

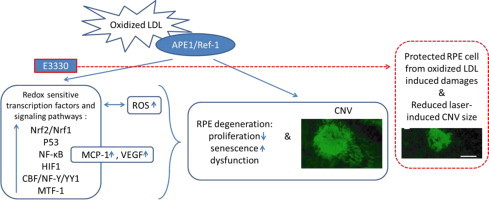
Abstract
The effectiveness of current treatment for age related macular degeneration (AMD) by targeting one molecule is limited due to its multifactorial nature and heterogeneous pathologies. Treatment strategy to target multiple signaling pathways or pathological components in AMD pathogenesis is under investigation for better clinical outcome. Inhibition of the redox function of apurinic endonuclease 1/redox factor-1 (APE1) was found to suppress endothelial angiogenesis and promote neuronal cell recovery, thereby may serve as a potential treatment for AMD. In the current study, we for the first time have found that a specific inhibitor of APE1 redox function by a small molecule compound E3330 regulates retinal pigment epithelium (RPEs) cell response to oxidative stress. E3330 significantly blocked sub-lethal doses of oxidized low density lipoprotein (oxLDL) induced proliferation decline and senescence advancement of RPEs. At the same time, E3330 remarkably decreased the accumulation of intracellular reactive oxygen species (ROS) and down-regulated the productions of monocyte chemoattractant protein-1 (MCP-1) and vascular endothelial growth factor (VEGF), as well as attenuated the level of nuclear factor-κB (NF-κB) p65 in RPEs. A panel of stress and toxicity responsive transcription factors that were significantly upregulated by oxLDL was restored by E3330, including Nrf2/Nrf1, p53, NF-κB, HIF1, CBF/NF-Y/YY1, and MTF-1. Further, a single intravitreal injection of E3330 effectively reduced the progression of laser-induced choroidal neovascularization (CNV) in mouse eyes. These data revealed that E3330 effectively rescued RPEs from oxidative stress induced senescence and dysfunctions in multiple aspects in vitro, and attenuated laser-induced damages to RPE–Bruch׳s membrane complex in vivo. Together with its previously established anti-angiogenic and neuroprotection benefits, E3330 is implicated for potential use for AMD treatment.
Abbreviations: AhR, aryl hydrocarbon receptor; AMD, age related macular degeneration; AP-1, activator protein 1; APE1, apurinic endonuclease 1/redox factor-1; ApoE, apolipoprotein E; CBF/NF-Y/YY1, CCAAT binding factor/nuclear factor-Y/Yin Yang 1; CECs, choroidal endothelial cells; CNV, choroidal neovascularization; DCFH-DA, dichlorodihydrofluorescin diacetate; DMSO, dimethylsulphoxide; Fluc, firefly luciferase; HIF-1α, hypoxia inducible factor-1α; HSF1, heat-shock factor 1; IκB-α, inhibitory NF-κB-α; MCP-1, monocyte chemoattractant protein-1; MTF1, metal regulatory transcription factor 1; NF-κB, nuclear factor-κB; Nox, NADPH oxidase; Nrf, nuclear factor erythroid-2-related factor; oxLDL, oxidized low density lipoprotein; redox, reduction/oxidation; Rluc, renilla luciferase; RNV, retinal neovascularization; ROS, reactive oxygen species; RPE, retinal pigment epithelium; RVECs, retinal vascular endothelial cells; SA-β-gal, senescence associated β-gal; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; TUNEL, TdT mediated dUTP-fluorescein nick end-labeling; VEGF, vascular endothelial growth factor
Keywords: APE1/Ref-1redox function, E3330, Oxidative stress, Retinal pigment epithelial cell, Transcription factor, Age-related macular degeneration.
Graphical abstract
Highlights
-
•
Specific inhibition of APE1/Ref-1 redox function with E3330 blocked RPE proliferation decline and senescence-like phenotype advancement induced by oxLDL.
-
•
E3330 suppressed intracellular ROS, down-regulated the MCP-1 and VEGF production, and reduced nuclear NF-κB p65 in RPEs.
-
•
E3330 repressed the redox sensitive transcription factors Nrf2/Nrf1, p53, NF-κB, HIF1, CBF/NF-Y/YY1, and MTF-1 that stimulated by oxLDL in RPEs.
-
•
Intravitreal injection of E3330 markedly reduced the laser-induced CNV in mouse eyes.
-
•
E3330 holds great potential for the management of AMD.
1. Introduction
Age-related macular degeneration (AMD) is the leading cause of central vision loss in the elderly in developed world, and an increasing public health problem due to population aging [1], [2]. There are two forms of AMD, the dry form (or non-exudative; atrophic) and the wet form (or exudative; neovascular). Currently the most widely used therapies for AMD are limited to laser photocoagulation and intravitreal anti-vascular endothelial growth factor (VEGF) agents for wet AMD [3]. However, these treatments have various limitations such as insufficient effectiveness or requirement of repetitive intravitreal injections [4], [5], [6]. In addition, no treatment is approved so far for dry AMD which could develop into irreversible vision loss as severe as neovascular AMD. Although the mechanism of AMD is not fully understood, it is well known that AMD affects different cell types in the retina with a broad spectrum of corresponding pathologies involving photoreceptor and RPE degeneration caused by cumulative oxidative stress and local inflammation, as well as endothelial cell activation and angiogenesis (for in-depth review, see Refs. [7], [8]). Therefore, strategies targeting a single molecule are not sufficient to control the disease process. Agents that regulate multiple signaling pathways or multiple pathological components are more likely to produce a greater therapeutic effect for AMD.
In the past decade, the reduction/oxidation (redox) regulation function of apurinic endonuclease 1/redox factor-1 (APE1/Ref-1, here referred to as APE1) has been studied for its extended repertoire in controlling cellular response to oxidative stress. APE1 has two major functions in mammalian cells: DNA repair [9], [10] and redox regulation of gene transcription [11], [12], [13]. The DNA repair function of APE1 dominants the base excision repair pathway that responsible for repairing DNA damaged by oxidative stress, alkylating agents, and ionizing radiation [14]. Equally important, the redox function of APE1 enhances the DNA binding of numerous redox-sensitive transcription factors via reduction of disulfide bonds [13], [15], [16]. APE1 thereby represents an interesting therapeutic target in different mechanistic contexts. While the DNA repair function was a underlying mechanism of resistance to chemotherapy in various cancers [9], the redox function may serve as a target for modulating cell stress response, proliferation, angiogenesis, and processes such as inflammation.
Many stress responsive transcription factors including nuclear factor-κB (NF-κB) [17], hypoxia inducible factor-1α (HIF-1α) [10], [18], cAMP response element binding protein (CREB) [19], activator protein-1 (AP-1) [17], [20], p53 [21], and others have been reported to be activated by APE1 dependent redox activation. Theoretically, specific inhibition of the APE1 redox function may influence multiple signaling pathways and downstream molecules that could lead to various pathologies, without affecting the genomic integrity as long as the DNA repair function is left intact. In fact, it is possible to precisely eliminate the redox function of APE1 without interfering with its DNA repair function, because the two functions of APE1 have distinctively independent domains and mechanisms [10], [22]. Previous studies have identified a small molecule inhibitor, E3330 (also called APX3330) which is specific to APE1 redox activity [10], [23], [24], [25], [26]. E3330 significantly reduced the transcriptional activities of NF-κB [25] and HIF-1α [10], [18] in various cancer cells and endothelial cells without affecting the DNA repair function [10], [25], [27]. These factors and their downstream effectors have been closely associated with the etiology and pathogenesis of AMD [5], [6], [28], [29], [30], [31], [32]. We have recently reported that E3330 effectively suppressed retinochoroidal angiogenesis [33], [34] and reduced neuronal cell loss [35]. These evidences indicate that E3330 may affect multiple signaling pathways and rectify multiple pathologies relevant to AMD pathogenesis through inhibition of APE1 redox function. However, the role of APE1 redox activity in retinal pigment epithelium cells (RPEs) is not known.
