

Abstract
Aging has been associated with pathological vascular remodeling and increased neointimal hyperplasia. The understanding of how aging exacerbates this process is fundamental to prevent cardiovascular complications in the elderly. This study proposes a mechanism by which aging sustains leukocyte adhesion, vascular inflammation, and increased neointimal thickness after injury. The effect of aging on vascular remodeling was assessed in the rat balloon injury model using microarray analysis, immunohistochemistry, and LINCOplex assays. The injured arteries in aging rats developed thicker neointimas than those in younger animals, and this significantly correlated with a higher number of tissue macrophages and increased vascular IL-18. Indeed, IL-18 was 23-fold more abundant in the injured vasculature of aged animals compared with young rats, while circulating levels were similar in both groups of animals. The depletion of macrophages in aged rats with clodronate liposomes ameliorated vascular accumulation of IL-18 and significantly decreased neointimal formation. IL-18 was found to inhibit apoptosis of vascular smooth muscle cells (VSMC) and macrophages, thus favoring both the formation and inflammation of the neointima. In addition, injured arteries of aged rats accumulated 18-fold more fibrinogen-γ than those of young animals. Incubation of rat peritoneal macrophages with immobilized IL-18 increased leukocyte adhesion to fibrinogen and suggested a proinflammatory positive feedback loop among macrophages, VSMC, and the deposition of fibrinogen during neointimal hyperplasia. In conclusion, our data reveal that concentration changes in vascular cytokine and fibrinogen following injury in aging rats contribute to local inflammation and postinjury neointima formation.
Keywords: inflammation, vascular injury, aging, neointima, vascular smooth muscle cells
aging is recognized as a major and independent risk factor for the development of vascular restenosis (29). This risk can be explained by profound, age-dependent changes in vascular structure and physiology that lead to endothelial dysfunction (18) and a chronic inflammatory stage (53), which impair healing and render a vascular system prone to develop occlusive neointimas in response to injury. However, even when this notion is largely accepted, the mechanisms by which aging alters vascular physiology and repair have remained understudied to date.
The process of aging increases and sustains inflammation within the vascular wall (52). Macrophages are commonly recognized as the most important cell type in the chronicity of this process (37). Indeed, inflammation in arteries of aged individuals is evidenced by an excessive accumulation of macrophages in the inner layer of the arterial wall (intima), and this occurs in the presence or absence of lipid-rich lesions (26, 39). Additionally, the number of macrophages in the neointima predicts restenosis after coronary intervention (37), an end result that is associated with the development of neointimal hyperplasia (NIH). Similarly, vascular inflammation is also exacerbated after mechanical injury in aged animals. Eghbalieh et al. (8) have shown that arteries of aged rats retain more macrophages after injury than those of young animals. Altogether, these studies suggest an important cross talk between inflammatory cells and vascular smooth muscle cells (VSMC) in the pathogenesis of vascular aging. The molecular mechanism as to why aged arteries recruit and retain more macrophages after injury is not known, but a change in the vascular wall composition is a logical culprit. Moreover, the infiltrated macrophages actively produce proinflammatory cytokines and a variety of growth factors that contribute to the activation of “resting” VSMC and thickness of the intima.
Aging could also enhance VSMC proliferative capacity, significantly increasing the number of cells undergoing the S phase of the cell cycle (16). Stemerman et al. (48) demonstrated in the rat balloon injury model that cells in arteries of aged rats proliferate faster than those of their younger counterparts. More recently, these results have been reproduced in mice (51) and rabbits (45). Interestingly, VSMC from aging animals also produce more monocyte chemoattractant protein-1 and interlukin-6 (IL-6) than those from young animals (44, 46), factors that just as in humans contribute to the formation of an age-related proinflammatory substrate in the intima (53). In addition, aged VSMC produce more reactive oxygen species after mitogenic stimulation compared with their younger counterparts (35), and are more resistant to apoptosis (51). Nevertheless, proliferation alone is not enough to sustain a thicker neointima. Instead, age-related changes in the vascular wall composition further promote VSMC migration to the subendothelial space, recruitment of circulating monocytes and adhesion of platelets. Fibrinogen accumulation is also pronounced in inflammatory vascular processes (47), and age-related increments in circulating levels in humans are associated with a number of cardiovascular complications (24).
The present study explores the effects of age-related changes in vascular cytokine concentrations and the vascular wall composition after injury. We demonstrate that 1) aging increases postinjury accumulation of arterial fibrinogen to favor monocyte adhesion and extravasation; 2) tissue macrophages infiltrated in the aged vasculature are responsible for the excessive accumulation of IL-18 and neointima formation; and 3) IL-18 in aged injured vessels increases macrophage adhesion, exacerbates inflammation, and protects VSMC from apoptosis. In conclusion, this study suggests the existence of a novel proinflammatory positive feedback loop that contributes to the exaggerated neointima formation observed in blood vessels of aged animals after injury.
MATERIALS AND METHODS
Balloon injury model.
All animal care was in compliance with the Principles of Laboratory Animal Care formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals prepared by the National Institutes of Health (NIH Publication No. 85–23, revised 1996). All studies were previously approved by the Institutional Committee for Use and Care of Laboratory Animals at the University of Miami. Aged male Fisher rats (21- to 23-mo-old, F344) were purchased from the National Institute on Aging (Bethesda, MD). Young male Fisher (2–4 mo-old) rats were obtained from Harlan Laboratories (Indianapolis, IN). All operative procedures were performed while the animals were under isoflurane anesthesia (Baxter, Irvine, CA). A balloon injury was inflicted in the right iliac artery with a 2F Fogarty catheter (Baxter) adapted to a custom angiographic kit (Boston Scientific, Scimed) (11). The balloon catheter was always inflated to yield a constant pressure between 1.5 and 1.6 atmospheres. Arterial specimens were collected 30 min, 3 h, and 1, 3, 7, 14 and 30 days after injury from formalin-perfused-fixed animals (12). Alternatively, animals were PBS perfused and arterial tissue was snap-frozen for protein and RNA analysis.
Systemic and local depletion of macrophages.
Systemic and local depletion of macrophages/monocytes in young and aged F344 rats was accomplished as described previously (4, 5). Clodronate and control liposomes were obtained from the Clodronate Liposomes Foundation (http://clodronateliposomes.org). Liposomal clodronate (5 mg/kg) was intravenously injected on days −2 and +6 after arterial balloon injury. Iliac arteries, spleens, and heparinized blood were harvested from injured and noninjured treated and control animals at days 3, 7, and 30 after injury for histopathological analysis and cell blood counts. Monocyte/macrophage deletion was assessed in blood and spleen sections by immunohistochemistry from treated and control animals at day 3 postsurgery. Rat cell blood count parameters were determined in the Comparative Pathology Laboratory at the University of Miami.
Histology and morphometric analysis.
Paraffin embedding and sectioning were performed by American Histolabs (Bethestha, MD). Iliac arteries were cut in three pieces that were embedded together in the same block. Eight to ten sections spaced every 30 μM were cut from each block and stained with hematoxylin and eosin for morphometric analysis. The areas inside the lumen (L), and internal and external elastic lamina (IEL and EEL, respectively) were directly measured in each slide. The media ([M = EEL − IEL]), neointimal area ([N = IEL − L]) and the neointima to neointima-media ratio {[N/NM = N/(N + M)]} were calculated. Morphometric measurements were performed on digital images using Image Pro Plus (Media Cybernetics, Bethesda, MD) by an investigator blinded to the experimental group. The final N/NM ratio per animal represents the average of all measurements obtained from 8–10 slides.
Immunohistochemistry.
