

Abstract
Phenylthiobutanoic acids (PTBAs) are a new class of histone deacetylase (HDAC) inhibitors that accelerate recovery and reduce postinjury fibrosis after ischemia-reperfusion-induced acute kidney injury. However, unlike the more common scenario in which patients present with protracted and less clearly defined onset of renal injury, this model of acute kidney injury gives rise to a clearly defined injury that begins to resolve over a short period of time. In these studies, we show for the first time that treatment with the PTBA analog methyl-4-(phenylthio)butanoate (M4PTB) accelerates recovery and reduces postinjury fibrosis in a progressive model of acute kidney injury and renal fibrosis that occurs after aristolochic acid injection in mice. These effects are apparent when M4PTB treatment is delayed 4 days after the initiating injury and are associated with increased proliferation and decreased G2/M arrest of regenerating renal tubular epithelial cells. In addition, there is reduced peritubular macrophage infiltration and decreased expression of the macrophage chemokines CX3Cl1 and CCL2. Since macrophage infiltration plays a role in promoting kidney injury, and since renal tubular epithelial cells show defective repair and a marked increase in maladaptive G2/M arrest after aristolochic acid injury, these findings suggest M4PTB may be particularly beneficial in reducing injury and enhancing intrinsic cellular repair even when administered days after aristolochic acid ingestion.
Keywords: HDAC inhibitor, aristolochic acid, kidney injury, fibrosis
acute kidney injury (AKI) is a risk factor for progression to chronic kidney disease (27, 42). However, no therapies have been proven to reduce postinjury fibrosis in patients with AKI (16). Part of the reason is that patients often present late to the hospital, and many of the drug candidates have to be administered early in the course of their injury. For this reason, therapeutic strategies targeting the repair mechanism after AKI are attractive since they may be effective when given days after the initial insult occurred. Using zebrafish as an experimental model system, we discovered a new class of histone deacetylase (HDAC) inhibitors (HDACi), the phenylthiobutanoic acids (PTBAs), that accelerate recovery and enhance regenerative repair of injured renal tubular epithelial cells (RTECs) when administered 48 h after toxin-induced AKI in zebrafish larvae (7, 10). The esterified analog of the parent compound PTBA, 4-methyl-thiobutanate (M4PTB), also accelerates recovery and decreases renal fibrosis when administered 24 h after the initiating injury in a model of ischemia-reperfusion-induced severe AKI (IR-AKI) in mice (7). These findings suggest that treatment with PTBA analogs can be used to reduce post-AKI fibrosis when administered late after the initiating injury. However, unlike the more common clinical scenario of patients presenting with a more protracted and less clearly defined onset of renal injury, this model of IR-AKI gives rise to a clearly defined renal injury that induced rapidly and over a short time period. Therefore, we sought to determine whether PTBA analogs are effective in a model of AKI in which there is a more prolonged and persistent cause of renal injury.
Aristolochic acid nephropathy (AAN) is a largely underdiagnosed cause of chronic kidney disease (CKD) resulting from ingestion of the plant extracts from Aristolochia species commonly used in Chinese herbal remedies (12). It has also been implicated as the causative agent in Balkan Endemic Nephropathy, and for an epidemic of CKD in Belgium resulting from contamination of slimming pills with Aristolochia extracts. Patients usually present with asymptomatic, progressive CKD, but ∼5% have AA-induced AKI (AA-AKI), and of these, the majority go onto develop end-stage renal disease (ESRD)(46). A significant proportion of these patients also develop urothelial malignancies. Pathologically AA-AKI is characterized by extensive proximal tubular epithelial cell (PTEC) injury, followed by progressive tubular atrophy, interstitial macrophage infiltration, and fibrosis (45). Experimental models of AAN recapitulate the features of AAN in humans (30, 39). With the use of these models it has been shown that renal injury results from selective uptake of AA metabolites by organic anion transporters that are enriched in the S3 segment PTECs (4, 13). These give rise to mutagenic, aristolactam-DNA adducts which either activate cell cycle arrest and apoptosis, or they give rise to survivable mutations that are inherited by daughter cells with each new cell division (2). For this reason, a single dose of AA gives rise to progressive PTEC injury that accumulates over time.
There are currently no established therapies for AAN (17). We now show that M4PTB initiated 4 days after injury accelerates functional recovery, reduces long-term tubular atrophy and interstitial fibrosis, and enhances regenerative repair of injured RTECs in a mouse model of AA-AKI. These findings establish that M4PTB is effective in reducing post-AKI fibrosis in a model of AKI with progressive tissue injury, and they establish a new disease-specific therapeutic approach for AA-AKI that can be delayed days after AA ingestion.
MATERIALS AND METHODS
Experimental model of AA-AKI.
For AA-AKI, male BALB/c mice (8–10 wk, 22–26 g, purchased from Charles River) were given a one-time intraperitoneal injection of 1 AA (Sigma Chemicals) at 4.7 mg/kg body wt in PBS. M4PTB, which is liquid oil, was solubilized in 20% (2-hydroxypropyl)-?-cyclodextrin (β-cyclodextrin; Sigma Chemicals) and injected intraperitoneally at 100 mg/kg daily starting 4 days after AA injection. Since initial injury with this model is variable, mice were randomly allocated vehicle or M4PTB treatment limbs on day 4 after serum creatinine values were first evaluated. Mice were randomized to receive vehicle or M4PTB if serum creatinine values ranged between 0.8 and 1.2 mg/dl on day 4. About 30% of AA-treated mice were excluded as their serum creatinine values lay outside this range before the study was started. Since 20–30% of mice treated with AA die after treatment, largely due to AKI and gastrointestinal tract toxicity (30, 39), we monitored daily weights and euthanized mice if there was >20% weight loss from baseline. Mouse numbers and attrition rates are documented at each time point in the figures. Serum creatinine was evaluated in duplicate samples using a commercially available enzymatic cascade assay (Pointe Scientific, which requires only ∼7 μl of serum). Unlike picric acid-based creatinine assays, this assay closely parallels HPLC measurements of serum creatinine in mice (22). Blood urea nitrogen (BUN) was determined in duplicate samples using a colorimetric enzymatic assay kit (Infinity Urea, Thermo Scientific), and spot urinary albumin/creatinine ratios were determined using a mouse albumin ELISA and urinary creatinine assay kit (Albuwell and Creatinine Companion, Exocell). All mouse work was performed in accordance with the animal use protocol approved by the Institutional Animal Care and User Committee at Vanderbilt University.
