

Abstract
AMP-activated protein kinase (AMPK) is a key cellular energy sensor and regulator of metabolic homeostasis. Activation of AMPK provides beneficial outcomes in fighting against metabolic disorders such as insulin resistance and type 2 diabetes. Currently, there is no allosteric AMPK activator available for the treatment of metabolic diseases, and limited compounds are available to robustly stimulate cellular/tissue AMPK in a specific manner. Here we investigated whether simultaneous administration of two different pharmacological AMPK activators, which bind and act on different sites, would result in an additive or synergistic effect on AMPK and its downstream signaling and physiological events in intact cells. We observed that cotreating primary hepatocytes with the AMP mimetic 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) and a low dose (1 μM) of the allosteric activator A769662 produced a synergistic effect on AMPK Thr172 phosphorylation and catalytic activity, which was associated with a more profound increase/decrease in phosphorylation of downstream AMPK targets and inhibition of hepatic lipogenesis compared with single-compound treatment. Mechanistically, we found that cotreatment does not stimulate LKB1, upstream kinase for AMPK, but it protects against dephosphorylation of Thr172 phosphorylation by protein phosphatase PP2Cα in an additive manner in a cell-free assay. Collectively, we demonstrate that AICAR sensitizes the effect of A769662 and promotes AMPK activity and its downstream events. The study demonstrates the feasibility of promoting AMPK activity by using two activators with distinct modes of action in order to achieve a greater activation of AMPK and downstream signaling.
Keywords: AMP-activated protein kinase, LKB1, A769662, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside, lipogenesis
amp-activated protein kinase (AMPK) is a major regulator of cellular energy homeostasis that coordinates metabolic pathways in order to balance nutrient supply with energy demand (14, 15, 27, 34). AMPK is activated by various physiological and pathological energy stresses that increase intracellular ADP-to-ATP and AMP-to-ATP ratios, either by accelerating ATP consumption (e.g., muscle contraction) or by decreasing ATP generation (e.g., hypoxia, ischemia, mitochondrial poisoning). Once activated, AMPK acts to restore energy homeostasis by promoting ATP production pathways such as glucose uptake and fatty acid oxidation while simultaneously inhibiting ATP utilization pathways such as lipid and protein synthesis.
AMPK is a heterotrimeric complex containing a catalytic subunit (α) and two regulatory subunits (β, γ). Two isoforms of the α- and β-subunits exist (α1, α2, β1, β2), whereas there are three isoforms of the γ-subunit (γ1, γ2, γ3). Isoform expression varies among cells/tissues and also species (36, 41), with α1, β1, and γ1 appearing in general to be ubiquitously expressed isoforms. Some isoforms are known to be expressed in a cell/tissue-specific/restricted manner; for example, α2 is more predominantly expressed in skeletal muscle (22) and also γ3 is exclusively found in skeletal muscle (2, 44).
AMPK is activated by upstream kinases LKB1 and Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) via phosphorylation of Thr172 on the α catalytic subunit (1, 34). AMP binding to the γ-subunit allosterically activates AMPK, and ADP and/or AMP binding promotes Thr172 phosphorylation and protects it against dephosphorylation by protein phosphatases (e.g., PP2Cα), while ATP competes for binding at the same sites and antagonizes the effects of AMP (12, 25).
AMPK has emerged as a key drug target for metabolic disorders such as insulin resistance, cardiovascular disease, and type 2 diabetes (19). This is based on numerous reports demonstrating that activation of AMPK by physiological (e.g., exercise/muscle contraction) or pharmacological means brings about desirable effects that ameliorate metabolic dysfunctions by improving glycemic control and plasma lipid profiles (13, 28). Although a variety of compounds including several xenobiotics (which are known to elicit anti-obesity/diabetic effects) stimulate AMPK, it has been demonstrated that almost all of them [with some exceptions (3, 11, 16)] activate AMPK by an indirect mechanism (17). For instance, antidiabetic drugs like metformin (and its analog phenformin) as well as resveratrol, which display anti-obesity/diabetes effects [although controversial (38, 43)], activate AMPK indirectly by lowering ATP levels (i.e., increase in AMP-to-ATP ratio via suppression of mitochondrial respiration) (17, 26). 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) is a very widely used AMPK activator that is transported into the cell and phosphorylated to form ZMP, which mimics the effect of AMP by directly binding to the γ-subunit. There are a few direct AMPK activators reported in the literature, of which the small molecule A769662 is the only well-characterized/validated AMPK activator that stimulates AMPK via allosteric action and also protects active (phosphorylated) AMPKα at Thr172 from dephosphorylation through interacting with the β1-subunit.
Given that there are few compounds available to robustly stimulate cellular/tissue AMPK in a specific manner, we explored here whether simultaneous administration of two different pharmacological AMPK activators (AICAR and A769662), which are proposed to act on different sites/regions (10, 11, 31, 33), would result in an additive or synergistic effect on AMPK and its downstream events compared with single-compound treatment (either AICAR or A769662) in intact cells.
MATERIALS AND METHODS
Materials.
λ-Phosphatase was from New England Biolabs (Ipswich, MA). PP2Cα was from the Division of Signal Transduction Therapy (DSTT), University of Dundee (UK). AICAR was from Toronto Research Chemicals (Toronto, ON, Canada). A769662 was from Selleckchem (Houston, TX) or DSTT. Horseradish peroxidase-conjugated secondary antibodies were from Jackson ImmunoResearch Europe (Newmarket, UK). C2C12 myoblasts were obtained from Sigma (Buchs, Switzerland). General and specific cell culture reagents, including Williams medium E (WME) and horse serum, were obtained from Life Technologies (Zug, Switzerland). [γ-32P]ATP was obtained from PerkinElmer (Schwerzenbach, Switzerland).
