

Abstract
Improvements in health care and lifestyle have led to an elevated lifespan and increased focus on age-associated diseases, such as neurodegeneration, cardiovascular disease, frailty and arteriosclerosis. In all these chronic diseases protein, lipid or nucleic acid modifications are involved, including cross-linked and non-degradable aggregates, such as advanced glycation end products (AGEs). Formation of endogenous or uptake of dietary AGEs can lead to further protein modifications and activation of several inflammatory signaling pathways. This review will give an overview of the most prominent AGE-mediated signaling cascades, AGE receptor interactions, prevention of AGE formation and the impact of AGEs during pathophysiological processes.
Abbreviations: ADAMST, a disintegrin and metalloproteinase with a thrombospondin type 1 motif; AGE, advanced glycation end products; E, from embryonic day; EGFR, epidermal growth factor receptor; ERK, extracellular-signal regulated kinase; F3NK, fructosamine 3-phosphokinase; FKHRL1, forkhead transcription factor; HMGB1, high-mobility-group-protein B1; HNE, 4-hydroxy-trans-2-nonenal; Jak1/2, Janus kinase 1/2; LDL, low density lipoprotein; HDL, high density lipoprotein; MDA, malondialdehyde; MEKK, mitogen-activated protein/ERK kinase kinases; MnSOD, manganese superoxide dismutase; Nf-κB, nuclear factor-light-chain-enhancer of activated B; PIK3, phosphoinositol 3 kinase; RAGE, receptor of AGEs; RCC, reactive carbonyl compounds; S100B, S100 calcium binding protein B; SIRt1, NAD+-dependent deacetylase and survival factor 1; SR-A, scavenger receptor class A; Stat 1/2, signal transducers and activators of transcription 1/2; VSMC, vascular smooth muscle cells
Keywords: Advanced glycation end products, RAGE, Signaling, NF-κB, Aging, Oxidative stress, AGE-receptors, Reactive carbonyl compounds, Aggregates, Age-associated diseases
Introduction
The pursuit of immortality and eternal youth has fascinated people for thousands of years and led to a growing interest in aging and clinical research. Since 1840 life expectancy has increased enormously and according to statistics will increase further to 90 years until 2050 [1]. Over the past few decades improvements in health care (including managements of infectious diseases) and supply of nutritional compounds are the main reasons for the increasing lifespan. But the older people get, the higher the risk of acquirement of age-related diseases is. As a result, the number of age-associated and chronic diseases has increased constantly [2]. In addition to the rising age, a worldwide rapid increase in Western lifestyle—including elevated consumption of highly processed food and sugar, smoking as well as little physical activity already in the young generation—increases the incidence in chronic diseases [3], [4], [5], [6]. In particular, neurodegenerative and cardiovascular diseases play the most important roles in limiting quality of life of the aging population. Furthermore, the main problem is that such diseases are no longer limited to elderly people. More often, young and middle-aged people are affected by such diseases, resulting in reduced working capacity, loss of life quality and a financial burden for the healthcare system. A lot of chronic diseases are associated with accumulation of age-related modified biomolecules. Normally, cell constituents which are no longer functional are repaired or removed by cellular defense mechanisms. Aggravated by the fact that endogenous repair and degradation systems become increasingly impaired during the aging process, damaged and dysfunctional cell components can accumulate and become non-degradable. In the process of normal aging, a gradual accumulation of modified proteins, lipids and nucleic acids in body fluids or cells is not unusual. Elevated levels of damaged and accumulated proteins are responsible for forming bulky aggregates and plaques that are common for neurodegenerative and vascular diseases [7], [8]. An increase of oxidative stress during aging promotes further protein modifications and leads to the impairment of defense mechanisms over time [9], [10], [11], [12], [13]. Additionally, it is believed that high intake of glucose and fat, due to the Western diet, accelerate the development of chronic diseases, such as arteriosclerosis, sarcopenia, cataracts, Parkinson's disease, vascular dementia and diabetes [14], [15], [16], [17], [18]. Furthermore, genetic predisposition and increasing amounts of environmental reactive substances, such as xenobiotics, pharmaceuticals and toxins also have the ability to accelerate chronic diseases [19] Consequently, a lot of different causes may be responsible for the modification of biomolecules. Protein glycation, oxidation and nitration are the most important non-enzymatic protein modifications involved in the formation of endogenous protein aggregates.
At the moment one of the best studied substance classes is the heterogeneous group of advanced glycation end products. The spontaneous discovery of AGEs goes back to the investigation of the non-enzymatic browning reaction by Maillard in 1912 [20]. He was the first to observe and describe brown-colored end products after the reaction of glucose with the amino acid glycin. Today, AGEs are widely used by the food industry to improve taste, safety and bioavailability of food [21]. The formation of AGEs during food preparations is a very fast procedure, given that formation of AGEs rises in parallel to the increasing temperature and sugar concentration. Due to the lower temperature, AGE formation in vivo is a prolonged process. It is in general believed that the long-term formation of AGEs in vivo affects especially long-lived proteins, such as hemoglobin, alkaline phosphatase, lysozyme, collagen or elastin [22], [23]. In the case of collagen, AGE cross-links are responsible for stiffening of the extracellular matrix (ECM) and often involved in organ and vessel dysfunction [24]. In addition to their long biological half-life, they are also directly exposed to the high extracellular glucose levels and therefore an appropriate target. This glycation results in alteration of protein structure and makes them more resistance to degradation processes, ending in the accumulation of cross-linked products in cells and body tissues [23]. Due to the amount of reactive substances that can cause cellular modifications, this review focuses primarily on role of AGEs in cellular signaling. Different precursors, arising out of glycation or lipid peroxidation are responsible for the formation of endogenous advanced glycation end products in vivo.