Histological analysis for renal injury and fibrosis.
Kidney sections were stained using periodic acid Schiff (PAS) to assess tubular injury, Sirius red (which stains collagens), and collagen IV immunostaining to assess interstitial fibrosis. Semiquantitative tubular injury scoring (0–4) was performed by a blinded observer, as described (7). In addition, we quantified interstitial fibrosis by determining the percent area staining positive for Sirius red from polarized light images using AxioVision software, and collagen IV from immunofluorescence images. For both of these studies, 10 randomly selected ×40 images from within the outer medulla or the cortex of the kidney were captured digitally by a blinded observer, and the percentage of the total surface area staining positive for Sirius red or collagen IV was quantified objectively using Image J software.
Immunohistochemistry.
Immunohistochemisty was performed on formalin-fixed, paraffin-embedded kidney sections. The following primary antibodies were used: mouse monoclonal anti-proliferating cell nuclear antigen (PCNA; 1:50, Clone F2, Santa Cruz Biotechnology), rat monoclonal F4/80 antibodies (1:200, Abd Serotec), rabbit polyclonal anti-Ki67 (1:100, AbCam), anti-phosphorylated Ser10 histone H3 (1:100, pH3, Cell Signaling), and anti-collagen IV (1:500, Abcam) antibodies. Citrate antigen retrieval was used for all antibodies except F4/80 for which sections were treated with 1 mg/ml trypsin (porcine pancreatic, Sigma) for 30 min at 37°C. Primary antibodies were detected using donkey anti-mouse Dylight 488 or anti-rabbit Dylight 549 antibodies (both 1:600, Jackson ImmunoResearch Laboratories). Rat anti-F4/80 antibody was detected using biotinylated anti-rat antibodies and avidin horseradish peroxidase (HRP; ABC kit, Vector Labs) and 3,3′-diaminobenzidine substrate (Sigma). Images were captured using an Olympus BX51 epifluorescence microscope and DP72 digital camera. To quantify cell proliferation, PCNA, Ki67, and pH3 counts were obtained from digitally acquired images by an observer blinded to the treatment group. Colabeling with collagen IV, which stains basement membranes and interstitial collagen IV, was used to identify tubular structures. Four ×400 fields were acquired, and the number of Ki67-, PCNA-, and pH3-positive tubular epithelial cells was counted and expressed as ratio to RTEC DAPI nuclei. To assess F4/80 immunostaining, four ×400 fields were obtained from each outer medulla and cortex and the percent area staining positive for F4/80 (excluding dilated tubules) was also quantified using Image J software.
Quantitative RT-PCR.
Whole kidney RNA was extracted using RNA-Bee (TEL-TEST) and cleaned using RNeasy Mini Kits (Qiagen), according to the manufacturer's protocols. For real-time RT-PCR, cDNA was prepared using a reverse transcriptase cDNA synthesis kit (Life Technologies), and the cDNA was amplified and labeled using SYBR Green Supermix PCR (Life Technologies). We used the following primer pairs: mCollagen3a1 FWD: CTGTAACATGGAAACTGGGGAAA, mCollagen3α1 REV: CCATAGCTGAACTGAAAACCACC; miNOS FWD: GTTCTCAGCCCAACAATACAAGA, miNOS REV: GTGGACGGGTCGATGTCAC; mArg1 FWD: CTCCAAGCCAAAGTCCTTAGAG, mArg1 REV: AGGAGCTGTCATTAGGGACATC; mCCL2 FWD: TTAAAAACCTGGATCGGAACCAA, mCCL2 REV: GCATTAGCTTCAGATTTACGGGT; mCX3CL1 FWD: ACGAAATGCGAAATCATGTGC, mCX3CL1 REV: CTGTGTCGTCTCCAGGACAA; mCCL3 FWD: TGCCCTTGCTGTTCTTCTCT, mCCL3 REV: GATGAATTGGCGTGGAATCT; mMR FWD: CAAGGAAGGTTGGCATTTGT, mMR REV: CCTTTCAGTCCTTTGCAAGC; mGapdh, mCollagen1α1, mα-smooth muscle actin (αSMA), mLoxL2, and mKim1 primers, as described (7). mGapdh was used as loading control as we saw no changes in Gapdh mRNA in AA-AKI (T.N., data not shown).
Western blots.
Tissue lysates were prepared by homogenizing snap-frozen whole kidneys directly in RIPA lysis buffer to which we added protease (Thermo Scientific, Pittsburgh, PA) and phosphatase inhibitor cocktails (Sigma Chemical, St. Louis, MO), as described (34). Western blots were performed on these tissue lysates using rat monoclonal anti-Kim1 (1:1,000, MAB1817 R&D Systems) and rabbit anti-Gapdh polyclonal antibodies (1:5,000, Cell Signaling Technology), and secondary HRP-conjugated chicken anti-rat (1:2,000, Santa Cruz Biotechnology) or goat anti-rabbit antibodies (1:5,000, Cell Signaling Techology). Blots were scanned using a UVP Epichem Darkroom scanner, and individual bands were identified and quantified using ImageJ software. Results were normalized to Gapdh as a loading control.
Statistical analysis.
Statistical analyses were performed by two-tailed Student's t-test for between-group comparisons with the minimal level of significance set at P < 0.05 using Graph Pad Prism 5 software.