Antibodies.
Total acetyl-CoA carboxylase (ACC; no. 3676), phospho-ACC1 (Ser79; no. 3661), total Raptor (no. 2280), phospho-Raptor (Ser792; no. 2083), total AMPKα (no. 2532), phospho-AMPKα (Thr172 and Ser485/Ser491; no. 2535 and no. 4185), total AMPKβ1 (no. 4182), phospho-AMPKβ1 (Ser108 and Ser182; no. 4181 and no. 4186), total LKB1 (no. 3047), total salt-inducible kinase (SIK)2 (no. 6919), total p70 S6 kinase (p70S6K; no. 2708), and phospho-p70S6K (Thr389; no. 9234) antibodies were from Cell Signaling Technology (Danvers, MA). The antibody against α-tubulin (no. T6074) was from Sigma. Total AMPKα1 and AMPKα2 isoform-specific antibodies were raised against the previously validated peptide sequences (30) TSPPDSFLDDHHLTR (α1) and MDDSAMHIPPGLKPH (α2) custom-generated by DSTT, MRC Protein Phosphorylation and Ubiquitylation Unit, University of Dundee and affinity purified in house. Total SIK2 antibody for immunoprecipitation was from DSTT (S227B, 3rd bleed).
Animals.
Wild-type C57BL/6 mice were obtained from Jackson Laboratories. Generation and breeding of AMPKβ1−/− and littermate AMPKβ1+/+ mice were described previously (6). All protocols for animal use and euthanasia were approved by the McMaster University Animal Research Ethics Board.
C2C12 cell culture.
C2C12 myoblasts were cultured in DMEM supplemented with 20% FBS. Myoblasts were differentiated to myotubes during 7 days in DMEM supplemented with 1% horse serum and treated for 30 min with compounds as indicated in Fig. 1, D and E.
Fig. 1.

Cotreatment of mouse primary hepatocytes with 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) and A769662 exhibits a synergistic effect on AMP-activated protein kinase (AMPK) activity. Primary hepatocytes were isolated from C57BL/6 (12–15 wk old) mice and cultured overnight. Cells were serum-starved for 2–3 h and then treated with vehicle, AICAR, and/or A769662 for 45 min before cell lysis. A: immunoblotting was performed with 20 μg per lane and with the indicated antibodies. B: AMPKα1 complexes were immunoprecipitated from 30 μg of extract with 1 μg of anti-AMPKα1 and protein G Sepharose. Immune complexes were assayed for AMPK activity (in duplicate) as described in materials and methods. Results are expressed as mean ± SE pmol Pi incorporated·min−1·mg−1. *Significance (AICAR vs. respective AICAR + A769662), P < 0.05; n = 3/condition. C: AMPKα1 complexes were immunoprecipitated (IP) from lysate (30 μg) of hepatocytes treated with 1 mM AICAR and 10 μM A769662 with 1 μg of anti-AMPKα1 and protein G Sepharose. Immune complexes were incubated in the absence or presence of 400 U of λ-phosphatase for 15 min at 30°C. Reactions were terminated with Laemmli buffer and analyzed by immunoblotting with anti-AMPKα1. D and E: differentiated C2C12 cells were treated with vehicle, AICAR, and/or A769662 for 30 min before cell lysis. D: immunoblots were performed with 30 μg of protein per lane and with the indicated antibodies. E: AMPKα1 complexes were immunoprecipitated from 30 μg of extract with 1 μg of anti-AMPKα1 and protein G Sepharose. Immune complexes were assayed for AMPK activity (in duplicate). Results are expressed as mean ± SE pmol Pi incorporated·min−1·mg−1. *Significance (AICAR vs. respective AICAR + A769662), P < 0.05; n = 3/condition.
Primary mouse hepatocytes.
Primary hepatocytes were isolated by collagenase perfusion and prepared as previously described (9). Briefly, cells were plated on collagen-coated six-well plates in WME containing 10% FBS and 1% antibiotic-antimycotic and allowed to adhere for 4 h before being washed with PBS and provided with fresh medium. For all protocols, hepatocytes were incubated overnight at 37°C and experiments were performed the following morning. [3H]acetate lipogenesis was executed as described previously (8) with minor modifications. Cells were washed with PBS and incubated in serum-free WME for 2–3 h. Serum-free medium containing [3H]acetate (10 μCi/ml; PerkinElmer) and 0.5 mM unlabeled sodium acetate (Sigma) was then applied, and cells were incubated in the presence or absence of AICAR and/or A769662 for 45 min. Cells were washed twice with PBS, and lipids were extracted in chloroform-methanol 1:2 for determination of radiolabel incorporation into total lipids as previously described (6). For protein experiments, hepatocytes were washed and incubated in serum-free WME for 2–3 h and then exposed to no drug, AICAR, and/or A769662 for 45 min. Medium was quickly removed, and cells were lysed on ice in 4°C lysis buffer [in mM: 50 HEPES, 150 NaCl, 100 NaF, 10 Na-pyrophosphate, 5 EDTA, 250 sucrose, 1 DTT, and 1 Na-orthovanadate, with 1% Triton X and 1 tablet/50 ml Complete protease inhibitor cocktail (Roche)]. Plates were scraped, and lysates were stored at −80°C for later analyses.