Formation of advanced glycation end products
During the aging process but also increasingly in young generations, a rise in blood glucose levels due to the Western lifestyle is no longer an uncommon feature. Over the years, elevated blood glucose levels will lead to insulin deficiency, resulting in hyperglycemia, kidney disease and eye disorders, the main secondary diseases of diabetes type 2. Apart from increased blood glucose levels, an increase in intra- and extracellular stress is also crucial for such diseases.
In addition, there are several studies which demonstrating a positive effect on metabolism by mild oxidative stress [25], [26], [27]. Throughout phagocytosis ROS play a crucial role in the defense of pathogens, suggesting the protective effect of mild doses of oxidative stress [28]. However, elevated levels of ROS lead to oxidation of proteins, lipids and nucleic acids increases. Normally, there is a balance between oxidants and antioxidant defense. During aging, the equilibrium of prooxidants and antioxidants shifts to the former, leading to a marked rise in reactive oxygen species (ROS). Usually, oxidized proteins are degraded by the 20S proteasome of the Ubiquitin–Proteasome-System (UPS) [29], [30], [31]. Located in the cytosol and nucleus of each cell, it is responsible for the degradation of short-lived and unfolded oxidized proteins. The cylindrical proteasome consists of two main parts, two outer α-rings for binding the substrate and two inner β-rings responsible for the proteolytic activity. Formation of cross-linked proteins, can cause inhibition of the UPS [31]. Due to their bulky structure, AGEs are able to block the entry of the proteasomal core [20], [32], [33]. The resulting decrease in proteolytic activity leads to an increase in oxidized and damaged proteins [21], [34], [35], which can promote further protein modification [2]. Further, elevated levels of oxidants promote the oxidation of lipids and glucose, resulting in the accelerated formation of AGEs. Alterations of extracellular proteins, such as collagen and elastin as well as the activation of different signaling pathways after intracellular uptake are the consequences of increasing AGE levels. Besides the proteasome, other enzymes of the cellular repair mechanisms are also affected by excessive ROS levels, resulting in a decline of antioxidative and repair potential of the cells. Consequently increasing levels of advanced glycation end products support the formations of reactive oxygen and nitrogen species which in turn induce further formation of AGEs. Thus, first reaction in the formation of AGEs is the condensation of the carbonyl group of a reducing sugar or aldehyde with a free amino group of a protein (Fig. 1). In the following, a reversible Schiff' base (aldimin) is formed, which can be in turn transformed into a covalently-bound Amadori product. The rearrangement of aldimins depends on the amount of glucose, free amino groups of the proteins as well as an alkaline pH value. In contrast, Amadori rearrangements proceed at an acidic pH value and have a slower reaction rate. The glycated hemoglobin (HbA1c) was one of the first known Amadori products. It is used as a suitable marker for average blood glucose levels and measurable in the blood up to three weeks. For the most long-lived proteins, as hemoglobin, the Amadori reaction is still reversible.
Fig. 1.
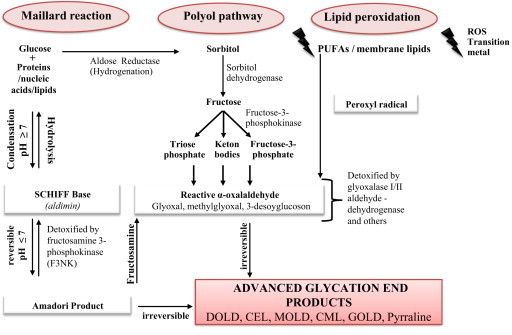
Formation of advanced glycation end products in vivo. Endogenous formation of advanced glycation end products has been described by three different paths in vivo: the non-enzymatic Maillard reaction, the Polyol-Pathway and lipid peroxidation. During all three reactions, formation of AGEs occurs over formation of reactive carbonyl compounds, such as glyoxal, methylglyoxal and 3-desoxyglucoson. If detoxification is impaired, they are able to react further until the formation of irreversible AGEs (modified from [3], [37], [41], [42], [216], [218]).