Sections were deparaffinized and rehydrated by serially immersing them in xylene, alcohol, and water. After tissue rehydration, endogenous peroxidase was blocked with 3% hydrogen peroxide. Epitope retrieval was performed by boiling slides in citrate buffer (10 mM sodium citrate, pH 6.0) for 25 min. Nonspecific binding was blocked with 0.5% blocking solution (DAKO, Carpinteria, CA). Subsequently, the following primary antibodies were added for 1 h at room temperature: mouse IgG anti-rat fibrinogen-γ (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-rat CD68 (1:50; AbD Serotech), rabbit IgG anti-rat IL-18 (1:50; Santa Cruz Biotechnology), and mouse anti-human (rat cross-reactivity) smooth muscle actin (1:400; DAKO). Bound antibodies were detected using the DAKO Universal link kit (DAKO). Color was developed with a DAB chromogenic solution (DAKO). Nuclei were counterstained with Meyer's hematoxylin and mounted in Entellan mounting medium (EMD, Gibbstown, NJ). Images were taken with an Olympus 1X71 camera fitted to an Olympus BX 40 microscope (Olympus America, Center Valley, PA).
Immunofluorescence.
Sections were deparaffinized, rehydrated, and antigen was retrieved as described above. Nonspecific binding sites were blocked with 10% goat serum (Chemicon International, Temecula, CA) in PBS for 1 h at room temperature (RT). Primary monoclonal antibodies mouse anti-rat CD68 (1:50) and rabbit IgG anti-rat IL-18 (1:50, Santa Cruz Biotechnology) were diluted with the blocking solution and applied onto sections overnight at 4°C. After being washed twice with PBS for 3 min, tissue sections were incubated with Alexa Fluor 546 goat anti-mouse and Alexa Fluor goat anti-rabbit 488 (Invitrogen, Carlsbad, CA) for 90 min at RT. Sections were mounted in Vectashield w/DAPI (Vector Laboratories, Burlingame, CA) and examined with a Olympus IX71 in an inverted configuration (Olympus Imaging America Inc, Central Valley, PA). Data were captured and analyzed with the Image Pro Plus.
Double-labeling assay for detecting apoptotic VSMC.
Apoptosis was detected in paraffin sections of arterial tissues using the ApopTag Peroxidase In Situ Oligo Ligation (ISOL) Kit (Millipore, Billerica, MA) according to the manufacturer's guidelines with some modifications. Epitope retrieval was performed by boiling slides in citrate buffer. Sections were twice rinsed in PBS-Tween 2% (Sigma-Aldrich) after developing color in peroxidase substrate for detection of apoptotic nuclei. Then, sections were incubated with either an anti-smooth muscle actin clone 1A4 (1:400; DAKO) or anti-CD68 (1:50; AbD Serotech) monoclonal antibody for 1 h at RT. The Envision G/2 System/AP (permanent red) kit (DAKO) was used to visualize VSMC. Sections were mounted in Faramount aqueous mounting medium (DAKO). Apoptotic cells were identified by a dark brown nucleus surrounded by red cytoplasm. The percentage of positively stained apoptotic cells was determined by randomly counting 10 fields per section.
Apoptosis in cell lines.
Rat VSMC were preincubated in the presence or absence of IL-18 (0–100 ng/ml; Peprotech, Rocky Hill, NJ) for 4 h before addition of 64 μM of camptothecin (CAM) for 24 h to induced cell death by apoptosis (38). Attached cells were dissociated using mild trypsinization and double-stained with annexin V-FITC and propidium iodide (PI) according to the manufacturer's instructions (Biovision, Milpitas, CA). Stained cells were detected with an LSRII flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo 7.6.1 software (Ashland, OR).
Cytokine quantification.
The screening for vascular cytokines was performed with a LINCOplex Rat Cytokine Assay (24-Plex; EMD Millipore). Briefly, frozen arterial tissues were minced with a BioPulverizer (Biospec, Bartlesville, OK) and homogenized with an Ultra-Turrax T8 homogenizer (IKA Works, Wilmington, NC) in 200 μl of RIPA buffer (Millipore) supplemented with 10 μg/ml leupeptin, 20 mU/ml aprotinin, 10 μM PMSF, 10 μM NaF, and 10 μM NaVO4. Proteins were clarified by centrifugation and quantified with the Bio-Rad DC Protein Assay (Bio-Rad Laboratories, Hercules, CA). A total of 100 μg of proteins were loaded per well that already contained the precoated color coded beads. The cytokines bound to the fluorescent beads were detected using protein-specific, biotinylated detector antibodies followed by incubation with streptavidin-PE, both provided with the kit. Multiplex were resolved in a Bio-Plex 100 (Bio-Rad) bead sorter. Values were expressed as fold of noninjured arteries. The average of each cytokine expression per time point was represented as a heat map generated with the GeneSpring GX 11 software (Silicon Genetics, Redwood City, CA). Plasma levels of rat IL-18 and interleukin-18 binding protein (IL-18BP) were measured using commercial ELISA systems from Life Technologies (Grand Island, NY) and MyBiosource (San Diego, CA), respectively, according to manufacturer instructions.
Gene expression.
Total RNAs were extracted from injured and noninjured (control) iliac arteries from aged and young rats using TRI Reagent (Molecular Research Center, Cincinnati, OH), followed by an extra-cleaning step with the RNeasy kit (Qiagen, Valencia, CA). The RNAs were validated in the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) before reverse-transcribing them in the presence of aminoallyl-dUTP (Life Technologies, Grand Island, NY). Obtained cDNAs were labeled in carbonate buffer (pH 9.0–9.3) supplemented either with Cy3- or Cy5-NHS ethers. Two-color Agilent Whole Rat Genome Arrays (Agilent Technologies) were used to profile differentially expressed genes in young vs. aged arteries before and after injury. Data extracted from array images were transferred to the software package Acuity 4.0 (Axon Instruments, Union City, CA) for normalization and statistical analysis. Each array was normalized for signal intensities across the whole array and locally using Lowess normalization. The resulting data were imported to the GeneSpring GX 11 software (Silicon Genetics, Redwood City, CA). Two criteria were used to determine whether a gene was differentially expressed in response to injury: fold change of ± 2.0 and P < 0.01 using a t-test for microarray analysis. No correction for multiple comparisons was done. Microarray results were confirmed using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA). Real-time PCR was performed on an ABI Prism 7500 Fast Real-Time PCR System (96-well plate). Relative gene expression was determined using the ΔΔCT method (31).
Rat macrophage isolation.
Five milliliters of Brewer's thioglycolate broth (Sigma-Aldrich) were intraperitoneally injected in Fisher 344 rats 7 days before death (13). Macrophages were isolated by lavage of the peritoneal cavity with 50 ml of sterile RPMI-5% FBS. After the medium was withdrawn, cells were counted and separated by centrifugation for 10 min at 250 g. Macrophages were resuspended in cold PBS, and purity was directly analyzed by single channel flow cytometry using ED1 (AbD Serotec, Raleigh, NC), WT.5 (BD Biosciences), and His36 (BD Biosciences) monoclonal antibodies that specifically recognize rat macrophages. Purity was typically >85%. Macrophages were then used in adhesion experiments.
Macrophage adhesion assay.
Wells of 384-well high binding plates were coated with fibrinogen (15 mg/ml in PBS) and the presence or absence of recombinant IL-18 (10–100 ng/ml) in triplicate for 1 h. The ligand coating was then blocked with 1% gelatin in TBS for 1 h. Macrophage cells obtained from the peritoneal lavage were washed and incubated with the ligand-coated wells for 10–15 min at 37°C to allow adhesion in the presence or absence of OX42 (BD Biosciences), a rat Mac-1 blocking antibody. The plate was then kept inverted for 30 min at room temperature to detach the nonadherent cells, after which the cells were fixed by addition of a drop of formaldehyde to each well. The plate was then analyzed for the number of cells adhered using the High Content Screening System.
Statistical analysis.
Data are expressed as means ± SE of at least three independent experiments. Two-group comparisons were performed using either t-test or Mann-Whitney U-test for nonparametrically distributed data. Multiple group statistical analysis was performed by one-way ANOVA followed by the Bonferroni correction for multiple comparisons. Statistics were calculated with GraphPad Prism 5 (GraphPad Software, La Jolla, CA).
RESULTS
Aging increases postinjury neointimal hyperplasia in rats.