Immunoblotting.
Lysates were denatured in SDS sample buffer, separated by SDS-PAGE, and transferred to nitrocellulose membrane. Membranes were blocked for 1 h in 10 mM Tris (pH 7.6), 137 mM NaCl, 0.1% (vol/vol) Tween 20 (TBST) containing 5% (wt/vol) skimmed milk. Membranes were incubated in primary antibody prepared in TBST containing 1% (wt/vol) BSA overnight at 4°C. Detection was performed with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence reagent.
Cloning and recombinant AMPK expression, purification, and activation.
The coding regions of AMPKα2 (NM_006252), AMPKβ1 (NM_006253), and AMPKγ1 (NM_002733) were amplified from brain RNA (Agilent) with a SuperScript III RT-PCR kit (Invitrogen). The resulting PCR products were ligated into intermediate vectors with a Strataclone PCR cloning kit (Agilent). Site-directed mutagenesis was carried out according to the QuikChange method (Stratagene) using KOD polymerase (Novagen). The sequences of all constructs were verified with the BigDyeR Terminator 3.1 kit and 3500XL Genetic analyzer (Applied Biosystems) in house.
Active heterotrimeric AMPK complexes were prepared by a modification of the method described previously (23). Briefly, BL21(DE3) were transformed with the polycistronic vector pET15B-6HIS-α2β1γ1 or pET15B-6HIS-α2β1(S108A)γ1 and cultured in 4l autoinduction medium (overnight, 18°C) (37). AMPK complexes were isolated from clarified lysates with His60 Ni Superflow Resin (Clontech). Imidazole was removed by buffer exchange over Sephadex G-25, and AMPK complexes were activated by in vitro phosphorylation with GST-CaMKKβ. GST-CaMKKβ was removed with glutathione Sepharose 4B (GE Healthcare), and AMPK complexes were further purified over Superdex200 10/300 (GE Healthcare). Preparations were stored in 12.5 mM HEPES pH 7.4, 75 mM NaCl, 0.5 mM DTT, 50% (vol/vol) glycerol at −20°C.
AMPK and SIK2 activity assay.
AMPK was immunoprecipitated from 30 μg of lysate with antibodies against α1- or α2-subunit with protein G Sepharose. The immune complex (endogenous AMPK), or recombinant AMPK was assayed for phosphotransferase activity toward AMARA peptide (AMARAASAAALARRR) with [γ-32P]ATP, as previously described (21). SIK2 was immunoprecipitated from 250 μg of lysate with 2 μg of anti-SIK2 antibody and protein G Sepharose. The assay was performed as previously described (18) with Sakamototide peptide (ALNRTSSDSALHRRR) as substrate.
Phosphatase protection assay.
The assay was performed as previously described (5) with some modifications. Briefly, fully phosphorylated recombinant AMPK α2β1γ1 complex (200 ng) (as described above) was incubated for 15 min at 30°C in the presence of PP2Cα (20 ng) and the indicated concentrations of AICAR and/or A769662 in 50 mM HEPES pH 7.4, 10 mM MgCl2, 100 μM EGTA. The dephosphorylation reaction was stopped by 20-fold dilution in 50 mM HEPES pH 7.4, 100 mM NaCl, 0.03% (vol/vol) Brij 35, 1 mM DTT. AMPK activity was then measured in the presence of 200 μM AMP as described above.
Statistical analysis.
Data are expressed as means ± SE or SD as indicated. Statistical analysis was performed by unpaired, two-tailed Student's t-test or two-way ANOVA with Bonferroni post hoc test. Differences between groups were considered statistically significant when P < 0.05.
RESULTS
Cotreatment of mouse primary hepatocytes with AICAR and A769662 exhibits synergistic effect on AMPK activity.
We first assessed whether coincubation of two different pharmacological AMPK activators (AICAR and A769662), which are proposed to act on different sites/regions (10, 11, 31, 33), would result in an additive or synergistic effect on AMPK activity compared with single-compound treatment (either AICAR or A769662) in intact cells. Mouse primary hepatocytes were treated with an increasing dose of AICAR (0, 0.01, 0.03, 0.1, 0.3, and 1 mM) in the absence or presence of a fixed concentration of A769662 (10 μM), and AMPK phosphorylation (Thr172) was assessed by immunoblotting using a phospho-specific antibody. As illustrated in Fig. 1A, A769662 (10 μM) alone showed no detectable increase in AMPK phosphorylation whereas AICAR stimulated Thr172 phosphorylation in a dose-dependent manner. Strikingly, when hepatocytes were coincubated with AICAR and A769662, AMPK phosphorylation, as judged by phospho-Thr172 signal and also upper shifting of band mobility of total AMPKα, was robustly enhanced compared with the respective single AICAR dose (Fig. 1A). As anticipated, the AMPK phosphorylation was tightly associated with AMPK (α1) phosphotransferase activity measured in vitro, demonstrating that the cotreatment results in a significant increase in AMPK activity at AICAR doses ranging from 0.1 to 1 mM compared with the respective single AICAR dose (Fig. 1B). We next assessed cellular AMPK activity by measuring phosphorylation of bona fide AMPK substrates such as ACC and Raptor in mouse primary hepatocytes. ACC phosphorylation (Ser79 on ACC1) was readily saturated at relatively low concentrations of AICAR (0.03 mM), whereas Raptor phosphorylation (Ser792) exhibited a pattern (dose-dependent increase) similar to AMPK phosphorylation (Thr172) and activity (Fig. 1A). This phosphorylation on Raptor, which has been proposed to inhibit mammalian target of rapamycin complex 1 (mTORC1), was associated with a decrease in phosphorylation (Thr389) of p70S6K, an established mTORC1 substrate.
