

Abstract
Thioredoxin reductase (TR) catalyzes the reduction of thioredoxin (TRX), which in turn reduces mammalian typical 2-Cys peroxiredoxins (PRXs 1–4), thiol peroxidases implicated in redox homeostasis and cell signaling. Typical 2-Cys PRXs are inactivated by hyperoxidation of the peroxidatic cysteine to cysteine-sulfinic acid, and regenerated in a two-step process involving retro-reduction by sulfiredoxin (SRX) and reduction by TRX. Here transient exposure to menadione and glucose oxidase was used to examine the dynamics of oxidative inactivation and reactivation of PRXs in mouse C10 cells expressing various isoforms of TR, including wild type cytoplasmic TR1 (Sec-TR1) and mitochondrial TR2 (Sec-TR2) that encode selenocysteine, as well as mutants of TR1 and TR2 in which the selenocysteine codon was changed to encode cysteine (Cys-TR1 or Cys-TR2). In C10 cells endogenous TR activity was insensitive to levels of hydrogen peroxide that hyperoxidize PRXs. Expression of Sec-TR1 increased TR activity, reduced the basal cytoplasmic redox state, and increased the rate of reduction of a redox-responsive cytoplasmic GFP probe (roGFP), but did not influence either the rate of inactivation or the rate of retro-reduction of PRXs. In comparison to roGFP, which was reduced within minutes once oxidants were removed reduction of 2-Cys PRXs occurred over many hours. Expression of wild type Sec-TR1 or Sec-TR2, but not Cys-TR1 or TR2, increased the rate of reduction of PRXs and improved cell survival after menadione exposure. These results indicate that expression levels of TR do not reduce the severity of initial oxidative insults, but rather govern the rate of reduction of cellular factors required for cell viability. Because Sec-TR is completely insensitive to cytotoxic levels of hydrogen peroxide, we suggest TR functions at the top of a redox pyramid that governs the oxidation state of peroxiredoxins and other protein factors, thereby dictating a hierarchy of phenotypic responses to oxidative insults.
Keywords: Redox signaling, Menadione, Hydrogen peroxide, Peroxiredoxins
Graphical abstract
Highlights
-
•
Sensitivity of TR1 and TR2 vs. PRXs to oxidative inactivation.
-
•
Role of Sec vs. Cys in the active site of TR in cell viability.
-
•
Reduction of roGFP as compared to PRX after oxidation.
-
•
Effect of TR1 and TR2 on oxidation state of cellular compartments.
Introduction
Cellular oxidants modulate the strength and duration of signaling through redox-responsive pathways that govern cell proliferation and survival [1], [2]. In many disease states management of endogenous and/or exogenous oxidants may be compromised, leading to oxidative stress, damage to macromolecules and loss of cell viability. Phenotypic adaptation to chronic oxidative stress, which occurs in many cancers, often involves up-regulation of stress response proteins and antioxidant enzymes, leading to improved cell survival [3]. Among the families of antioxidant enzymes that are often over-expressed in cancer and other disease states are thioredoxin reductases (TRs) and peroxiredoxins (PRXs), both of which are involved in the metabolism of hydrogen peroxide [4]. Mammalian TRs are a family of selenocysteine-containing oxidoreductases that act as the primary reductases for thioredoxin (TRX) in a NADPH dependent reaction [4], [5]. Recent studies show that selenocysteine in the C-terminal redox center renders mitochondrial TR insensitive to inactivation from oxidants [6], suggesting TR and other selenoenzymes may mediate resistance to oxidative stress.
Malignant mesotheliomas express elevated levels of TR1 [7], [8] and mesothelioma cells propagated in vitro have higher levels of TR activity than untransformed mesothelial cells do [9]. Given that this phenotype has been observed in many other tumor types, TR has emerged as an attractive therapeutic target in human malignancies [10], [11]. Because of the limited number of cellular proteins that contain selenocysteine, one approach is to use compounds (such as aurothioglucose and auranofin) that selectively target selenocysteine [12], [13], [14]. However, the effects of inhibition or over-expression of TR on cell growth and survival are not straightforward, as expression of TR in which the activity of selenium is compromised may induce cell death due to pro-oxidant activity [15]. TR and TRX have been shown to influence the activity of transcription factors, kinases and other factors in cell signaling circuits, and in some contexts the pro-proliferative role of its primary substrate thioredoxin (TRX) has been ascribed to the regulation of the ERK pathway leading to the expression of cyclin D1 [16].
Peroxiredoxins are a family of thiol peroxidases that are ubiquitously expressed throughout eukaryotes. In mammals there are six family members, with peroxiredoxins 1–4 representing typical 2-Cys PRXs; these enzymes function as head-to-tail homodimers with two reaction sites, each consisting of a peroxidatic cysteine in one monomer and a resolving cysteine in the opposing subunit [17]. During the metabolism of hydrogen peroxide the initial step in the peroxiredoxin reaction cycle is oxidation of the peroxidatic cysteine to sulfenic acid (–SOH), which then spontaneously reacts with the resolving cysteine in the opposing subunit to produce an intermolecular disulfide bond. The intermolecular disulfide bond then is reduced by thioredoxin, which in turn is reduced by TR, using reducing equivalents from NADPH. The peroxidatic cysteines of 2-Cys PRXs are among the most reactive cysteines in the cell [18], in part due to the stabilization of the thiolate in the active site and structural interactions positioning peroxide for attack [19], [20], [21]. Curiously, an evolutionarily conserved carboxy-terminal extension on PRXs 1–4 stabilizes the peroxidatic −SOH intermediate, making it susceptible to further attack by hydrogen peroxide, thereby leading to the formation of sulfinic acid (–SOOH). This process, which has been termed “over-oxidation” or “hyperoxidation”, precludes disulfide bond formation and would be expected to permanently inactivate the enzyme, for sulfinic acid residues in proteins generally are not repaired. PRXs are a notable exception, for with the aid of ATP the reductase sulfiredoxin (SRX) is capable of “retro-reduction” of PRX-SOOH and regeneration of enzyme activity [22], [23]. In addition to homodimers, the oxidation state of PRXs influences their relative distribution in dimers, decamers and higher order oligomers that have chaperone and other functions [24]. The unusual features of PRX catalysis and oligomerization, as well as their interaction with many cellular regulatory factors, have led to proposals that PRXs act as peroxide sensors in which structural transitions and/or changes in protein–protein interactions with cellular regulators modulate responses to oxidative stress [25], [26]. In yeast, the TR system is devoted to metabolism of hydrogen peroxide [27], and regulation of hierarchical responses to oxidative insults by thiol peroxidase Tsa1 has been elegantly dissected [28], [29]. However, detailed descriptions of similar hierarchical pathways in mammalian cells have yet to be reported.