The impact of aging on vascular remodeling was evaluated using the rat balloon injury model (11). First, we proved that arterial damage was inflicted to the same degree in arteries of aged and young rats regardless of the size of the vessel. We found similar numbers of elastic lamina ruptures and medial lacerations in arteries harvested early after surgery (30 min) from both groups of animals (data not shown). Then, we searched for changes in neointima formation with age and time after injury. We observed that neointimas in aged rats turned significantly thicker than their younger counterparts at days 14 and 30 (30-day N/NM: 0.8 ± 0.2 vs. 0.54 ± 0.15; U = 0.008); however, no changes in the neointima formation were observed between groups at day 7 (Fig. 1, A–C). Neointimas in aged rats also contained twofold more cells than those in young animals (610 ± 100 vs. 320 ± 120 cell/mm2), which suggests that aging does influence the accumulation of cells in the injury site (Fig. 1D). Nevertheless, a similar number of medial VSMC populated the media of aged and young rats at any given time point (data no shown). The contralateral uninjured arteries revealed no apparent histological differences between aged and young animals.
Fig. 1.

Arteries in aged rats exhibited significantly more neointimal hyperplasia in response to injury than those of young ones. Balloon injury was inflicted in the right iliac artery of young (3–4 mo) and aged (21–24 mo) Fischer rats (n = 7/group). A and B: representative hematoxylin and eosin (H&E) sections of injured arteries from aged (A) and young (B) rats at day 30 postsurgery. Arrows mark the neointima between the internal elastic lamina and the lumen. Neointima (N), media (M), and adventitia (A) layers are noted. Scale bar = 50 μM. C: neointima formation in injured vessels from both groups at day 30 postsurgery as represented by the neointima to neointima-media thickness ratio (N/NM ratio). Statistical differences were calculated by the Mann-Whitney U-test for nonparametrically distributed data. D: temporal accumulation of cells in the neointima of injured arteries of aged and young rats. The number of cells in the noninjury, contralateral artery is labeled as control (sham). A two-tailed t-test with unequal variances (n = 4–7) was performed; *P < 0.05, significant differences.
Aging exacerbates inflammation after vascular injury.
Since it is well accepted that the inflammatory response to vascular injury determines neointimal thickness after angioplasty and stenting (25, 34), we examined the involvement of T lymphocytes, macrophages, and dendritic cells in the pathological remodeling of the aging vasculature. Table 1 presents inflammatory cell counts in injured arteries in aged and young animal groups. The most abundant inflammatory cell type in injured arteries was CD68+ macrophages (Fig. 2, A and B and Table 1). The temporal variations in the number and location of macrophages in injured arteries from aged and young rats are depicted in Figs. 2C. Macrophages appeared in the adventitia of both groups as early as 72 h after injury. The number of these cells significantly decreased after day 7 in young animals but remained elevated in the aged ones until the end of the experiment (day 30; Table 1). As in the adventitia, macrophage infiltration in the media was more intense and sustained in injured arteries of aged animals than in those from their younger counterparts (14 days, 2,801 ± 80 vs. 189 ± 12 cells/mm2; P = 0.00023; Table 1), where they localized close to the neointima, mostly below the internal elastic lamina (Fig. 2A). Numbers in the neointima became detectable only after day 7 and were also slightly higher in aged vs. young injured arteries (Fig. 2, A–C). Interestingly, even though the adventitia of injured arteries in young animals showed two times more CD8+ lymphocytes than those in aged rats at day 3 (Table 1), the former also suffered the most pronounced drop in the number of CD8+ cells after this time point, suggesting a less favorable environment for their survival. No differences in the number of adventitial dendritic cells were observed between groups (Table 1).
Table 1.
Inflammatory cells in the media and adventitia layers of injured arteries from aged and young rats
| Media |
Adventitia |
|||||||
|---|---|---|---|---|---|---|---|---|
| 3 | 7 | 14 | 30 | 3 | 7 | 14 | 30† | |
| Aged | ||||||||
| CD8+ | – | – | – | – | 264 ± 79 | 129 ± 49 | 153 ± 62 | 97 ± 39 |
| CD4+ | – | – | – | – | 139 ± 44 | 96 ± 36 | 81 ± 36 | – |
| CD68+ | 327 ± 90 | 1,370 ± 39* | 2,801 ± 80* | 820 ± 23* | 993 ± 27 | 690 ± 19* | 634 ± 18* | 302 ± 87* |
| CD11c+ | – | – | – | – | 63 ± 23 | 92 ± 63 | 77 ± 54 | 63 ± 24 |
| Young | ||||||||
| CD8+ | – | – | – | – | 405 ± 117 | 79 ± 32 | 129 ± 57 | 107 ± 53 |
| CD4+ | – | – | – | – | – | – | – | – |
| CD68+ | 259 ± 78 | 450 ± 13 | 189 ± 12 | 265 ± 83 | 712 ± 21 | 272 ± 78 | 225 ± 55 | 143 ± 45 |
| CD11c+ | – | – | – | – | 36 ± 32 | 75 ± 45 | 79 ± 65 | 12 ± 60 |
Values are expressed as the means ± SE; n = 10–12. †Days after injury.
Statistically different values with respect to the younger counterpart (U <0.01). CD8, CD4, CD68, and CD11c are cluster differentiation antigens used to detect cytotoxic and helper T cells, macrophages, and dendritic cells.
Fig. 2.
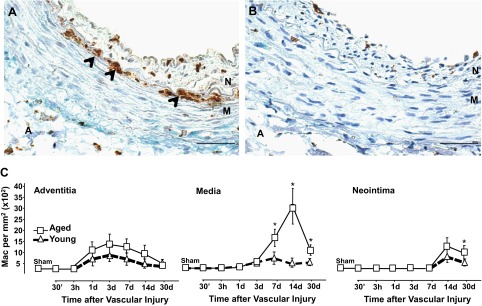
Injured arteries accumulated more macrophages (Mac) with aging. A and B: representative microphotographs of injured arteries from aged (A) and young rats (B) harvested at 7 days postsurgery. The arrows point to CD68+-infiltrated macrophages that are stained in brown. Scale bar = 50 μM. Neointima, media, and adventitia layers are noted. C: temporal accumulation of macrophages in the neointima, media, and adventitia of injured arteries from aged and young rats. Each point in graphs represents the mean of CD68+ cell per mm2 ± SE (n = 5–10). Significant differences between cell counts in injured arteries from aged and young animals at each time point were calculated with a two-tailed t-test with unequal variances; *P < 0.05, significant values.
Next, the impact of aging on the accumulation of 22 cytokines in the vascular wall after injury was measured using a multiplex bead assay. Aging increased in a time-dependent manner the postinjury accumulation of four proinflammatory cytokines [growth-regulated oncogene-keratinocyte chemoattractant (GRO KC), IL-6, IL-18, and leptin] that can promote neointima formation. The levels of these proinflammatory cytokines peaked between days 3 and 14 and stayed elevated in aged animals until the end of the experiment (Fig. 3A). Their concentrations oscillated between 2- and 10-fold over noninjured levels, with the exception of IL-18 whose expression was increased over 20-fold by day 3 (Fig. 3B). Conversely, aging reduced the temporal accumulation of the anti-inflammatory cytokine IL-10 and the immunoregulatory factor IL-13 (Fig. 3A). IL-10 is a known monocyte deactivator and vascular protective factor (10), while IL-13 is involved in the phenotypic differentiation of macrophages (15). The concentrations of IL-9 and IL-17 were also lower in injured arteries of aged rats (Fig. 3A).
Fig. 3.

