

Abstract
Excessive reactive oxygen/nitrogen species have been associated with the onset, progression, and outcome of sepsis, both in preclinical and clinical studies. However, the signaling pathways regulating oxidative/nitrative stress in the pathogenesis of sepsis-induced acute lung injury and acute respiratory distress syndrome are not fully understood. Employing the novel mouse model with genetic deletions of both caveolin-1 (Cav1) and adiponectin (ADPN) [double knockout (DKO) mice], we have demonstrated the critical role of Cav1 and ADPN signaling cross talk in regulating oxidative/nitrative stress and resulting inflammatory lung injury following LPS challenge. In contrast to the inhibited inflammatory lung injury in Cav1−/− mice, we observed severe lung inflammation and markedly increased lung vascular permeability in DKO mice in response to LPS challenge. Accordingly, the DKO mice exhibited an 80% mortality rate following a sublethal dose of LPS challenge. At basal state, loss of Cav1 and ADPN resulted in a drastic increase of oxidative stress and resultant nitrative stress in DKO lungs. Scavenging of superoxide by pretreating the DKO mice with MnTMPYP (a superoxide dismutase mimetic) restored the inflammatory responses to LPS challenge including reduced lung myeloperoxidase activity and vascular permeability. Thus oxidative/nitrative stress collectively modulated by Cav1 and ADPN signalings is a critical determinant of inflammatory lung injury in response to LPS challenge.
Keywords: adiponectin, caveolin-1, inflammatory lung injury, LPS, oxidative/nitrative stress
acute lung injury (ali) and its severe form, acute respiratory distress syndrome (ARDS), is a complex multifactorial syndrome featuring persistent pulmonary inflammation and increased pulmonary microvascular permeability, leading to plasma leakage into lung tissue and excessive fluid accumulation in air spaces (25, 33). Mounting evidence has demonstrated that pronounced oxidative/nitrative stress is a prominent characteristic of ALI/ARDS (5, 19, 24, 29, 35, 46). High levels of reactive oxygen/nitrogen species (ROS/RNS), resulting in oxidative/nitrative stress, not only can directly cause tissue damage, but can also affect the molecular mechanisms controlling lung inflammation. Excessive ROS/RNS have been associated with the onset, progression, and outcome of sepsis, in both preclinical and clinical studies (3, 5, 15, 19, 24, 29, 35, 46). However, the signaling pathways regulating oxidative/nitrative stress in the pathogenesis of ALI/ARDS are not fully understood, and hence antioxidant therapy remains unsuccessful (11, 31).
Caveolin-1 (Cav1), the scaffolding protein of caveolae in many nonmuscle cell types, such as endothelial cells (9, 32), plays a critical role in regulating signaling transduction by orchestrating multiple signaling molecules at its scaffolding microdomain (6, 9, 32). Cav1-deficient mice exhibit multiple phenotypes, including pulmonary hypertension, cardiomyopathy, and pulmonary hypercellularity (30, 44, 45). In response to systemic LPS challenge, Cav1 deficiency attenuates the inflammatory responses (13, 26). Cav1−/− lungs exhibit decreased NF-κB activation and inflammation. LPS-induced increases of lung vascular permeability and edema formation are also inhibited in Cav1−/− mice. Consistently, the mortality rate following lethal dose of LPS challenge is drastically decreased in Cav1−/− mice compared with wild-type (WT) mice. These data demonstrate an important role of Cav1 in regulating innate immune responses and inflammatory lung injury following LPS challenge.
Adiponectin (ADPN) is an abundant adipocyte-derived plasma protein (5–20 μg/ml in normal human subjects) with insulin-sensitizing metabolic effects and anti-inflammatory properties (2, 18, 21, 42). ADPN treatment attenuates monocyte attachment by reducing the expression of adhesion molecules on endothelial cells, suppresses expression of proinflammatory cytokines, such as TNF-α and IL-6, upregulates anti-inflammatory cytokines IL-10 and IL-1RA, and inhibits NF-κB activation (27, 28, 41). Plasma ADPN levels are inversely correlated with obesity and obesity-associated complications, such as type 2 diabetes, metabolic syndrome, and cardiovascular disease in humans (2, 34, 40). It has been shown that body mass index is associated with increased risk of ARDS in a weight-dependent manner and with increased length of stay in the Intensive Care Unit, but not with mortality (8, 14). Other studies also show that plasma ADPN in sepsis patients is significantly lowered (17, 37). These clinical observations suggest a role of impaired ADPN signaling in the pathogenesis of ALI/ARDS.