Both TR and PRXs may be over-expressed in cancer, and studies in which TR has been down-regulated by RNA interference strongly support the hypothesis that elevated expression of TR promotes tumor cell proliferation in culture and tumor progression in animal models [30], [31]. To mimic the situation often encountered in malignancies, we have expressed cytoplasmic TR1 and mitochondrial TR2 in mouse lung epithelial C10 cells that are immortalized but not tumorigenic. Previously we described the relationship between the oxidation state of PRX2 and C10 cell cycle progression in response to fluxes of hydrogen peroxide, and showed that expression of cyclin D1 and cell cycle progression was not recovered after oxidative insult until PRX2 was completely reduced [32]. However, in contrast to PRXs, mammalian TRs with selenocysteine in the C-terminal catalytic site are remarkably resistant to inactivation by hydrogen peroxide and other oxidants in vitro [6], raising the possibility that TR sits at the top of a redox pyramid that includes peroxiredoxins as exquisitely sensitive detectors of cellular peroxide.
To test this possibility, we compared the ability of TRs encoding selenocysteine in the C-terminal redox center to TRs in which the codon for selenocysteine had been changed to that for cysteine to mediate recovery from oxidative insults. Our data suggest that TR levels do not directly influence the extent of the initial insult, but rather dictate the rate of recovery and cell viability, restoring proliferative capacity over time. Moreover, only wild type TRs had the capacity to improve cell survival, indicating that the insensitivity of selenocysteine to inactivation by hydrogen peroxide is an important feature of mammalian TRs.
Materials and methods
Cell culture
C10 mouse lung epithelial cells were maintained in CMRL cell culture media (Corning Cellgro, Manassas, VA), supplemented with 10% fetal bovine serum (FBS), 200 mM glutamine, and 0.5% penicillin streptomycin and propagated in a humidified incubator at 37 °C and 5% CO2. Cells were trypsinized and re-plated to obtain 75% confluence on the following day for all subsequent experiments.
Molecular cloning
Full length human TR1 was amplified from pCMV6-XL4 vector by PCR using specific forward (TR1: 5'-GAAAGTCGAGGAGACAGTTAAGCATG-3') and reverse (TR1: 5'-CACAAGGAAAGGTCATGCTAAAACTG-3') primers and subsequently cloned into pcDNA 3.1 mammalian expression vector (Promega, Madison WI). Full length cDNA for human TR2 was recovered from pCMV6-XL4 by digestion with XbaI and EcoRI, purified by gel electrophoresis, and ligated into linear pcDNA 3.1 using T4 DNA Ligase. Insertion of wild type Sec-TR1 and Sec-TR2 full-length cDNAs, including the 3' SECIS elements, into pcDNA 3.1 was confirmed by sequencing using the appropriate forward and reverse primer sets (T7 Forward: BGH Reverse). PCR-based mutagenesis was performed by Mutagenex (Piscataway, NJ) to replace the Sec codon with Cys and was confirmed by sequencing. Expression vectors for human TRX1 and TRX 2 were purchased from Open Biosystems (Thermo Scientific, Waltham, MA). Expression vectors for cytosolic and mitochondrial targeted roGFPs were a kind gift from J.A. Melendez (College of Nanoscale Science and Engineering, Albany, NY).
Plasmid transfection and siRNA silencing
Plasmid expression vectors were introduced into C10 cells by transfection or co-transfection using Lipofectamine 2000™ following the manufacturer׳s guidelines (Invitrogen, Grand Island, NY). Pilot experiments with GFP and RFP expression vectors showed co-transfection efficiency with Lipofectamine 2000™ in C10 cells was 90% or better (data not shown). Specific siRNA to TR1 (si-TR1, 5'-CCAUAGAGGGCGAAUUUAAUU-3') was introduced into C10 cells using Dharmafect™ following the manufacturer׳s guidelines (Dharmacon, Thermo Scientific).
Live cell imaging with roGFP
Cells were transfected with indicated expression vectors in combination with either cytosolic (Cyto-roGFP) or mitochondrial targeted (Mito-roGFP) dynamic oxidant sensors [33]. Eighteen hours after transfection cells were re-plated into 35-mm glass bottom imaging dishes (MatTek™, Ashland, MA). The following day media was changed to imaging media (20 mM HEPES, 134 mM NaCL, 5.4 mM KCL, 1 mM MgSO4, 1.8 mM CaCl2, 5 mM glucose pH 7.4) and dishes were mounted on a Nikon Ti-E inverted microscope. Images were collected and oxidation state was determined as previously described [34]. Briefly, fluorescence emission was collected at 525 nm after sequential excitation at ~405 nm and ~488 nm. The ratio of oxidized (emission after excitation at ~405) to reduced (emission after excitation at ~488 nm) roGFP probe, after subtraction of background for each channel, is presented as a measure of relative redox status of the cytosol (Cyto-roGFP) or the mitochondria (Mito-roGFP). The change in oxidation (ΔOx) of roGFP was determined by calculating the change in the ratio 10 min after removal of menadione.
Detection of oxidant levels by DCF
Post-transfection, cells were plated into flat-bottom 96 well tissue culture plates, allowed to adhere and treated as indicated in the text in complete media. The following day cells were washed 2× with pre-warmed HBSS plus calcium and magnesium and incubated with HBSS containing 20 uM 2',7'-dichlorodihydrofluorescein diacetate (DCF). After a 30 min incubation, DCF was removed and the cells were washed 2× with HBSS and fluorescence intensity was determined using a Synergy HT Microplate Reader™ with excitation at 495 nm and emission at 525 nm.
Thioredoxin reductase activity assay
Thioredoxin reductase activity was determined in whole cell lysates using the SC-TR assay as previously described [35]. Briefly, cells were grown to ~75% confluence, treated as indicated in the text, and lysed on ice in NP-40 lysis buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40, 1 mg/mL leupeptin, 1 mg/mL aprotinin, 1 mM NaF, 1 mM NaVO3, 1 mM PMSF). Lysates were cleared by centrifugation at 14,000 rpm and total protein amount was determined by reading the absorbance of Bradford reagent at 595 nm. Equal protein amounts (25 μg) were added to individual wells of a clear round bottom 96 well microplate. Master mix containing 1 mM NADPH, and 2 mM selenocystine in ddH2O were added to each well, yielding a final concentration of 400 μM and 800 μM, respectively. The conversion of NADPH to NADP+ was monitored in 30 s intervals over a 20 min time period at 340 nm on a Synergy HT Microplate Reader™ (BioTek). TR activity was determined by comparing the initial slopes of NADPH oxidation for each experimental condition and expressed as nmoles NADPH×min−1×mg−1 of protein lysate using an extinction coefficient of 6220 M−1 cm−1.