To generate the final and irreversible AGEs, further reactions must follow [36]. Amadori rearrangement not only occurs via oxidative processes but also in non-oxidative reactions, like reordering of functional groups and hydrolysis [37]. The formation of AGEs from autoxidation of Amadori products is known as Hodge-pathway (Fig. 2). The combination of increased blood glucose, oxidative and non-oxidative processes result in rearrangements of the Amadori products into the irreversible advanced glycation end products [3], [17]. But not only Amadori products are responsible for the AGE-formation. In addition, lipid and amino acid degradation, cleavage of dicarbonyl compounds from aldimins (Namiki pathway) as well as the formation of carbonyl-compounds after autoxidation of monosaccharaides; such as glucose, ribose, fructose and glyceraldehyde (Wolff pathway) [38]; can lead to the development of AGE compounds [39], [40] (Fig. 2). In the definition of in vivo-formed AGEs, these compounds are divided into two groups according to their origin: advanced glycation end products deriving from protein–carbonyl reactions (AGEs) or advanced lipoxidation end products (ALEs). While former arise from non-enzymatic glycation, autoxidation of glucose or from the sorbitol-pathway, ALEs are generated by reactive dicarbonyl compounds (RCC) arising from lipid peroxidation. In further course of this review both will be summarized under the term AGE. In vivo lipid peroxidation is the main source of carbonyl compounds called α-oxoaldehydes (Fig. 1). The reaction takes places when polyunsaturated fatty acids, main components of biological membranes, are attacked by ROS. The resulting products, such as the peroxyl radical are highly reactive and lead to further oxidation, generating lipid hydroperoxides and hydrogen peroxide [41]. Resulting products of this further oxidation are reactive carbonyl species (RCS) such as malondialdehyde (MDA), 4-hydroxy-trans-2-nonenal (HNE) and α-oxoaldehydes, such as glyoxal, methylglyoxal, 3-deoxyglucuson and acrolein [3], [37], [42]. In addition to further rearrangements, RCS can lead to AGEs such as Nε-carboxyethyllysine (CEL), arginine pyrimidine, pentosidine, pyrralin, methylglyoxal, glyoxal lysine and Nε-carboxymethyllysine (CML). Due to the different AGE-precursors, AGEs are a very complex and heterogeneous group of compounds of which not all structures are known yet [43]. For the partial characterization of AGEs, a number of fluorometric and colorimetric methods exist to examine the level of protein alteration. Modifications on arginine and lysine side chains, aggregation state of proteins, formation of protein carbonyls, the fructosamine content as well as autofluorescence of some AGE compounds are widely used parameters (27). Also measuring of pentosidine or CML via high-performance liquid chromatography (HPLC) [32], [35], immunoblot and enzyme-linked immunosorbent assay (ELISA) [44], and mass spectroscopy [33] allow the detection and provide an insight in the structures of different AGEs. Immunohistochemistry of arteriosclerotic plaques showed increased intracellular AGE accumulation in foam cells [45]. Due to various methods and protocols for measuring and detecting AGEs, it is challenging to compare published results. For the effect of AGEs on cellular reactions different ways of AGE binding and uptake have been described.
Fig. 2.
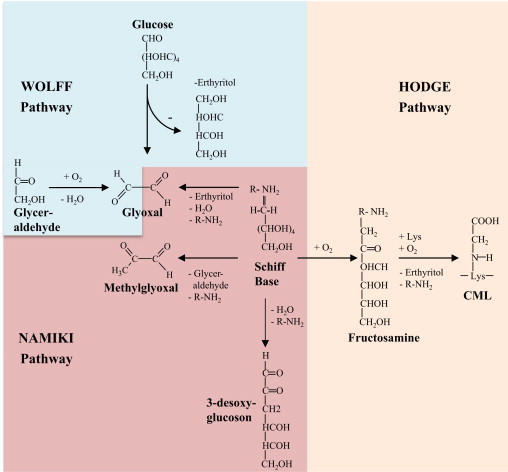
Formation of AGEs via Wolff'-, Namiki- and Hodge-pathway. Autoxidation of monosaccharides or carbonyl compounds (Wolff' pathway), aldimins (Namiki-pathway) or Amadori products (Hodge-pathway) via transition metals or ROS can lead directly to the formation of RCCs and in further reaction to AGEs (modified from [3], [17], [38], [39], [40]).
Receptors for advanced glycation end products—structure and function
Formation of AGEs leads to the activation of different signaling pathways mediated by a series of cell surface receptors. The most studied AGE-receptor is the multi-ligand receptor for advanced glycation end products (RAGE). Today several other AGE-receptors was also identified consisting of the AGE-receptor complex (AGE-R1/OST-48, AGE-R2/80K-H, AGE-R3/galectin-3) [46], [47] and some members of the scavenger receptor family (SR-A [48]; SR-B: CD36 [49], [50], SR-BI [51] SR-E: LOX-1 [52]; FEEL-1; FEEL-2 [53]) which are all illustrated in Fig. 3. The expression of these AGE-receptors depends on the cell/tissue-type and is regulated in response to metabolic changes for instances during aging, diabetes and hyperlipidemia [54] (Table 1).
Fig. 3.
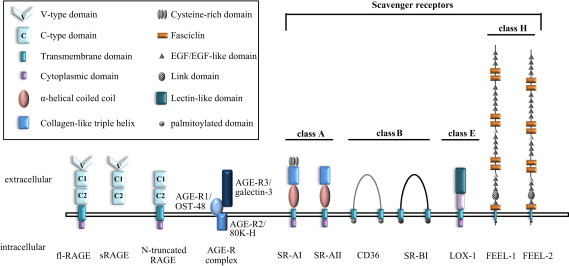
Structure of AGE receptors. Scavenger receptor family and AGE receptors. The scavenger receptor family is divided into class A, class B, class C and other not classified receptors. Some of these receptors are able to recognize AGEs. The AGE receptor complex (OST48; 80 K-H; Galectin-3) and RAGE are also identified as AGE-binding receptors but they do not belong to the scavenger receptor family (modified from [49]).
Table 1.