Since obesity and resultant hypoadiponectinemia are only associated with increased length of stay in the Intensive Care Unit, but not with mortality, other genetic and/or environmental factors interacting with ADPN signaling are also important for the development and poor prognosis of ALI/ARDS. Given that Cav1 deficiency promotes oxidative/nitrative stress (26, 45), and ADPN deficiency has also been linked to increased oxidative stress (10, 36), we hypothesized that loss of Cav1 and ADPN induces excessive oxidative/nitrative stress, resulting in severe inflammatory lung injury in response to LPS challenge. Here, employing the novel mouse model with genetic deletion of both Cav1 and ADPN [double knockout (DKO) mice], we have demonstrated the critical role of Cav1-ADPN signaling cross talk in regulating inflammatory lung injury in response to LPS challenge. Despite the protective effects of Cav1 deficiency per se on the inflammatory response, the DKO mice exhibited severe lung inflammation and vascular injury and markedly increased mortality following LPS challenge. We also show that severe inflammatory lung injury is the result of excessive oxidative/nitrative stress in the DKO lungs. These data suggest that activating ADPN signaling represents a novel therapeutic approach for the prevention and treatment of ALI/ARDS in patients with Cav1 and ADPN deficiency, such as the population with lipoatrophic diabetes (16, 23).
MATERIALS AND METHODS
Mice.
Mice deficient in either Cav1 or ADPN were purchased from the Jackson Laboratory and bred together to produce the DKO mice. To eliminate any background effects from either Cav1−/− or ADPN−/− line on the observed phenotypes of DKO mice, F5 or higher generations were used for these studies. WT, Cav1−/−, ADPN−/−, and DKO mice with matched genetic background at age of 3–5 mo old were used for all studies. All animals were housed in accordance with guidelines from the American Association for Laboratory Animal Care. Approval for animal care and use in these experiments was granted by the Animal Care and Use Committees of the University of Illinois at Chicago.
Molecular analysis.
Western blot analyses were performed using antibodies against Cav1 (1:1,000, Santa Cruz, CA) and nitrotyrosine (1:1,000, EMD, Millipore, MA). The same blots were reprobed with antibody against α-tubulin (1:3,000, BD Biosciences, San Jose, CA) as loading control. Plasma ADPN levels were quantified with a mouse ADPN ELISA kit following manufacture instructions (Linco Research, St. Charles, MO).
RNA was isolated using an RNeasy Mini kit including DNase I digestion (Qiagen, Valencia, CA) and quantitative real-time RT-PCR analysis was performed in an ABI Prism 7000 Sequence Detection System (Life Technologies, Grand Island, NY) with SYBR Green RT-PCR Kit (Life Technologies). The following primer sets were used for analyses: mouse inducible nitric oxide synthase primers, 5′-ACATCAGGTCGGCCATCACT-3′ and 5′-CGTACCGGATGAGCTGTGAATT-3′; mouse monocyte chemoattractant protein-1 (MCP-1) primers, 5′-CTCACTGAAGCCAGCTCTCTCT-3′ and 5′-TCCAGCCTACTCATTGGGATCA-3′; mouse macrophage inflammatory protein (MIP)-1α primers, 5′-GGCTCTCTGCAACCAGTTCTCT-3′ and 5′-TGGCTGCTCGTCTCAAAGTAGT-3′; and mouse cyclophilin primers, 5′-CTTGTCCATGGCAAATGCTG-3′ and 5′-TGATCTTCTTGCTGGTCTTGC-3′. The mouse gene expression was normalized to cyclophilin.
Myeloperoxidase assay.
Myeloperoxidase (MPO) activity in lung tissues was determined as described previously (26). Briefly, lung tissues were collected following perfusion free of blood with PBS and homogenized in 50 mM phosphate buffer. Homogenates were centrifuged at 15,000 g for 20 min at 4°C. Thereafter, the pellets were resuspended in phosphate buffer containing 0.5% hexadecyl trimethylammonium bromide (Sigma-Aldrich, St. Louis, MO) and subjected to a cycle of freezing and thawing. Subsequently, the pellet was homogenized, and the homogenates were centrifuged again. The supernatants were assayed for MPO activity using kinetics readings for 3 min, and absorbance was measured at 460 nm. The results were presented as change in 460-nm optical density per minute per gram lung tissue.
Pulmonary microvascular permeability.
Capillary filtration coefficient (Kf,c) was measured to determine pulmonary microvascular permeability to liquid, as described previously (12, 43). Briefly, after the standard 30-min equilibration perfusion, the outflow pressure was rapidly elevated by 10 cmH2O for 20 min and then was returned to normal. The lung wet weight changed in a ramplike fashion, reflecting net fluid extravasation. At the end of each experiment, lungs were dissected free of nonpulmonary tissue, and lung dry weight was determined. Kf,c (ml·min−1·cmH2O−1·dry g−1) was calculated from the slope of the recorded weight change normalized to the pressure change and to lung dry weight.
TBARS determination.