Immunoblotting
Equal amounts of protein sample (15–35 ug/lane) were separated on 12% gels by SDS-PAGE and transferred to PVDF membrane. Membranes were blocked in 5% milk (Tris-buffered saline with 0.2% Tween-20, TBST) for 1 h at room temperature. Primary antibodies were diluted in 5% milk/TBST and incubated for 1 h at room temperature. Anti-TR1 (1:2000), anti-Trx1 (1:2000), anti-Trx2 (1:2000) were purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-TR2 (1:2000), anti-PRX3 (1:2000), anti-PRX2 (1:2000), and anti-PRX-SO2/SO3H (1:2000) were purchased from ABFrontier (Seoul, Korea). Anti-actin antibody (1:5000) was from Millipore (Billerica, MA). The membranes were washed for 5 min with fresh TBST and washed in this manner 5×, after which the membranes were incubated with appropriate species of horseradish peroxidase conjugated secondary antibody (Millipore) diluted in 5% milk/TBST for 1 h at room temperature. The membranes were again washed 5× (5 min each) with TBST and protein bands were visualized with Enhanced Chemiluminescent™ detection system (Millipore).
Cell viability assays
Post transfection, cells were plated into flat bottom 96-well tissue culture plates, allowed to adhere, and treated as indicated in the text in complete media. The following day wells were rinsed 2x with phosphate buffered saline (PBS) and fixed in 3.4% paraformaldehyde for 10 min and washed 1× in PBS. Viable cells were stained with 0.1% crystal violet dissolved in ddH2O for 30 min and then rinsed sufficiently with water and allowed to dry. Crystal violet was solubilized by addition of 100% methanol and absorbance was read at 540 nm on a Synergy HT Microplate Reader™.
Statistics
Statistical significance was determined using the Students t-test (two-tailed, unpaired). The data within is presented as the mean +/−S.D. of 3 replicates unless otherwise stated and the level of significance is described in the figure legends.
Results
Previously we characterized the responses of mouse lung epithelial C10 cells to oxidative insults in regard to cell cycle arrest and recovery [1], [36], [37] and demonstrated that after arrest expression of cyclin D1 and cell proliferation does not resume until hyper-oxidized PRXs are reduced and high-molecular oligomers of PRX2 are resolved to decamers and dimers [32]. C10 cells were chosen as model for these experiments since airway epithelial cells are exposed to ambient levels of oxygen in vivo, and while C10 cells are immortalized, they do not form tumors in syngeneic mice. Given that TR and TRX play important roles in the reduction of PRXs, and that over-expression of TR is often associated with resistance to oxidative stress and cell survival in tumor cells, we examined the effects of expression of TR1 and TR2, with or without expression of the cognate TRX, on recovery of C10 cells from oxidative insults and cell survival. To examine the role of selenocysteine in TR-mediated cell survival, expression of wild type TR mRNAs were compared to TR mRNAs in which the codon for the catalytic selenocysteine was replaced with the codon for cysteine. To control for effects of non-coding sequences, plasmid cDNA vectors expressing full-length mRNAs with native 5' and 3' untranslated regions, including the cognate selenocysteine insertion element (SECIS element), were used.
Cellular TR activity is insensitive to oxidative insults
After exposure of C10 cells to concentrations of glucose oxidase (GOx) greater than 2.5 mU/mL for 3 h, expression of cyclin D1 ceases and cells arrest in the cell cycle [32]. Exposure of C10 cells to increasing concentration of GOx showed that concentrations of 1.25 mU/mL or greater induced hyperoxidation of PRXs (Fig. 1B, lanes 1–6), whereas under the same exposure conditions GOx caused no significant reduction of total cellular TR activity (Fig. 1A). In contrast, exposure of C10 cells to the reactive aldehyde acrolein, which attacks the selenocysteine moiety and thereby inhibits TR activity [38], completely abolished cellular TR activity (Fig. 1A). These data indicate that cellular TR activity, identical to purified TR enzyme [6], is highly resistant to inactivation by hydrogen peroxide, and that the vast majority of TR activity detected in C10 cell lysates is sensitive to acrolein.
Fig. 1.
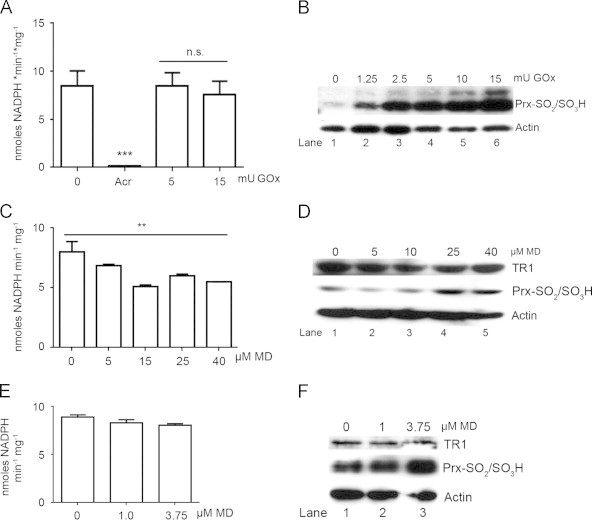
Cellular TR activity is insensitive to oxidative insults. (A) TR activity in C10 cell lysates after treatment with 30 µM acrolein (Acr) for 1 h or the indicated concentrations of glucose oxidase (GOx) for 3 h (n=3, ***p<0.001, ns=not significant). (B) Immunoblot analysis of hyper-oxidized PRXs in C10 cell lysates after treatment of cells with indicated concentrations of GOx for 3 h. (C) TR activity was quantified in C10 cell lysates after acute treatment with the indicated concentrations of menadione (MD) for 45 min (n=3, **p<0.01). (D) Immunoblot analysis of TR1 expression levels and hyper-oxidized PRXs in cell lysates from (C). (E) TR activity was quantified in cell lysates treated with low levels of MD overnight (n=3). (F) Immunoblot analysis of TR1 expression levels and hyper-oxidized PRXs in cell lysates from (E). In panels B, D and F actin was used as a loading control.
Under similar culture conditions exposure of C10 cells to highly cytotoxic concentrations of menadione (MD), a quinone that produces oxidative stress in both the cytoplasmic and mitochondrial compartments [39], [40], resulted in increased levels of hyperoxidized PRX and modest reductions in TR activity, even at the highest concentrations (Fig. 1C and D). At lower but cytostatic doses of MD, increased hyperoxidation of PRXs was still evident (Fig. 1E), but TR activity was completely unaffected (Fig. 1F). These initial studies showed that under conditions in which PRXs are inactivated by hyperoxidation, cellular TR activity is largely unaffected, supporting the supposition that selenocysteine contributes to the resistance of TR to inactivation by reactive oxygen species [41].