An interesting observation of the AMPK immunoblot signal (both phospho and total) mentioned above is the band shift upon cotreatment, which is attributable to both AMPKα1 and AMPKα2 isoforms (as shown by immunoblots using isoform-specific antibodies) (Fig. 1A). Band shifts on SDS-PAGE are typically observed when proteins are phosphorylated at multiple residues. To test whether this was the case for AMPKα, we immunoprecipitated endogenous AMPKα1 from extracts of hepatocytes (which had been stimulated with 1 mM AICAR and 10 μM A769662), and the resulting immunoprecipitants were incubated with a recombinant protein phosphatase (λ-phosphatase) for 15 min in vitro. We observed that the upper band shift induced by the cotreatment was abolished after the phosphatase treatment (Fig. 1C). To determine whether the cotreatment promoted phosphorylation of sites on AMPKα apart from the Thr172 in the activation T-loop residue, we checked another known phosphorylation site (Ser485 on α1 and Ser491 on α2), which is outside of the kinase domain and is proposed to be regulated by Akt (also known as PKB) (20) and p70S6K (4). AICAR alone stimulated Ser485/Ser491 phosphorylation in a dose-dependent manner, while there was no further increase in phosphorylation of this site with coincubation with A769662 (10 μM) (Fig. 1A). This suggests that the Ser485/Ser491 site is unlikely to be responsible for the band shift observed in the total and phospho-Thr172 AMPKα signals. It would be interesting to perform phosphopeptide mapping of adenovirally expressed AMPKα in primary hepatocytes treated with AICAR and A769662 and then identify/determine whether there are novel phosphorylation sites that are uniquely regulated by the cotreatment. Given that the effect of A769662 in activating AMPK requires the β1 isoform in the trimeric complex and the Ser108 residue is implicated to play a key role (16, 31, 33), we examined whether two known autophosphorylation sites on AMPKβ1 (Ser108 and Ser182) were regulated by a single or dual treatment with AMPK-activating compound(s) in primary hepatocytes. Ser108 phosphorylation was increased in a dose-dependent manner upon AICAR stimulation alone but was not further increased when cotreated with A769662 (10 μM). Ser182 phosphorylation was detectable in unstimulated cells, which was not altered by either AICAR or A769662 treatment (Fig. 1A).
To determine whether the synergistic effect of the cotreatment on AMPK phosphorylation/activation can be observed in other cell types apart from primary hepatocytes, C2C12 (mouse skeletal muscle cell line) myotubes were treated with an increasing dose of AICAR (0, 0.2, 0.5, and 1 mM) with or without a fixed concentration of A769662 (30 μM). We chose to use 30 μM, as opposed to 10 μM used in hepatoyctes, because our previous work (10) and pilot experiments (data not shown) revealed that the dose required to activate de novo AMPK (by monitoring ACC phosphorylation) is different among cell types. While 1–10 μM is sufficient to activate AMPK in hepatocytes, 10–30 μM is required to stimulate AMPK in C2C12 (and also mouse embryonic fibroblasts) (unpublished data). Similar to the results observed in primary hepatocytes, AICAR increased AMPK T-loop phosphorylation (Thr172) in a dose-dependent fashion, whereas A769662 (30 μM) alone did not, and the cotreatment promoted further phosphorylation and activity of AMPK as well as phosphorylation of AMPK substrates (Fig. 1D) compared with AICAR treatment alone (at 0.5 and 1 mM) (Fig. 1, D and E). We also observed that the cotreatment (AICAR + A769662) promotes phosphorylation of AMPK and its downstream targets (i.e., ACC and Raptor) in other cell types such as NIH-3T3 fibroblasts (data not shown).
Low concentration (1 μM) of A769662 is sufficient to significantly increase effect of AICAR on AMPK activity.
To determine whether an A769662 dose lower than 10 μM would elicit the synergistic effect of the AICAR-A769662 cotreatment on AMPK catalytic activity, primary mouse hepatocytes were treated with increasing concentrations of A769662 (0, 1, 3, 10, 30, and 100 μM) combined with a fixed AICAR dose (100 μM). While 1 μM A769662 had no effect on AMPK phosphorylation, this dose was readily sufficient to robustly increase the effects of AICAR on phosphorylation of AMPK and its substrates (ACC and Raptor) as well as the inhibition of p70S6K phosphorylation (Fig. 2A). Given that Thr172 and its surrounding residues are conserved between AMPKα1 and AMPKα2 isoforms and thus the phospho-specific antibody detects both isoforms, we examined whether the cotreatment selectively activates one isoform or both. Individual AMPKα isoforms were immunoprecipitated from the hepatocyte lysates (with isoform-specific antibodies) followed by an in vitro kinase assay. We observed that the cotreatment resulted in an increase in AMPK activity at A769662 doses ranging from 1 to 30 μM for AMPKα1-containing complexes and 1 and 10 μM for AMPKα2-containing complexes (Fig. 2B). Cotreatment with higher doses of A769662 (100 μM for AMPKα1, 30 and 100 μM for AMPKα2) failed to increase AMPK activity above AICAR treatment alone, most likely because of a toxic effect of the compound on hepatocytes.