RESULTS
M4PTB enhances functional recovery and reduces postinjury fibrosis after AA-induced AKI.
We used an established model in which BALB/c mice are injected with a single dose of AA intraperitoneal (43). Preliminary studies indicated that there was an unacceptably high mortality (∼60%) and inconsistent results at the published dose of 5mg/kg AA, so the dose of AA was reduced to 4.7 mg/kg. At this dose, 36% of mice died over 21 days follow up, but they also developed more consistent results with moderate AKI and long-term renal fibrosis (Fig. 1A). Increased mortality was observed 10 days after AA treatment in control mice. M4PTB was administered intraperitoneally daily at an established dose that has been shown to accelerate recovery and reduce postinjury fibrosis after IR-AKI (7). However, since renal tubular repair is delayed in this model compared with IR-AKI (43), we initiated M4PTB treatment on the 4th day after AA injury. Under these conditions, there was a decrease in mortality in M4PTB-treated mice 21 days after injury, and renal functional recovery (serial serum creatinine values over time) in surviving mice was accelerated by M4PTB treatment (Fig. 1B). By 21 days after AA injury, mice treated with M4PTB showed a significant reduction in serum creatinine [vehicle (n = 14) means ± SE: 0.70 ± 0.08 vs. M4PTB (n = 12): 0.52 ± 0.04 mg/dl, t-test, P < 0.05] and reduced BUN (Fig. 1C). In addition, low-level albuminuria in vehicle-treated mice was significantly reduced compared with M4PTB-treated mice at 21 days (Fig. 1D), suggesting lower levels of renal tubular injury. Consistent with these findings, there was also marked chronic renal tubular injury in the control arm of the study after AA-AKI, and M4PTB treatment reduced tubular injury scores but only in the outer medulla after 21 days (Fig. 1E). This was associated with a marked reduction in Kim1 protein expression, a marker of renal tubular injury (20, 40), in M4PTB-treated mouse kidneys (Fig. 1, F and G). Despite these changes in functional recovery and tubular injury, we were unable to detect any change in the expression fibrosis markers Col1a1, αSMA, LoxL2, or Col3 mRNA in whole kidney extracts from M4PTB-treated mice after AA-AKI (Fig. 2, A–D). However, M4PTB treatment significantly reduces regional deposition of interstitial collagen in the outer medulla as detected by Sirius red staining (Fig. 2, E–G) and by collagen IV immunostaining 21 days after AA injury (Fig. 2, H–J). These findings indicate that M4PTB accelerates functional recovery and has regional effects in reducing tubular atrophy and postinjury fibrosis in the outer medulla after AA-induced AKI.
Fig. 1.
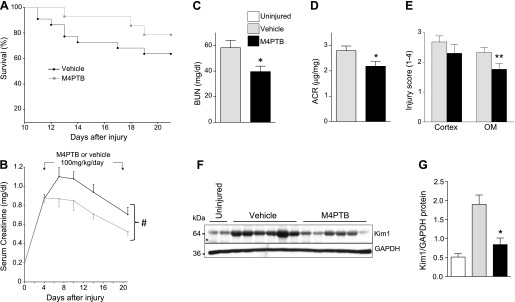
Effect of methyl-4-(phenylthio)butanoate (M4PTB) on survival and kidney function after aristolochic acid-induced acute kidney injury (AA-AKI). Male BALB/c mice received a single dose of 4.7 mg/kg ip AA on day 0. Those with serum creatinine values of 0.8–1.2 mg/dl on day 4 were randomized to receive 100 mg·kg−1·day−1 of M4PTB or vehicle for 17 days. A: survival curves for 22 vehicle- and 15 M4PTB-treated mice with AA-AKI induced at day 0. B: serial serum creatinine in mg/dl on the 14 vehicle- and 12 M4PTB-treated mice that survived 21 days. For statistical analyses, we calculated areas under the curve (AUC) for serial serum creatinine values in vehicle- and M4PTB-treated mice from days 4–21. t-test, #P < 0.05 M4PTB vs. vehicle control AUC. C–G: other renal function tests and injury markers at day 21. C: blood urea nitrogen (BUN; 11 vehicle, 8 M4PTB-treated). D: urinary albumin/creatinine ratios (ACR; 5 vehicle, 6 M4PTB-treated). E: tubular injury scores in the cortex and outer medulla (OM; 0–4 arbitrary units). Fourteen vehicle- and 12 M4PTB-treated mice. F and G: renal Kim1 protein expression. F: Western blot showing renal Kim1 protein expression 21 days after AA-AKI in uninjured mice, and vehicle- and M4PTB-treated mice 21 days after AA-AKI. G: quantification of Kim1 band densitometry relative to Gapdh loading. Results expressed as means ± SE. t-test, *P < 0.05 vs. vehicle control. **P < 0.05 vs. vehicle control in the OM.
Fig. 2.
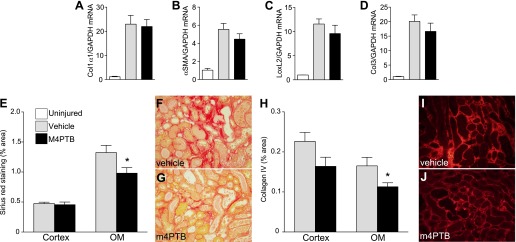
Renal fibrosis after AA-AKI. Kidneys evaluated 21 days after AA injury. A–D: expression of fibrosis markers after AA-AKI. QRT-PCR performed on whole kidney RNA extracted in uninjured kidneys and at 21 days after injury expressed as the ratio of gene expression to Gapdh mRNA. A: collagen 1 α1 chain (Col1α1). B: α-smooth muscle actin (αSMA). C: LoxL2. D: collagen 3 α1 chain (Col3). Uninjured controls (n = 3), vehicle-treated mice after injury (n = 12), M4PTB treatment (n = 10). No significant differences between vehicle and M4PTB treatment groups. E–G: interstitial fibrosis (Sirius red staining). E: quantification of the surface area of Sirius red staining cells in the cortex and OM of mice treated with vehicle or M4PTB. 10 mice per group. F and G: representative images of Sirius red stain showing renal fibrosis in OM of vehicle- and M4PTB-treated mice. H–J: interstitial fibrosis (collagen IV staining) in the cortex and OM in mice treated with vehicle or M4PTB. 10 mice per group. I and J: representative images of Col IV stain in OM of vehicle- and M4PTB-treated mice. 4–6 mice per group. Results expressed as means ± SE. t-test, *P < 0.05 vs. vehicle control in the OM.