Receptors for advanced glycation end products—structure and function.
| AGE-binding protein | Molecular weight | Cell types | Structural features | Ligands | Function |
|---|---|---|---|---|---|
| RAGE | Sequence: | Monocytes/Macrophages, T-lymphocytes (CD4+, CD8+), endothelial cells, mesangial cells, fibroblasts, smooth muscle cells, neuronal cells | Highly acidic cytoplasmic domain, a single transmembrane domain, extracellular domain consisting of VC1-ligand binding domain and C2 domain | S100 proteins, HMGB-1, β-amyloid, amyloid fibrils, β-integrin Mac-1, AGEs | Endocytosis, signaling (cell activation)→generation of ROS, inflammatory response |
| fl-RAGE: ~43 kDa | |||||
| sRAGE: ~36 kDa | |||||
| N-truncated RAGE: ~32 kDa | |||||
| Western Blot (WB): | |||||
| fl-RAGE: ~55 kDa | |||||
| sRAGE: ~46–50 kDa | |||||
| N-truncated RAGE: ~42 kDa | |||||
| AGE-R1/OST-48/p60 | ~50 kDa (Sequence +WB) | Monocytes/macrophages, T-lymphocytes (CD4+, CD8+), Endothelial cells, mesangial cells, fibroblasts, smooth muscle cells, neuronal cells | Integral plasma membrane protein with a single transmembrane domain, formation of a complex with ribophorin I, II and DAD1 in the RER | - | Endocytic uptake and degradation of AGE-modified proteins, protective against oxidative stress |
| AGE-R2/80K-H/p90 | Sequence: ~59 kDa | Monocytes/macrophages, T-lymphocytes (CD4+, CD8+), endothelial cells, mesangial cells, fibroblasts, smooth muscle cells, neuronal cells | No transmembrane domain, phosphorylation on tyrosine residues, formation of a complex with PKCζ and munc18c | AGEs | Signaling (cell activation), regulatory subunit of glucosidase II |
| WB: ~80 kDa | |||||
| AGE-R3/galectin-3 | Sequence: ~26 kDa | Monocytes/macrophages, T-lymphocytes (CD4+, CD8+), endothelial cells, mesangial cells, fibroblasts, smooth muscle cells, neuronal cells | Formerly known as Mac-2 or CBP-35, consists of a carbohydrate-binding C-terminal domain, PGAY-rich repeating domain, N-terminal domain, no transmembrane domain | Cell surface: IgE receptor, colon cancer mucin, CD66, LPS, Mac-1, Mac-3, heavy chain of CD98, myelin-associated glycoprotein, lysosomal-membrane-associated glycoproteins (LAMPs) 1 and 2; extracellular environment: AGEs, IgE, laminin, tenascin, fibronectin, collagen IV, gp90/Mac-2 binding protein | Signaling (cell activation) |
| WB: ~32 kDa | |||||
| SR-A (I/II) | Sequence: ~50 kDa (monomeric subunit) | Monocytes/macrophages, dendritic cells, endothelial cells | Domain I: cytoplasmic N-terminus, Domain II: transmembrane domain, Domain III: spacer domain, Domain IV: α-helical coiled-coil, Domain V: collagen triple helix, Domain VI: C-terminal domain | Endogenous ligands: AcLDL, OxLDL, AGE, β-amyloid; Pathogen recognition: Gram-negative bacteria (E. coli), Gram-positive bacteria (S. aureus), bacterial DNA, LTA | Endocytic uptake and degradation of modified LDL and AGE-modified proteins |
| WB: ~77 kDa (monomer), ~220 kDa (homotrimer) | |||||
| SR-B/CD36 | Sequence: ~53 kDa | Platelets, endothelial cells, epithelial cells, adipocytes, B-lymphocytes | Two transmembrane domains with an extracellular loop, palmitoylated on cysteine residues, two on each N- and C-terminus | Endogenous ligands: AcLDL, OxLDL, HDL, LDL, VLDL, collagen, thrombospondin, long chain fatty acids, maleylated BSA, anionic phospholipids, AGE, apoptotic cells; Pathogen recognition: Microbial diacylglycerides, S. aureus | Endocytic uptake and degradation of AGEs, cell adhesion, regulator of fatty acid transport |
| WB: ~88 kDa | |||||
| SR-BI | Sequence: ~60 kDa | Tissues which are active in selective uptake of HDL (liver, steroidogenic) | Two transmembrane domains with an extracellular loop, palmitoylated on C-terminal Cys462 and Cys470 | AcLDL, OxLDL, LDL, HDL, VLDL, maleylated BSA, anionic phospholipids, AGE | Selective uptake of HDL, endocytic uptake and degradation of AGEs |
| WB: ~76 kDa | |||||
| SR-E/LOX-1 | Sequence: ~31 kDa | Endothelial cells, macrophages, smooth muscle cells | Group V C-type lectin, short N-terminal cytoplasmic domain, single transmembrane domain, extracellular NECK domain with a subsequent CTLD | Endogenous ligands: OxLDL, Hsp70, AGE, apoptotic cells, activated platelets; Pathogen recognition: S. aureus, E. coli | Endocytic uptake and degradation of modified oxLDL, signaling |
| WB: ~50 kDa | |||||
| FEEL-1/FEEL-2 | FEEL-1: ~275 kDa | Endothelial cells, monocytes/macrophages | Type I transmembrane protein, consists of 7 fasciclins, 16 EGF-like, 2 laminin-type EGF-like and 1 link domain transmembrane region | Endogenous ligands: AcLDL, AGE, SPARC, hyaluronic acid; pathogen recognition: S. aureus, E. coli | Endocytic uptake and degradation of AGEs, Hyaluronic acid and AcLDL |
| FEEL-2: ~277 kDa (sequence) | |||||
| 190 kDa form stabilin-2 (by proteolytic cleavage): ~190 kDa (WB), ~154 kDa (sequence) | |||||
Receptor for AGEs (RAGE)