Lipid oxidation in mouse lungs was quantified with the thiobarbituric acid-reactive substances (TBARS) using the TBARS assay kit (ZeptoMetrix, Buffalo, NY). Briefly, lung tissue was homogenized in 10 volumes of 20 mM phosphate buffer (pH 7.4) at 4°C, and the resulting homogenate was centrifuged at 500 g for 10 min at 4°C. One hundred microliters of either sample or the standard were added to 50 μl of 8.1% (wt/vol) SDS, 375 μl of 20% (vol/vol) acetic acid, and 375 μl of 0.8% (wt/vol) thiobarbituric acid. The samples were heated for 60 min in a boiling water bath, followed by incubation on an ice bath for 10 min, and centrifuged at 3,000 g for 15 min. The supernatant was removed, and the absorbance of the solution was monitored at 532 nm. Malondialdehyde was used as a standard, and the level of TBARS was reported in nanomoles of malondialdehyde formed per mg of protein.
Statistics.
Differences between groups were examined for statistical significance using Student's t-test (Figs. 1 and 7) or ANOVA with Bonferroni post hoc tests (Figs. 2–4 and 6). Statistical analysis in the mortality study following LPS challenge was performed with the Peto-Peto-Wilcoxon test (Fig. 5). P < 0.05 denoted the presence of a statistically significant difference.
Fig. 1.
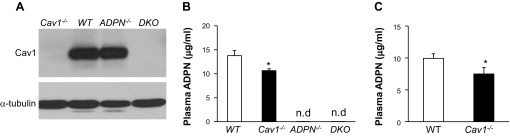
Generation of double knockout (DKO) mice. A: Western blotting demonstrating loss of caveolin-1 (Cav1) in DKO lungs. Lung lysates (15 μg/lane) were loaded and blotted with anti-Cav1 antibody. The same membrane was blotted with anti-α-tubulin as loading control. The experiment was repeated twice with similar results. B and C: plasma adiponectin (ADPN) levels. Plasma was collected from 3-mo-old mice at basal (B) and 6 h post-LPS (7.5 mg/kg ip; C) for ELISA analysis of ADPN levels. Values are means ± SD. WT, wild type; nd, not detected. *P < 0.05 vs. WT (n = 3–4).
Fig. 7.
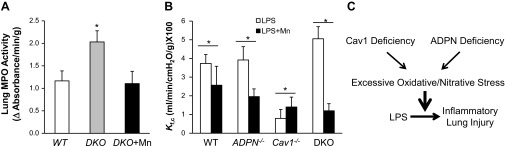
Normalized inflammatory lung injury in DKO mice pretreated with reactive oxygen species scavenger. A: normalized MPO activity in lungs of DKO mice pretreated with Mn. DKO mice were pretreated with either saline (DKO) or Mn (5 mg/kg ip) (DKO+Mn) for 4 h and then challenged with LPS (7.5 mg/kg BW ip). Lung tissues were collected at 22 h post-LPS challenge for assessment of MPO activity. Values are means ± SD (n = 4/group). *P < 0.01, DKO vs. WT or DKO+Mn. B: markedly reduced lung microvascular permeability in DKO mice treated with Mn. Mice were treated with either saline (LPS) or Mn (LPS+Mn) for 4 h and then challenged with LPS (7.5 mg/kg ip). At 5 h post-LPS challenge, Kf,c was measured. Values are means ± SD (n = 3–4/group). *P < 0.05, LPS vs. LPS+Mn. C: model of the role of Cav1 and ADPN signaling cross talk in modulating oxidative/nitrative stress and LPS-induced inflammatory lung injury. Deficiency in Cav1 and ADPN collectively augments oxidative/nitrative stress. The resulting excessive oxidative/nitrative stress exaggerates the inflammatory responses following LPS challenge and induces severe inflammatory lung injury and marked increase of mortality.
Fig. 2.

Marked increase of myeloperoxidase (MPO) activity in DKO lungs following LPS challenge. A: MPO activity in mouse lungs. Lung tissues were collected for determination of MPO activity at basal or 6 or 22 h post-LPS challenge [7.5 mg/kg body weight (BW) ip]. Values are means ± SD (n = 4/group). *P < 0.05, AD vs. WT. **P < 0.05, CV vs. WT. #P < 0.01, DKO vs. WT. B: increased MPO activity in ADPN−/− (AD) and DKO lungs following a lower dose of LPS challenge. Lung tissues were collected from 3-mo-old mice at basal or 22 h post-LPS challenge (1.5 mg/kg ip) for MPO activity assay. Values are means ± SD (n = 4/group). Δ, Change; CV, Cav1−/−. *P < 0.05, AD vs. WT. **P < 0.05, CV vs. WT. #P < 0.01 vs. the other three genotypes.
Fig. 4.

Marked increase of lung microvascular permeability in DKO mice following LPS challenge. Capillary filtration coefficient (Kf,c) was measured at basal or 5.5 h post-LPS challenge (7.5 mg/kg ip). Values are means ± SD (n = 4/group). *P < 0.001, CV-LPS vs. WT-LPS or AD-LPS. **P < 0.05, DKO-LPS vs. WT-LPS or AD-LPS.