Aging exacerbated the accumulation of proinflammatory cytokine IL-18 at the site of vascular injury. A: heat map representing temporal cytokine variations after vascular injury in arteries from aged and young rats. Cytokines were quantified with a multiplex rat cytokine assay. Cytokine levels are expressed as fold control with respect to the cytokine concentration in the noninjured contralateral artery. GRO KC, growth-regulated oncogene-keratinocyte chemoattractant; GMCSF, granulocyte-macrophage colony-stimulating factor; MCP-1, monocyte chemotactic protein-1; G-CSF, granulocyte colony-stimulating factor; IP-10, interferon-inducible protein 10; RANTES, regulated on activation normal T-expressed and secreted. B: temporal accumulation of IL-18 in injured arteries of aged and young rats. The presurgical levels of IL-18 in both groups are labeled as control (Co). C: bar graph representing the amount of double positive macrophages for CD68 and IL-18 found in injured arteries from aged and young rats at 3 days after surgery. D–G: representative microphotographs showing IL-18 (green) and CD68 (yellow) macrophages in injured arteries from aged (D) and young (G) rats harvested at 3 days after surgery. Nuclei were counterstained with DAPI (blue). Arrows point to double positive macrophages in the media and adventitia that are shown magnified in E and F, respectively. Scale bars = 50 μM (D and G) or 25 μM (E and F). Media and adventitia layers are noted. H: IL-18BP gene expression in injured arteries of aged and young rats at 3 days postsurgery determined by TaqMan qRT-PCR. I–J: plasma concentrations of IL-18 and IL-18BP in aged and young rats as determined by ELISA (n = 4/group). *P < 0.05, significant difference between groups; ns, no significant difference using a two-tailed t-test with unequal variances.
Age-related differences in IL-18 concentrations are local and macrophage-derived.
IL-18 promotes inflammation (7) and vascular diseases in humans (1) and multiple animal models (9, 32). Taking into consideration its marked elevation in the aged vasculature after injury (Fig. 3, A and B) and the significantly higher numbers of macrophages in the media of aged injured arteries, we hypothesized that macrophages were the likely source of this cytokine. Costaining of injured arteries with anti-CD68 and anti-IL-18 antibodies confirmed our prediction and showed a significantly higher number of double-positive macrophages in aged vs. young injured vessels (Fig. 3C). Colocalization images show that these double-positive cells can be found in both the adventitia and media layers of aged injuries arteries (Fig. 3, D–F) and are less easily detectable in young animals (Fig. 3G). As expected, IL-18 is abundant throughout the media in aged rats, with intense concentration pockets below the internal elastic lamina.
In addition, we monitored the local mRNA expression of IL-18BP in injured arteries from aged and young animals. IL-18BP inhibits IL-18 signaling by preventing this cytokine to bind to its receptor (28). Therefore, the expression of IL-18BP can be potentially upregulated in injured arteries to compensate for the noxious effect of IL-18 after injury. Instead, the expression of IL-18BP was decreased by 40% in the aged vasculature with respect to their younger counterparts (Fig. 3H) and likely unable to fully modulate the inflammatory effect of its ligand. These results suggest that lower levels of the natural inhibitor IL-18BP in the vascular wall of aged animals contribute to the exacerbated proinflammatory activity of local IL-18. Furthermore, an assessment of circulating IL-18 and IL-18BP serum levels in both aged and young injured animals indicated that systemic concentrations are similar in both groups (Fig. 3, I–J) confirming therefore that the observed differences in aged vs. young injured arteries are local inflammatory signatures related to aging.
Lastly, depletion of monocytes/macrophages with liposomal clodronate decreased local IL-18 gene expression by 75% in injured aged arteries and reduced the thickness of their neointimas to a N/NM ratio level that is indistinguishable from that of young injured arteries (Fig. 4, A and B). Previous studies have shown that this treatment is innocuous to VSMC and endothelial cells within the vascular wall (8). Moreover, the injection of empty liposomes did not modify neointima formation in aged nor in young animals. Table 2 also shows that with the exception of white blood cells and monocytes, whose numbers decreased with liposomal clodronate as previously described (5), the levels of other circulating immune cells were unaffected by this treatment. In addition, clodronate liposomes did selectively deplete splenic macrophages in both young and aged injured animals (data not shown). These results demonstrate the essential role of infiltrated macrophages in the production of vascular IL-18 and the pathological remodeling of aged arteries.
Fig. 4.

Systemic administration of clodronate liposomes decreased IL-18 accumulation in the injured vasculature while compromising neointima formation. A: gene expression of IL-18 in injured arteries of aged animals injected with either clodronate or vehicle liposomes (control) at 3 days postsurgery. Values are expressed as fold of noninjured arteries (control) ± SE. **P < 0.01, significant values; n = 4–6. B: neointima development in aged and young rats injected with either clodronate or vehicle-loaded liposomes at days −1 and +6 (n = 6–8/group). Differences between aged and young groups were calculated using a two-tailed t-test with unequal variances: *P < 0.05, significant differences; ns, nonsignificant values.
Table 2.
Effect of clodronate liposomes on blood circulating cells
| Aged |
Young |
|||
|---|---|---|---|---|
| Clodronate | Vehicle | Clodronate | Vehicle | |
| WBC | 4.15 ± 0.21* | 7.75 ± 3.88 | 5.55 ± 0.91* | 9.60 ± 2.33 |
| RBC | 8.58 ± 1.06 | 9.10 ± 0.43 | 7.26 ± 1.32 | 7.31 ± 0.96 |
| Lymphocytes | 42.5 ± 6.36 | 48.00 ± 9.89 | 80.00 ± 5.65* | 65.00 ± 1.41 |
| Monocytes | 7.00 ± 2.80* | 8.00 ± 0.00 | 5.00 ± 2.82* | 7.50 ± 0.70 |
| Eosinophils | 2.00 ± 1.41 | 3.00 ± 0.00 | 1.50 ± 0.70 | 1.50 ± 0.70 |
Values are expressed as the means ± SE; n = 3. WBC, white blood cells; RBC, red blood cells.
P < 0.05, using a t-test. Comparison was done between treatments inside each group.
IL-18 enhances VSMC survival after injury.
Since VSMC express the IL-18 receptors (IL-18R) α- and β-subunits (14), we hypothesized that high levels of IL-18 could promote neointima formation after injury in aged arteries by stimulating VSMC proliferation and/or by making these cells more resistant to apoptosis. Our results showed that adding IL-18 to VSMC in culture failed to stimulate growth in a dose-dependent manner given that no difference in DNA synthesis ([3H]deoxythymidine incorporation) was observed between treated and control cells (data not shown). This is in line with previous studies that showed no growth-promoting effects of IL-18 on arterial VSMC (41, 42). Conversely, IL-18 did have a clear anti-apoptotic effect on camptothecin-treated cells (Fig. 5, A and B). CAM is a topoisomerase I inhibitor and a potent proapoptotic agent (30). Cultures of VSMC treated with CAM and IL-18 (>25 ng/ml) for 24 h had significantly fewer apoptotic cells (annexin V+) than those treated with CAM alone (Fig. 5B). In fact, the number of late apoptotic events (annexin V+ PI+) was lower in cells incubated with IL-18 and CAM simultaneously (Fig. 5A).
Fig. 5.

IL-18 inhibited VSMC apoptosis. A: number of apoptotic cells in rat VSMCs treated with or without IL-18 (100 ng/ml) before addition of 50 μM of camptothecin (CAM) to induce cell death. PI, propidium iodide. Early (annexin V+ PI−) and late (annexin V+ PI+) apoptosis were detected by FACS analysis. Cells cultured in the absence of both IL-18 and CAM that were used as negative control for FACS analysis. B: quantitative assessment of CAM-induced apoptosis in VSMC treated with increasing concentrations of rat IL-18 (*P < 0.05; n = 4). C–F: aging decreased the incidence of apoptosis during vascular remodeling. Representative microphotographs of apoptotic VSMC (C and D) and macrophages (E and F) in arteries of aged (C and E) and young (D and F) rats at 3 days after injury. Apoptotic VSMC and macrophages were detected with In Situ Oligo Ligation and immunohistochemistry for smooth muscle actin (SMA) or CD68. Apoptotic nuclei are shown in brown and cytoplasmic SMA or CD68 in red. Representative apoptotic VSMC are marked with arrowheads. Scale bars = 50 μM. G: temporal changes in the number of apoptotic cells in the vascular wall of injured arteries were represented in the plot; **P < 0.01, significant difference. The number of apoptotic cells in the noninjury, contralateral artery is labeled as control (Co).