M4PTB reduces injury and enhances regenerative capacity of RTECs after AA-AKI.
To explore the mechanism by which M4PTB accelerates recovery after AA-AKI, we evaluated tubular injury at early time points after injury. There was no change in tubular injury score in the outer medulla of M4PTB-treated mouse kidneys at day 7 or 14 after AA injection (Fig. 3A). However, Kim1 mRNA, which is a more sensitive marker of subtle changes in tubular injury than histological tubular injury scores in experimental AKI (40), is significantly decreased in M4PTB-treated mouse kidneys 7 days after AA injury (Fig. 3B). Consistent with Kim1 protein expression data (Fig. 1, F and G), Kim1 mRNA levels are persistently increased 21 days after AA-induced renal injury and are also reduced in M4PTB-treated kidneys. These data indicate that M4PTB has subtle effects in reducing renal tubular injury at early time points after AA-AKI and that these effects persist.
Fig. 3.

Quantification of renal injury after AA-AKI. A: tubular injury scores. Tubular injury scores (04 arbitrary units) obtained from PAS-stained kidney sections at 7 and 14 days after AA injury in mice treated with vehicle or M4PTB. 4–5 mice per group. B: renal Kim-1 mRNA expression. QRT-PCR performed on whole kidney RNA extracted before injury and at 7, 14, and 21 days after injury expressed as the ratio of gene expression to Gapdh mRNA. Uninjured controls (n = 3); day 7 (n = 4 per group); day 14 (n = 8 per group); day 21 (n = 14 vehicle, n = 12 M4PTB treatment). Results expressed as means ± SE. t-test, *P < 0.05 vs. M4PTB-treated kidneys at the same time points.
To determine whether M4PTB also enhances tubular repair after AA-AKI, we evaluated RTEC proliferation at day 14, corresponding to the peak RTEC proliferation in this model (43). Ki67, PCNA, and phospho-Ser10 on histone H3 (pH3) antibodies mark actively cycling cells, cells in S-phase, and in cells in G2/M phases of the cell cycle, respectively (8, 48). All three markers are increased in the outer medulla vs. cortex after AA-AKI (Fig. 4). Ki67 and PCNA are increased in cortical RTECs in M4PTB-treated mice (Fig. 4, A and B). There is also an increase in Ki67- and PCNA-positive RTECs in the outer medulla but this is not statistically significant. In contrast, there is a marked decrease in pH3-positive RTECs that is restricted to outer medulla of M4PTB-treated mice after AA-AKI (Fig. 4C). Morphological analysis of these cells indicates that the majority are arrested in M-phase. Taken together, these data suggest that M4PTB enhances RTEC cell cycle progression in the cortex and reduces RTECs in G2/M arrest in the outer medulla after AA-AKI.
Fig. 4.

Proliferation of renal tubular epithelial cells 14 days after AA injury. A and B: Ki67 (marker of actively cycling cells). A: representative images of renal cortex costained with Ki67 (green) and Col IV (red) in vehicle- and M4PTB-treated mice. Ki67-positive renal tubular epithelial cells (RTECs) can be identified from their relationship with interstitial Col IV staining. B: quantification of Ki67 proliferative index in cortex and OM RTECs. C and D: proliferating cell nuclear antigen (PCNA; marker of S-phase cells). C: representative images of renal cortex costained with PCNA (green) and Col IV (red) in vehicle- and M4PTB-treated mice. D: quantification of PCNA proliferative index in cortex and OM RTECs. E and F: phosphorylated Ser10 histone H3 (pH3, marker of G2 and M phase cells). E: representative images of renal OM costained with pH3 (yellow) and Col IV (red) in vehicle- and M4PTB-treated mice. F: quantification of pH3 proliferative index in cortex and OM RTECs. Results expressed as means ± SE, 5 mice per group. t-test, *P < 0.05 vs. vehicle in the cortex. **P < 0.05 vs. vehicle in the OM.
M4PTB reduces numbers of infiltrating macrophages postinjury.
Macrophages are recruited to sites of injury and play an important role in promoting early, proinflammatory, and late reparative responses after different forms of AKI (32). We therefore sought to determine whether M4PTB influences macrophage recruitment in AA-AKI. Consistent with previous studies in a rat model of AA-induced kidney injury (37), there is a progressive increase in peritubular F4/80-positive cells [a marker of resident and infiltrating macrophages and dendritic cells (3)] in the renal cortex and outer medulla of AA-treated mice. M4PTB reduces macrophage numbers in both compartments at early (day 7) and late (day 21) time points after AA injury (Fig. 5). To explore the mechanism by which M4PTB decreases renal macrophages after AA injury, we evaluated expression of chemokines that play an important role in recruiting circulating macrophages to the kidney post-AKI, CX3CL1/Fractalkine, and CCL2/MCP1 (24, 35). CCL2, and to a lesser extent CX3CL1, expression is increased 7–21 days after AA injury. M4PTB reduces CX3CL1 and CCL2 expression at early and late time points after the initiating injury, respectively (Fig. 6, A and B). This suggests that M4PTB reduces macrophage recruitment to the kidney by decreasing macrophage chemokine expression after injury. Infiltrating macrophage functions are associated with broad changes in macrophage phenotypes and expression profiles from proinflammatory M1 to regenerative M2 macrophages in different models of AKI (14). M4PTB treatment reduces expression of both the M1 marker CCL3/MIP-1α and the M2 markers arginase-1 (Arg-1) and mannose receptor (MR) after AA injury (Fig. 6, C–E). Inducible nitric oxide synthase (iNOS) expression, which is a classical M1 marker, progressively increases over time after AA injury and is unaffected by M4PTB (Fig. 6F). However, unlike CCL3 which is largely restricted to macrophages and neutrophils (28), iNOS is expressed in tubular epithelium and glomeruli after injury (5, 6, 36), so changes in iNOS may not reflect changes in macrophage polarity. Taken together, these findings suggest that M4PTB reduces macrophage numbers after AA-AKI but does not have a selective effect on proinflammatory vs. proregenerative macrophage phenotypes.