Expression of Sec-TR and Cys-TR on total cellular TR activity
To mimic over-expression of TR that occurs in malignant mesothelioma and other tumor cell types, plasmid expression vectors were constructed for wild type human cytoplasmic TR (Sec-TR1) and mitochondrial TR2 (Sec-TR2), as well as mutants in which the codon for selenocysteine was replaced by site-directed mutagenesis with a codon for cysteine (here referred to as Cys-TRs, or TR1-U498C and TR2-U523C) (Fig. 2A). In transfection experiments ectopic expression of immunoreactive TR1 polypeptides was dependent on the concentration of plasmid DNA (Fig. 2B and data not shown), and in C10 cells transfected with the WT TR1 expression vector (1 ug DNA/ 60 mm dish) total TR activity increased 2-fold (Fig. 2C). In C10 cells transfected with the expression vector for TR1-U498C levels of TR activity were not significantly greater than in C10 cells transfected with the pCDNA3.1 vector alone (Fig. 2B and C). The increased TR1 protein and activity levels after transfection of C10 cells with WT TR1 reflect those in malignant mesothelioma and other tumor cell types [9]. Transfection of siRNAs targeted to TR1 reduced expression of TR1 by 90% (data not shown) and reduced TR activity to 10% of endogenous levels (Fig. 2C). Expression of WT-TR2 and TR2-U523C at relatively high levels did not result in increased total cellular TR activity (Fig. 2D), a finding in agreement with previous studies that showed mitochondrial TR activity represents less than 10% of the total cellular TR activity [42], as suggested here by the residual TR activity after silencing TR1 (Fig. 2C). Approximately 50% of the TR activity in control cells transfected with WT-TR1 or TR1-U498C was sensitive to the TR inhibitor auranofin (Fig. 2E). We recognize that the amount of WT-TR1 protein expressed after 24 h was significantly higher compared to endogenous levels, and while the total TR activity also increased, it did not increase more than 2 to 3-fold. The reason for this apparent discrepancy is not known.
Fig. 2.
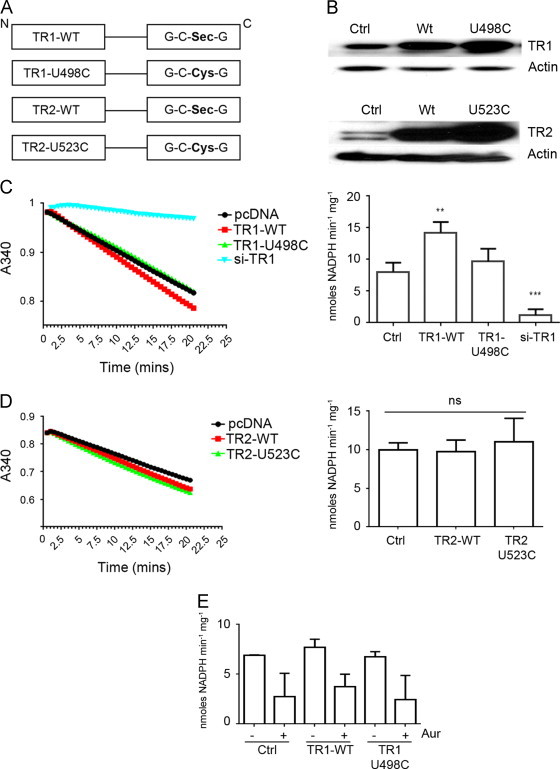
Effects of expression of Sec and Cys containing TR1 and TR2 on total cellular TR activity. (A) Schematic depiction of Sec-TR (WT-TR1, WT-TR2) and Cys-TR (TR1-U498C and TR2-U523C) expression vectors used in subsequent studies. U indicates selenocysteine and C cysteine. (B) Immunoblot analysis of cell lysates from C10 cells transfected with the indicated expression vectors and blotted for TR1 (upper panel) or TR2 (lower panel). (C) Rate of NADPH consumption (A340, see Materials and methods) in lysates from C10 cells transfected with the indicated TR1 expression vectors or with TR1-siRNA (left panel). TR activity from 3 replicate samples is summarized in the right panel (**p<0.01). (D) Rate of NADPH consumption measured at A340 of lysates from C10 cells transfected with the TR2 expression vectors (left panel); TR activity from 3 replicate samples is summarized in the right panel. (E) TR activity in the indicated cell lysates with or without Auranofin (Aur) (n=3).
These transfection studies established that the plasmid vectors for WT-TR1 and Cys-TR1 mutants produced polypeptides with apparent molecular weights indistinguishable from endogenous TR, and that expression of Sec-TR1 increased cellular TR activity. Expression of TR1-U498C, in contrast, failed to significantly increase cellular TR activity. Similarly expression of Sec-TR2 and Cys-TR2 resulted in increased expression of immunoreactive TR2 polypeptides with apparent molecular weights identical to endogenous TR2, but neither Sec-TR2 nor TR2-U523C resulted in statistically significant increases to total cellular TR activity. However, when mitochondrial fractions were prepared that contained the ectopically expressed Sec-TR2, increased TR activity was observed (data not shown). Co-transfection of TRX1 or TRX2 with or without the cognate TR did not increase total TR activity above that observed with transfection of TR alone (data not shown).
Effects of TR expression of reduction of roGFP
Using genetically-encoded GFP probes that are sensitive to oxidation–reduction (roGFP) [33], the effects of expression of Sec and Cys-TRs on the basal redox state of the cytoplasm and mitochondria were investigated with ratiometric live cell imaging (Fig. 3A). For examining the redox state of the cytoplasm, C10 cells were co-transfected with vectors for roGFP targeted to the cytoplasm (cyto-roGFP) and either Sec-TR1 or Cys-TR1, with or without an expression vector for TRX1. For mitochondria, C10 cells were transfected with vectors for roGFP targeted to the mitochondria (mito-roGFP) and either Sec-TR2 or Cys-TR2, with or without an expression vector for TRX2. On the day following transfection, the ratio of oxidized to reduced roGFP was evaluated by live cell fluorescence microscopy (Fig. 3A). Expression of Sec-TR1 but not Cys-TR1 reduced the basal redox state of the cell cytoplasm (Fig. 3B), and in a similar fashion expression of Sec-TR2 but not Cys-TR2 caused a significant reduction in the basal redox state of mitochondria (Fig. 3C and D). These results correlated well with the effects of expression of Sec and Cys-TRs on cellular TR activity (Fig. 2).
Fig. 3.