Then, we sought to confirm the existence of this protective effect on local VSMC of aged vs. young injured arteries. We used double immunohistochemistry to simultaneously stain apoptotic nuclei (ISOL) and cytoplasmic α-actin, a marker for SMC. An anti-CD68 antibody was also included to explore whether this protection was extended to macrophages as well. Figure 5, C–F, demonstrates a significantly higher number of both apoptotic VSMC and macrophages at day 3 in young injured vessels compared with aged animals. In fact, the number of total apoptotic cells in young arteries was ∼35% between days 3 and 7 postsurgery and then significantly dropped by day 14 (Fig. 5G). In contrast, apoptotic cells in injured arteries of aged animals were barely detected at any given time point (Fig. 5G).
Increased fibrinogen in aged injured arteries enhances macrophage adhesion in the presence of IL-18.
The protective effects of IL-18 on VSMC survival and the major apoptotic changes occurring in young arteries at day 7 postinjury suggest that this is a critical time point for the modulation of the remodeling process. Therefore, we used whole genome microarray to analyze differential gene expression in young and aged vasculature at day 7 postsurgery when inflammation was maximal in both groups. We identified 32 upregulated (Table 3) and 26 downregulated genes (Table 4), including several platelet-related mRNAs among the overexpressed factors. Overexpressed genes in aged injured arteries were mostly inflammatory (denoted as Fgg, End1, Saa4, Alb, Pdgfr1, and Col1a1 in Table 3) as well as proteins related to lipid metabolism (listed as Lrat, Fasn, and Alox15 in Table 3). Other platelet-related abundant mRNAs in this group were Hbb, Add2, Car2, and Stfa2l1. No distinguishing patterns were noted among downregulated genes.
Table 3.
Differentially upregulated arterial genes in aged and young rats after injury
| Gene Symbol | Genbank Accession | Fold Change (Aged vs. Control) | Regulation | Gene Name |
|---|---|---|---|---|
| Lrat | NM_022280 | 13.861616 | Up | Lecithin-retinol acyltransferase (phosphatidylcholine-retinol-O-acyltransferase) |
| Fgg | NM_012559 | 7.355132 | Up | Fibrinogen gamma chain |
| Cyp2c6 | NM_001013904 | 5.028335 | Up | Cytochrome P450-like |
| Fasn | NM_017332 | 4.3083034 | Up | Fatty acid synthase |
| Add2 | NM_012491 | 4.128852 | Up | Adducin 2 (beta) |
| Hbb | NM_033234 | 4.0617003 | Up | Hemoglobin, beta” |
| RGD1566313 | XM_213006 | 3.915119 | Up | Similar to Murinoglobulin 1 homolog |
| Fam46b | NM_001107907 | 3.6325316 | Up | Family with sequence similarity 46, member B |
| Cyp2c7 | NM_017158 | 3.5658238 | Up | Cytochrome P450, family 2, subfamily c, polypeptide 7” |
| Mug1 | NM_023103 | 3.288283 | Up | Murinoglobulin 1 |
| Tmem63c | NM_001108045 | 3.124968 | Up | Transmembrane protein 63c |
| Alox15 | NM_031010 | 3.0448697 | Up | Arachidonate 15-lipoxygenase |
| Car2 | NM_019291 | 2.9950109 | Up | Carbonic anhydrase II |
| S100vp | NM_176076 | 2.9130733 | Up | S100 calcium-binding protein, ventral prostate” |
| Tmem74 | XM_001063530 | 2.8362632 | Up | Transmembrane protein 74 |
| Ifrd1 | NM_019242 | 2.8061323 | Up | Interferon-related developmental regulator 1 |
| Cpeb1 | NM_001106276 | 2.7688448 | Up | Cytoplasmic polyadenylation element binding protein 1 |
| Alb | NM_134326 | 2.6524928 | Up | Albumin |
| Slc7a1 | NM_013111 | 2.6295495 | Up | Solute carrier family 7 (cationic amino acid transporter, y+ system), member 1” |
| Slc4a1 | NM_012651 | 2.6126096 | Up | Solute carrier family 4 (anion exchanger), member 1” |
| Actc1 | NM_019183 | 2.5835407 | Up | Actin, alpha, cardiac muscle 1” |
| Col1a1 | NM_053304 | 2.563527 | Up | Collagen, type I, alpha 1” |
| Saa4 | NM_001009478 | 2.553154 | Up | Serum amyloid A4, constitutive” |
| MGC72973 | NM_198776 | 2.4776847 | Up | Beta-glo |
| Lsg1 | AF452729 | 2.4240317 | Up | Large subunit gtpase 1 homolog (S. Cerevisiae) |
| Stmn2 | NM_053440 | 2.388565 | Up | Stathmin-like 2 |
| Cyp2c37 | XM_001063361 | 2.352945 | Up | Cytochrome P450, 2c37” |
| Edn1 | NM_012548 | 2.3310828 | Up | Endothelin 1 |
| Pdgfrl | NM_001011921 | 2.311612 | Up | Platelet-derived growth factor receptor-like |
| Kcne3 | NM_022235 | 2.249047 | Up | Potassium voltage-gated channel, Isk-related subfamily, gene 3” |
| Ret | NM_001110099 | 2.171445 | Up | Ret proto-oncogene |
| Ambp | NM_012901 | 2.1551707 | Up | Alpha-1-microglobulin/bikunin precursor |
| Stfa2l2 | XM_221411 | 2.0045373 | Up | Stefin A2-like 2 |
Table 4.
Differentially downregulated arterial genes in aged and young rats
| Gene Symbol | Genbank Accession | Fold Change (Aged vs. Control) | Regulation | GeneName |
|---|---|---|---|---|
| Hbe1 | NM_001008890 | −2.0025373 | Down | Hemoglobin, epsilon 1” |
| Bdh2 | NM_001106473 | −2.0112457 | Down | 3-hydroxybutyrate dehydrogenase, type 2” |
| Rtp4 | NM_001108321 | −2.0225835 | Down | Receptor (chemosensory) transporter protein 4 |
| Pcdh24 | XM_214434 | −2.023506 | Down | Protocadherin 24 |
| Ppfia3 | XM_341856 | −2.0373125 | Down | Protein tyrosine phosphatase, receptor type, PTPRF, interacting protein (liprin), alpha 3” |
| Hbe1 | NM_001008890 | −2.06978 | Down | Hemoglobin, epsilon 1” |
| Xpa | NM_001106656 | −2.117625 | Down | Xeroderma pigmentosum, complementation group A” |
| Prlr | NM_012630 | −2.1328924 | Down | Prolactin receptor |
| Pipox | NM_001012009 | −2.1369357 | Down | Pipecolic acid oxidase |
| Nrep | AY724475 | −2.1691911 | Down | Neuronal regeneration related protein |
| Olr463 | NM_001000934 | −2.2017255 | Down | Olfactory receptor 463 |
| Ddx58 | NM_001106645 | −2.251468 | Down | DEAD (Asp-Glu-Ala-Asp) box polypeptide 58 |
| Ddit4l | NM_080399 | −2.3486383 | Down | DNA-damage-inducible transcript 4-like |
| Dbndd1 | NM_001014156 | −2.3826697 | Down | Dysbindin (dystrobrevin binding protein 1) domain containing 1 |
| Slc40a1 | NM_133315 | −2.386447 | Down | Solute carrier family 39 (iron-regulated transporter), member 1” |
| Rnf128 | XM_236533 | −2.4151244 | Down | Ring finger protein 128 |
| Hao2 | NM_032082 | −2.418837 | Down | Hydroxyacid oxidase 2 (long chain) |
| Esr1 | NM_012689 | −2.4310546 | Down | Estrogen receptor 1 |
| RGD1306636 | XM_215083 | −2.434963 | Down | Hypothetical LOC293498 |
| Slco1a4 | NM_131906 | −2.5215487 | Down | Solute carrier organic anion transporter family, member 1a4” |
| Cdkn1c | NM_001033757 | −2.5289268 | Down | Cyclin-dependent kinase inhibitor 1C (P57) |
| E230034O05Rik | NR_002154 | −2.5897272 | Down | E230034O05Rik gene |
| Olr1164 | NM_001000984 | −2.6794624 | Down | Olfactory receptor 1164 |
| Upk3a | NM_001130507 | −3.337711 | Down | Uroplakin 3A |
| Mlana | NM_001106348 | −3.3447795 | Down | Melan-A |
| Slc40a1 | NM_133315 | −4.7855835 | Down | Solute carrier family 39 (iron-regulated transporter), member 1” |
Fibrinogen deposition plays a key role in the initial recruitment of leukocytes after vascular injury and in the inflammatory response of the vessel wall. Its cleavage products regulate leukocyte adhesion and extravasation and are mitogens for several cell types (21). Based on these previous findings and knowing Fgg-gamma chain (Fgg-γ) is among the most differentially upregulated genes in injured arteries when aged and young rats are compared (Table 3), we further examined the role of Fgg-γ, one of the fibrinogen subunits and essential for fibrin deposition (50). In this way, we confirmed overexpression of this gene by quantitative RT-PCR as well as protein accumulation by immunohistochemistry (Fig. 6, A and B). The mRNA expression of Fgg-γ was 17.9-fold higher in aged vs. young injured animals (Fig. 6A). In addition, fibrinogen deposition was 7.2-fold more abundant on the denudated arteries of aged rats than in those of young animals (Fig. 6B).