Fig. 6.

Excessive oxidative/nitrative stress in DKO lungs. A: quantification of thiobarbituric acid-reactive substances (TBARS) in mouse lung tissues. Lung tissues at basal condition were collected for quantification of TBARS, an indicator of oxidative stress. Values are means ± SD (n = 4/group). *P < 0.05, AD and CV vs. WT. **P < 0.001, DKO vs. WT. #P < 0.05, DKO vs. AD or CV. B: representative Western blotting demonstrating marked increase of nitrative stress (tyrosine nitration) in DKO lungs at basal. Mouse lung tissues were collected and lysed for Western blotting analysis of protein tyrosine nitration using anti-nitrotyrosine (NT) antibody. The same blot was reprobed with anti-β-actin (actin) for loading control. Lungs from Cav1−/− (Cav1−/−+Mn) and DKO (DKO+Mn) mice treated with manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride [MnTMPyP (Mn); 5 mg/kg ip] for 4 h were also collected for Western blotting analysis. Mn treatment inhibited tyrosine nitration in these lungs.
Fig. 5.
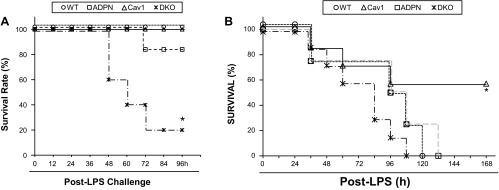
Survival rates following LPS challenge. A: increased mortality rate of DKO mice following a sublethal dose of LPS challenge. Mice were challenged with LPS (15 mg/kg ip) and housed under normal conditions. At 96 h post-LPS challenge, 80% of DKO mice died, whereas <20% of ADPN mice died. At the same time period, all WT and CV mice survived. *P < 0.001 vs. other three genotypes (n = 10 mice/group). B: decreased mortality rate of CV mice following a lethal dose of LPS challenge. Mice were challenged with 25 mg/kg of LPS (ip). Mortality rate was monitored for 7 days. *P < 0.001 vs. other three genotypes (n = 5 WT or ADPN mice, 7 CV or DKO mice).
RESULTS
Severe lung inflammation in DKO mice following LPS challenge.
To delete Cav1 and ADPN in mice, Cav1−/− mice were bred into the genetic background of ADPN−/−. DKO mice were born normally, had no gross developmental defects, survived to adulthood, and were indistinguishable from WT mice. As shown in Fig. 1, DKO mice did not express Cav1 and ADPN. Interestingly, we also observed an ∼25% decrease of plasma ADPN levels in Cav1−/− mice compared with WT mice at basal and 6 h post-LPS.
To determine the inflammatory response of DKO mice following LPS challenge, we assessed lung inflammation by determining lung MPO activity (a measure of neutrophil infiltration) (26, 43) and expression of proinflammatory molecules. As shown in Fig. 2A, DKO lungs exhibited minimal MPO activity similar to WT and other genotypes at basal. At 6 and 22 h post-LPS challenge [7.5 mg/kg body weight (BW) ip], Cav1−/− lungs exhibited less MPO activity compared with WT and ADPN−/− lungs. However, MPO activity in DKO lungs was reversed compared with that in Cav1−/− lungs and even greater than that in WT lungs. In response to a lower dose of LPS challenge (1.5 mg/kg BW ip), ADPN−/− lungs exhibited greater MPO activity compared with WT at 22 h post-LPS challenge (Fig. 2B), indicating ADPN−/− mice are more sensitive to LPS-induced lung inflammation. Consistent with a higher dose of LPS challenge, MPO activity in DKO lungs was also markedly increased compared with the other three genotypes at 22 h following a lower dose of LPS challenge.
Quantitative RT-PCR analysis revealed no marked difference in expression of proinflammatory cytokine MCP-1 in mouse lungs of the four genotypes at basal (Fig. 3A). At 6 h post-LPS challenge, DKO lungs exhibited greater level of MCP-1 expression compared with WT and ADPN−/− lungs, whereas MCP-1 induction was markedly inhibited in Cav1−/− lungs (Fig. 3A). Similarly, there was significant decrease of both MIP-1α and inducible nitric oxide synthase expression in Cav1−/− lungs compared with WT lungs at 6 h post-LPS challenge. However, the expression of these proinflammatory molecules was restored in DKO lungs to levels similar to those in WT lungs (Fig. 3, B and C).
Fig. 3.

Expression of proinflammatory genes in lung tissues. At basal or 6 h post-LPS challenge (7.5 mg/kg BW ip), lung tissues were collected, and the expression levels of monocyte chemoattractant protein-1 (MCP-1; A), macrophage inflammatory protein (MIP)-1α (B), and inducible nitric oxide synthase (iNOS; C) were quantified by quantitative RT-PCR analysis. Values are means ± SD (n = 4/group). *P < 0.05, CV vs. WT. **P < 0.05, DKO vs. WT or ADPN−/−. #P < 0.05, DKO and ADPN−/− vs. WT.