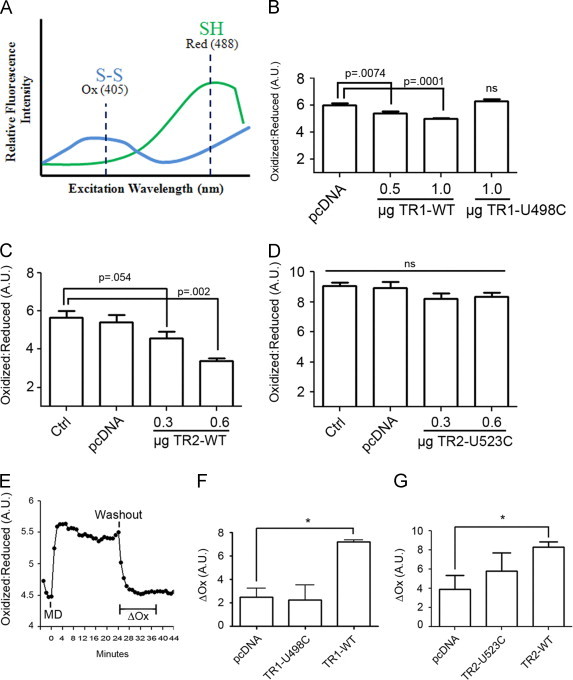
Sec-TRs but not Cys-TRs accelerate roGFP reduction after oxidative insult. (A) Schematic of the dynamic response of the roGFP probe to changes in intracellular thiol status. In the presence of oxidants a disulfide bond is formed in roGFP that increases fluorescence intensity after excitation of the probe at 405 nm (Ox). Under reduced conditions, fluorescence intensity is increased after excitation at 488 nm (Red). The ratio of oxidized to reduced roGFP reflects cellular redox status. (B) Basal cytosolic redox state in cells co-transfected with Cyto-roGFP, TRX1, and the indicated TR1 expression vectors (n=20 cells). (C and D) Basal mitochondrial oxidation state in cells transfected with Mito-roGFP, TRX2, and the indicated amount of TR2 expression vectors (n=20, p value determined by students t-test). (E) The basal cellular redox state in C10 cells expressing Cyto-roGFP was assessed by ratiometric imaging prior to adding 40 uM menadione (MD) for 20 min. After washing out MD by 5 exchanges with fresh imaging media the same cells were re-examined over time to quantify change in oxidation (ΔOx, see Materials and methods) of the cyto-roGFP probe. (F) The experiment in panel E was repeated in cells co-expressing the indicated TR1 expression vector and Cyto-roGFP, and the ΔOx was calculated post menadione washout after 10 min (n=10, *p<0.05). (G) The experiment in E was repeated using cells co-transfected with Mito-roGFP and the indicated TR2 expression vectors and the ΔOx was calculated post menadione washout after 10 min (n=10, *p<0.05).
Cells co-transfected with vectors for roGFP and WT-TR1 or Cys-TR1 (with or without expression of TRX1) were then exposed to high concentrations of menadione for 30 min, followed by a recovery period after the menadione was removed by 5 successive changes of imaging medium, and ratiometric imaging was used to assess changes in the roGFP oxidation state over time. As shown in a representative experiment in Fig. 3E, roGFP was rapidly oxidized in response to menadione, and the probe remained oxidized for at least 25 min. Once the menadione was removed, the reduction of the roGFP probe was rapid, with a return to basal state within 10–15 min. In cells transfected with Sec-TR1 the change in the oxidation (ΔOx) of the roGFP probe from oxidized to reduced was increased over cells transfected with vector control or Cys-TR1 (Fig. 3F). In a similar fashion, expression of Sec-TR2 increased the ΔOx of mito-roGFP after washout of menadione over cells transfected with pcDNA (Fig. 3G). Expression of Cys-TR2 tended to increase the ΔOx of the mito-roGFP, but the increases were not statistically significant across several experiments (Fig. 3G). The ratiometric imaging data confirmed that acute menadione exposure induces both cytoplasmic and mitochondrial oxidative stress, as reported previously [40], and that expression of Sec-TR1 or Sec-TR2 reduces the basal redox state of C10 cells and improves reduction of the roGFP probes targeted to the cytoplasm or mitochondria, respectively.
TR and hyper-oxidation and reduction of PRXs
The peroxidatic cysteines of PRXs are among the most reactive thiols in the cell [18]. To compare the oxidative modifications of PRXs to oxidation of roGFP in response to menadione, cells were treated with 40 µM menadione for 1 h, then the menadione was washed out, and cells were allowed to recover for 4 h. Cell extracts were prepared at various time points and assessed for the presence of hyperoxidized PRXs by blotting with an antibody specific to PRX-SO2/SO3H. A strip-reprobe blotting method was used to estimate the relative amount of a specific PRX that was inactivated by hyperoxidation [32]. To assess the levels of PRX-S-S-PRX dimers, which can only be generated during the PRX catalytic cycle, extracts were resolved under denaturing conditions in the absence of reducing agent (i.e. with SDS but no DTT) [32]. Under no exposure condition was a significant reduction in the total levels of PRX2-3 observed (Fig. 4, Supplementary Fig. 1).
Fig. 4.
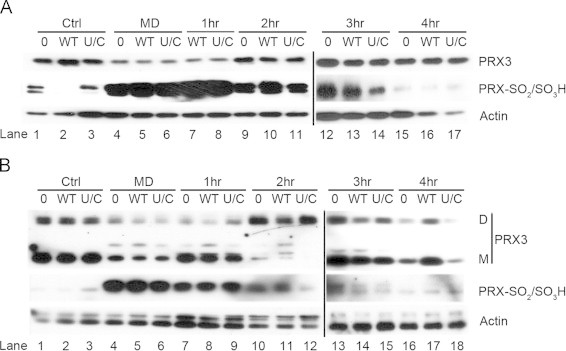
Hyper-oxidation and reduction of PRXs in response to ectopic expression of TR2. (A) C10 cells were transfected with indicated TR2 expression vectors (WT=TR2, U/C=TR2-U523C) and the following day were treated with menadione (MD) for 1 h before exchange with fresh culture media. Cell lysates were prepared from control cells (Ctrl), cells treated with MD for 1 h (MD), and cells allowed to recover for 1, 2, 3 or 4 h after removal of menadione. Cell lysates were prepared and separated by SDS-PAGE under reducing conditions for immunoblot analysis to evaluate the levels of hyper-oxidized PRXs (PRX-SO2/SO3H). The blot was then stripped and reprobed with antibody to PRX3 to specifically reveal the amount of hyper-oxidized PRX3 (see reference [27]). Note expression of TR2 did not influence retro-reduction of PRX3-SO2/SO3H by SRX. (B) The extracts in (A) were resolved under denaturing but non-reducing conditions to evaluate PRX3-S-S-PRX3 dimers (D) and PRX3 monomers (M). All hyper-oxidized PRX3 migrates as a monomer under both electrophoretic conditions because PRX3-SO2/SO3H cannot form disulfide bonds.
The response of PRX3 to oxidative inactivation and reduction during exposure and recovery is shown in Fig. 4, and this response was representative of that for cytoplasmic PRX2 (Supplementary Fig. 1). Within 1 h of menadione treatment, markedly increased levels of hyper-oxidized PRXs were observed and expression of WT-TR2 or TR2-U523C had no effect on oxidative inactivation of PRXs, and reprobing the blot with antibody to PRX3 indicated a significant fraction of PRX3 was inactivated under these conditions (Fig. 4A, lanes 4–6). Hyper-oxidized 2-Cys PRXs are repaired by SRX, which is recruited to mitochondria in response to inactivation of PRX3 [43]. Over time hyperoxidized PRXs were repaired, and after 4 h the cellular pools of hyperoxidized PRXs had returned to baseline, as did the signal for total PRX3 (Fig. 4A, lanes 7–17). Although expression of WT-TR2 reduced the basal oxidation state of cellular mitochondria (Fig. 3C), the change in basal redox state did not affect either the inactivation of PRX3 by menadione or the apparent rate of retro-reduction of hyperoxidized PRX3 by SRX. The redox status of roGFP probes as compared to cellular proteins is not well understood and may contribute to the interpretation of this result [33].