Fig. 6.
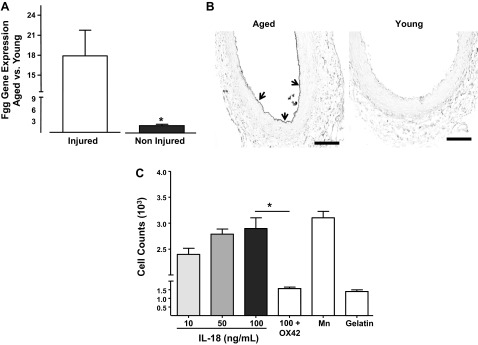
-Immobilized IL-18 increases macrophage adhesion to fibrinogen in a Mac-1-dependent manner. A: fibrinogen γ (Fgg-γ) gene expression in aged vs. young injured arteries as measured by TaqMan RT-PCR. *P < 0.05, significant difference when n = 4. B: representative microphotography of Fgg- γ deposition in the injured artery of aged and young rats at 3 days after injury using immunohistochemistry. Fibrinogen deposition is marked with arrows. C: bar graph showing adhesion of rat peritoneal macrophages to fibrinogen-coated wells in the presence of increasing concentrations of rat IL-18. Some macrophages were allowed to adhere in the presence of OX-42, a rat Mac-1 blocking antibody. Manganese (Mn) was used as a positive control as a nonselective activator of integrin-mediated adhesion. Cells incubated in gelatin-treated wells were used as negative controls. *P < 0.05, significant differences were calculated by one-way ANOVA followed by the Bonferroni correction for multiple comparisons.
Finally, to determine whether IL-18 and fibrinogen cooperate to exacerbate inflammation after vascular injury, we measured the ability of immobilized IL-18 to increase adhesion of rat peritoneal macrophages to fibrinogen, its physiological ligand. The incubation of rat peritoneal macrophages with immobilized IL-18 increased leukocyte adhesion to fibrinogen in a dose-dependent manner (Fig. 6C). IL-18 (100 ng/ml) was capable of increasing adhesion comparable to the levels of the nonselective integrin agonist Mn2+ (Fig. 6C). Interestingly, the proadhesive effects of IL-18 depended on the activation of the Mac-1 integrin (CD11b/CD18), since addition of OX-42 (an anti-CD11b blocking antibody) prevented macrophage adhesion to fibrinogen/IL-18-coated wells (Fig. 6C).
DISCUSSION
Our incomplete understanding of how aging contributes to vascular inflammation explains the inability of current treatments to prevent restenosis after cardiovascular interventions. Herein, we propose a novel mechanism by which aging prolongs the vascular inflammatory reaction to injury and increases the thickness of the vascular wall. This study describes for the first time the association among macrophage infiltration, accumulation of IL-18, and fibrinogen deposition in injured arteries of aged animals. Interestingly, we have found a feedback mechanism by which IL-18 enhances leukocyte adhesion while increasing VSMC resistance to apoptosis in aged animals. Thus our study reveals new potential targets to treat and prevent vascular inflammation and subsequent age-related vascular diseases.
First, we extended previous findings to confirm the negative effects of aging on postinjury vascular remodeling and showed that neointimas in aged rats are thicker than those in their younger counterparts. While our results are in line with multiples studies approaching the effects of aging on vascular remodeling (45, 48, 51), they are in disagreement with some recently published data that showed similar neointimal formation in young and aged Wistar rats after balloon injury (49). We used Fischer 344 instead of Wistar rats because the former strain is a validated animal model of aging (43). In addition, aged F344 animals have previously shown aggravated NIH after endothelial denudation, which provided an opportunity to study age-related pathological remodeling. Without a doubt, the rat genetic background can modify the healing response to injury and the degree of NIH after balloon angioplasty (2).
Our results indicate that the exaggerated response to injury in arteries of aged rats is concerted, at least in part, by proinflammatory factors secreted by infiltrated leukocytes and changes in the vascular wall composition. We found a prominent accumulation of fibrinogen/fibrin on the denudated endothelium of aged rats. Increased fibrinogen levels in the vascular wall after injury could reflect excessive deposition of plasma fibrinogen, slow resolution of arterial thrombi, and/or local production and secretion of fibrinogen by platelets. Nonetheless, the fact that we found elevated levels of both fibrinogen protein and its mRNA suggests that fibrinogen in the aged vasculature may come from platelets. Interestingly, the number of fibrinogen-positive platelets predicts restenosis after stent implantation in acute myocardial infarction patients (20). Furthermore, it is known that platelets accumulate intracellular fibrinogen with age (17) and that increased fibrinogen concentration is significantly associated with the risk of vascular proliferative diseases (47). The role of platelets in aged-related NIH is understudied and warrants further investigations that go beyond the scope of this present study. However, we claim our findings as the primary cause for the onset of inflammation in injured arteries of aged rats because fibrinogen/fibrin can facilitate rolling, firm adhesion, and infiltration of leukocytes into the vascular wall (23).
Infiltrated leukocytes actively produce proinflammatory cytokines and a variety of growth factors that contribute to the activation of resting VSMC (3, 27). In support of this hypothesis, we revealed that arteries in aged rats contained an elevated number of macrophages compared with those from their younger counterparts. Eghbalieh et al. (8) recently published that aged rats have increased numbers of monocytes incorporated into the vessel wall following balloon angioplasty and that elderly humans with carotid stenosis have increased numbers of circulating monocytes compared with younger patients. As previously done for both rats and rabbits (7, 8), we demonstrated that the ablation of monocytes/macrophages with pharmacological compounds efficiently reduced inflammation and NIH in aged rats. Interestingly, CD8+ T cells were the only cell type found at higher numbers in young arteries at day 3 postinjury. It is possible that this observation is related to the protection of the young vascular wall against chronic inflammation and NIH. Dimayuga et al. (6) found that adoptive transfer of CD8+ T cells inhibited neointima formation in mice after arterial injury.
Another critical finding in our study was to identify macrophage-derived IL-18 as a crucial inflammatory mediator in the injured vascular wall of aged rats. This proinflammatory cytokine is a member of the IL-1 cytokine family that amplifies inflammation by promoting TNF-α, IL-1β, and IL-6 gene expression in leukocytes, while inducing VSMC adhesion and migration (7, 40). IL-18 is a crucial player in atherosclerosis (9), neointima formation (32), and atherosclerotic plaque destabilization (33). Interestingly, the expression of IL-18BP, which functions as natural inhibitor of IL-18, was downregulated in aged vs. young arteries, indicating an unbalanced proinflammatory activity of this cytokine in the vascular wall with aging. The importance of IL-18BP in preventing the noxious effects of IL-18 in the vasculature was recently published by Li et al. (28). These authors significantly decreased NIH after balloon injury of the superficial femoral of young rats with single bolus injections of IL-18BP.