The type I cell surface receptor for AGEs (RAGE) belongs to the immunoglobulin (Ig) superfamily and has been described as a pattern recognition receptor [55], [56]. Based on molecular cloning studies, RAGE was first identified as a polypeptide with a molecular weight of 35 kDa in bovine lung endothelial cells. According to the protein sequence of human RAGE that consists of 404 amino acids, the calculated molecular weight is 42.8 kDa. Western Blot analysis of RAGE-transfected 293 cells showed major bands with a molecular mass of ~50 kDa due to post-translational processing [55]. RAGE is expressed on several cell types (monocytes/macrophages [57], [58], [59], T-lymphocytes [60], [61], [62], endothelial cells [63], dendritic cells [64], [65], fibroblasts [66], smooth muscle cells [59], [67], neuronal cells, glia cells, chondrocytes [68], keratinocytes [69]) and recognizes a large number of different ligands (AGEs, amyloid β peptide, S100/calgranulin protein, HMGB1, LPS). Ligand binding is followed by activation of different signaling pathways (for review see [70]). The full-length RAGE (fl-RAGE) protein consists of a large extracellular domain, a single transmembrane-spanning helix and a short cytoplasmic domain. This intracellular domain of fl-RAGE was shown to be essential for downstream signaling and activation of NF-κB [71]. The extracellular region is composed of one N-terminal V-type (variable) domain and two C-type (constant) domains [72]. The ligand-binding site is reported to be located on the V-type domain which secondary structure consists of two β-sheet, a short α-helix and random-coil and is quite similar to the structure of the immunoglobulin V-type domains [73]. In contrast to the C2-type domain, the V- and C1-type domain act as structural unit (VC1) and form not an independent structure. Besides, it was also shown that the VC1 domain is attached to the C2 domain by a flexible linker [72]. Due to alternative splicing 20 different variants of RAGE are known [74]. Two major splicing variants of fl-RAGE mRNA are characterized. One of these variants (N-truncated RAGE) is lacking the N-terminal V-type domain, but is also located in the cytoplasmic membrane. The second splicing variant is the soluble form of RAGE which is lacking the C-terminal domain, but contains all of the immunoglobulin domains of fl-RAGE [75]. The soluble form of RAGE is secreted extracellularly and two different types are identified. The first is known as sRAGE or esRAGE and is a result of alternative splicing of RAGE mRNA. Furthermore, fl-RAGE undergoes proteolytic cleavage through the metalloproteinase ADAM10 and MMP9 and leads to the release of sRAGE [76]. It was reported that sRAGE as a decoy prevents ligands to interact with RAGE or with other cell surface receptors [77]. In patients with coronary artery disease lower plasma levels of sRAGE were demonstrated in an acute event suggesting an increased AGE–RAGE interaction leading to increased production of ROS and inflammatory response [74]. RAGE signaling induced by AGEs and its physiological role will be discussed in the next sections.
The AGE-receptor complex (AGE-R)
The AGE-R complex consists of three different components and is involved in AGE signaling and endocytotic uptake of AGE-modified proteins. In 1991 the first component of the AGE-R complex was published initially as the cell surface receptor p60 in connection with AGE-modified proteins on monocytes/macrophages [78]. Later it was shown that p60 is homologous to OST-48, which was identified as a 48 kDa subunit of the oligosaccharyl-transferase complex [46], [79]. OST-48 forms a complex with the two integral membrane glycoproteins ribophorin I and II and also with the defender against apoptotic cell death (DAD1) in the rough endoplasmic reticulum (RER) [80], [81], [82]. It has been shown that the homologous to OST-48, also termed AGE-R1, is an integral plasma membrane protein with a single transmembrane segment and a long extracellular domain, which represents the N-terminal domain of OST-48 located luminal in the RER [46]. This identified protein is known to be active in binding of AGE-modified proteins on the cell surface and also as the subunit of the oligosaccharyl-transferase complex which catalyzes the transfer of high mannose oligosaccharides onto asparagine-acceptor sites of proteins inside the RER [46], [79]. AGEs induce an enhanced expression of AGE-R1/OST-48, which activation suppresses RAGE, prevent the generation of cellular oxidative stress and also blocks the activation of the oxidative stress dependent signaling of the EGF-receptor [83], [84], [85]. Vlassara et al. have also shown that the AGE-R1/OST-48 level in PBMCs positively correlates with plasma and urine AGEs as well as with oxidative stress markers in healthy participants and negatively in patients with stage 3 chronic kidney disease (CKD-3) [86]. These data support the hypothesis that AGE-R1/OST-48 has a protective role against the formation of ROS, oxidative stress dependent cell damage and tissue injury.