RPEs are highly specialized pigmented epithelium lining between the neural retina and Bruch׳s membrane, that plays critical roles in maintaining the homeostasis of retinal neurons and choroid [36], [37]. RPEs are subject to lifelong chronic oxidative injury due to the constant exposure to light, a high level of metabolic activity, and accumulation of oxidized lipoprotein component in the cell [38], [39]. Oxidative stress induced senescence and dysfunction of the macular RPEs is believed to herald AMD [8], [40]. This study focused on the role of E3330 in regulating RPE responses to the oxidative stimulus elicited by oxidized low-density lipoprotein (oxLDL) in vitro and laser-induced RPE–Bruch׳s membrane damage in vivo. We found that specific inhibition of APE1 redox activity with E3330 significantly rescued human RPEs from proliferation decline and senescence advancement induced by oxLDL. These effects were associated with reduced intracellular ROS, down-regulated secretion of monocyte chemoattractant protein-1 (MCP-1) and VEGF, and suppressed nuclear accumulation of NF-κB p65. The DNA binding activities of multiple redox-sensitive transcription factors including Nrf2/Nrf1, p53, NF-κB, HIF1, CBF/NF-Y/YY1, and MTF-1 were directly repressed by E3330. A single intravitreal injection of E3330 markedly reduced the laser-induced lesion size in RPE–Bruch׳s membrane complex in mouse eyes. These findings suggest that E3330 holds great therapeutic promise for AMD by rescuing RPEs from oxidative stress in multiple aspects and reducing in vivo RPE–Bruch׳s membrane complex damage. APE1 redox function could serve as a potential therapeutic target in the management of AMD.
2. Results
2.1. E3330 blocks sub-lethal doses of oxLDL-induced proliferation decline of RPEs
OxLDL has been widely used as an experimental stimulant to induce pathological stresses in RPE cells and mimic oxidative damages seen in AMD [41], [42]. In a TUNEL assay to exclude the potential cell toxicity induced by oxLDL, no significant RPE apoptosis was observed at the doses of less than 450 μg/mL oxLDL (p>0.05 when compared with the negative control group; Fig. 1A). Notable apoptotic cells that accounted for 8.11±0.87% and 36.62±6.18% of the total cells were found respectively when 450 μg/mL and 600 μg/mL oxLDL were applied (p<0.05 when compared with the negative control group; Fig. 1A). A DNase I treated group was included as positive control which induced a 100% cell apoptosis. Based on these data, previous reports [33], [34], and our previous studies on retino-choroidal angiogenesis in vitro and in vivo [33], [43], we adopted a sub-lethal dose range of 20–50 μM E3330, mostly 30 μM, which do not induce significant apoptosis in ARPE-19 in the rest of the experiments of the study.
Fig. 1.
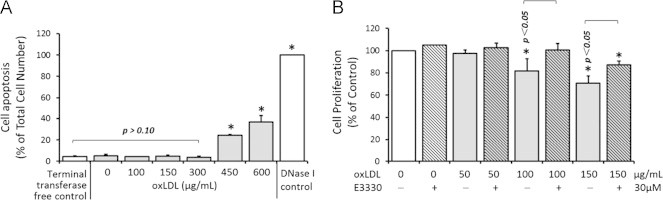
Effects of oxLDL and E3330 on RPE apoptosis (A) and proliferation (B) in vitro. A, TUNEL assay of RPEs exposed to various doses of oxLDL. A negative control without terminal transferase and a positive control with DNase I was included. All data points were normalized to the total number of the cells seeded in each well. OxLDL at or under 300 μg/mL did not induced significant cell apoptosis when compared with the negative control group. Significant apoptosis was only seen at 450 μg/mL and 600 μg/mL oxLDL treated groups. Bars represent mean±SEM of three independent experiments. ⁎p<0.05 versus the negative control. B, MTS assay of RPE proliferation in response to 0, 50, 100, or 150 μg/mL oxLDL alone or combined with 30 μM E3330. All treatment groups were normalized to be a percentage of the control group. OxLDL inhibited the RPE proliferation dose dependently. 30 μM E3330 remarkably reversed the suppression effect of 100 μg/mL and 150 μg/mL oxLDL on RPE proliferation, but had no effect on the cell growth by itself. Bars represent mean±SEM of three independent experiments. ⁎p<0.05 versus the no treatment control.
To investigate the effect of E3330 on oxLDL stressed RPEs, we first examined RPE cell proliferation using a MTS assay. The RPE growth was inhibited in a dose-dependent manner by 0–150 μg/mL oxLDL. While 50 μg/mL oxLDL showed minimal effect, both 100 and 150 μg/mL oxLDL exposure significantly reduced the cell viability by 18.22±10.9% and 29.3±6.73%, respectively (p<0.05 when compared with the control group; Fig. 1B). Concomitant addition of 30 μM E3330 completely prevented the suppression by 100 μg/mL oxLDL and restored the cell viability to 100±5.99% of the control level (p<0.05 when compared with 100 μg/mL oxLDL challenged group; Fig. 1B). A partial proliferation restoration of 86.9±7.87% by E3330 was observed when the RPEs were challenged with a higher dosage of 150 μg/mL oxLDL (p<0.05 when compared with 150 μg/mL oxLDL challenged group; Fig. 1B). Application of 30 μM E3330 alone did not affect the ARPE-19 cell viability in the absence of oxLDL challenge (Fig. 1B).
2.2. E3330 protects RPEs from oxLDL-induced senescence-like phenotype
RPE cell senescence is a biological aging process associated with physiological decline of the cellular function, and a key contributor to AMD [44]. Using a SA-β-gal assay, we examined the cellular senescence status in ARPE-19 cells under the challenge of various doses of oxLDL with or without the treatment of E3330. As shown in Fig. 2, both 100 and 150 μg/mL oxLDL promoted senescence-like phenotype in RPEs by increasing the SA-β-gal stain (p<0.05 when compared with the control group). Addition of 30 μM E3330 significantly abolished the increment of SA-β-gal activity induced by oxLDL (p<0.05 when compared with the same dose of oxLDL challenged group; Fig. 2). Among all dosage groups, it was striking that E3330 was able to divert the entire upsurge of over 70% increase of SA-β-gal accumulation in RPEs challenged by 150 μg/mL oxLDL. E3330 alone or with 50 μg/ml oxLDL did not affect the SA-β-gal level in RPEs. These results clearly show that E3330 effectively protected RPEs from oxLDL-induced stress without affecting the basal senescence-related function.
Fig. 2.
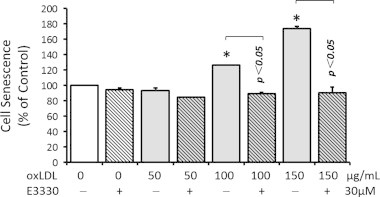
SA-β-gal stain of senescence-like phenotype in RPEs exposed to oxLDL and/or E3330. ARPE-19 cells were exposed to 0, 50, 100, or 150 μg/mL of oxLDL alone or combined with 30 μM E3330 for 48 h. Both 100 and 150 μg/mL oxLDL remarkably increased the SA-β-gal level in ARPE-19. The increments were completely abolished by concomitant treatment of 30 μM E3330. E3330 alone did not alter the basal level of the SA-β-gal accumulation. Bars represent mean±SEM of three independent experiments. ⁎p<0.05 versus the control group.
According to these above mentioned apoptosis, proliferation and senescence phenotype assays, 150 μg/mL oxLDL was adopted as a sub-lethal dose to challenge RPEs for all the subsequent experiments.
2.3. E3330 reduces the accumulation of intracellular ROS, secretion of MCP-1 and VEGF, and nuclear level of NF-κB p65 in oxLDL-challenged RPEs
Accumulation of intracellular ROS is a major source of oxidative stress in AMD [40], [45]. 150 μg/mL oxLDL significantly increased the intracellular ROS in RPEs by 20.23±8.03% (p<0.05 when compared with the control group; Fig. 3A). This elevated intracellular ROS was eliminated by 30 μM E3330 (p<0.05 when compared with 150 μg/mL oxLDL treated group; Fig. 3A). This is an interesting finding that may at least partly explain the protective effect of E3330 on oxLDL stressed RPEs.
Fig. 3.