Fig. 2.
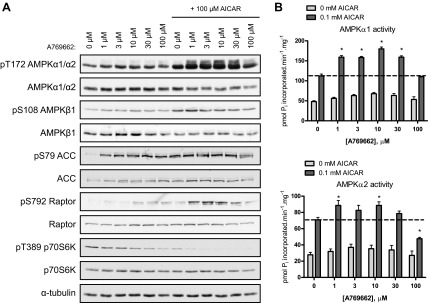
A low concentration of A769662 is sufficient to increase the effect of AICAR on AMPK activity. Primary hepatocytes were isolated from C57BL/6 mice (12–15 wk old) and cultured overnight. Cells were serum starved for 2–3 h and then treated with vehicle, AICAR, and/or A769662 for 45 min before cell lysis. A: immunoblotting was performed with 30 μg of protein and the indicated antibodies. B: AMPKα1 or α2 complexes were immunoprecipitated from 30 μg of lysate with anti-AMPKα1 or anti-AMPKα2. Immune complexes were assayed for AMPK activity (in duplicate). Results are expressed as mean ± SE pmol Pi incorporated·min−1·mg−1. *Significance (AICAR vs. respective AICAR + A769662), P < 0.05; n = 3/condition.
Synergistic effect of A769662 and AICAR cotreatment is ablated in AMPKβ1-deficient hepatocytes.
A769662 has been reported to be selective to act on β1-containing complexes of AMPK (16, 33). Therefore we investigated whether the synergistic effect of the cotreatment (AICAR and A769662) would be lost in primary hepatocytes lacking AMPKβ1. As previously described (33), in AMPKβ1−/− hepatocytes there is a profound loss of AMPKα catalytic subunits due to instability in the absence of the predominant scaffold β1 regulatory subunit in those mouse hepatocytes (Fig. 3A). β1-Null hepatocytes were treated with an increasing dose of AICAR (0, 0.01, 0.03, 0.1, 0.3, and 1 mM) and with a fixed dose of A769662 (10 μM). AMPK phosphorylation was still dose-dependently increased by AICAR treatment alone (Fig. 3B). In contrast, the elevation of AMPK phosphorylation upon cotreatment with A769662 observed in wild-type hepatocytes (Fig. 1A) was ablated in the β1-null hepatocytes (Fig. 3B), and therefore the activity of AMPK was not affected by the cotreatment either (Fig. 3C). This confirms that the β1 specificity of A769662 is retained/confirmed and that synergistic effect of the cotreatment (AICAR + A769662) can only take place in intact cells/tissues expressing β1-containing complexes.
Fig. 3.

The synergistic effect of A769662 and AICAR cotreatment is ablated in AMPKβ1-deficient hepatocytes. Primary hepatocytes were isolated from AMPKβ1−/− mice and wild-type (WT) littermates (12–15 wk old) and cultured overnight. Cells were serum starved for 2–3 h and then treated with vehicle, AICAR, and/or A769662 for 45 min before cell lysis. A: vehicle-treated hepatocyte lysates were analyzed by immunoblotting using 30 μg of protein and the indicated antibodies. B: hepatocyte lysates from AMPKβ1−/− mice were analyzed by immunoblotting using 30 μg of protein extracts and the indicated antibodies. C: AMPKα1 complexes were immunoprecipitated from 30 μg of extract with 1 μg of anti-AMPKα1 and protein G Sepharose. Immune complexes were assayed for AMPK activity (in duplicate). Results are expressed as mean ± SE pmol Pi incorporated·min−1·mg−1; n = 3.
Lipogenesis is robustly inhibited in hepatocytes treated with AICAR and A769662.
Next we investigated whether the AICAR-A769662 cotreatment would also have an effect on a downstream physiological outcome of AMPK activation, of which one established example is inhibition of lipogenesis in hepatocytes. AMPK inhibits lipogenesis through phosphorylation of its targets ACC1 and ACC2 (8). Primary mouse hepatocytes were left untreated or treated with AICAR (0.03 or 0.1 mM) in the presence or absence of A769662 (10 μM), and incorporation of [3H]acetate (a widely used lipogenic substrate) into the lipid pool was measured as a readout of de novo lipogenesis. AICAR treatment decreased lipogenesis to ∼60% or ∼40% (for 0.03 mM and 0.1 mM, respectively) of the basal rate (100%), while 10 μM A769662 decreased lipogenesis to ∼40%. Cotreatment of AICAR (0.03 and 0.1 mM) with A769662 (10 μM) further reduced lipogenesis to ∼30% and ∼25% of the basal rate, demonstrating that both compounds could inhibit lipogenesis in hepatocytes in an additive manner (Fig. 4).
Fig. 4.

Lipogenesis is robustly inhibited in hepatocytes treated with AICAR and A769662. Primary hepatocytes were isolated from C57BL/6 mice (12–15 wk old) and cultured overnight. Cells were serum starved for 2–3 h and labeled with [3H]acetate for 45 min in the presence of vehicle, AICAR, and/or 10 μM A769662. De novo lipogenesis was measured as described in materials and methods. Results are expressed as mean ± SE [3H]acetate incorporated into lipids (% of control). *P < 0.05 (comparing the effect of AICAR doses within [−A769662] group or [+A769662] group); #P < 0.05 (comparing the effect of A769662 to the respective AICAR doses); n = 12/condition.