Fig. 5.

Infiltrating macrophages after AA-AKI. A: quantification of F4/80 staining. Surface area of F4/80-stained cells in the cortex and OM at 7, 14, and 21 days after AA injury. Results expressed as means ± SE, 5 mice per group at day 7 and 14, 10 at day 21. t-test, *P < 0.05 vs. vehicle cortex. **P < 0.05 vs. vehicle OM. B: representative images of F4/80 staining (brown color) in vehicle- and M4PTB-treated mouse kidneys at 7 and 21 days after AA-induced renal injury.
Fig. 6.

Expression of macrophage-specific chemokines and polarity markers after AA-AKI. QRT-PCR performed on whole kidney RNA extracted in uninjured kidneys and at 7, 14, and 21 days after injury expressed as the ratio of gene expression to Gapdh mRNA. A and B: chemokine expression. A: CCl2/MCP1. B: CX3CL1/Fractalkine. C–F: macrophage polarity markers (M1 and M2). C: arginase-1 (M2). D: mannose receptor (M2). E: CCL3 (M1). F: inducible nitric oxide synthase (iNOS; M1). Results expressed as means ± SE, 4–7 mice per group. t-test, *P < 0.05 vs. M4PTB-treated kidneys at the same time points.
DISCUSSION
Our data show that M4PTB treatment initiated 4 days after injury reduces tubular injury, accelerates recovery, and reduces long-term postinjury fibrosis after AA-AKI. These effects are regional, localized to the outer medulla of the kidney, and associated with sustained improvement in renal function. M4PTB treatment also increases proliferation of surviving RTECs in the renal cortex and reduces numbers of RTECs in G2/M arrest in the outer medulla. There is also a marked reduction in infiltrating macrophages throughout the cortex and outer medulla in M4TPB-treated mouse kidneys over a 21-day period after injury. These findings establish that M4PTB is effective therapeutically in a progressive form of kidney injury resulting from accumulation of genotoxic damage over time, and they suggest that these effects may result from reduced injury, enhanced regenerative repair of RTECs, and reduced macrophage recruitment to the injured kidney. These observations may have clinical significance since M4PTB treatment can be delayed 4 days after AA injury and suggest that patients presenting with AA-induced AKI may respond to delayed M4PTB treatment.
M4PTB has regional effects on collagen deposition in the outer medulla that was detected using two independent, quantitative histological assays. Moreover, persistent improvement in renal function after M4PTB treatment suggests that these regional anti-fibrotic effects of M4PTB are functionally important. However, these findings contrast with the fact that we were unable to detect changes in expression of a panel of profibrotic marker mRNAs in whole kidney extracts from mice treated with M4PTB. Since AA induces widespread injury throughout the renal cortex and medulla (30, 39), the most likely explanation for this discrepancy is that anti-fibrotic effects of M4PTB in the outer medulla are masked in whole kidney extracts by the dominant fibrosis that persists throughout the rest of the kidney.
Apart from renal replacement therapy for ESRD, there is currently very little nephrologists can do to delay the inexorable progression of CKD after AA ingestion in humans (17). Unlike other models of CKD, AAN does not respond to renin-angiotensin blockade (11), and prednisolone therapy to suppress the inflammatory response in AAN has only been shown to be of benefit in anecdotal clinical reports (41). In addition, while preclinical studies have identified a number of potential approaches to address this, many of these treatments would have to be administered at the time of AA ingestion and are therefore of limited practical use in a clinical setting. For example, the growth factor erythropoietin, when administered at the time of AA ingestion, reduces tubular atrophy and interstitial fibrosis in a model of chronic AAN, but it is ineffective if treatment is delayed (19). One promising drug candidate that can be administered late after injury and still reduce post-AKI fibrosis is the p53 inhibitor pifithrin-α. Like M4PTB, pifithrin-α is thought to act by reducing the number of maladaptive RTECs in G2/M arrest post-AKI (44). Pifithrin-α reduces acute AA-induced RTEC injury when administered 3 days after AA injury (51), but the effects of p53 inhibition on renal fibrosis after AA-AKI have not been established. However, pifithrin-α given at 3 and 14 days after severe IR-AKI reduces long-term postinjury fibrosis (43). Nevertheless, unlike M4PTB, the temporal windows over which pifithrin-α can be used without exacerbating long-term fibrosis appear to be narrow since it has also been shown that short-term daily treatment with pifithrin-α for 7 days after injury increases postinjury fibrosis after IR-AKI (9).