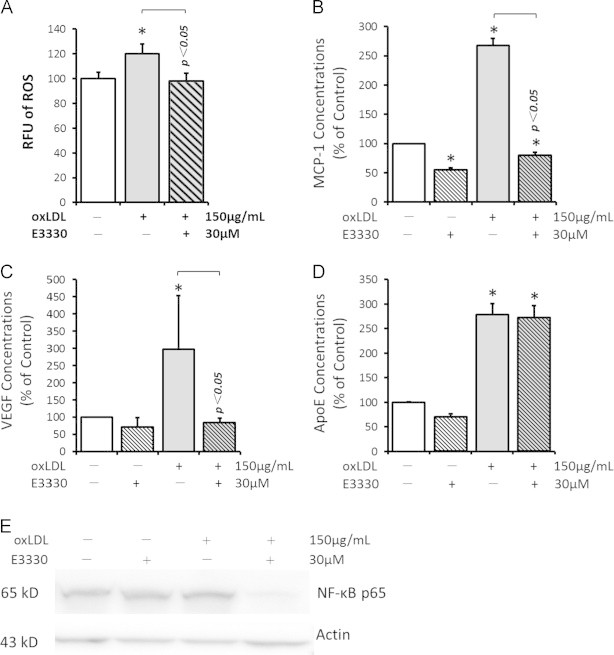
The effects of oxLDL and E3330 on intracellular ROS accumulation, cytokines production, and nuclear level of NF-κB p65 in RPEs. A, intracellular ROS level in ARPE-19 cells was measured by a fluorescent probe DCFH-DA and presented as relative fluorescent unit (RFU). OxLDL significantly increased the intracellular ROS, which was effectively abolished by concomitant E3330. B-D, ELISA assay of MCP-1 (B), VEGF (C), and ApoE (D) protein in APRE-19 conditioned media. The secretion of all the three proteins was prominently increased by oxLDL. Addition of E3330 effectively prevented oxLDL-induced MCP-1 and VEGF overproduction, but did not alter the level of ApoE. E3330 alone also reduced the production of MCP-1, but did not affect the VEGF and ApoE levels in un-challenged ARPE-19 cells. E, a representative gel image of NF-κB p65 and internal control protein actin in ARPE-19 nuclear fraction. Bars represent mean±SEM of three independent experiments. ⁎p<0.05 versus the control group.
RPEs derived MCP-1 [46], VEGF [47] and ApoE [48], [49] are critical contributors to local inflammation and angiogenesis in the development of AMD. Co-treatment of E3330 remarkably eliminated the striking upsurge secretion of both MCP-1 and VEGF stimulated by oxLDL (p<0.01 and p<0.05 respectively when compared with 150 μg/mL oxLDL challenged group; Fig. 3B and C). In addition, 30 μM E3330 significantly decreased MCP-1 production by 40% in the unchallenged RPEs (p<0.01; Fig. 3B), but did not affect much of the basal VEGF secretion from RPEs (Fig. 3C). However, the increase of ApoE production by oxLDL was not altered by the addition of 30 μM E3330 (Fig. 3D).
Several studies have reported that E3330 inhibits NF-κB mediated transcription via different mechanisms [25], [50], [51], [52], [53]. In this study, 150 μg/mL oxLDL significantly increased the NF-κB p65 level in RPE nucleus, which was prominently reduced by concomitant 30 μM E3330 (Fig. 3E). E3330 alone did not affect the level of p65 in un-challenged RPE nucleus.
2.4. E3330 blocks the activation of multiple transcription factors induced by oxLDL in RPEs
To identify the transcription factor(s) that the APE1 redox inhibitor E3330 regulates, a reporter gene assay array containing a set of ten known stress and toxicity responding genes in ten different pathways was applied. ARPE-19 cells were exposed to 150 μg/mL oxLDL alone or with 30 μM E3330. OxLDL remarkably upregulated all the ten transcriptional activities in RPEs (p-value varies when compared with the control group; Fig. 4 and Table 1). The presence of E3330 significantly repressed seven of the ten transcription factors that stimulated by oxLDL. When compared with the oxLDL challenged groups, E3330 decreased the transcriptional activities of Nrf2/Nrf1 (59.91±10.53%, p<0.01), p53 (59.35±6.80%, p<0.001), NF-κB (57.66±8.06%, p<0.05), HIF1 (62.05±11.06%, p<0.05), CBF/NF-Y/YY1 (44.24±9.03%, p<0.01), MTF1 (80.12±11.60%, p<0.05), and HSF-1 (63.18±2.95%, p<0.01) in RPEs. When compared with the un-treated control groups, six of the seven transcription factors had their DNA binding activities almost completely restored to the basal levels by E3330 (p>0.05 when compared with the control group; Fig. 4 and Table 1), with the exception of HSF1. Though HSF1 of heat shock pathway and AhR in xenobiotic drug metabolism were also markedly repressed by E3330, the end point levels of these two transcription factors were still significantly higher than that in the control groups (both p<0.05 when compared with the control group). The rest two transcription factors, glucocorticoid receptor and AP1, demonstrated a trend towards reduction by E3330 but did not reach statistic significant points (both p>0.05 when compared with 150 μg/mL oxLDL challenged group). The fold change and corresponding p-value are summarized in Table 1. These data clearly revealed that E3330 was able to restore multiple signaling pathways that stimulated by oxLDL which in turn may prevent a range of pathogenic responses in RPEs.
Fig. 4.
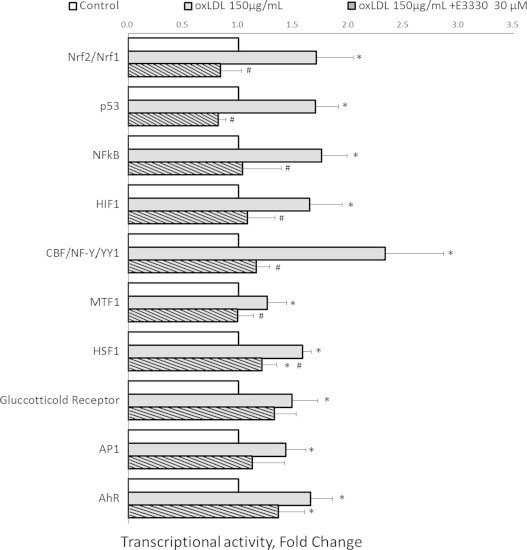
Dual-luciferase reporter gene assay of the stress responding transcription factors in RPEs. OxLDL exposure significantly up-regulated all the tested transcriptional activities. E3330 completely blocked the up surge of six transcription factors (Nrf2/Nrf1, p53, NF-κB, HIF1, CBF/NF-Y/YY1, and MTF1) and markedly attenuated the activities of HSF1 and AhR pathways. Glucocorticoid receptor exhibited a trend of reduction by E3330, but results were not statistically significant. Bars represent mean±SEM of three independent experiments. ⁎p<0.05 versus the control group. #p<0.05 versus the oxLDL challenged group.
Table 1.
Fold changes of the reporter gene activities in ARPE-19 cells in response to oxLDL alone or combined with E3330.
| Transcription factor | Reporter gene activity fold change | Pathway | |||||
|---|---|---|---|---|---|---|---|
| oxLDL/Control | p Value | oxLDL+E3330/oxLDL | p Value | oxLDL+E3330/Control | p Value | ||
| Nrf2/Nrf1 | 1.71 | 0.008 | 0.60 | 0.003 | 0.84 | 0.409 | Antioxidant response |
| p53 | 1.70 | 0.001 | 0.59 | 0.000 | 0.82 | 0.130 | DNA damage |
| NFκB | 1.76 | 0.008 | 0.58 | 0.011 | 1.04 | 0.850 | NFκB |
| HIF-1 | 1.65 | 0.012 | 0.63 | 0.021 | 1.08 | 0.658 | Hypoxia |
| CBF/NF-Y/YY1 | 2.33 | 0.002 | 0.44 | 0.004 | 1.16 | 0.549 | ER stress |
| MTF1 | 1.26 | 0.043 | 0.80 | 0.042 | 1.00 | 0.980 | Heavy metal stress |
| HSF-1 | 1.59 | 0.000 | 0.63 | 0.002 | 1.22 | 0.024 | Heat shock |
| Glucocorticoid Receptor (GR) | 1.49 | 0.015 | 0.68 | 0.312 | 1.33 | 0.065 | Glucocorticoid |
| AP-1 | 1.43 | 0.039 | 0.71 | 0.113 | 1.13 | 0.465 | MAPK/JNK |
| AhR | 1.65 | 0.004 | 0.61 | 0.094 | 1.36 | 0.047 | Xenobiotic |
OxLDL versus Con, 150 μg/mL oxLDL challenge versus vehicle control.