Coincubation with AMP and A769662 displays additive protection effect on AMPK phosphorylation against dephosphorylation by protein phosphatase.
We and others previously reported that not only does A769662 directly activate AMPK but its binding (independently of canonical AMP/ZMP-binding sites) also prevents Thr172 dephosphorylation in a cell-free assay (10, 33). To test whether the mechanism by which cotreatment with AMP and A769662 promotes AMPK phosphorylation in intact cells is due to protection against dephosphorylation by protein phosphatase, a recombinant AMPKα2β1γ1 (which had been purified from E. coli and phosphorylated/activated by recombinant CaMKKβ in vitro as described in materials and methods) was incubated with recombinant PP2Cα in the presence of AICAR, A769662, or both compounds together in vitro. As previously shown, both AMP (saturated ∼3–10 μM) and A769662 (0.1 μM) robustly protected against dephosphorylation by PP2Cα. As anticipated in the control experiment, there was no dephosphorylation/deactivation of AMPK in the absence of Mg2+, as it is an essential cofactor in the activation of PP2Cα. Interestingly, cotreatment with various doses of AMP and a fixed dose of A769662 (0.1 μM) showed an additive protective effect against dephosphorylation by PP2Cα (Fig. 5A). To more directly demonstrate that the observed additive effect is indeed due to A769662 interaction with β1-subunit, we used a recombinant AMPK complex containing a A769662-resistant mutant (β1-S108A). In the AMPKα2β1(S108A)γ1 complex, AMP was able to protect against dephosphorylation by PP2Cα, while the effect of A769662 was abolished (Fig. 5B). We examined whether the enhanced Thr172 phosphorylation by the cotreatment is due to activation of the upstream kinase of AMPK, LKB1, in mouse primary hepatoyctes and measured activity of a known LKB1-dependent substrate, SIK2, expressed in the liver (Fig. 5C). We observed that both SIK activity and LKB1 expression were not altered with or without cotreatment. These results may explain the effect of the cotreatment in vivo, where AICAR/ZMP would promote AMPK phosphorylation (Thr172) by LKB1 and in turn allow A769662 to exert its effect by protecting AMPK from dephosphorylation by protein phosphatases present in the cells.
Fig. 5.
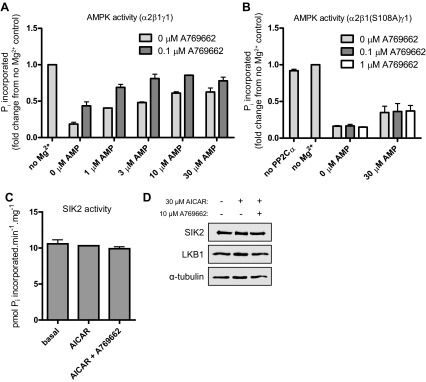
Coincubation with AICAR and A769662 displays an additive protection effect on AMPK phosphorylation against dephosphorylation by a protein phosphatase. Phosphatase protection assays were performed with recombinant AMPK complex and PP2Cα (20 ng) as described in materials and methods. A: prephosphorylated/activated recombinant AMPKα2β1γ1 was dephosphorylated by a recombinant PP2Cα in the presence or absence of its cofactor Mg2+ and assayed for AMPK phosphotransferase activity in vitro. Results are expressed as Pi incorporated (fold change from no Mg2+ control ± SE); n = 2. B: prephosphorylated/activated recombinant AMPKα2β1(Ser108Ala)γ1 was dephosphorylated by a recombinant PP2Cα in the presence or absence of its cofactor Mg2+ and assayed for AMPK phosphotransferase activity in vitro. Results are expressed as Pi incorporated (fold change from no Mg2+ control ± SE); n = 3. C and D: primary hepatocytes were isolated from C57BL/6 mice (12–15 wk old) and cultured overnight. Cells were serum starved for 2–3 h and then treated with vehicle, 30 μM AICAR, and/or 10 μM A769662 for 45 min. C: salt-inducible kinase (SIK)2 was immunoprecipitated from extracts and assayed as described in materials and methods. Results are expressed as mean ± SE pmol Pi incorporated·min−1·mg−1; n = 2/condition (assay in duplicate). D: immunoblotting was performed with 25 μg extract per lane and the indicated antibodies.
DISCUSSION
We here show that cotreatment of cells with two distinct AMPK activators (AICAR and A769662), which act on different sites/regions, exhibits a synergistic effect on AMPK and its downstream targets/physiological consequences compared with single-compound treatment in intact cells, including primary hepatocytes (7) and muscle cells (C2C12). We have also confirmed that the cotreatment displays similar results in other cell types such as NIH-3T3 fibroblasts (data not shown). A notable observation was that in the presence of AICAR a low concentration of A769662 (as low as 1 μM) produces a robust synergistic effect on Thr172 phosphorylation and activation of AMPK, which indicates that AICAR/ZMP binding profoundly sensitizes the effect of A769662. This was particularly striking because we previously observed that, although A769662 significantly increased ACC phosphorylation (presumably through direct/allosteric activation of AMPK) at 1–10 μM, it failed to display detectable increase in Thr172 phosphorylation up to ∼100–200 μM in mouse hepatocytes (7). We initially hypothesized that the conformational change of AMPK complex induced by ZMP binding to AMPK γ-subunit facilitates A769662 interaction not only to β1-containing complex but also to β2-containing complex (which is not normally activated by the compound when treated alone); therefore cotreatment would enhance global (β1/β2-complex) AMPK activation in cells. However, experiments performed employing AMPKβ1-deficient hepatocytes ruled out this hypothesis and involvement of the β2 isoform in the observed effects (Fig. 3).