Examination of the relative levels of PRX3 dimers by denaturing but not reducing gel electrophoresis showed that increases in hyperoxidized PRX3 (which cannot form disulfide-bonded dimers) was accompanied by reduction in the abundance of PRX3 dimers, as expected (Fig. 4B, lanes 4–6). After 2 h of recovery, levels of PRX-SO2/SO3H were reduced, and levels of dimeric PRX3 were increased as compared to baseline levels (Fig. 4B, compare lanes 1–3 to lanes 10–12). Expression of WT or TR2-U523C had no effect on the level of PRX3 dimers or monomers by this time, but after 4 h of recovery expression of WT-TR2 increased the levels of both dimers and monomers as compared to vector control or TR2-U523C (Fig. 4 lanes 16–18). Together these expression experiments showed that both mito-roGFP and PRX3 were rapidly oxidized in response to menadione, but mito-roGFP was reduced in the order of minutes whereas repair of hyperoxidized PRX3 occurred over several hours. Only Sec containing WT-TR2 had any perceptible effect on recovery from acute oxidative insult. Our results confirm the relative sensitivity of PRXs to inactivation by oxidants and reinforce the concept that oxidative modification of cellular proteins depends in part on the reactivity of specific cysteine residues, thereby providing a mechanistic basis for a hierarchy of modifications in regulatory factors that mediate phenotypic outcomes.
Sec-TRs but not Cys-TRs improve cell survival
Acute oxidative insults invoke stress responses that differ from chronic oxidative stress, and to model these conditions cells were exposed to lower doses (60 nM–7.5 µM) of menadione for 24 h. To evaluate the level of cellular oxidants after chronic exposure to a range of menadione concentrations, we incubated cells with the general oxidant probe DCF 24 h after addition of menadione to cells. DCF fluorescence showed that increasing concentrations of menadione caused a corresponding continuous increase in cellular oxidants for at least 24 h, and expression of either Sec-TR1 or TR1-U498C did not reduce the levels of DCF fluorescence (Fig. 5A). In contrast, viability assays showed that expression of Sec-TR1 promoted cell survival and most likely cell proliferation above control values for menadione concentrations up to 3.75 uM, above which concentration cell viability was markedly reduced (Fig. 5B). Note that low concentrations of menadione apparently stimulated cell growth as suggested by increases in total cellular mass, and that this effect was increased by expression of Sec-TR1, strengthening the possibility that enhanced production of oxidants and elevated expression of TR1 in tumor cells cooperate to promote cell proliferation.
Fig. 5.
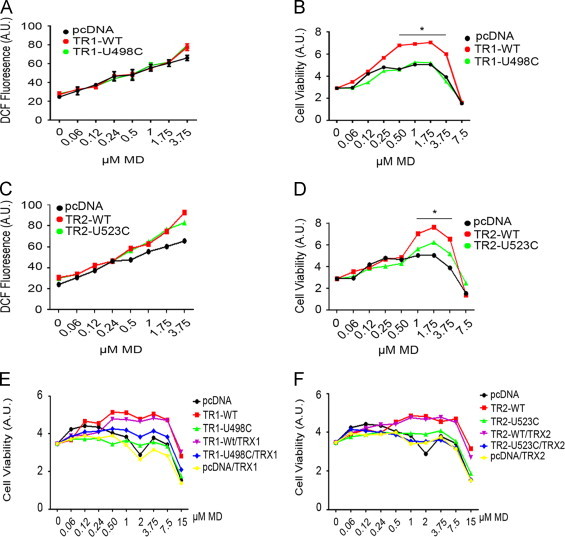
Overexpression of Sec-TRs but not Cys-TRs supports increased cell viability in response to low levels of oxidative stress. (A and C) C10 cells were transfected with the indicated constructs, treated overnight with subcytotoxic concentrations of MD, and oxidant production was evaluated by monitoring DCF fluorescence. Expression of any isoform of TR1 or TR2 had no statistically significant effect on the relative intensity of DCF fluorescence by 24 h. (B and D) C10 cells were transfected with the indicated expression vectors and cell viability after 24 h treatment with MD was quantified (*p<0.05, WT-TR1 compared to pcDNA). (E and F) C10 cells were transfected with the indicated TR expression vectors as in B and D, but in this instance with or without expression vectors for the cognate TRX. Cells were treated with the indicated concentrations of MD and after 24 h cell viability was quantified. Note that expression of TRX1 or TRX2 alone, or with the cognate TR, did not improve viability over that observed with expression of Sec-TR1 or Sec-TR2 alone.
Under similar conditions the expression of Sec-TR2 and TR2-U523C unexpectedly tended to increase DCF fluorescence (but not significantly) after 24 h (Fig. 5C), but only Sec-TR2 improved viability at menadione concentrations between 0.5 and 3.75 uM (Fig. 5D). Expression of TR2-U523C tended to increase viability, especially at higher concentrations of menadione, but this effect was not statistically significant from experiment to experiment (Fig. 5D). Co-expression of TRX1 with Sec-TR1 or TR1-U498C did not improve viability above that observed with expression of WT-TR1 alone (Fig. 5E), nor did co-expression of TRX2 with WT-TR2 or TR2-U523C improve viability above that observed by expression of WT-TR2 alone (Fig. 5F).
Discussion
The thioredoxin system comprised of TR, TRX and NADPH, plays a complex role in cancer [12], [44]. In normal cells TR is thought to impede neoplastic transformation because it counteracts oxidative stress, but once cells are transformed, over-expression of TR promotes tumor progression by protecting tumor cells from elevated levels of endogenous oxidants [45]. We sought to mimic these conditions by expressing TR1 and TR2 in non-transformed but immortal mouse lung epithelial cells. To evaluate the contribution of selenocysteine in TR to cell survival, we compared the effects of expression of wild type cDNAs for TR1 and TR2 to cDNAs in which the codons for selenocysteine had been changed to codons for cysteine. Gladyshev and colleagues have previously demonstrated that even with normal levels of dietary selenium, cysteine is inserted at the Sec codon in approximately 10% of cellular TR1 [46]. How often Cys is incorporated instead of Sec in TR1 in human malignancies is not known, and although here the culture medium contained selenium, we did not investigate the ratio between Sec-TR and Cys-TR in untransfected C10 cells, or in cell transfected with various TR1 and TR2 expression vectors. Rather we evaluated the relative expression levels of various TR1 and TR2 polypeptides, their relationship to total TR activity with and without oxidative insult, and their relative ability to support cell viability under oxidative stress.