OxLDL+E3330 versus oxLDL, concomitant treatment of 150 μg/mL oxLDL plus 30 μM E3330 versus 150 μg/mL oxLDL challenge.
OxLDL+E3330 versus Con, concomitant treatment of 150 μg/mL oxLDL plus 30 μM E3330 versus vehicle control.
2.5. E3330 reduces laser-induced CNV lesion in mouse eyes
A laser-induced CNV model is a classic animal model of wet AMD which is an ocular wound-healing response to damage of the RPE–Bruch׳s membrane–choroidal complex [7]. We adopted this model to determine the in vivo effect of E3330. A single intravitreal injection of either E3330 or vehicle was given right after the laser photocoagulation. The treatment dosage of 1 μL 200 μM E3330 was equivalent to a final concentration of approximately 20 μM in the retinochoroidal tissue. After 2 weeks survival, a remarkable 40% reduction of the laser-induced CNV size was found in E3330 treated eyes when compared with the control eye (Fig. 5 A, B). Mean value of the CNV size treated by E3330 was 10,568 μm2 while that of vehicle control treated was 16,531 μm2. The difference was statistically significant (n=6; p=0.0200). These results are consistent with our previous finding that a single intravitreal treatment of E3330 inhibited the retinal angiomatous proliferation (RAP)-like neovascularization in Vldlr−/− mice [33].
Fig. 5.
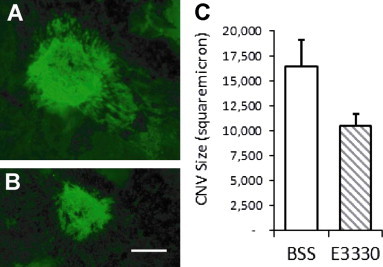
Single intravitreal injection of E3330 significantly reduced laser-induced CNV area in mouse eyes. Laser-induced CNV mice received a single intravitreal injection of 1 μL of 200 μM E3330 (equivalent to intravitreal centration of approximately 20 μM) in one eye and 1 μL BSS in the fellow eye immediately following laser photocoagulation of RPE–Bruch׳s membrane. Isolectin stain was performed after two weeks survival. (A and B) Representative images of isolectin staining of CNV lesions. The area of CNV lesion in E3330 treated eye (B) was obviously reduced when compared to BSS treated eyes (A). Scale bar: 100 μm. (C) Quantitative analysis showed that the mean value of the CNV area treated by E3330 was significantly lower than that of vehicle control treated (n=6; p=0.0200).
3. Discussion
Age-related macular degeneration (AMD) is a progressive disease encompassing multifactorial pathogenesis involving metabolic, functional, genetic and environmental factors, and affecting multiple components in the retina including photoreceptors, retinal pigment epithelium (RPE), Bruch׳s membrane and choriocapillaries. Current therapies still have limitations in effectively controlling this complex disease. The oxidative stress induced degeneration and dysfunction of macular RPEs are early and crucial changes contributing to local drusogenesis, inflammation and neovascularization [8], [40]. Protecting RPEs from oxidative damage is thereby an essential consideration for AMD treatment. In this study, we demonstrated for the first time that specific inhibition of APE1 redox function with E3330 effectively protected RPEs from oxLDL-induced impairment and reduced the area of laser-induced damage in RPE–Bruch׳s–choroidal complex. Taken together with our previous findings that E3330 significantly suppressed in vitro angiogenesis of retinal and choroidal endothelial cells [33], [34], and reduced retinal neovascularization in vivo [33], these data strongly suggest E3330 is a promising therapeutic agent for AMD by rescuing RPE degeneration and dysfunction from oxidative stress and reducing endothelial angiogenesis at the same time.
3.1. APE1 redox function and its role in the retina and choroid
The presence of APE1 in the retina and choroid has been established due to its critical roles in mammalian cells. We have reported high levels of APE1 in the neural retina, purified retinal vascular endothelial cells (RVECs) and retinal pericytes in mice [33]. We also found abundant expression of APE1 in human ARPE-19, RVECs and choroidal endothelial cells (CECs) [34]. Chiarini et al. [54], [55] demonstrated that APE1 was mainly located in the nuclear of all retinal cells and positively associated with the retina cell differentiation during postnatal development in rats. APE1 was postulated to accomplishes both functions of DNA repair and reductively activation of transcription factors in the nuclear [56].
In previous studies, the function of APE1 was evaluated by approaches such as gene knockout [57], [58], DNA antisense [59], [60] and siRNA [61]. While valid, these approaches are not suitable for the current study as they affect the entire APE1 molecule regardless of its distinctive dual functions. The subsequent changes are the mixture of multiple effects in which the specific role of APE1 redox activity cannot be depicted. In addition, such approaches could lead to cell cycle arrest [58], cell death [61] and early embryonic lethality [57]. Recent identification of the novel small molecule inhibitor that specifically suppresses APE1 redox function, namely E3330, makes it possible to investigate the redox activity individually [11], [16], [23]. The specificity of E3330 has been well documented by several studies. E3330-fixed latex beads specifically bind to APE1 protein [24], and E3330 effectively blocks multiple redox sensitive transcription factors without interfering with the DNA repair function of APE1 [10], [23], [25], [26], [51]. By using E3330, we have previously reported that APE1 redox function was required for retinal and choroidal angiogenesis [33], [34], and involved in neural tissue recovery after stroke in diabetic rats [35]. These and our current findings in RPEs clearly indicate that inhibition of APE1 redox function have multiple beneficial effects in the retinochoroidal region including anti-angiogenesis, anti-inflammation and neural protection.
3.2. Inhibition of APE1 redox function rescued degeneration and dysfunction of RPEs induced by oxidative stress
OxLDL is known as a major contributor of oxidative stress and inflammation in AMD pathology. Previous studies showed that oxLDL inhibits RPE viability, promotes RPE senescence and apoptosis in a dose dependent manner [42], and disrupts multiple functions of RPE cells at sub-lethal dosages [42], [62]. OxLDL-stimulated intracellular accumulation of ROS plays an important role in linking oxidative stimulus with early dysfunctions of RPEs [40], [45]. The biological effects of oxLDL are mainly mediated through signaling pathways, especially via activation of transcription factors such as NF-κB [41], [63]. In this study, E3330 effectively attenuated sub-lethal oxLDL induced viability decline and senescence advancement of RPEs, at the same time blocked intracellular accumulation of ROS in the cell. It is quite possible that these events occurred through the suppression of multiple transcription factors via inhibition of APE1 redox function, since the ROS generation mediated by NADPH oxidase (Nox) was actually controlled by transcription factors including NF-κB [64], HIF-1 [65] and Nrf2 [66], [67]. Intracellular ROS reduction may subsequently relieve the cellular macromolecules such as DNA, proteins, and cell membrane lipids from oxidative stress [40], [68], and suppress the ROS triggered “activation signals” and the cross talk between ROS and redox-sensitive transcription factors such as NF-κB [69] and HIF-1 [70]. These changes presented as the regain of RPE proliferation and reduction of senescence-like phenotype in RPEs in our study. The fact that we saw such a broad spectrum of effects by E3330 on RPE functions strongly indicated that all of these aspects were affected in various degrees.
By examining the production of MCP-1 and VEGF by RPEs, we found that the inhibitory effect of E3330 was translated from redox sensitive transcription factors to downstream effector molecules capable of mediating chronic inflammation and angiogenesis in the retina and choroid. These findings supported our previous reports that intravitreal application of E3330 significantly reduced retinal and choroidal neovascularization in mice [33], [34], as RPEs is a considerable source of VEGF and MCP-1 in the retinochoroidal tissue. ApoE [49], another AMD related molecule that synthesized by RPEs, was minimally regulated by E3330. The transcriptional regulation of ApoE gene expression is known to be cell-type specific [71]. Ishida et al. reported that ApoE secretion by human RPEs was transcriptionally regulated by ligands for nuclear hormone receptors such as thyroid hormone receptor, liver X receptor and retinoid X receptor [48]. While little is known so far about APE1 redox regulation on these transcription factors, our data indicated that oxLDL-induced ApoE production in RPEs was not regulated by APE1 redox signals.