Similar to the AGE-R1/OST-48, the second component of the AGE-R complex was known first as 80K-H protein, which is an effective phosphorylation substrate for the protein kinase C (PKC). This acidic 80 kDa protein was first purified from the human carcinoma Ca9-22 cell line and is located in the cytosol and membrane with no apparent transmembrane domain [87], [88]. A few years later it was shown that 80K-H is identical to the AGE-binding protein p90, which was identified as specific cell surface binding protein on the membrane of murine macrophage cell line RAW 264.7 for the AGE 2-(2-furoyl)-4(5)-(2-furanyl)-1H-imidazole [46], [89], [90]. The biological function of AGE-R2/80K-H is poorly understood. It was demonstrated that phosphorylation of this protein occurs on tyrosine residues and it is involved in the intracellular receptor signaling, like FGF receptor signaling [90], [91], [92]. The AGE-R2/80K-H protein is also linked to intracellular trafficking in connection with AGEs and it is closely related to VASAP-60, which is a regulator of vesicle trafficking [46], [93]. Furthermore, it was suggested that the 80K-H phosphoprotein was an interactor with both, the PKCζ and munc18c and plays a role in the GLUT4 vesicle trafficking pathway, whereby insulin triggers the formation of a PKCζ–80K-H–munc18c complex which leads to an enhanced glucose transport through GLUT4 [94].
The third component of the AGE-R complex is a member of the lectin family. It was first described on the cell surface of activated macrophages with a size of 32 kDa and termed Mac-2 [95] This protein has been rediscovered in different species (human, dog, rat and mouse tissues) and also in different contexts, leading to multiple names like Mac-2, CBP-35, CBP-30 and L-29 [96]. Thus, a generally accepted nomenclature was established and this protein was named galectin-3 [97]. In 1995, galectin-3 was identified as a part of the AGE-R complex with a high binding affinity of AGE ligands [47]. AGE-R3/galectin-3 consists of a carbohydrate-binding C-terminal domain composed of 130 amino acid residues, a proline–glycine–alanine–tyrosine-rich repeating domain (100 residues) and a short N-terminal domain (~30 residues) [96]. AGE-binding occurs also in the C-terminal carbohydrate recognition domain and promotes the formation of a high molecular weight complex with other receptor molecules and the AGE ligands [47], [91]. The lack of a transmembrane domain supports the assumption that this protein is located on the cell surface linked to other AGE receptor molecules [47]. The AGE-R3/galectin-3 protein is mainly located in the cytoplasma, but was also found in the nucleus, on the cell surface and the extracellular environment. It has been shown that the endogenous lectin is predominantly phosphorylated on the N-terminal Ser6 residue and the phosphorylated form could be detected also in the cytoplasm and the nucleus of 3T3 fibroblasts and colon cancer cells [98], [99], [100]. The phosphorylation affected the ligand binding significantly and leads to a reduced saturation binding greater than 85% which was tested with the two galectin-3 ligands laminin and mucin. Dephosphorylation can completely restore the ligand binding activity and suggests that phosphorylation on Ser6 works as an “on/off” switch for its sugar binding capabilities and seems to be important for the understanding of the biological function of the AGE-R3/galectin-3 protein [100]. However, it remains unknown if the extracellular AGE-R3/galectin-3 can also be phosphorylated and if this affects the extracellular binding of AGE-modified proteins as well. Upregulation of AGE-R3/galectin-3 leads to an increased metastatic potential in cancer. Phosphorylation of AGE-R3/galectin-3 regulates the anti-apoptotic signaling activity resulting to an enhanced metastatic potential [101]. Furthermore, the biological function of the AGE-R3/galectin-3 protein has been implicated also in the processes of cell migration and adhesion, immune response modulation, growth and differentiation [47], [96], [102] and in addition it has been shown that AGE-R3/galectin-3 and RAGE participates in vascular osteogenesis by modulating Wnt/β-catenin signaling with the two receptors showing diverging effects on VSMC osteogenic differentiation [103]. This assists in understanding the mechanism of the development of atherosclerosis and the role of the AGE receptors in this process.
Macrophage scavenger receptor family
The scavenger receptor family describes a group of receptors with a common affinity to recognize modified proteins like oxidized low-density lipoprotein (OxLDL) or acetylated LDL (AcLDL). The scavenger receptor was first discovered in 1979 by Goldstein and Brown. They demonstrated a high affinity binding site on mouse peritoneal macrophages that recognizes AcLDL but not native LDL. In addition to AcLDL also other negatively charged ligands including maleyl-LDL, malelyl-BSA and sulfated polysaccharides were shown to bind this receptor [104]. One contemporary problem at that time was the physiological relevance because the uptake of AcLDL occurred in vitro and was not known in vivo. Two years later it was shown that native human LDL incubated with rabbit aortic endothelial cells was modified into a form of LDL that was also recognized by the macrophage receptor for AcLDL [105]. The generation of this modified LDL molecules can occur in the absence of cells using a medium containing redox metals, such as copper [106] and/or by free SH-groups [107], [108]; in the presence of smooth muscle cells, endothelial cells and macrophages themselves [108] and it could be proved that oxidation of LDL also occurs in vivo [109]. The definition of this type of receptors as scavenger receptors is due to the initial described function to mediate the uptake and degradation of modified proteins. Today, a large number of different scavenger receptors have been identified and are classified according to their structural properties (Fig. 3).