Finally, we revealed two novel pleiotropic effects of IL-18 in the aged vasculature. First, we demonstrated that IL-18 protects VSMC from chemically induced apoptosis and found a reduced number of apoptotic cells in injured arteries of aged rats with respect to those of young animals. The role of IL-18 on regulating apoptosis is controversial and seems to be driven by cell type-specific mechanisms. IL-18 inhibits neutrophil apoptosis (19) while it accelerates cell death in chondrocytes (22) and renal tubular cells (54). Secondly, we found that immobilized IL-18 stimulates macrophage adhesion to fibrinogen in a Mac-1-dependent fashion. The mechanism by which immobilized IL-18 increases adhesion is largely undefined; yet, it is plausible to speculate on the cross talk between IL-18 signaling and those mechanisms leading to de novo production of integrins. For example, IL-18 significantly enhanced ICAM-1 and VCAM-1 expression on endothelial cells to promote adhesion of HL-60 cells (36). These results explain the prolonged inflammatory reaction of arteries in aged rats and, more importantly, identify IL-18 as the molecular link between inflammation and increased NIH.
The main limitation of this study is the use of a vascular injury model in rats, an animal species with limited resources such as conditional knockout strains to selectively deplete the expression of IL-18 in macrophages and then measure the direct contribution of IL-18 to age-related NIH. Replication of the present work in an IL-18 knockout model would help further elucidate the role of IL-18 as well as the contribution of other factors. The highest fold increase in cytokine concentrations in aged vs. young animals does not necessarily have to correlate with the strongest effect on vascular inflammation. Therefore, the resolution of NIH achieved with the clodronate liposomes could have in fact been mediated by the reduction of other macrophage-derived proinflammatory factors. Another limitation is the lack of comparable data on human injured arteries, but we hope our study serves as a rationale to further investigate age-related changes in human vasculature postintervention.
In conclusion, this study proposes that aging activates an inflammatory loop in arteries after injury that can be linked to the initiation of various pathophysiological events (e.g., accumulation of IL-18, inhibition of VSMC apoptosis, and enhanced monocyte/macrophage adhesion) that together underlie the complex biological phenomenon described as “aged-related NIH.” Furthermore, the physiological responses of our experimental animals to vascular injury demonstrate differences dictated only by age and unaffected, to our knowledge, by any other cardiovascular risk factor. Altogether, our findings shed light on the mechanisms leading to the improper healing of blood vessels with age and hold promise for reducing cardiovascular mortality in an aging population.
GRANTS
This work was supported by National Institutes of Health Grants R01-HL-109582 (to R. I. Vazquez-Padron and V. Gupta), K01-HL-096413 (to R. I. Vazquez-Padron), and R25-GM-061347 (to A. Mesa).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.R.-M., M.H.F., J.C.D., Y.W., A.M., and A.P. performed experiments; L.R.-M., M.H.F., L.S., V.G., and R.I.V.-P. analyzed data; L.M. and R.I.V.-P. prepared figures; L.M. and R.I.V.-P. drafted manuscript; L.M., A.M., and S.M.P. edited and revised manuscript; A.M. and S.M.P. interpreted results of experiments; V.G., S.M.P., and R.I.V.-P. conception and design of research; V.G. and R.I.V.-P. approved final version of manuscript.
REFERENCES
- 1.Anderson JL, Carlquist JF. Cytokines, interleukin-18, and the genetic determinants of vascular inflammation. Circulation 112: 620–623, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Assadnia S, Rapp JP, Nestor AL, Pringle T, Cerilli GJ, Gunning WT, Webb TH, 3rd, Kligman M, Allison DC. Strain differences in neointimal hyperplasia in the rat. Circ Res 84: 1252–1257, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Chen L, Frister A, Wang S, Ludwig A, Behr H, Pippig S, Li B, Simm A, Hofmann B, Pilowski C, Koch S, Buerke M, Rose-John S, Werdan K, Loppnow H. Interaction of vascular smooth muscle cells and monocytes by soluble factors synergistically enhances IL-6 and MCP-1 production. Am J Physiol Heart Circ Physiol 296: H987–H996, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Danenberg HD, Fishbein I, Epstein H, Waltenberger J, Moerman E, Monkkonen J, Gao J, Gathi I, Reichi R, Golomb G. Systemic depletion of macrophages by liposomal bisphosphonates reduces neointimal formation following balloon-injury in the rat carotid artery. J Cardiovasc Pharmacol 42: 671–679, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Danenberg HD, Fishbein I, Gao J, Monkkonen J, Reich R, Gati I, Moerman E, Golomb G. Macrophage depletion by clodronate-containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation 106: 599–605, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Dimayuga PC, Chyu KY, Kirzner J, Yano J, Zhao X, Zhou J, Shah PK, Cercek B. Enhanced neointima formation following arterial injury in immune deficient Rag-1−/− mice is attenuated by adoptive transfer of CD8 T cells. PLoS One 6: e20214, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dinarello CA. Interleukin-18, a proinflammatory cytokine. Eur Cytokine Netw 11: 483–486, 2000 [PubMed] [Google Scholar]
- 8.Eghbalieh SD, Chowdhary P, Muto A, Ziegler KR, Kudo FA, Pimiento JM, Mirmehdi I, Model LS, Kondo Y, Nishibe T, Dardik A. Age-related neointimal hyperplasia is associated with monocyte infiltration after balloon angioplasty. J Gerontol A Biol Sci Med Sc i 67: 109–117, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, Bayard F, Hansson GK. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res 59: 234–240, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Feldman LJ, Aguirre L, Ziol M, Bridou JP, Nevo N, Michel JB, Steg PG. Interleukin-10 inhibits intimal hyperplasia after angioplasty or stent implantation in hypercholesterolemic rabbits. Circulation 101: 908–916, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Gabeler EE, van Hillegersberg R, Statius van Eps RG, Sluiter W, Gussenhoven EJ, Mulder P, van Urk H. A comparison of balloon injury models of endovascular lesions in rat arteries. BMC Cardiovasc Disord 2: 16, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. J Vis Exp 65: 3564, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia D, Delgado R, Ubeira FM, Leiro J. Modulation of rat macrophage function by the Mangifera indica L. extracts Vimang and mangiferin. Int Immunopharmacol 2: 797–806, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schonbeck U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med 195: 245–257, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon S. Alternative activation of macrophages. Nat Rev Immunol 3: 23–35, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Hariri RJ, Hajjar DP, Coletti D, Alonso DR, Weksler ME, Rabellino E. Aging and arteriosclerosis. Cell cycle kinetics of young and old arterial smooth muscle cells. Am J Pathol 131: 132–136, 1988 [PMC free article] [PubMed] [Google Scholar]
- 17.Heilmann E, Hynes LA, Friese P, George IN, Burstein SA, Dale GL. Dog platelets accumulate intracellular fibrinogen as they age. J Cell Physiol 161: 23–30, 1994 [DOI] [PubMed] [Google Scholar]