NF-κB is arguably the most studied transcription factor in the pathogenesis of AMD. It is known NF-κB can be activated by oxidative stress and regulates expressions of various pro-inflammatory and pro-angiogenesis factors in RPEs, including MCP-1, ICAM-1, ILs, and VEGF [28], [41], [72]. E3330 is able to suppress the NF-κB activity through different mechanisms. For example, Handa׳s team reported that E3330 inhibited the NF-κB DNA binding through reversing the APE1-dependent reduction of Cys-62 residue of p50, without affecting the IκB-α degradation and p65 translocation or phosphorylation in Jurkat cells [25], [50]. Goto et al. showed that E3330 impaired the nuclear translocation of NF-κB and degradation of IκB-α in human peripheral monocytes [51]. The differences noted in these reports are probably due to the usage of different methodology and cell-specific differences. In the current study, we examined the effect of E3330 on the nuclear level of NF-κB p65 as nuclear localization is required for both APE1 and NF-κB to accomplish their functions. We found E3330 markedly reduced the nuclear accumulation of p65 protein, which was complied with the inhibitory effect of E3330 on NF-κB DNA binding as is shown in luciferase reporter gene assay. As other NF-κB proteins such as p50, p52, RelB and cRel may also contribute to the transcriptional activity of NF-κB in RPEs, additional experiments investigating the underlying mechanism are necessary.
3.3. Inhibition of APE1 redox function represses multiple stress-responding transcription factors in RPEs
We have provided data in this study that E3330 significantly repressed the oxLDL-induced stimulation of at least six (Nrf2/Nrf1, p53, NFκB, HIF1, CBF/NF-Y/YY1, and MTF1) transcription factors in RPEs. Some of these transcription factors are directly implicated in dry and/or wet AMD pathogenesis. Nrf2/Nrf1 is critical in retinal antioxidant and detoxification responses [73], [74]. p53 stimulates RPE apoptosis and cell cycle arrest [75], [76]. NF-κB regulates the production of pro-inflammatory factors and growth factors in several stress sceneries including immune reactions, inflammation and hypoxia in RPEs [28], [29], [77]. HIF1 mainly controls the VEGF production induced by hypoxia [30]. It is noteworthy that although the effect of E3330 is quit potent affecting multiple transcription factors, at the given dose it only blocked the aberrantly up-regulated transcriptional activities and brought them back down to the levels comparable to their basal activities. These data demonstrated that targeting APE1 redox activity could modulate multiple signaling pathways closely related to AMD pathogenesis and may achieve a greater therapeutic effect than any mono-target therapies, without causing side effects from complete inhibition of any given transcription factor.
3.4. APE1 redox function in inhibiting choroidal neovascularization
Laser-induced CNV is an wound-healing response to the damage of RPE–Bruch׳s–choroidal complex [7]. The growth of CNV is largely promoted and directed by RPE derived signals, typically increased expression of VEGF via HIF-1 [78] and NF-κB [72] signaling pathways, and MCP-1 via NF-κB pathway [28], [79], [80]. Therefore, the finding that intravitreal E3330 significantly reduced the laser-induced CNV in mouse eyes can be at least partly attribute to the attenuated transcription activities of NF-κB and HIF-1, as well as decreased production of MCP-1 and VEGF in RPEs by E3330. However, E3330 also directly inhibits the angiogenic activities of endothelial cells [33], [34]. Due to the nature of laser-induced CNV animal model, it is difficult for us to distinguish whether the actual effect of APE1 redox inhibition is from CEC or RPE or both at this point. Further studies are underway for a more comprehensive understanding of this issue.
In summary, we demonstrated that specific inhibition of APE1 redox activity with E3330 significantly protected RPEs from oxLDL-induced stresses and reduced laser-induced CNV in mouse eyes. This effect was mediated by inhibiting ROS generation, repressing multiple redox sensitive transcription factors, and preventing upsurge of downstream pro-inflammatory and pro-angiogenic molecules. These findings in RPEs along with our previous reports of the anti-angiogenenic effect on retinochoroidal endothelial cells [33], [34], [79] suggested that specific inhibition of APE1 redox activity with E3330 could be a promising therapeutic strategy for both dry and wet AMD by targeting multiple signaling pathways in the context of hypoxia, choronic inflammation, and angiogenesis. Further studies are necessary for a better understanding of the underlying mechanisms for this novel AMD treatment.
4. Materials and methods
4.1. Materials
A RPE cell line from adult human, ARPE-19 (American Type Culture Collection, Manassas, VA), was cultured in DMEM (Gibco® Life Technologies, Grand Island, NY) supplemented with 10% FBS in a 37 °C incubator with 5% CO2. ARPE-19 cells near confluence were resuspended by 0.25% Trypsin–EDTA (Gibco® Life Technologies, Grand Island, NY) and used for sub-culture and various assays.
OxLDL, a widely used experimental paradigm for AMD [41], [42], was commercially purchased (Kalen Biomedical, Montgonery Village, MD) and used to induce pathological stress in ARPE-19 cells. E3330[(2E)-3-[5-2,3-dimethoxy-6-methyl-1,4-benzoquinoyl)]-2-nonyl-2-propenoic acid] was first dissolved in dimethylsulphoxide (DMSO, Sigma-Aldrich Co. Saint Louis, MO) at a concentration of 20 mM, then diluted in culture medium to reach various end point doses. The final concentration of DMSO in the medium did not exceed 0.01% (vol/vol). A dose of 30 μM of E3330 was applied throughout this study according to our previous observation that 20–50 μM E3330 significantly reduced retinochoroidal angiogenesis in vitro and in vivo in the absence of dose-related adverse effects [33], [34], as well as our pilot dose-response experiments in ARPE-19 cells. Cells were treated with vehicle control, 30 μM E3330 alone or in combination with various doses of oxLDL. A vehicle control with matched concentration of DMSO without E3330 was routinely included in each experiment.
4.2. Cell apoptosis and proliferation assay
The effects of oxLDL and/or E3330 on RPE apoptosis and proliferation was measured by a TdT mediated dUTP-fluorescein nick end-labeling (TUNEL) Reaction kit (In Situ Cell Death Detection Kit, Roche, Indianapolis, IN) and a CellTiter 96® AQueous One Solution Reagent (Promega Corporation, Madison, WI) respectively. ARPE-19 cells were seeded in 96-well plates at 2000 cell/well and exposed to various doses of oxLDL alone or combined with 30 μM E3330 for 48 h in a 37 °C incubator. These assays were performed following the manufacture׳s protocol.
4.3. Cell senescence associated β-gal (SA-β-gal) assay
A cellular senescence assay kit (Cell Biolabs, San Diego, CA) was used to determine the effect of E3330 on senescence-like phenotype of RPEs. ARPE-19 cells seeded at 2000 cells/well in 96-well plates were exposed to oxLDL alone or combined with 30 μM E3330 for 48 h in a 37 °C incubator. After centrifugation, 50 μL of the supernatant of each sample was collected and immediately incubated with 50 μL of freshly prepared fluorometric substrate for 2 h at 37 °C in darkness. The fluorescent intensity of each reaction mixture was determined by a SpectraMax M2e microplate reader (Molecular Devices LLC, Sunnyvale, CA) at an excitation wavelength of 360 nm and emission wavelength of 465 nm.
4.4. Intracellular ROS measurement
The intracellular ROS accumulation in RPEs was measured by 2׳, 7׳-dichlorodihydro fluorescin diacetate (DCFH-DA) using an intracellular ROS assay kit (Cell Biolabs, San Diego, CA). ARPE-19 cells were pre-loaded with DCFH-DA by exposure to freshly prepared 100 µM DCFH-DA in the culture medium for 30 min in a 37 °C incubator. Then the DCFH-DA loaded cells were treated with 150 μg/mL oxLDL alone or combined with 30 μM E3330 for 48 h. DCFH fluorescence of the cell lysate was measured with excitation and emission settings of 485 and 530 nm by a microplate reader.