Marked increase of lung vascular permeability in DKO mice in response to LPS challenge.
Given that persistent increase of pulmonary microvascular permeability leading to protein-rich lung edema is a hallmark of the pathology of ALI/ARDS, we next assessed lung vascular permeability by determining pulmonary microvascular filtration coefficient Kf,c (12, 43). Basal Kf,c values were similar among the four genotypes (Fig. 4). Our previous studies have shown that LPS at this concentration induces maximal increase of Kf,c at ∼6 h in WT lungs (26, 45). In the present study, we observed that Kf,c values of WT and ADPN−/− lungs were markedly increased at 5.5 h post-LPS challenge (7.5 mg/kg ip) compared with basal, whereas Cav1−/− lungs exhibited only a modest increase (Fig. 4). However, pulmonary microvascular permeability in DKO lungs was reversed compared with that in Cav1−/− lungs and even significantly greater than that in WT and ADPN−/− lungs, indicating severe lung vascular injury in DKO mice following LPS challenge.
Greater mortality rate in DKO mice following LPS challenge.
To determine whether marked increases of lung inflammation and vascular permeability in DKO mice are consistent with increased mortality rate, survival studies were made using a higher dosage of LPS. Strikingly, LPS challenge of 15 mg/kg resulted in a significantly greater mortality rate in DKO mice compared with mice of other three genotypes (Fig. 5A). We observed 80% of DKO mice died within 96 h of LPS challenge. In contrast, all WT and Cav1−/− mice survived, although ∼20% of ADPN−/− mice died within this time. Consistent with previous studies (13, 26), LPS challenge of 25 mg/kg induced only 40% mortality of Cav1−/− mice, whereas 100% mortality of the other three groups (Fig. 5B).
Modulation of oxidative/nitrative stress by Cav1 and ADPN signalings.
It has been shown that both Cav1 and ADPN are important regulators of oxidative stress (10, 36, 45). We next determined whether deficiency of both Cav1 and ADPN augments oxidative stress in lung tissues at basal. TBARS in lung tissues was measured as an indicator of oxidative stress (10, 20, 22). As shown in Fig. 6A, TBARS levels were increased in both ADPN−/− lungs and Cav1−/− lungs compared with WT lungs. Furthermore, we observed a marked increase of TBARS levels in DKO lungs compared with either Cav1−/− or ADPN−/− lungs.
We have recently shown that Cav1 deficiency results in a marked increase in nitrative stress due to overproduction of ROS and endothelial nitric oxide synthase-derived nitric oxide, which form peroxynitrite and thereby modify proteins through tyrosine nitration (26, 45). We employed Western blotting to determine protein tyrosine nitration. As shown in Fig. 6B, Cav1−/− lung tissue exhibited increased tyrosine nitration compared with those of WT and ADPN−/− at basal. Tyrosine nitration in DKO lungs was further markedly increased compared with Cav1−/− lungs, consistent with marked increase of oxidative stress in these lungs. These data suggest Cav1 and ADPN signalings collectively modulate oxidative/nitrative stress.
The causal role of excessive oxidative/nitrative stress in LPS-induced severe inflammatory lung injury in DKO mice.
To gain insights into the role of excessive oxidative/nitrative stress in the mechanism of severe inflammatory lung injury in DKO mice in response to LPS challenge, we determined whether scavenging superoxide by manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP; a superoxide dismutase mimetic) (45) would normalize the inflammatory responses. Mice were pretreated with MnTMPyP for 4 h and then challenged with LPS. As shown in Fig. 6B, MnTMPyP treatment inhibited protein tyrosine nitration in DKO lungs at basal, indicating efficient scavenging of ROS by MnTMPyP. Accordingly, MnTMPyP treatment reduced inflammatory lung injury in DKO mice in response to LPS challenge (Fig. 7). Lung MPO activity of DKO mice pretreated with MnTMPyP was returned to levels similar to those of WT mice at 22 h post-LPS challenge (Fig. 7A). More profoundly, pulmonary microvascular permeability (determined by Kf,c measurement) was drastically inhibited in MnTMPyP-pretreated DKO mice, which was even much less than that of WT mice (Fig. 7B). Intriguingly, MnTMPyP treatment resulted in decreased lung vascular permeability in WT and ADPN−/− mice, whereas there was increased permeability in Cav1−/− mice compared with untreated controls following LPS challenge (Fig. 7B). Taken together, our data provide unequivocal evidence that Cav1 and ADPN signaling interplay regulates LPS-induced inflammatory lung injury through modulation of oxidative/nitrative stress (Fig. 7C).