- 18.Higashi Y, Kihara Y, Noma K. Endothelial dysfunction and hypertension in aging. Hypertens Res 35: 1039–1047, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Hirata J, Kotani J, Aoyama M, Kashiwamura S, Ueda H, Kuroda Y, Usami M, Okamura H, Marukawa S. A role for IL-18 in human neutrophil apoptosis. Shock 30: 628–633, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Jaster M, Horstkotte D, Willich T, Stellbaum C, Knie W, Spencker S, Pauschinger M, Schultheiss HP, Rauch U. The amount of fibrinogen-positive platelets predicts the occurrence of in-stent restenosis. Atherosclerosis 197: 190–196, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Jennewein C, Tran N, Paulus P, Ellinghaus P, Eble JA, Zacharowski K. Novel aspects of fibrin(ogen) fragments during inflammation. Mol Med 17: 568–573, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.John T, Kohl B, Mobasheri A, Ertel W, Shakibaei M. Interleukin-18 induces apoptosis in human articular chondrocytes. Histol Histopathol 22: 469–482, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Kamath S, Lip GY. Fibrinogen: biochemistry, epidemiology and determinants. QJM 96: 711–729, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Kaptoge S, White IR, Thompson SG, Wood AM, Lewington S, Lowe GD, Danesh J. Associations of plasma fibrinogen levels with established cardiovascular disease risk factors, inflammatory markers, and other characteristics: individual participant meta-analysis of 154,211 adults in 31 prospective studies: the fibrinogen studies collaboration. Am J Epidemiol 166: 867–879, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Kornowski R, Hong MK, Tio FO, Bramwell O, Wu H, Leon MB. In-stent restenosis: contributions of inflammatory responses and arterial injury to neointimal hyperplasia. J Am Coll Cardiol 31: 224–230, 1998 [DOI] [PubMed] [Google Scholar]
- 26.Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation 107: 490–497, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Lee E, Grodzinsky AJ, Libby P, Clinton SK, Lark MW, Lee RT. Human vascular smooth muscle cell-monocyte interactions and metalloproteinase secretion in culture. Arterioscler Thromb Vasc Biol 15: 2284–2289, 1995 [DOI] [PubMed] [Google Scholar]
- 28.Li JM, Eslami MH, Rohrer MJ, Dargon P, Joris I, Hendricks G, Baker S, Cutler BS. Interleukin 18 binding protein (IL18-BP) inhibits neointimal hyperplasia after balloon injury in an atherosclerotic rabbit model. J Vasc Surg 47: 1048–1057, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Lindsay J, Jr, Reddy VM, Pinnow EE, Little T, Pichard AD. Morbidity and mortality rates in elderly patients undergoing percutaneous coronary transluminal angioplasty. Am Heart J 128: 697–702, 1994 [DOI] [PubMed] [Google Scholar]
- 30.Liu LF, Desai SD, Li TK, Mao Y, Sun M, Sim SP. Mechanism of action of camptothecin. Ann NY Acad Sci 922: 1–10, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[−delta delta C(T)] method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Maffia P, Grassia G, Di Meglio P, Carnuccio R, Berrino L, Garside P, Ianaro A, Ialenti A. Neutralization of interleukin-18 inhibits neointimal formation in a rat model of vascular injury. Circulation 114: 430–437, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Mallat Z, Corbaz A, Scoazec A, Graber P, Alouani S, Esposito B, Humbert Y, Chvatchko Y, Tedgui A. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ Res 89: E41–45, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Mickelson JK, Lakkis NM, Villarreal-Levy G, Hughes BJ, Smith CW. Leukocyte activation with platelet adhesion after coronary angioplasty: a mechanism for recurrent disease? J Am Coll Cardiol 28: 345–353, 1996 [DOI] [PubMed] [Google Scholar]
- 35.Moon SK, Thompson LJ, Madamanchi N, Ballinger S, Papaconstantinou J, Horaist C, Runge MS, Patterson C. Aging, oxidative responses, and proliferative capacity in cultured mouse aortic smooth muscle cells. Am J Physiol Heart Circ Physiol 280: H2779–H2788, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Morel JC, Park CC, Woods JM, Koch AE. A novel role for interleukin-18 in adhesion molecule induction through NF kappa B and phosphatidylinositol (PI) 3-kinase-dependent signal transduction pathways. J Biol Chem 276: 37069–37075, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Moreno PR, Bernardi VH, Lopez-Cuellar J, Newell JB, McMellon C, Gold HK, Palacios IF, Fuster V, Fallon JT. Macrophage infiltration predicts restenosis after coronary intervention in patients with unstable angina. Circulation 94: 3098–3102, 1996 [DOI] [PubMed] [Google Scholar]
- 38.Nieves-Neira W, Pommier Y. Apoptotic response to camptothecin and 7-hydroxystaurosporine (UCN-01) in the 8 human breast cancer cell lines of the NCI Anticancer Drug Screen: multifactorial relationships with topoisomerase I, protein kinase C, Bcl-2, p53, MDM-2 and caspase pathways. Int J Cancer 82: 396–404, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Orlandi A, Bochaton-Piallat ML, Gabbiani G, Spagnoli LG. Aging, smooth muscle cells and vascular pathobiology: implications for atherosclerosis. Atherosclerosis 188: 221–230, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Reddy P. Interleukin-18: recent advances. Curr Opin Hematol 11: 405–410, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Reddy VS, Valente AJ, Delafontaine P, Chandrasekar B. Interleukin-18/WNT1-inducible signaling pathway protein-1 signaling mediates human saphenous vein smooth muscle cell proliferation. J Cell Physiol 226: 3303–3315, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sahar S, Dwarakanath RS, Reddy MA, Lanting L, Todorov I, Natarajan R. Angiotensin II enhances interleukin-18 mediated inflammatory gene expression in vascular smooth muscle cells: a novel cross-talk in the pathogenesis of atherosclerosis. Circ Res 96: 1064–1071, 2005 [DOI] [PubMed] [Google Scholar]
- 43.Shimokawa I, Higami Y, Hubbard GB, McMahan CA, Masoro EJ, Yu BP. Diet and the suitability of the male Fischer 344 rat as a model for aging research. J Gerontol 48: B27–B32, 1993 [DOI] [PubMed] [Google Scholar]
- 44.Song Y, Shen H, Schenten D, Shan P, Lee PJ, Goldstein DR. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spagnoli LG, Sambuy Y, Palmieri G, Mauriello A. Age-related modulation of vascular smooth muscle cells proliferation following arterial wall damage. Artery 13: 187–198, 1985 [PubMed] [Google Scholar]
- 46.Spinetti G, Wang M, Monticone R, Zhang J, Zhao D, Lakatta EG. Rat aortic MCP-1 and its receptor CCR2 increase with age and alter vascular smooth muscle cell function. Arterioscler Thromb Vasc Biol 24: 1397–1402, 2004 [DOI] [PubMed] [Google Scholar]
- 47.Stec JJ, Silbershatz H, Tofler GH, Matheney TH, Sutherland P, Lipinska I, Massaro JM, Wilson PF, Muller JE, D'Agostino RB., Sr Association of fibrinogen with cardiovascular risk factors and cardiovascular disease in the Framingham Offspring Population. Circulation 102: 1634–1638, 2000 [DOI] [PubMed] [Google Scholar]
- 48.Stemerman MB, Weinstein R, Rowe JW, Maciag T, Fuhro R, Gardner R. Vascular smooth muscle cell growth kinetics in vivo in aged rats. Proc Natl Acad Sci USA 79: 3863–3866, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torella D, Leosco D, Indolfi C, Curcio A, Coppola C, Ellison GM, Russo VG, Torella M, Li Volti G, Rengo F, Chiariello M. Aging exacerbates negative remodeling and impairs endothelial regeneration after balloon injury. Am J Physiol Heart Circ Physiol 287: H2850–H2860, 2004 [DOI] [PubMed] [Google Scholar]
- 50.Uitte de Willige S, Standeven KF, Philippou H, Ariens RA. The pleiotropic role of the fibrinogen gamma' chain in hemostasis. Blood 114: 3994–4001, 2009 [DOI] [PubMed] [Google Scholar]
- 51.Vazquez-Padron RI, Lasko D, Li S, Louis L, Pestana IA, Pang M, Liotta C, Fornoni A, Aitouche A, Pham SM. Aging exacerbates neointimal formation, and increases proliferation and reduces susceptibility to apoptosis of vascular smooth muscle cells in mice. J Vasc Surg 40: 1199–1207, 2004 [DOI] [PubMed] [Google Scholar]
- 52.Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res 111: 245–259, 2012 [DOI] [PubMed] [Google Scholar]
- 53.Wang M, Zhang J, Jiang LQ, Spinetti G, Pintus G, Monticone R, Kolodgie FD, Virmani R, Lakatta EG. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension 50: 219–227, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Zhang H, Hile KL, Asanuma H, Vanderbrink B, Franke EI, Campbell MT, Meldrum KK. IL-18 mediates proapoptotic signaling in renal tubular cells through a Fas ligand-dependent mechanism. Am J Physiol Renal Physiol 301: F171–F178, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]