4.5. ELISA assay
The protein concentrations of MCP-1, VEGF and ApoE in the conditioned medium of ARPE-19 cells were determined using commercially available ELISA kits (R&D Systems, Minneapolis, MN) following the manufacturer׳s instructions.
4.6. Western blot
ARPE-19 cells were treated with 150 μg/mL oxLDL or 30 μM E3330 alone or in combination for 48 h. Nuclear proteins were extracted from ARPE-19 cells with a NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific, Rockford, IL) according to the manufacture׳s instruction. The nuclear proteins were separated by SDS-PAGE, and western blot was performed with an anti-NF-κB p65 antibody (Cell Signaling Technology, Beverly, MA) overnight at 4 °C. β-actin (Abcam, Cambridge, MA) was used as a loading control. Quantitative densitometry analysis was performed using an Image Lab™ Software (Bio-Rad, Hercules, CA). The ratio of NF-κB p65 and β-actin was presented.
4.7. Luciferase reporter gene assay
The reporter gene assay was performed using a Cignal Finder Stress & Toxicity 10-Pathway Reporter Array (Qiagen-SABiosciences, Valencia, CA). Briefly, ARPE-19 cells were exposed to 150 μg/mL oxLDL alone or combined with 30 μM E3330 for 48 h. The treated cells at 40–60% confluence were transfected with equal amounts of the mixed firefly luciferase (Fluc) and Renilla luciferase (Rluc) reporter vector for 24 h using Attractene transfection reagent (Qiagen, Valencia, CA). Then the fresh medium was added and incubated for another 6 h. Fluc and Rluc expressions were detected using a Dual-Luciferase Reporter Assay System (Promega Corporation, Madison, WI) with a Luminometer SpectraMax M2e according to the manufacturer׳s instruction. The transcription activities, as indicated by the Fluc reading, were normalized to Rluc. The ratio of Fluc and Rluc was divided by the ratio of the promoterless positive control luciferase to determine the fold change.
4.8. Animals
Male wild-type C57BL/6J mice of 6–8 weeks were used in the in vivo studies. Animals were housed on a 12-h light-dark cycle with free access to food and water. All procedures were performed with strict adherence to the guidelines for animal care and experimentation prepared by the Association for Research in Vision and Ophthalmology and approved by the Henry Ford Health System Institutional Animal Care and Use Committee.
4.9. Laser photocoagulation, E3330 intravitreal injection, and quantification of the CNV lesions
Mice were anesthetized by 50 mg/kg ketamine HCl and 10 mg/kg xylazine. Topical 1% tropicamide, and 2.5% phenylephrine were administered for pupil dilation. Animals were positioned before a slit-lamp with a laser-delivery system (Coherent Novus 2000 Argon Laser System, Santa Clara, CA, USA). The fundus was visualized using a cover slip with Cornea Coat® (Hydroxypropyl- methylcellulose, Insight Instruments Inc., Stuart, FL, USA). Laser photocoagulation (532 nm wavelength, 100 mW power, 50 μm spot size, and 0.1 s duration) was performed bilaterally in each mouse. A series of four laser spots were concentrically placed at approximately 9, 12, 3, and 6-o׳clock positions and 75–100 μm around the optic disk in each eye. All the laser burns had the appearance of a cavitation bubble. Right after laser photocoagulation, animals received a single intravitreal injection of 1 μl E3330 in one eye and the same volume of Balanced Saline Solution (BSS, Akorn Inc., Lake Forest, IL, USA) as a vehicle control in the fellow eye. Target concentration of E3330 in the retinochoroidal was approximately 20 μM. Two weeks after laser photocoagulation, mice were sacrificed and eyes were processed for CNV analysis.
Fluorescent labeled isolectin was used to stain CNV spots according to previous reports [33], [80]. Briefly, 4% PFA fixed eyecups were incubated with blocking solution containing 3% normal donkey serum, 0.2% Triton x-100 in PBS and then stained with a 1:100 diluted isolectin in blocking buffer at 4 °C overnight. With 4–6 relaxing radial cuts, the RPE-choroid-sclera complex was flattened and mounted with the RPE side upward. The area of CNV lesion in each sample was measured using SPOT Advanced software (Diagnostic Instruments Inc., Sterling Heights, MI, USA).
4.10. Statistical analysis
Results of quantitative studies were expressed as mean±SEM of at least three independent experiments. Differences were assessed using one-way analysis of variance (ANOVA) for repeated measures followed by Fisher׳s least significant difference test when appropriate. The statistical analyses were performed using IBM SPSS Statistics 19 (Armonk, NY). A value of p<<0.05 was considered statistically significant.
Acknowledgments
This work was supported by Grants from the International Retinal Research Foundation, Midwest Eye Bank, Reeves Foundation, Alliance for Vision Research, and Henry Ford Research Foundation to X.Q., Grants from the National Institutes of Health CA121168 and CA167291 and the Riley Children׳s Foundation to M.R.K and by Grants from the National Natural Science Foundation of China (Grant no. 81000390), Alliance for Vision Research to Y.L.
References
- 1.Tomany SC W.J., Van Leeuwen R., Klein R., Mitchell P., Vingerling J.R., Klein B.E., Smith W., De Jong P.T. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. 2004;111:1280–1287. doi: 10.1016/j.ophtha.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Ryskulova A. T.K., Makuc D.M., Cotch M.F., Klein R.J., Janiszewski R. Self-reported age-related eye diseases and visual impairment in the United States: results of the 2002 national health interview survey. Am. J. Public Health. 2008;98:454–461. doi: 10.2105/AJPH.2006.098202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferris FL F.S., 3rd, Hyman L. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch. Ophthalmol. 1984;102:1640–1642. doi: 10.1001/archopht.1984.01040031330019. [DOI] [PubMed] [Google Scholar]
- 4.Brown DM M.M., Kaiser P.K., Heier J.S., Sy J.P., Ianchulev T. ANCHOR Study Group. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR study. Ophthalmology. 2009;116:57–65.e55. doi: 10.1016/j.ophtha.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 5.CATT Research Group, M. D., Maguire M.G., Ying G.S., Grunwald J.E., Fine S.L., Jaffe G.J. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011;364:1897–1908. doi: 10.1056/NEJMoa1102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tufail A.P.P., Egan C., Hykin P., da Cruz L., Gregor Z., Dowler J., Majid M.A., Bailey C., Mohamed Q., Johnston R., Bunce C., Xing W. ABC Trial Investigators. Bevacizumab for neovascular age related macular degeneration (ABC Trial): multicentre randomised double masked study. Br. Med. J. 2010;340 doi: 10.1136/bmj.c2459. 10.1136/bmj.c2459. [DOI] [PubMed] [Google Scholar]
- 7.WR G. Histopathology of age-related macular degeneration. Mol. Vis. 1999;27 [PubMed] [Google Scholar]
- 8.JG. H. Age-related macular degeneration: the molecular link between oxidative damage, tissue-specific inflammation and outer retinal disease: the Proctor lecture. Invest. Ophthalmol. Vis. Sci. 2010;51:1275–1281. doi: 10.1167/iovs.09-4478. [DOI] [PubMed] [Google Scholar]