There is marked infiltration with CD68-positive macrophages in the cortex and outer medulla of AA-treated rat kidneys beginning 7 days after injury and persisting for up to 35 days (37). Our studies demonstrate a similar, progressive accumulation of peritubular F4/80-positive macrophages after AA injury in mice, and M4PTB abrogates this response at both early (7 days) and late time points (21 days) after injury. Since numbers of intrarenal macrophages reflect the severity of renal injury (38), these findings are consistent with our observation that M4PTB enhances repair after AA injury. However, the functional role of renal macrophage accumulation after AA-AKI is poorly understood. Our studies suggest that M4PTB treatment does not have a selective effect on proinflammatory vs. proregenerative macrophage phenotypes after AA-AKI. However, unlike IR-AKI in which expression of M1 macrophage markers is transient and appears early after injury (23), our studies also show that expression of the M1 markers CCL3 and iNOS increases progressively after AA injury. Since proinflammatory M1 macrophages promote tubular injury after AKI (32), this suggests there may be progressive macrophage-dependent renal tubular injury after AA-AKI. Therefore, by reducing macrophage numbers, M4PTB may also be exerting long-term beneficial effects in reducing macrophage-dependent renal tubular injury following AA-induced kidney injury. Our data also provide insight into how M4PTB decreases intrarenal macrophage recruitment after AA injury. M4PTB decreases expression of the chemokines CX3Cl1 and CCl2 at early (7 days) and later time points (14 and 21 days) after AA injury, respectively. Since these chemokines play a critical role in recruitment of circulating macrophages through activation of their cognate receptors on macrophages after AKI (24), it is possible that M4PTB reduces intrarenal macrophage recruitment by decreasing expression of CX3Cl1 and/or CCL2 by the kidney after injury. Further studies will be required to determine whether these changes in CX3Cl1 and CCL2 mRNA mediate M4PTB-dependent changes in renal macrophage recruitment after AA-AKI. Furthermore, the molecular mechanism by which M4PTB decreases CCL2 and CX3Cl1 expression is unclear. However, injured RTECs express CCl2, and CX3Cl1 after AKI (15, 33), so that this effect may result from enhanced repair of damaged tubular epithelial cells that we observe after M4PTB treatment post-AKI.
Long-term treatment with a variety of HDACis reduces progressive kidney disease in models of diabetic and HIV nephropathy (1, 50), and in immune-mediated glomerulonephritis (21, 31). In addition, short-term treatment with the selective HDACi, MS-275, reduces fibrosis after unilateral ureteric obstruction (26). PTBA analogs also have HDAC inhibitory activity in vitro and in vivo (7, 10), but it is unknown whether other HDACi would have similar anti-fibrotic effects in AA-AKI. Having said that, the mechanism of action of M4PTB post-AKI is likely to be different from that of long-term treatment with other HDACi in models of established CKD. We previously showed that short-term treatment (7 days) with M4TPB initiated 24 h after injury reduces long-term postinjury fibrosis after severe IR-AKI (7). One explanation for this effect is that M4PTB exerts cytoprotective effects on the kidney after IR injury, so that improved long-term outcome results from amelioration of the initial injury. This hypothesis is consistent with evidence that the HDAC inhibitors have cytoprotective effects on cultured neuronal and myocardial cells exposed to hypoxia (18, 29) and that they can reduce cerebral and myocardial infarct size following cerebral and coronary artery occlusion, respectively, in mice (18, 47). However, we saw no change in histological tubular injury or expression of the tubular injury marker Kim1 mRNA in M4PTB-treated kidneys at an early time point (3 days) after IR-AKI (7), suggesting that the beneficial effects of M4PTB on post-AKI recovery after IR-AKI do not result from a reduction in the initial phases of renal injury. In contrast, while our data also show that M4PTB treatment does not reduce histological evidence of acute tubular injury after AA-AKI, there is a significant reduction in renal Kim1 mRNA expression 7 days after AA injury, 3 days after initiating M4PTB treatment. Since changes in Kim1 mRNA can occur without histological changes in renal tubular cell injury after AKI (40), these data suggest that unlike IR-AKI, M4PTB treatment after AA-induced AKI has subtle effects in reducing tubular injury.
We previously demonstrated an early upregulation of genes promoting cell cycle progression in M4PTB-treated kidneys after IR-AKI, particularly in genes promoting transition through the G2/M phase of the cell cycle (7). This was associated with an increase in actively cycling RTECs during the peak regenerative phase 72 h after IR injury, along with a reduction in RTECs arrested in G2/M. In our current studies, we show that M4TPB exerts similar effects on RTEC proliferation and on G2/M arrest after AA-AKI. However, these effects are regional, since M4PTB only significantly increases numbers of proliferating RTECs in the cortex and decreases numbers of RTECs in G2/M arrest in the outer medulla. Since maladaptive RTECs in the G2/M arrest secrete profibrotic factors that drive postinjury fibrosis in different AKI models (25, 43, 49), we speculate that M4PTB reduces local postinjury fibrosis by reducing numbers of maladaptive RTECs in G2/M arrest in the outer medulla post-AKI. While there was a parallel increase in actively proliferating RTECs in the outer medulla with M4PTB, these changes were not statistically significant, suggesting that the observed anti-fibrotic effects of M4PTB in the outer medulla do not substantially result from increased cellular repair in this model. This contrasts with the observation that M4PTB accelerates recovery and increases PTEC proliferation after toxin-induced AKI in zebrafish (7), and it does not exclude the possibility that increased RTEC proliferation in other models, such as IR-AKI (7), may also contribute to repair. It is unclear why effects of M4PTB on G2/M arrest are regional. AA is concentrated in the S3 segment PTECs in the outer medulla (4), so there is likely to be increased DNA damage-induced G2/M arrest in this region of the kidney. Our data indicate this may be the case since most of cells in G2/M arrest are restricted to the outer medulla. Therefore, it is possible that the effect of M4PTB on this maladaptive injury response is only apparent when and where G2/M arrest is strongly activated. These effects may be particularly beneficial in reducing postinjury fibrosis after AA-AKI since surviving RTECs show a marked increase in G2/M arrest after AA injury (43, 45).