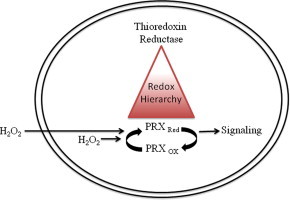
TR with selenocysteine in the active site is resistant to inactivation by a wide range of cellular oxidants in vitro [6]. In response to extracellular fluxes of hydrogen peroxide generated by glucose oxidase, total cellular TR activity was unaffected (Fig. 1A), whereas hyperoxidation of PRXs was readily evident (Fig. 1B). Treatment of cells with menadione results in the production of intracellular oxidants (Fig. 5A), but TR activity was diminished only after acute exposure to very high, cytotoxic concentrations over short periods of time (Fig. 1C). Overnight exposure to lower concentrations of menadione that affected cell viability (e.g. 3.75 μM) did not affect total cellular TR activity while hyperoxidation of PRXs was readily evident (Fig. 1E and F). These data show that cellular TR is insensitive to levels of intracellular and extracellular oxidants that influence redox responsive signaling pathways and PRX oxidation state and function. While in yeast inactivation of thiol peroxidase by hyper-oxidation is required to conserve pools of reduced thioredoxin for the repair of other oxidized proteins [47], hyper-oxidation of PRXs may have more complex functions in mammalian cells. For example, the oxidation state of PRXs is known to govern interactions with regulatory factors [17]. In our previous studies we observed that expression of cyclin D1 and recovery from cell cycle arrest did not occur until hyperoxidized PRXs were reduced [32]. Because inactivation of PRXs in response to oxidative insult is rapid and recovery is slow, whereas TR activity is unaffected, we suggest that TR sits at the top of a protein redox pyramid that controls a hierarchy of responses to oxidative stress (Fig. 6).
Fig. 6.
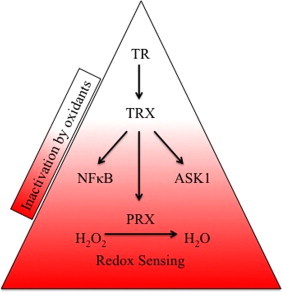
TR sits at the top of a protein redox pyramid. We suggest selenocysteine renders thioredoxin reductase insensitive to inactivation by oxidative insult and permits it to regulate a hierarchy of responses via regulation of the oxidation state of peroxiredoxins and other regulatory protein factors. In contrast, typical 2-Cys mammalian peroxiredoxins have acquired evolutionary adaptations that render them highly sensitive to inactivation by oxidation, and therefore are well suited to function as intracellular redox sensors. The levels of TR affect the rate of reduction of PRXs, but not the rate of inactivation, thereby contributing to survival in response to chronic oxidative insults.
The C-terminal extension of 2-Cys PRXs is a structural feature in eukaryotes that stabilizes the sulfenic acid intermediate and promotes hyperoxidation. In stark contrast, we have proposed that the chemistry of selenocysteine in the C-terminal redox center is the structural feature of mammalian TR that renders the enzyme resistant to inactivation by oxidative insult [48], thereby providing an evolutionary advantage that permits TR to regulate a hierarchy of responses to oxidative stress. While cellular TR contains a pool of TR1 with cysteine in the C-terminal redox center, in viability assays only Sec-TR1 or SecTR2 provided a survival advantage (Fig. 5). The Cys mutant of TR1 has lower enzymatic activity than WT TR1 containing selenocysteine [49], and may or may not be more sensitive to oxidative inactivation. It is interesting to note that over-expression of Cys-TR2 tended to enhance viability under low menadione concentrations, yet this effect was not statistically significant. Two factors may contribute to the ability of Cys-TR2 to support viability: (1) mitochondrial and cytosolic redox potentials differ [50], and (2) the substrate specificity for TR1 and TR2 differs [51]. Moreover, co-expression of TRX1 or TRX2 with its cognate reductase did not enhance survival, suggesting the expression levels of TRX1 and TRX2 were not rate limiting survival factors under these experimental conditions.
TR functions as a homodimer, and in the absence of stress we observed no adverse effects from expressing Cys-TR1 or Cys-TR2 on cell viability, indicating these polypeptides do not exert a dominant-negative phenotype or induce cell death via a SecTRAP mechanism [15]. Together these studies support the hypothesis that tumor cells maintain a pro-proliferative state under increased oxidative stress through up-regulation of antioxidant networks, and that over-expression of TR potentiates cell growth under increased endogenous and exogenous oxidant levels [3]. Further, selenocysteine in TR1 and TR2 may play an important role in cell viability in response to oxidative insult by rendering both enzymes insensitive to inactivation by oxidation [6].
Acknowledgments
This work was supported by NIH Grant GM094172, and BC was supported by a graduate fellowship from the University of Vermont Department of Pathology.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2014.01.021.
Contributor Information
Robert J. Hondal, Email: Robert.Hondal@uvm.edu.
Nicholas H. Heintz, Email: Nicholas.Heintz@uvm.edu.
Appendix A. Supplementary materials
Supplementary material
References
- 1.Burch P.M., Heintz N.H. Redox regulation of cell-cycle re-entry: cyclin D1 as a primary target for the mitogenic effects of reactive oxygen and nitrogen species. Antioxid. Redox Signal. 2005;7(5–6):741–751. doi: 10.1089/ars.2005.7.741. [DOI] [PubMed] [Google Scholar]
- 2.Burhans W.C., Heintz N.H. The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radic. Biol. Med. 2009;47(9):1282–1293. doi: 10.1016/j.freeradbiomed.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 3.Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 4.Lu J., Holmgren A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2013 doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 5.Mustacich D., Powis G. Thioredoxin reductase. Biochem. J. 2000;346(Pt 1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 6.Snider G.W. Selenocysteine Confers Resistance to Inactivation by Oxidation in Thioredoxin Reductase: comparison of Selenium and Sulfur Enzymes. Biochemistry. 2013 doi: 10.1021/bi400462j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kahlos K. Up-regulation of thioredoxin and thioredoxin reductase in human malignant pleural mesothelioma. Int. J. Cancer. 2001;95(3):198–204. doi: 10.1002/1097-0215(20010520)95:3<198::aid-ijc1034>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 8.Kinnula V.L., Paakko P., Soini Y. Antioxidant enzymes and redox regulating thiol proteins in malignancies of human lung. FEBS Lett. 2004;569(1–3):1–6. doi: 10.1016/j.febslet.2004.05.045. [DOI] [PubMed] [Google Scholar]
- 9.Newick K. Peroxiredoxin 3 is a redox-dependent target of thiostrepton in malignant mesothelioma cells. PLoS One. 2012;7(6):e39404. doi: 10.1371/journal.pone.0039404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng H.H., Wang L.H. Targeting thioredoxin reductase: anticancer agents and chemopreventive compounds. Med. Chem. 2010;6(5):286–297. doi: 10.2174/157340610793358864. [DOI] [PubMed] [Google Scholar]
- 11.Pennington J.D. Thioredoxin and thioredoxin reductase as redox-sensitive molecular targets for cancer therapy. Curr. Pharm. Des. 2007;13(33):3368–3377. [PubMed] [Google Scholar]