- 9.Fishel ML K.M. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol. Asp. Med. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Luo M D.S., Jiang A., Reed A., He Y., Fishel M., Nyland R.L. 2nd, Borch R.F., Qiao X., Georgiadis M.M., Kelley M.R. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid. Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tell G Q.F., Tiribelli C., Kelley M.R. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid. Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhakat KK M.A., Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid. Redox Signal. 2009;11:621–638. doi: 10.1089/ars.2008.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans AR L.-F.M., Kelley M.R. Going APE over ref-1. Mutat. Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 14.Wilson DM B.D., 3rd The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 15.Tell G D.G., Caldwell D., Kelley M.R. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid. Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 16.Kelley M.R. G.M., Fishel M.L. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr. Mol. Pharmacol. 2012;5:36–53. doi: 10.2174/1874467211205010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ando K H.S., Kabe Y., Ogura Y., Sato I., Yamaguchi Y., Wada T., Handa H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36:4327–4336. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou GM K.C., Kabe Y., Handa H., Anders R.A., Maitra A. The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: therapeutic implications in tumor angiogenesis. J. Cell. Physiol. 2009;219:209–218. doi: 10.1002/jcp.21666. [DOI] [PubMed] [Google Scholar]
- 19.Xanthoudakis S M.G., Wang F., Pan Y.C., Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xanthoudakis S C.T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jayaraman L M.K., Zhu C., Curran T., Xanthoudakis S., Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev. 1997;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- 22.Xanthoudakis S M.G., Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl. Acad. Sci. U. S. A. 1994;91:23–27. doi: 10.1073/pnas.91.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J., Marasco L.M., Logsdon D., LaFavers D., Chen K.A., Reed Q., Kelley A., Gross M.R., Georgiadis MM. M.L. Inhibition of apurinic/apyrimidinic endonuclease I׳s redox activity revisited. Biochemistry. 2013;52:2955–2966. doi: 10.1021/bi400179m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimizu N., Tang S.K., Nishi J., Sato T., Hiramoto I., Aizawa M., Hatakeyama S., Ohba M., Hatori R., Yoshikawa H., Suzuki T., Oomori F., Tanaka A., Kawaguchi H., Watanabe H., Handa H. H. High-performance affinity beads for identifying drug receptors. Nat. Biotechnol. 2000;18:877–881. doi: 10.1038/78496. [DOI] [PubMed] [Google Scholar]
- 25.Hiramoto M., Sugimoto S.N., Tang K., Kawakami J., Ito Y., Aizawa M., Tanaka S., Makino H., Handa H. I. Nuclear targeted suppression of NF-κB activity by the novel quinone derivative E3330. J. Immunol. 1998;160:810–819. [PubMed] [Google Scholar]
- 26.Nyland R.L., Kelley L.M., Borch RF. M.R. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1) J. Med. Chem. 2010;53:1200–1210. doi: 10.1021/jm9014857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X.C., Allen J.C., Roberts J.B., Jaffe GJ. W.L. Suppression of NF-kappaB-dependent proinflammatory gene expression in human RPE cells by a proteasome inhibitor. Invest. Ophthalmol. Vis. Sci. 1999;40:477–486. [PubMed] [Google Scholar]
- 28.Kaarniranta K S.A. NF-kappaB signaling as a putative target for omega-3 metabolites in the prevention of age-related macular degeneration (AMD) Exp. Gerontol. 2009;44:685–688. doi: 10.1016/j.exger.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Arjamaa O., Salminen N.M., Kaarniranta K. A. Regulatory role of HIF-1alpha in the pathogenesis of age-related macular degeneration (AMD) Ageing Res. Rev. 2009;8:349–358. doi: 10.1016/j.arr.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Jonas J.B., Neumaier T.Y., Findeisen P. M. Monocyte chemoattractant protein 1, intercellular adhesion molecule 1, and vascular cell adhesion molecule 1 in exudative age-related macular degeneration. Arch. Ophthalmol. 2010;128:1281–1286. doi: 10.1001/archophthalmol.2010.227. [DOI] [PubMed] [Google Scholar]
- 31.Raoul W., Camelo A.C., Guillonneau S., Feumi X., Combadière C., Sennlaub F. C. CCL2/CCR2 and CX3CL1/CX3CR1 chemokine axes and their possible involvement in age-related macular degeneration. J. Neuroinflamm. 2010;87 doi: 10.1186/1742-2094-7-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang A., Kelley G.H., Qiao X. M.R. Inhibition of APE1/Ref-1 redox activity with APX3330 blocks retinal angiogenesis in vitro and in vivo. Vis. Res. 2011;51:93–100. doi: 10.1016/j.visres.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiao X., Zhou L.Y., Liu T., Kelley X., Edwards M.R., Gao H. P. Selective blocking of APE1/Ref-1 redox function by a novel compound, APX3330 inhibits choroidal endothelial cells in vitro and choroidal neovascularization in vivo. Abstr. Assoc. Res. Vis. Ophthalmol. 2012;342 [Google Scholar]
- 34.Yan T., C.M., Zacharek A., Ning R., Qiao X., Kelley M.R., Roberts C., Chen J., Neurorestorative therapy for stroke in type one diabetic rats using APX3330, JCBFM Meeting, 2013.
- 35.O. S. The retinal pigment epithelium in visual function. Physiol. Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 36.Simó R., Corraliza V.M., Hernández L., Garcia-Ramírez M. C. The retinal pigment epithelium: something more than a constituent of the blood-retinal barrier – implications for the pathogenesis of diabetic retinopathy. J. Biomed. Biotechnol. 2010;190724 doi: 10.1155/2010/190724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roth F., Holz FG. B.A. Key pathophysiologic pathways in age-related macular disease. Graefes Arch. Clin. Exp. Ophthalmol. 2004;242:710–716. doi: 10.1007/s00417-004-0976-x. [DOI] [PubMed] [Google Scholar]
- 38.Ruberti J.W., Millican C.C., Menco C.L., Huang B.P., Johnson M. J.D. Quick-freeze/deep-etch visualization of age-related lipid accumulation in Bruch׳s membrane. Invest. Ophthalmol. Vis. Sci. 2003;44:1753–1759. doi: 10.1167/iovs.02-0496. [DOI] [PubMed] [Google Scholar]
- 39.Mettu P.S., Ong W.A., Cousins SW. S.S. Retinal pigment epithelium response to oxidant injury in the pathogenesis of early age-related macular degeneration. Mol. Asp. Med. 2012;33:376–398. doi: 10.1016/j.mam.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 40.Yamada Y., Yang T.J., Cutler Y., Wu R.G., Telljohann T., Mattson R.S., Handa JT. M.P. Oxidized low density lipoproteins induce a pathologic response by retinal pigmented epithelial cells. J. Neurochem. 2008;105:1187–1197. doi: 10.1111/j.1471-4159.2008.05211.x. [DOI] [PubMed] [Google Scholar]
- 41.Kim J.H., Kim L.S., Yu K.W., Kim JH. Y.S. Oxidized low density lipoprotein-induced senescence of retinal pigment epithelial cells is followed by outer blood-retinal barrier dysfunction. Int. J. Biochem. Cell Biol. 2012;44:808–814. doi: 10.1016/j.biocel.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 42.M.R. K. RPE cell senescence: a key contributor to age-related macular degeneration. Med. Hypotheses. 2012;78:505–510. doi: 10.1016/j.mehy.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 43.Beatty S., Phil K.H., Henson M., Boulton M. D. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 44.Penfold P.L., Gillies M.M., Provis JM. M.C. Immunological and aetiological aspects of macular degeneration. Prog. Retin. Eye Res. 2001;20:385–414. doi: 10.1016/s1350-9462(00)00025-2. [DOI] [PubMed] [Google Scholar]
- 45.Witmer A.N., Van Noorden V.G., Schlingemann RO. C.J. Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retin. Eye Res. 2003;22:1–29. doi: 10.1016/s1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 46.Ishida B.Y., Duncan B.K., Chalkley K.G., Burlingame R.J., Kane A.L., Schwartz DM. J.P. Regulated expression of apolipoprotein E by human retinal pigment epithelial cells. J. Lipid Res. 2004;45:263–271. doi: 10.1194/jlr.M300306-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Johnson L.V., Banna F.D., Radeke C.D., Maloney C.M., Hu M.A., Spencer J., Walker C.N., Tsie A.M., Bok M.S., Radeke D., Anderson DH. M.J. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 2011;108:18277–18282. doi: 10.1073/pnas.1109703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishi T., Hiramoto S.N., Sato M., Yamaguchi I., Hasegawa Y., Aizawa M., Tanaka S., Kataoka H., Watanabe K., Handa H. H. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J. Biol. Chem. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 49.Goto M., Katayama Y.K., Tanaka I. K. Inhibitory effect of E3330, a novel quinone derivative able to suppress tumor necrosis factor-alpha generation, on activation of nuclear factor-kappa B. Mol. Pharmacol. 1996;49:860–873. [PubMed] [Google Scholar]
