar]
- 58. Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197 [DOI] [PubMed] [Google Scholar]
- 60. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barrie SE, Potter GA, Goddard PM, Haynes BP, Dowsett M, Jarman M. Pharmacology of novel steroidal inhibitors of cytochrome P450(17) α (17 α-hydroxylase/C17–20 lyase). J Steroid Biochem Mol Biol. 1994;50:267–273 [DOI] [PubMed] [Google Scholar]
- 64. Chang KH, Sharifi N. Prostate cancer-from steroid transformations to clinical translation. Nat Rev Urol. 2012;9:721–724 [DOI] [PubMed] [Google Scholar]
- 65. Locke JA, Guns ES, Lubik AA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–6415 [DOI] [PubMed] [Google Scholar]
- 66. Gupta MK, Guryev OL, Auchus RJ. 5α-Reduced C21 steroids are substrates for human cytochrome P450c17. Arch Biochem Biophys. 2003;418:151–160 [DOI] [PubMed] [Google Scholar]
- 67. Auchus RJ, Lee TC, Miller WL. Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J Biol Chem. 1998;273:3158–3165 [DOI] [PubMed] [Google Scholar]
- 68. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab. 2004;15:432–438 [DOI] [PubMed] [Google Scholar]
- 70. Russell DW, Wilson JD. Steroid 5 α-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63:25–61 [DOI] [PubMed] [Google Scholar]
- 71. Sharifi N, McPhaul MJ, Auchus RJ. “Getting from here to there”—mechanisms and limitations to the activation of the androgen receptor in castration-resistant prostate cancer. J Investig Med. 2010;58:938–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, Melner MH. Molecular biology of the 3β-hydroxysteroid dehydrogenase/Δ5-Δ4 isomerase gene family. Endocr Rev. 2005;26:525–582 [DOI] [PubMed] [Google Scholar]
- 73. Lorence MC, Murry BA, Trant JM, Mason JI. Human 3 β-hydroxysteroid dehydrogenase/Δ 5—-4isomerase from placenta: expression in nonsteroidogenic cells of a protein that catalyzes the dehydrogenation/isomerization of C21 and C19 steroids. Endocrinology. 1990;126:2493–2498 [DOI] [PubMed] [Google Scholar]
- 74. Cai C, Chen S, Ng P, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–6513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Evaul K, Li R, Papari-Zareei M, Auchus RJ, Sharifi N. 3β-Hydroxysteroid dehydrogenase is a possible pharmacological target in the treatment of castration-resistant prostate cancer. Endocrinology. 2010;151:3514–3520 [DOI] [PubMed] [Google Scholar]
- 76. Li R, Evaul K, Sharma KK, et al. Abiraterone inhibits 3β-hydroxysteroid dehydrogenase: a rationale for increasing drug exposure in castration-resistant prostate cancer. Clin Cancer Res. 2012;18:3571–3579 [DOI] [PubMed] [Google Scholar]
- 77. Chang K-H, Li R, Kuri B, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tomkins GM. The enzymatic reduction of Δ 4–3-ketosteroids. J Biol Chem. 1957;225:13–24 [PubMed] [Google Scholar]
- 79. Andersson S, Russell DW. Structural and biochemical properties of cloned and expressed human and rat steroid 5 α-reductases. Proc Natl Acad Sci USA. 1990;87:3640–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Andersson S, Berman DM, Jenkins EP, Russell DW. Deletion of steroid 5 α-reductase 2 gene in male pseudohermaphroditism. Nature. 1991;354:159–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Titus MA, Gregory CW, Ford OH, 3rd, Schell MJ, Maygarden SJ, Mohler JL. Steroid 5α-reductase isozymes I and II in recurrent prostate cancer. Clin Cancer Res. 2005;11:4365–4371 [DOI] [PubMed] [Google Scholar]
- 82. Mitsiades N, Sung CC, Schultz N, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012;72:6142–6152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825 [DOI] [PubMed] [Google Scholar]
- 84. Thigpen AE, Cala KM, Russell DW. Characterization of Chinese hamster ovary cell lines expressing human steroid 5 α-reductase isozymes. J Biol Chem. 1993;268:17404–17412 [PubMed] [Google Scholar]
- 85. Chang KH, Li R, Papari-Zareei M, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci USA. 2011;108:13728–13733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Prehn C, Möller G, Adamski J. Recent advances in 17β-hydroxysteroid dehydrogenases. J Steroid Biochem Mol Biol. 2009;114:72–77 [DOI] [PubMed] [Google Scholar]
- 87. Andersson S, Russell DW, Wilson JD. 17β-Hydroxysteroid dehydrogenase 3 deficiency. Trends Endocrinol Metab. 1996;7:121–126 [DOI] [PubMed] [Google Scholar]
- 88. Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liedtke AJ, Adeniji AO, Chen M, et al. Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem. 2013;56:2429–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen M, Adeniji AO, Twenter BM, Winkler JD, Christianson DW, Penning TM. Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg Med Chem Lett. 2012;22:3492–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sharifi N. The 5α-androstanedione pathway to dihydrotestosterone in castration-resistant prostate cancer. J Investig Med. 2012;60:504–507 [DOI] [PMC free article] [PubMed] [Google Scholar]