GRANTS
M. P. de Caestecker was supported by National Institutes of Health (NIH) Grants RC4DK090770, P30DK079341, R03HL115112. N. A. Hukriede was supported by NIH Grants R01DK069403, RC4DK090770, P30DK079307, and R01HD053287.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: T.N., N.H., and M.P.d.C. conception and design of research; T.N., L.M., K.X.Z., T.C., and P.P. performed experiments; T.N. and T.C. analyzed data; T.N. and M.P.d.C. interpreted results of experiments; T.N. and M.P.d.C. prepared figures; T.N., L.M., P.P., N.H., and M.P.d.C. edited and revised manuscript; T.N., L.M., K.X.Z., P.P., N.H., and M.P.d.C. approved final version of manuscript; M.P.d.C. drafted manuscript.
ACKNOWLEDGMENTS
We thank Haichun Yang for assistance setting up quantitative assays for renal fibrosis.
REFERENCES
- 1.Advani A, Huang Q, Thai K, Advani SL, White KE, Kelly DJ, Yuen DA, Connelly KA, Marsden PA, Gilbert RE. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol 178: 2205–2214, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arlt VM, Stiborova M, Schmeiser HH. Aristolochic acid as a probable human cancer hazard in herbal remedies: a review. Mutagenesis 17: 265–277, 2002 [DOI] [PubMed] [Google Scholar]
- 3.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol 11: 805–815, 1981 [DOI] [PubMed] [Google Scholar]
- 4.Baudoux TE, Pozdzik AA, Arlt VM, De Prez EG, Antoine MH, Quellard N, Goujon JM, Nortier JL. Probenecid prevents acute tubular necrosis in a mouse model of aristolochic acid nephropathy. Kidney Int 82: 1105–1113, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Broadbelt NV, Chen J, Silver RB, Poppas DP, Felsen D. Pressure activates epidermal growth factor receptor leading to the induction of iNOS via NFκB and STAT3 in human proximal tubule cells. Am J Physiol Renal Physiol 297: F114–F124, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi JY, Nam SA, Jin DC, Kim J, Cha JH. Expression and cellular localization of inducible nitric oxide synthase in lipopolysaccharide-treated rat kidneys. J Histochem Cytochem 60: 301–315, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cianciolo Cosentino C, Skrypnyk NI, Brilli LL, Chiba T, Novitskaya T, Woods C, West J, Korotchenko VN, McDermott L, Day BW, Davidson AJ, Harris RC, de Caestecker MP, Hukriede NA. Histone deacetylase inhibitor enhances recovery after AKI. J Am Soc Nephrol 24: 943–953, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol 22: 874–885, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dagher PC, Mai EM, Hato T, Lee SY, Anderson MD, Karozos SC, Mang HE, Knipe NL, Plotkin Z, Sutton TA. The p53 inhibitor pifithrin-α can stimulate fibrosis in a rat model of ischemic acute kidney injury. Am J Physiol Renal Physiol 302: F284–F291, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Groh ED, Swanhart LM, Cosentino CC, Jackson RL, Dai W, Kitchens CA, Day BW, Smithgall TE, Hukriede NA. Inhibition of histone deacetylase expands the renal progenitor cell population. J Am Soc Nephrol 21: 794–802, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debelle FD, Nortier JL, Husson CP, De Prez EG, Vienne AR, Rombaut K, Salmon IJ, Deschodt-Lanckman MM, Vanherweghem JL. The renin-angiotensin system blockade does not prevent renal interstitial fibrosis induced by aristolochic acids. Kidney Int 66: 1815–1825, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Debelle FD, Vanherweghem JL, Nortier JL. Aristolochic acid nephropathy: a worldwide problem. Kidney Int 74: 158–169, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Dickman KG, Sweet DH, Bonala R, Ray T, Wu A. Physiological and molecular characterization of aristolochic acid transport by the kidney. J Pharmacol Exp Ther 338: 588–597, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duffield JS. Macrophages and immunologic inflammation of the kidney. Semin Nephrol 30: 234–254, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durkan AM, Alexander RT, Liu GY, Rui M, Femia G, Robinson LA. Expression and targeting of CX3CL1 (fractalkine) in renal tubular epithelial cells. J Am Soc Nephrol 18: 74–83, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Faubel S, Chawla LS, Chertow GM, Goldstein SL, Jaber BL, Liu KD. Ongoing clinical trials in AKI. Clin J Am Soc Nephrol 7: 861–873, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Gokmen MR, Cosyns JP, Arlt VM, Stiborova M, Phillips DH, Schmeiser HH, Simmonds MS, Cook HT, Vanherweghem JL, Nortier JL, Lord GM. The epidemiology, diagnosis, and management of aristolochic acid nephropathy: a narrative review. Ann Intern Med 158: 469–477, 2013 [DOI] [PubMed] [Google Scholar]
- 18.Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, Epstein JA, Gruber PJ. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J 22: 3549–3560, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamano Y, Aoki T, Shirai R, Hatano M, Kimura R, Ogawa M, Yokosuka O, Ueda S. Low-dose darbepoetin alpha attenuates progression of a mouse model of aristolochic acid nephropathy through early tubular protection. Nephron Exp Nephrol 114: e69–e81, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is upregulated in renal cells after injury. J Biol Chem 273: 4135–4142, 1998 [DOI] [PubMed] [Google Scholar]