It has been shown that coincubation of the fully phosphorylated (Thr172 residue by CaMKKβ in vitro) AMPKαβγ complex (prepared from bacteria) with AMP and A769662 does not display an additive allosteric effect on kinase activity in a cell-free assay (31), while in intact mammalian cells we observed that the coincubation promoted AMPK activity, which was associated with an increase in Thr172 phosphorylation in the activation T loop of AMPKα catalytic subunit. Therefore, it is reasonable to assume that the effect of cotreatment on AMPK Thr172 phosphorylation is due to an increase in the activity of the upstream kinase of AMPK (i.e., LKB1), protection against dephosphorylation via protein phosphatase(s), or the combination of both. We initially measured de novo activity of LKB1 by assaying its substrate, SIK2 (also known as QIK) (1, 18), and observed that cotreatment had no effect (Fig. 5C). This observation is consistent with our previous finding that LKB1 is a constitutively active kinase (29) and AMPK activity is uniquely regulated among other related kinases by its ability to be controlled through adenine nucleotide (i.e., ADP and AMP) binding to the γ-subunit (24, 32). Consequently, we showed that the cotreatment displayed a significant additive effect on protection from dephosphorylation via protein phosphatase in a cell-free assay (Fig. 5A). However, currently we are not able to demonstrate this in a cellular context/assay, as the identity of AMPK phosphatase(s) is still unknown/elusive [although there are some reported candidates (35, 40)].
Another key observation was that the enhanced AMPK activation by cotreatment resulted in a more profound inhibition of lipogenesis compared with single-compound treatment in primary hepatocytes (Fig. 4). It would be interesting to see whether the cotreatment strategy also works efficiently in the whole animal and elicits more pronounced physiological effects downstream of AMPK, for example, in reversing steatosis/fatty liver caused by high-fat diet feeding or leptin/leptin receptor deficiency (i.e., ob/ob or db/db mice). This cotreatment strategy might be attractive from the pharmaceutical point of view, as it would potentially be feasible to reduce the concentration of the compounds by promoting sensitivity by dual treatment. However, one caveat is that it only works in cells/tissues expressing the AMPKβ1 isoform. It has been reported that the β2 isoform is predominantly expressed in skeletal muscle and that there is very little β1 isoform, resulting in insufficient AMPK activation to elicit glucose uptake in isolated rodent skeletal muscles in response to A769662 (33, 39). However, this cotreatment strategy is still a useful tool to study the function of AMPK in various cell systems given that the majority of cultured cell lines express the β1 isoform and may be resistant to AICAR. Interestingly, a recent paper has demonstrated that a patented AMPK activator termed “991” possesses structural similarity and shares a common binding site with A769662 in the carbohydrate-binding domain of the AMPKβ subunit, as evidenced by a cocrystallization study (42). However, 991 is more effective in stimulating AMPK in both cell-based and cell-free assays that include AMPK complexes containing β2. It would be interesting to test whether 991 also elicits synergistic activation of AMPK at very low concentration when cotreated with AICAR.
In summary, we demonstrated that coincubation of cells with AICAR and a low concentration of A769662 resulted in a synergistic effect on AMPK Thr172 phosphorylation, which appears to be at least partially due to the compound binding to distinct sites causing conformation changes favorable to protection against dephosphorylation by phosphatase(s). Interestingly, we have recently observed in a parallel study that A769662 treatment of cardiac myocytes enhanced the activation of AMPK and the subsequent stimulation of glucose uptake induced by various indirect AMPK activators that raise cellular AMP-to-ATP ratio, including phenformin, oligomycin, and hypoxia (Timmermans A and Bertrand L, unpublished observations). All in all, these results suggest that it is feasible to promote AMPK activity by using two activators with distinct modes of action in order to achieve a greater resulting physiological effect of AMPK in intact cells.
GRANTS
These studies were supported by grants and fellowships from the Australian Research Council (B. E. Kemp), the Australian National Health and Medical Research Council (B. E. Kemp and G. R. Steinberg), the Canadian Diabetes Association (G. R. Steinberg), and the Canadian Institutes of Health Research (CIHR) (G. R. Steinberg) and supported in part by the Victorian Government's OIS Program (B. E. Kemp) and the Canadian Foundation for Innovation (G. R. Steinberg). G. R. Steinberg is a Canada Research Chair in Metabolism and Obesity. L. Bertrand is Research Associate of the Fonds National de la Recherche Scientifique (FNRS), Belgium.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.D., R.J.F., L. Bultot, and M.D. performed experiments; S.D., R.J.F., L. Bultot, G.R.S., and K.S. analyzed data; S.D., R.J.F., L. Bultot, G.R.S., and K.S. interpreted results of experiments; S.D. prepared figures; S.D. and K.S. drafted manuscript; S.D., R.J.F., L. Bultot, M.D., L. Bertrand, B.E.K., G.R.S., and K.S. approved final version of manuscript; R.J.F., L. Bultot, M.D., L. Bertrand, B.E.K., and G.R.S. edited and revised manuscript; L. Bertrand and K.S. conception and design of research.
ACKNOWLEDGMENTS
We thank D. Grahame Hardie and Simon Hawley (University of Dundee) for valuable technical advice on protein phosphatase protection assay.