DISCUSSION
We show here that excessive oxidative/nitrative stress secondary to loss of Cav1 and ADPN induces severe inflammatory lung injury following LPS challenge. We have demonstrated that loss of Cav1 and ADPN resulted in marked increases of lung inflammation and vascular permeability and greater mortality rate following LPS challenge. We observed a marked increase of oxidative/nitrative stress in DKO lungs at basal. Inhibition of oxidative stress and resultant nitrative stress resulted in normalization of the inflammatory responses of DKO mice following LPS challenge. It has been shown that Cav1 deficiency dampens the inflammatory responses to LPS challenge (13, 26). Cav1−/− mice exhibit less inflammatory lung injury and reduced mortality compared with WT mice. Our present study has shown that loss of ADPN in DKO mice not only completely abolished the Cav1 deficiency-mediated protection from LPS-induced lung injury, but also induced more severe inflammatory lung injury and greater mortality in response to a sublethal dose of LPS challenge (15 mg/kg). The DKO lungs exhibited marked increases of MPO activity, expression of proinflammatory cytokines, and vascular permeability compared with WT lungs following LPS challenge. However, ADPN deficiency per se did not induce severe inflammatory lung injury in ADPN−/− mice following LPS challenge (7.5 mg/kg BW ip) compared with WT lungs. Taken together, our data suggest that Cav1 and ADPN signaling cross talk is a critical regulator of the inflammatory responses following LPS challenge.
Our data have shown that DKO lungs exhibited marked increases of oxidative stress and resultant nitrative stress at basal compared with the other three genotypes, although oxidative stress was also increased in Cav1−/− as well as ADPN−/− lungs compared with WT lungs. These data suggest Cav1 and ADPN signaling collectively modulate oxidative/nitrative stress. Previous studies have shown that Cav1 deficiency promoted oxidative stress (26, 45). ADPN deficiency has also been linked to increased oxidative stress (15, 22). To the best of our knowledge, our study for the first time has demonstrated the role of Cav1 and ADPN signaling cross talk in modulating oxidative/nitrative stress. Recently, Wang et al. (39) have shown that Cav3, the muscle form of caveolins forms a complex with ADPN receptor 1 and thereby mediates the protective effects of ADPN on myocardial ischemia-reperfusion injury. Future studies would be interesting to investigate the potential interaction of Cav1 and ADPN receptors in other cell types, e.g., endothelial cells in modulating oxidative stress.
The most important finding of the present study is that Cav1 and ADPN signalings collectively modulate oxidative stress, and resultant nitrative stress thereby regulates the severity of inflammatory lung injury and mortality following LPS challenge. Our data have demonstrated the causal role of excessive oxidative/nitrative stress in DKO lungs in the pathogenesis of severe inflammatory lung injury. Scavenging the excessive oxidative stress in DKO mice before LPS challenge normalized the inflammatory responses, including lung MPO activity and vascular permeability. Intriguingly, ROS scavenging by MnTMPyP pretreatment also decreased lung vascular permeability in WT and ADPN−/− mice, although it was more efficient in DKO lungs. These data are consistent with the general concept that oxidative stress is detrimental in mediating inflammatory lung injury. The more efficient decrease of lung vascular permeability in MnTMPyP-treated DKO mice may be attributed to a large amount of available nitric oxide from Cav1 deficiency-induced endothelial nitric oxide synthase activation (26, 45). However, MnTMPyP pretreatment induced increased lung vascular permeability in Cav1−/− mice. Consistent with our published data that Cav1 deficiency inhibits LPS-induced lung vascular injury through increased nitrative stress (26), which is mediated by increased oxidative stress and nitric oxide production, forming the strong oxidizing RNS, such as peroxynitrite, MnTMPyP treatment scavenged ROS and inhibited nitrative stress in Cav1−/− mice and thereby led to increased vascular permeability compared with control Cav1−/− mice. Similarly, MnTMPyP treatment also inhibited nitrative stress in DKO mice seen in MnTMPyP-treated Cav1−/− mice. Thus these mice exhibited similar lung vascular permeability. Although both ADPN−/− and Cav1−/− lungs exhibited similar increase of oxidative stress, only Cav1−/− lungs exhibited increased nitrative stress, which is responsible for the protective effects from LPS challenge in Cav1−/− mice. Taken together, these data suggest that modest nitrative stress seen in Cav1−/− mice is protective from LPS-induced lung vascular injury, possibly through IL-1 receptor-associated kinase-4 nitration and resultant inhibition of its kinase activity (26), whereas excessive nitrative stress secondary to excessive oxidative stress seen in DKO mice is detrimental, as it induces massive protein tyrosine nitration, which, in turn, results in severe lung injury in response to LPS challenge. Future study is warranted to determine the proteins targeted for tyrosine nitration and the extent of tyrosine nitration in DKO lungs compared with Cav1−/− lungs, thereby delineating the molecular basis of the distinct pathophysiology seen in these mice following LPS challenge. Consistently, it has been demonstrated that pronounced oxidative/nitrative stress is a prominent characteristic of ALI/ARDS in patients, which induces nitration of many proteins and impairs their functions, such as surfactant protein A (5, 19, 24, 29, 35, 46).