- 21.Imai N, Hishikawa K, Marumo T, Hirahashi J, Inowa T, Matsuzaki Y, Okano H, Kitamura T, Salant D, Fujita T. Inhibition of histone deacetylase activates side population cells in kidney and partially reverses chronic renal injury. Stem Cells 25: 2469–2475, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Keppler A, Gretz N, Schmidt R, Kloetzer HM, Groene HJ, Lelongt B, Meyer M, Sadick M, Pill J. Plasma creatinine determination in mice and rats: an enzymatic method compares favorably with a high-performance liquid chromatography assay. Kidney Int 71: 74–78, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, Huang L, Sung SS, Vergis AL, Rosin DL, Rose CE, Jr, Lobo PI, Okusa MD. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int 74: 1526–1537, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin SL, Li B, Rao S, Yeo EJ, Hudson TE, Nowlin BT, Pei H, Chen L, Zheng JJ, Carroll TJ, Pollard JW, McMahon AP, Lang RA, Duffield JS. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc Natl Acad Sci USA 107: 4194–4199, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu N, He S, Ma L, Ponnusamy M, Tang J, Tolbert E, Bayliss G, Zhao TC, Yan H, Zhuang S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLos One 8: e54001, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lo LJ, Go AS, Chertow GM, McCulloch CE, Fan D, Ordonez JD, Hsu CY. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int 76: 893–899, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23: 549–555, 2002 [DOI] [PubMed] [Google Scholar]
- 29.Meisel A, Harms C, Yildirim F, Bosel J, Kronenberg G, Harms U, Fink KB, Endres M. Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem 98: 1019–1031, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Mengs U. Acute toxicity of aristolochic acid in rodents. Arch Toxicol 59: 328–331, 1987 [DOI] [PubMed] [Google Scholar]
- 31.Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest 111: 539–552, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J. The renal mononuclear phagocytic system. J Am Soc Nephrol 23: 194–203, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishihara K, Masuda S, Shinke H, Ozawa A, Ichimura T, Yonezawa A, Nakagawa S, Inui K, Bonventre JV, Matsubara K. Urinary chemokine (C-C motif) ligand 2 (monocyte chemotactic protein-1) as a tubular injury marker for early detection of cisplatin-induced nephrotoxicity. Biochem Pharmacol 85: 570–582, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novitskaya T, Baserga M, de Caestecker MP. Organ-specific defects in insulin-like growth factor and insulin receptor signaling in late gestational asymmetric intrauterine growth restriction in Cited1 mutant mice. Endocrinology 152: 2503–2516, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh DJ, Dursun B, He Z, Lu L, Hoke TS, Ljubanovic D, Faubel S, Edelstein CL. Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am J Physiol Renal Physiol 294: F264–F271, 2008 [DOI] [PubMed] [Google Scholar]
- 36.Pfeilschifter J. Nitric oxide triggers the expression of proinflammatory and protective gene products in mesangial cells and the inflamed glomerulus. Nephrol Dial Transplant 17: 347–348, 2002 [DOI] [PubMed] [Google Scholar]
- 37.Pozdzik AA, Salmon IJ, Husson CP, Decaestecker C, Rogier E, Bourgeade MF, Deschodt-Lanckman MM, Vanherweghem JL, Nortier JL. Patterns of interstitial inflammation during the evolution of renal injury in experimental aristolochic acid nephropathy. Nephrol Dial Transplant 23: 2480–2491, 2008 [DOI] [PubMed] [Google Scholar]
- 38.Rees AJ. Monocyte and macrophage biology: an overview. Semin Nephrol 30: 216–233, 2010 [DOI] [PubMed] [Google Scholar]
- 39.Sato N, Takahashi D, Chen SM, Tsuchiya R, Mukoyama T, Yamagata S, Ogawa M, Yoshida M, Kondo S, Satoh N, Ueda S. Acute nephrotoxicity of aristolochic acids in mice. J Pharm Pharmacol 56: 221–229, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Vaidya VS, Ozer JS, Dieterle F, Collings FB, Ramirez V, Troth S, Muniappa N, Thudium D, Gerhold D, Holder DJ, Bobadilla NA, Marrer E, Perentes E, Cordier A, Vonderscher J, Maurer G, Goering PL, Sistare FD, Bonventre JV. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol 28: 478–485, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vanherweghem JL, Abramowicz D, Tielemans C, Depierreux M. Effects of steroids on the progression of renal failure in chronic interstitial renal fibrosis: a pilot study in Chinese herbs nephropathy. Am J Kidney Dis 27: 209–215, 1996 [DOI] [PubMed] [Google Scholar]
- 42.Wald R, Quinn RR, Luo J, Li P, Scales DC, Mamdani MM, Ray JG. Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA 302: 1179–1185, 2009 [DOI] [PubMed] [Google Scholar]
- 43.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L, Humphreys BD, Bonventre JV. Pathophysiology of acute kidney injury to chronic kidney disease: maladaptive repair. Contrib Nephrol 174: 149–155, 2011 [DOI] [PubMed] [Google Scholar]
- 45.Yang L, Li X, Wang H. Possible mechanisms explaining the tendency towards interstitial fibrosis in aristolochic acid-induced acute tubular necrosis. Nephrol Dial Transplant 22: 445–456, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Yang L, Su T, Li XM, Wang X, Cai SQ, Meng LQ, Zou WZ, Wang HY. Aristolochic acid nephropathy: variation in presentation and prognosis. Nephrol Dial Transplant 27: 292–298, 2012 [DOI] [PubMed] [Google Scholar]
- 47.Yildirim F, Gertz K, Kronenberg G, Harms C, Fink KB, Meisel A, Endres M. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp Neurol 210: 531–542, 2008 [DOI] [PubMed] [Google Scholar]
- 48.Yu CC, Woods AL, Levison DA. The assessment of cellular proliferation by immunohistochemistry: a review of currently available methods and their applications. Histochem J 24: 121–131, 1992 [DOI] [PubMed] [Google Scholar]
- 49.Zahedi K, Revelo MP, Barone S, Wang Z, Tehrani K, Citron DP, Bissler JJ, Rabb H, Soleimani M. Stathmin-deficient mice develop fibrosis and show delayed recovery from ischemic-reperfusion injury. Am J Physiol Renal Physiol 290: F1559–F1567, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Zhong Y, Chen EY, Liu R, Chuang PY, Mallipattu SK, Tan CM, Clark NR, Deng Y, Klotman PE, Ma'ayan A, He JC. Renoprotective effect of combined inhibition of angiotensin-converting enzyme and histone deacetylase. J Am Soc Nephrol 24: 801–811, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou L, Fu P, Huang XR, Liu F, Lai KN, Lan HY. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J Am Soc Nephrol 21: 31–41, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]