REFERENCES
- 1.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem 75: 137–163, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J 346: 659–669, 2000 [PMC free article] [PubMed] [Google Scholar]
- 3.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 3: 403–416, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, Kahn BB. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin's effect on food intake. Cell Metab 16: 104–112, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett 377: 421–425, 1995 [DOI] [PubMed] [Google Scholar]
- 6.Dzamko N, van Denderen BJ, Hevener AL, Jorgensen SB, Honeyman J, Galic S, Chen ZP, Watt MJ, Campbell DJ, Steinberg GR, Kemp BE. AMPK beta1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J Biol Chem 285: 115–122, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest 120: 2355–2369, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O'Neill HM, Ford RJ, Palanivel R, O'Brien M, Hardie DG, Macaulay SL, Schertzer JD, Dyck JR, van Denderen BJ, Kemp BE, Steinberg GR. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19: 1649–1654, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fullerton MD, Hakimuddin F, Bonen A, Bakovic M. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J Biol Chem 284: 25704–25713, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem 282: 32549–32560, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guigas B, Sakamoto K, Taleux N, Reyna SM, Musi N, Viollet B, Hue L. Beyond AICA riboside: in search of new specific AMP-activated protein kinase activators. IUBMB Life 61: 18–26, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci 36: 470–477, 2011 [DOI] [PubMed] [Google Scholar]
- 13.Hardie DG, Ross FA, Hawley SA. AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol 19: 1222–1236, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology 21: 48–60, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, Kemp BE, Sakamoto K, Steinberg GR, Hardie DG. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336: 918–922, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 11: 554–565, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henriksson E, Jones HA, Patel K, Peggie M, Morrice N, Sakamoto K, Goransson O. The AMPK-related kinase SIK2 is regulated by cAMP via phosphorylation at Ser358 in adipocytes. Biochem J 444: 503–514, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horman S, Beauloye C, Vanoverschelde JL, Bertrand L. AMP-activated protein kinase in the control of cardiac metabolism and remodeling. Curr Heart Fail Rep 9: 164–173, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 281: 5335–5340, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Hunter RW, Treebak JT, Wojtaszewski JF, Sakamoto K. Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 60: 766–774, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musi N, Fujii N, Hirshman MF, Ekberg I, Froberg S, Ljungqvist O, Thorell A, Goodyear LJ. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 50: 921–927, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Neumann D, Woods A, Carling D, Wallimann T, Schlattner U. Mammalian AMP-activated protein kinase: functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expr Purif 30: 230–237, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc Natl Acad Sci USA 107: 19237–19241, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oakhill JS, Scott JW, Kemp BE. AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol Metab 23: 125–132, 2012 [DOI] [PubMed] [Google Scholar]
- 26.Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia 56: 1898–1906, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J 418: 261–275, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest 123: 2764–2772, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol Endocrinol Metab 287: E310–E317, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Sakamoto K, Zarrinpashneh E, Budas GR, Pouleur AC, Dutta A, Prescott AR, Vanoverschelde JL, Ashworth A, Jovanovic A, Alessi DR, Bertrand L. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am J Physiol Endocrinol Metab 290: E780–E788, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem 282: 32539–32548, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 403: 139–148, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, Iseli TJ, Koay A, Gooley PR, Stapleton D, Kemp BE. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol 15: 1220–1230, 2008 [DOI] [PubMed] [Google Scholar]
- 34.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev 89: 1025–1078, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Steinberg GR, Michell BJ, van Denderen BJ, Watt MJ, Carey AL, Fam BC, Andrikopoulos S, Proietto J, Gorgun CZ, Carling D, Hotamisligil GS, Febbraio MA, Kay TW, Kemp BE. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab 4: 465–474, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, Viollet B, Guigas B. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 54: 3101–3110, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41: 207–234, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Timmers S, Konings E, Bilet L, Houtkooper RH, van de Weijer T, Goossens GH, Hoeks J, van der Krieken S, Ryu D, Kersten S, Moonen-Kornips E, Hesselink MK, Kunz I, Schrauwen-Hinderling VB, Blaak EE, Auwerx J, Schrauwen P. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 14: 612–622, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JF. A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol Cell Physiol 297: C1041–C1052, 2009 [DOI] [PubMed] [Google Scholar]
- 40.Voss M, Paterson J, Kelsall IR, Martin-Granados C, Hastie CJ, Peggie MW, Cohen PT. Ppm1E is an in cellulo AMP-activated protein kinase phosphatase. Cell Signal 23: 114–124, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Wu J, Puppala D, Feng X, Monetti M, Lapworth AL, Geoghegan KF. Chemoproteomic analysis of inter-tissue and inter-species isoform diversity of AMP-activated protein kinase (AMPK). J Biol Chem 288: 35904–35912, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, Patel BR, Heath RB, Walker PA, Hallen S, Giordanetto F, Martin SR, Carling D, Gamblin SJ. Structural basis of AMPK regulation by small molecule activators. Nat Commun 4: 3017, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshino J, Conte C, Fontana L, Mittendorfer B, Imai S, Schechtman KB, Gu C, Kunz I, Rossi Fanelli F, Patterson BW, Klein S. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab 16: 658–664, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu H, Fujii N, Hirshman MF, Pomerleau JM, Goodyear LJ. Cloning and characterization of mouse 5′-AMP-activated protein kinase gamma3 subunit. Am J Physiol Cell Physiol 286: C283–C292, 2004 [DOI] [PubMed] [Google Scholar]