The scavenger receptor class A (SR-A) are trimeric transmembrane glycoproteins with a relative molecular weight of 220 kDa (monomeric subunit: 77 kDa) and consist of five (SR-AII) or six (SR-AI) different domains. The N-terminal cytoplasmic domain contains 50 amino acids, followed by a 25-amino acid transmembrane spanning domain; a 75-amino acid spacer region; a 121-amino acid α-helical coiled coil motif, a 69-amino acid collagen-like triple helix and the C-terminal domain [110]. The C-terminal domain of the SR-AI receptor contains a cysteine-rich motif which is absent in the SR-AII receptor [111]. The ligand binding occurs at the collagenous domain and leads to the endocytic uptake of OxLDL or AcLDL and further to the intracellular accumulation of cholesterol and the formation of foam cells from macrophages in the early state of atherosclerosis [112]. The SR-A was also described as AGE-binding receptor, mediating the endocytotic uptake and degradation of AGE-modified proteins in competition to modified LDL [48]. Due to the fact that foam cells are formed from macrophages and the SR-A are the mainly expressed receptors on these cells, the AGE-binding on these receptors could play a crucial role in the development of atherosclerosis.
Two other receptors, CD36 and SR-BI, belong to the scavenger receptor class B (SR-B) and are concentrated in caveolae-like domains on the cell surface. Both receptors are glycoproteins with two transmembrane domains, a large extracellular loop and short N- and C-terminal domains with a significant sequence homology in the extracellular domain [113]. However, this sequence homology between the two receptors is not present in the N- and C-terminus. It was also shown that CD36 and SR-BI are fatty-acylated. In the human CD36 four cysteine residues could be identified as major site of palmitoylation, two at both the N-terminal and C-terminal domain [114]. Studies on murine SR-BI indicates a potential site of palmitoylation at Cys462 and Cys470 which are located in the C-terminus [115]. In CHO cells overexpressing human CD36 or hamster SR-BI a specific cell association to AGE-modified BSA was reported, leading to endocytic uptake of AGEs and identified CD36 and SR-BI as AGE-receptors [49], [51]. As shown in murine SR-BI-transfected ldlA cells (LDL receptor-negative CHO cells) SR-BI recognizes HDL and mediates selective transfer from HDL to cells without endocytic uptake of HDL [116]. Binding of AGE ligands to SR-BI can inhibit the selective HDL uptake and involves AGEs in the regulation of the SR-BI mediated cholesterol metabolism [51]. Related to the SR-A receptors, CD36 is as well highly expressed on macrophages in atherosclerotic lesions and recognizes OxLDL as ligand. Nakata et al. detected CD36 mainly in the core region of an atherosclerotic plaque by immunohistochemical staining, whereas SR-A is mainly localized around the core region which suggests different functions of both CD36 and SR-A in the process of atherogenesis [117]. CD36 was also described as AGE-receptor on mouse 3T3-L1 cells and human subcutaneous adipocytes which recognized AGE-BSA modified by glycolaldehyde (GA) and methylglyoxal (MG) undergoing endocytic degradation [118]. Incubation of these cells with GA-BSA induces intracellular oxidative stress and leads to leptin downregulation which is related to exacerbation of insulin sensitivity in vivo [119]. This indicates a pathological role of CD36 as AGE-receptor in diabetic complications and atherogenesis.
Furthermore, some other members of the scavenger receptor family were reported to bind AGEs. One of them is the lectin-like oxidized LDL receptor-1 (LOX-1). This receptor was identified in endothelial cells as a receptor for OxLDL and has a molecular weight of 50 kDa [120]. LOX-1 is a type II transmembrane glycoprotein and belongs to the group V C-type lectins [121]. It is expressed with a short N-terminal cytoplasmic domain, a single transmembrane domain and a stalk region (NECK domain) with a subsequent C-terminal lectin-like domain (CTLD) in the extracellular region. The CTLD of the human LOX-1 has an inter-chain disulfide bridge at Cys140 to form the homodimeric ligand-binding domain and the NECK domain as a homodimer consists of two α-helices that wrap in a left-handed helix to build a coiled coil [122]. LOX-1 is localized mainly in vascular tissues and is highly expressed in patients or animals with atherosclerosis or diabetic complications. It is reported that NF-κB is a downstream signal of LOX-1 and its expression is affected by testosterone that can regulate the atherosclerotic plaque progression in the rabbit thoracic aorta [123]. In addition to OxLDL, LOX-1 also binds MG- or GA-modified BSA. Cross-competition experiments show that the binding of OxLDL or AGE-BSA was not affected by the other ligands. Similar results have been obtained with HDL or AGE-BSA binding on SR-BI in CHO cells overexpressing SR-BI. A possible reason for these results might be the presence of multiple ligand binding sites in both receptors, LOX-1 and SR-BI. In these studies it was also shown that in contrast to the receptor LOX-1 which mediates the endocytic uptake and degradation of OxLDL but not of AGE-BSA, the SR-BI receptor mediates the endocytic uptake of AGE-modified proteins but not of HDL [52]. Further studies indicate that AGE-BSA induces LOX-1 expression in endothelial cells in vitro that is not mediated by activation of NF-κB but rather through RAGE-mediated activation of the mTOR pathway [124]. Due to the fact that mTOR plays a role in cardiovascular-related disorders, the induced LOX-1 expression by AGEs may also contribute to diabetic complications such as atherosclerosis [124], [125].