- 50.Fishel M.L., Rajeshkumar J.Y., Scandura N.V., Sinn G., He A.L., Shen Y., Jones C., Pollok D.R., Ivan K.E., Maitra M., Kelley M.R. A. Impact of APE1/Ref-1 redox inhibition on pancreatic tumor growth. Mol. Cancer Ther. 2011;10:1698–1708. doi: 10.1158/1535-7163.MCT-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han W., Cai L.H., Gleaves J., Polosukhin L.A., Segal V.V., Yull B.H., Blackwell TS. F.E. NADPH oxidase limits lipopolysaccharide-induced lung inflammation and injury in mice through reduction-oxidation regulation of NF-κB activity. J. Immunol. 2013;190:4786–4794. doi: 10.4049/jimmunol.1201809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiarini L.B., Petrs-Silva F.F., Linden R. H. Evidence that the bifunctional redox factor / AP endonuclease Ref-1 is an anti-apoptotic protein associated with differentiation in the developing retina. Cell Death Differ. 2000;7:272–281. doi: 10.1038/sj.cdd.4400639. [DOI] [PubMed] [Google Scholar]
- 53.Chiarini LB L.R. Tissue biology of apoptosis. Ref-1 and cell differentiation in the developing retina. Ann. N. Y. Acad. Sci. 2000:64–78. doi: 10.1111/j.1749-6632.2000.tb05599.x. [DOI] [PubMed] [Google Scholar]
- 54.Wu H.H., Chang C.Y., Wu J.T., Liu T.C., Chen W.S., Lee H. C.Y. Subcellular localization of apurinic endonuclease 1 promotes lung tumor aggressiveness via NF-kappaB activation. Oncogene. 2010;29:4330–4340. doi: 10.1038/onc.2010.178. [DOI] [PubMed] [Google Scholar]
- 55.Xanthoudakis S., Wallace S.R., Curran T. J.D. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. U. S. A. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fishel M.L., Reed H.Y., Chin-Sinex A.M., Hutchins H., Mendonca G.D., Kelley M.R. M.S. Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair (Amst.) 2008;7:177–186. doi: 10.1016/j.dnarep.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ono Y., Ohmoto F.T., Akiyama T., Seki S. K. Stable expression in rat glioma cells of sense and antisense nucleic acids to a human multifunctional DNA repair enzyme, APEX nuclease. Mutat. Res. 1994;315:55–63. doi: 10.1016/0921-8777(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 58.Walker L.J., Harris C.R., Hickson ID. A.L. A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res. 1994;22:4884–4889. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang Y., Sandusky Z.S., Kelley G.E., Fishel ML. M.R. Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Invest. 2010;28:885–895. doi: 10.3109/07357907.2010.512816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoppe G., Pennock M.A., Hoff HF. E.A. Oxidized low density lipoprotein-induced inhibition of processing of photoreceptor outer segments by RPE. Invest. Ophthalmol. Vis. Sci. 2001;42:2714–2720. [PubMed] [Google Scholar]
- 61.Robbesyn F., Negre-Salvayre A. S.R. Dual role of oxidized LDL on the NF-kappaB signaling pathway. Free Radic. Res. 2004;38:541–551. doi: 10.1080/10715760410001665244. [DOI] [PubMed] [Google Scholar]
- 62.O׳Leary D.P., Woolley B.L., Gough J.F., Wang D.R., Cotter J.H., Redmond HP. T.G. TLR-4 signalling accelerates colon cancer cell adhesion via NF-κB mediated transcriptional up-regulation of Nox-1. PLoS One. 2012;7:e44176. doi: 10.1371/journal.pone.0044176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Diebold I., Hess P.A., Görlach A. J. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell. 2010;21:2087–2096. doi: 10.1091/mbc.E09-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pendyala S N.V. Redox regulation of Nox proteins. Respir. Physiol. Neurobiol. 2010;174:265–271. doi: 10.1016/j.resp.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pendyala S., Kalari M.J., Kleeberger S., Zhao S.R., Reddy Y., Garcia S.P., Natarajan V. J.G. Nrf2 regulates hyperoxia-induced Nox4 expression in human lung endothelium: identification of functional antioxidant response elements on the Nox4 promoter. Free Radic. Biol. Med. 2011;50:1749–1759. doi: 10.1016/j.freeradbiomed.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Decanini A., Feng N.C., Ferrington X., Olsen TW. D.A. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am. J. Ophthalmol. 2007;143:607–615. doi: 10.1016/j.ajo.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bonello S., BelAiba Z.C., Djordjevic R.S., Hess T., Michiels J., Kietzmann C., Görlach A. T. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler. Thromb. Vasc. Biol. 2007;27:755–761. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- 68.Cakir Y.B.S. Reactive species-mediated regulation of cell signaling and the cell cycle: the role of MAPK. Antioxid. Redox Signal. 2005;7:726–740. doi: 10.1089/ars.2005.7.726. [DOI] [PubMed] [Google Scholar]
- 69.Rossello X.S., Weisman I.U., Sun G.A., Wood WG. G.Y. AP-2β regulates amyloid beta-protein stimulation of apolipoprotein E transcription in astrocytes. Brain Res. 2012:87–95. doi: 10.1016/j.brainres.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma W., Guo L.S., Qu J., Hudson W., Schmidt B.I., Barile GR. A.M. RAGE ligand upregulation of VEGF secretion in ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 2007;48:1355–1361. doi: 10.1167/iovs.06-0738. [DOI] [PubMed] [Google Scholar]
- 71.Zhao Z., Wang C.Y., Sternberg J., Freeman P., Grossniklaus M.L., Cai J. H.E. Age-related retinopathy in NRF2-deficient mice. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rezaei K.A., Cai C.Y., Sternberg P. J. Modulation of Nrf2-dependent antioxidant functions in the RPE by Zip2, a zinc transporter protein. Invest. Ophthalmol. Vis. Sci. 2008;49:1665–1670. doi: 10.1167/iovs.07-0959. [DOI] [PubMed] [Google Scholar]
- 73.Bhattacharya S., Johnson C.E., Johnson LR. D.A. Age-related susceptibility to apoptosis in human retinal pigment epithelial cells is triggered by disruption of p53–Mdm2 association. Invest. Ophthalmol. Vis. Sci. 2012;53:8350–8366. doi: 10.1167/iovs.12-10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kwok A.K., Lai Y.C., Chan T.Y., Pang CP. K.P. Effects of trypan blue on cell viability and gene expression in human retinal pigment epithelial cells. Br. J. Ophthalmol. 2004;88:1590–1594. doi: 10.1136/bjo.2004.044537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagineni C.N., William K.V., Detrick A., Hooks JJ. B. Regulation of VEGF expression in human retinal cells by cytokines: implications for the role of inflammation in age-related macular degeneration. J. Cell. Physiol. 2012;227:116–126. doi: 10.1002/jcp.22708. [DOI] [PubMed] [Google Scholar]
- 76.Smith G.W., Prentice D.C., Blanks J. H. The importance of hypoxia-regulated, RPE-targeted gene therapy for choroidal neovascularization. Adv. Exp. Med. Biol. 2012:269–277. doi: 10.1007/978-1-4614-0631-0_35. [DOI] [PubMed] [Google Scholar]
- 77.Chen Y., Chen K.A., Yang P. Y. IL-17A stimulates the production of inflammatory mediators via Erk1/2, p38 MAPK, PI3K/Akt, and NF-κB pathways in ARPE-19 cells. Mol. Vis. 2011:3072–3077. [PMC free article] [PubMed] [Google Scholar]
- 78.Ping D., Rogers B.G., Boss J.M. E.M. Nuclear factor-kappa B p65 mediates the assembly and activation of the TNF-responsive element of the murine monocyte chemoattractant-1 gene. J. Immunol. 1999;162:727–734. [PubMed] [Google Scholar]
- 79.Qiao X., Liu L.Y., Zhou X., Kelley T., Edwards M.R., Gao H. P. APE1/Ref-1 redox inhibitor APX3330 modulates choroidal endothelial cells by transcriptional regulation of NF-κB and STAT3 activity. Abstr. Assoc. Res. Vis. Ophthalmol. 2013;274 [Google Scholar]
- 80.Mullins R.F., Skeie J.M. G.M. Glycoconjugates of choroidal neovascular membranes in age-related macular degeneration. Mol. Vis. 2005:509–517. [PubMed] [Google Scholar]