Our study may help to explain the clinical observation that obesity and resultant hypoadiponectinemia are only associated with increased length of stay in the Intensive Care Units, but not with mortality of ARDS patients (8, 14). Although hypoadiponectinemia is associated with sepsis-induced ALI and ARDS (17, 37), other genetic and environmental factors interacting with ADPN signaling are important for the development and poor prognosis of ALI/ARDS. The present study demonstrates that Cav1 is a critical factor cross talking with ADPN signaling to regulate the inflammatory responses and mortality following sepsis challenge. It has been shown that plasma ADPN levels decrease in patients with metabolic syndrome, including obesity, coronary artery disease, and type 2 diabetes (18, 21, 42). Cav1 deficiency has also been shown in various patient populations, including idiopathic pulmonary artery hypertension (1, 45), idiopathic pulmonary fibrosis (38), and systemic sclerosis (7). Furthermore, deficiency in both Cav1 and ADPN has been demonstrated in patients with lipoatrophic diabetes (16, 23). Thus, on the basis of our findings in the DKO mice, it is plausible that patients deficient in both Cav1 and ADPN have a greater risk to develop ARDS and exhibit poor prognosis following sepsis.
In conclusion, we have demonstrated, for the first time, the critical role of Cav1 and ADPN signaling interplay in modulating oxidative/nitrative stress in vivo and thereby regulating the inflammatory responses to LPS challenge. Our findings suggest that hypoadiponectinemia coupled with other genetic and/or environmental factors, such as Cav1 deficiency, is a critical risk factor for the development and poor prognosis of ALI/ARDS following sepsis.
GRANTS
This work was supported in part by National Heart, Lung, and Blood Institute Grants R01-HL-085462, R01-HL-085462–3S1, R56-HL-085462, and P01-HL-077806 (project 4) to Y. Y. Zhao. L. Cai was supported in part by the China Scholarship Council.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.C., F.Y., Z.D., X.H., Y.D.Z., M.K.M., J.X., and S.M.V. performed experiments; L.C., F.Y., Z.D., X.H., Y.D.Z., M.K.M., J.X., S.M.V., and Y.-Y.Z. analyzed data; L.C., Y.D.Z., M.K.M., S.M.V., and Y.-Y.Z. interpreted results of experiments; L.C. and Y.-Y.Z. prepared figures; L.C. drafted manuscript; L.C., F.Y., Z.D., X.H., Y.D.Z., M.K.M., J.X., S.M.V., and Y.-Y.Z. approved final version of manuscript; Y.-Y.Z. conception and design of research; Y.-Y.Z. edited and revised manuscript.
REFERENCES
- 1.Achcar RO, Demura Y, Rai PR, Taraseviciene-Stewart L, Kasper M, Voelkel NF, Cool CD. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest 129: 696–705, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257: 79–83, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Boueiz A, Hassoun PM. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc Res 77: 26–34, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Brandes RP, Weissmann N, Schroder K. NADPH oxidases in cardiovascular disease. Free Radic Biol Med 49: 687–706, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Cochrane CG, Spragg R, Revak SD. Pathogenesis of the adult respiratory distress syndrome. Evidence of oxidant activity in bronchoalveolar lavage fluid. J Clin Invest 71: 754–761, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev 84: 1341–1379, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Del Galdo F, Lisanti MP, Jimenez SA. Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol 20: 713–719, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dossett LA, Heffernan D, Lightfoot M, Collier B, Diaz JJ, Sawyer RG, May AK. Obesity and pulmonary complications in critically injured adults. Chest 134: 974–980, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol 23: 1161–1168, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–1761, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gadek JE, DeMichele SJ, Karlstad MD, Pacht ER, Donahoe M, Albertson TE, Van Hoozen C, Wennberg AK, Nelson JL, Noursalehi M. Effect of enteral feeding with eicosapentaenoic acid, gamma-linolenic acid, and antioxidants in patients with acute respiratory distress syndrome. Enteral Nutrition in ARDS Study Group. Crit Care Med 27: 1409–1420, 1999 [DOI] [PubMed] [Google Scholar]
- 12.Gao X, Xu N, Sekosan M, Mehta D, Ma SY, Rahman A, Malik AB. Differential role of CD18 integrins in mediating lung neutrophil sequestration and increased microvascular permeability induced by Escherichia coli in mice. J Immunol 167: 2895–2901, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Garrean S, Gao XP, Brovkovych V, Shimizu J, Zhao YY, Vogel SM, Malik AB. Caveolin-1 regulates NF-kappa B activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol 177: 4853–4860, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Gong MN, Bajwa EK, Thompson BT, Christiani DC. Body mass index is associated with the development of acute respiratory distress syndrome. Thorax 65: 44–50, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal 9: 1991–2002, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, Park YE, Nonaka I, Hino-Fukuyo N, Haginoya K, Sugano H, Nishino I. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest 119: 2623–2633, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hillenbrand A, Knippschild U, Weiss M, Schrezenmeier H, Henne-Bruns D, Huber-Lang M, Wolf AM. Sepsis induced changes of adipokines and cytokines–septic patients compared with morbidly obese patients. BMC Surg 10: 26, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hopkins TA, Ouchi N, Shibata R, Walsh K. Adiponectin actions in the cardiovascular system. Cardiovasc Res 74: 11–18, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133: 235–249, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jayet PY, Rimoldi SF, Stuber T, Salmon CS, Hutter D, Rexhaj E, Thalmann S, Schwab M, Turini P, Sartori-Cucchia C, Nicod P, Villena M, Allemann Y, Scherrer U, Sartori C. Pulmonary and systemic vascular dysfunction in young offspring of mothers with preeclampsia. Circulation 122: 488–494, 2010 [DOI] [PubMed] [Google Scholar]
- 21.Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev 26: 439–451, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Kakoki M, Kizer CM, Yi X, Takahashi N, Kim HS, Bagnell CR, Edgell CJ, Maeda N, Jennette JC, Smithies O. Senescence-associated phenotypes in Akita diabetic mice are enhanced by absence of bradykinin B2 receptors. J Clin Invest 116: 1302–1309, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, Semple RK, O'Rahilly S, Dugail I, Capeau J, Lathrop M, Magre J. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab 93: 1129–1134, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Lang JD, McArdle PJ, O'Reilly PJ, Matalon S. Oxidant-antioxidant balance in acute lung injury. Chest 122: 314S–320S, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 122: 2731–2740, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirza MK, Yuan J, Gao XP, Garrean S, Brovkovych V, Malik AB, Tiruppathi C, Zhao YY. Caveolin-1 deficiency dampens Toll-like receptor 4 signaling through eNOS activation. Am J Pathol 176: 2344–2351, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, Hotta K, Nishida M, Takahashi M, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation 100: 2473–2476, 1999 [DOI] [PubMed] [Google Scholar]
- 28.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation 102: 1296–1301, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Quinlan GJ, Lamb NJ, Tilley R, Evans TW, Gutteridge JM. Plasma hypoxanthine levels in ARDS: implications for oxidative stress, morbidity, and mortality. Am J Respir Crit Care Med 155: 479–484, 1997 [DOI] [PubMed] [Google Scholar]
- 30.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276: 38121–38138, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Rinaldi S, Landucci F, De Gaudio AR. Antioxidant therapy in critically septic patients. Curr Drug Targets 10: 872–880, 2009 [DOI] [PubMed] [Google Scholar]
- 32.Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell 68: 673–682, 1992 [DOI] [PubMed] [Google Scholar]
- 33.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Shetty S, Kusminski CM, Scherer PE. Adiponectin in health and disease: evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol Sci 30: 234–239, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Sittipunt C, Steinberg KP, Ruzinski JT, Myles C, Zhu S, Goodman RB, Hudson LD, Matalon S, Martin TR. Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 163: 503–510, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation 115: 1408–1416, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Venkatesh B, Hickman I, Nisbet J, Cohen J, Prins J. Changes in serum adiponectin concentrations in critical illness: a preliminary investigation. Crit Care 13: R105, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA, Liu F, Ifedigbo E, Xu X, Oury TD, Kaminski N, Choi AM. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med 203: 2895–2906, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Wang X, Jasmin JF, Lau WB, Li R, Yuan Y, Yi W, Chuprun K, Lisanti MP, Koch WJ, Gao E, Ma XL. Essential role of caveolin-3 in adiponectin signalsome formation and adiponectin cardioprotection. Arterioscler Thromb Vasc Biol 32: 934–942, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 86: 1930–1935, 2001 [DOI] [PubMed] [Google Scholar]
- 41.Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun 323: 630–635, 2004 [DOI] [PubMed] [Google Scholar]
- 42.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7: 941–946, 2001 [DOI] [PubMed] [Google Scholar]
- 43.Zhao YY, Gao XP, Zhao YD, Mirza MK, Frey RS, Kalinichenko VV, Wang IC, Costa RH, Malik AB. Endothelial cell-restricted disruption of FoxM1 impairs endothelial repair following LPS-induced vascular injury. J Clin Invest 116: 2333–2343, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J, Jr, Chien KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A 99: 11375–11380, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, Brovkovych V, Yuan JX, Wharton J, Malik AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest 119: 2009–2018, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu S, Ware LB, Geiser T, Matthay MA, Matalon S. Increased levels of nitrate and surfactant protein a nitration in the pulmonary edema fluid of patients with acute lung injury. Am J Respir Crit Care Med 163: 166–172, 2001 [DOI] [PubMed] [Google Scholar]