- 12.Arner E.S., Holmgren A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006;16(6):420–426. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 13.Gromer S. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 1998;273(32):20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 14.Rigobello M.P. Gold complexes inhibit mitochondrial thioredoxin reductase: consequences on mitochondrial functions. J. Inorg. Biochem. 2004;98(10):1634–1641. doi: 10.1016/j.jinorgbio.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 15.Anestal K. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PLoS One. 2008;3(4):e1846. doi: 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mochizuki M. Thioredoxin regulates cell cycle via the ERK1/2-cyclin D1 pathway. Antioxid. Redox Signal. 2009;11(12):2957–2971. doi: 10.1089/ars.2009.2623. [DOI] [PubMed] [Google Scholar]
- 17.Poole L.B., Hall A., Nelson K.J. Overview of peroxiredoxins in oxidant defense and redox regulation. Curr. Protoc. Toxicol. 2011;7:9. doi: 10.1002/0471140856.tx0709s49. (Chapter 7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baty J.W., Hampton M.B., Winterbourn C.C. Proteomic detection of hydrogen peroxide-sensitive thiol proteins in Jurkat cells. Biochem. J. 2005;389(Pt 3):785–795. doi: 10.1042/BJ20050337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall A. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J. Mol. Biol. 2010;402(1):194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poole L.B., Hall A., Nelson K.J. Overview of peroxiredoxins in oxidant defense and redox regulation. Curr Protoc Toxicol. 2011;7:9. doi: 10.1002/0471140856.tx0709s49. (Chapter 7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall A., Karplus P.A., Poole L.B. Typical 2-Cys peroxiredoxins--structures, mechanisms and functions. FEBS J. 2009;276(9):2469–2477. doi: 10.1111/j.1742-4658.2009.06985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biteau B., Labarre J., Toledano M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425(6961):980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 23.Chang T.S. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004;279(49):50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 24.Rhee S.G., Woo H.A. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H(2)O(2), and protein chaperones. Antioxid. Redox Signal. 2011;15(3):781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- 25.Barranco-Medina S., Lazaro J.J., Dietz K.J. The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 2009;583(12):1809–1816. doi: 10.1016/j.febslet.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 26.Kumsta C., Jakob U. Redox-regulated chaperones. Biochemistry. 2009;48(22):4666–4676. doi: 10.1021/bi9003556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toledano M.B. The system biology of thiol redox system in Escherichia coli and yeast: differential functions in oxidative stress, iron metabolism and DNA synthesis. FEBS Lett. 2007;581(19):3598–3607. doi: 10.1016/j.febslet.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Lee J. Yap1 and Skn7 control two specialized oxidative stress response regulons in yeast. J. Biol. Chem. 1999;274(23):16040–16046. doi: 10.1074/jbc.274.23.16040. [DOI] [PubMed] [Google Scholar]
- 29.Tachibana T. A major peroxiredoxin-induced activation of Yap1 transcription factor is mediated by reduction-sensitive disulfide bonds and reveals a low level of transcriptional activation. J. Biol. Chem. 2009;284(7):4464–4472. doi: 10.1074/jbc.M807583200. [DOI] [PubMed] [Google Scholar]
- 30.Yoo M.H. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 2006;281(19):13005–13008. doi: 10.1074/jbc.C600012200. [DOI] [PubMed] [Google Scholar]
- 31.Yoo M.H. Targeting thioredoxin reductase 1 reduction in cancer cells inhibits self-sufficient growth and DNA replication. PLoS One. 2007;2(10):e1112. doi: 10.1371/journal.pone.0001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phalen T.J. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J. Cell Biol. 2006;175(5):779–789. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dooley C.T. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 2004;279(21):22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 34.Cunniff B. Mitochondrial-targeted nitroxides disrupt mitochondrial architecture and inhibit expression of peroxiredoxin 3 and FOXM1 in malignant mesothelioma cells. J. Cell Physiol. 2013;228(4):835–845. doi: 10.1002/jcp.24232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cunniff B. A direct and continuous assay for the determination of thioredoxin reductase activity in cell lysates. Anal. Biochem. 2013;443(1):34-40. doi: 10.1016/j.ab.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ranjan P. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid. Redox Signal. 2006;8(9–10):1447–1459. doi: 10.1089/ars.2006.8.1447. [DOI] [PubMed] [Google Scholar]
- 37.Ranjan P., Heintz N.H. S-phase arrest by reactive nitrogen species is bypassed by okadaic acid, an inhibitor of protein phosphatases PP1/PP2A. Free Radic. Biol. Med. 2006;40(2):247–259. doi: 10.1016/j.freeradbiomed.2005.08.049. [DOI] [PubMed] [Google Scholar]
- 38.Spiess P.C. Proteomic profiling of acrolein adducts in human lung epithelial cells. J. Proteomics. 2011;74(11):2380–2394. doi: 10.1016/j.jprot.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chuang Y.Y. Gene expression after treatment with hydrogen peroxide, menadione, or t-butyl hydroperoxide in breast cancer cells. Cancer Res. 2002;62(21):6246–6254. [PubMed] [Google Scholar]
- 40.Loor G. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic. Biol. Med. 2010;49(12):1925–1936. doi: 10.1016/j.freeradbiomed.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hondal R.J., Ruggles E.L. Differing views of the role of selenium in thioredoxin reductase. Amino Acids. 2011;41(1):73–89. doi: 10.1007/s00726-010-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patenaude A., Ven Murthy M.R., Mirault M.E. Mitochondrial thioredoxin system: effects of TrxR2 overexpression on redox balance, cell growth, and apoptosis. J. Biol. Chem. 2004;279(26):27302–27314. doi: 10.1074/jbc.M402496200. [DOI] [PubMed] [Google Scholar]
- 43.Noh Y.H. Sulfiredoxin translocation into mitochondria plays a crucial role in reducing hyperoxidized peroxiredoxin III. J. Biol. Chem. 2009;284(13):8470–8477. doi: 10.1074/jbc.M808981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hatfield D.L. Selenoproteins that function in cancer prevention and promotion. Biochim. Biophys. Acta. 2009;1790(11):1541–1545. doi: 10.1016/j.bbagen.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trachootham D. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10(3):241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Xu X.M. Targeted insertion of cysteine by decoding UGA codons with mammalian selenocysteine machinery. Proc. Natl. Acad. Sci. USA. 2010;107(50):21430–21434. doi: 10.1073/pnas.1009947107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Day A.M. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol. Cell. 2012;45(3):398–408. doi: 10.1016/j.molcel.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 48.Hondal R.J., Ruggles E.L. Differing views of the role of selenium in thioredoxin reductase. Amino Acids. 2010;41(1):73–89. doi: 10.1007/s00726-010-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong L., Holmgren A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 2000;275(24):18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]
- 50.Jones D.P., Go Y.M. Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 2010;12(Suppl. 2):S116–S125. doi: 10.1111/j.1463-1326.2010.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rackham O. Substrate and inhibitor specificities differ between human cytosolic and mitochondrial thioredoxin reductases: implications for development of specific inhibitors. Free Radic. Biol. Med. 2011;50(6):689–699. doi: 10.1016/j.freeradbiomed.2010.12.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material