Fasciclin, EGF-like-, laminin-type EGF-like, and link domain-containing scavenger receptor-1 (FEEL-1) and FEEL-2 was cloned from human umbilical vein endothelial cells and first reported as a receptor for AcLDL [126]. FEEL-2 is a paralogous protein of FEEL-1 (2570 amino acids) with a sequence homology of 39.8%. Both receptors are type I transmembrane proteins and are composed of seven fasciclins, 16 EGF-like, two laminin-type EGF-like and one link domain near the transmembrane region [53], [126]. First, FEEL-1/FEEL-2 were known as stabilin-1 and stabilin-2, and additionally FEEL-1 was also identified as the common lymphatic endothelial and vascular endothelial receptor-1 (CLEVER-1) [127], [128]. In CHO cells overexpressing FEEL-1 or FEEL-2 an endocytic uptake and degradation of AGE-BSA was shown. The AGE-binding by FEEL-1/FEEL-2 was competed with other ligands like AcLDL or OxLDL suggesting the same binding site on the receptors [53]. It was also shown that AGE-BSA is endocytosed by choriocapillaris endothelial cells located in Bruch's membrane via a FEEL-1/FEEL-2 mediated pathway [129].
Receptor trafficking and intracellular proteolysis of AGEs
Whether AGEs are originating from exogenous or endogenous sources these compounds have to be removed to prevent additional protein modifications and protein cross-linking. Prior to the disposal of AGEs they have to be further processed to be eliminated by the renal systems. Thereby, the degradation of AGEs occurs intracellular, meaning that they first need to be taken up by the cells. Processing of AGE-modified proteins into AGE-modified peptides makes them eliminable for the renal system. As already mentioned, some of the AGE-receptors are able to bind AGEs for subsequent intracellular processing. AGE-receptors which are believed to be involved in detoxification are the AGE-R1/OST-48, AGE-R3/galectin-3 as well as scavenger-receptors (MSR-AII, MSR-BI, CD36) [48], [50], [130]. There exist strong evidence that under basal conditions AGE-R1/OST-48 and AGE-R3/galectin-3 have a high affinity to bind AGEs [131]. Cells constantly express a majority of cell surface receptors to internalize signaling molecules leading to an activation of specific intracellular signaling cascades in response to changes of the extracellular environment. Thereby, it is assumed that they inhibit the AGE-mediated signaling and oxidative stress by competing with RAGE to AGEs. Uptake of AGEs by AGE- and scavenger receptors is described through receptor-mediated endocytosis [48], [132], [133]. Ligand-binding activates the receptor through phosphorylation or ubiquitinylation of the cytoplasmic side of the receptor stimulating the receptor endocytosis. The internalization of the ligand-receptor complex undergoes a regulated mechanism that is either clathrin-dependent or -independent. The best-studied mechanism is the clathrin-mediated endocytosis through clathrin-coated vesicles. These vesicles are transported to early endosomes and undergo a not yet well-understood series of endosomal sorting processes. Inside the newly formed endosomes AGEs are dissociated from the receptor, triggered by a decrease of pH within the endosomes (for review see [134], [135], [136]). After separating the receptor can be recycled and transferred back to the cell surface. In case of RAGE, it was reported that in endothelial cells the receptor can be rapidly transferred back from intracellular storage pools to the plasma membrane [137]. Meanwhile endosomes with AGEs fuse with lysosomes, following by processing of AGEs by lysosomal proteases. Grimm et al. demonstrated that lysosomal proteases, such as cathepsins D, L and B play a crucial role in AGE disposal [138], [139]. After processing, small soluble AGE modified peptides are probably released by unknown mechanisms from the cells and transported to the renal system by processes which are not known yet. Impaired function of the kidney would result in accumulation of AGEs. In addition Sano et al. demonstrated that insulin can also lead to AGE elimination over enhanced uptake via macrophage scavenger receptors. Thereby, it comes to an activation of the phosphatidyl-inositol-3-OH kinase (PI3) pathway, following by increase in nitric oxide and insulin-mediated glucose transport in skeletal muscles and adipocytes [140]. Sevillano et al. have reported the internalization of RAGE after AGE binding and its necessity for the generation of intracellular responses [141].
AGE-mediated signaling via RAGE
As mentioned above, AGEs can be formed exo- and endogenously and afterwards influence cellular metabolism. The main sources of exogenous AGEs are heat-treated protein rich foods containing lipids and/or sugar and compounds formed during smoking of cigarettes [142]. Even though several studies were already able to show an increase of plasma-AGE-levels after intake of AGE-rich food, the intestinal uptake of AGEs as well as the intake in lung alveoli play only a minor role [143], [144], [145]. Nevertheless, exogenous AGEs were shown to have pro-inflammatory and pro-oxidant effects and that a dietary restriction of AGE-rich food was associated with significantly reduced inflammation [143] and protein modifications [146], [147], [148]. How and to which amount dietary AGEs are taken up is not known yet. Geissler et al. detected that peptide-transporter PEPT1 is possibly able to absorb pyrallin [149]. Furthermore, Forster et al. have demonstrated in a study that after intake of pentosidine- and pyrraline-rich food, approximately 50% of the AGE were measurable in the urine until the third day after consumption [150]. As mentioned in the previous section, many different receptors are discussed to play a crucial role in the cellular uptake or effects of AGEs. Thereby, RAGE is currently the best studied. It is assumed that the AGE–RAGE interactions play a major role in many signaling pathways (Table 2). In the following section some of them as well as the elimination of AGEs will be described.
Table 2.
Effect of advanced glycation end products on intracellular signaling.
| AGE-precursor/AGE | Cell line | Pathways/activated transcription factors | Effective concentrations | Outcome | Ref. |
|---|---|---|---|---|---|
| BSA–AGE (glucose, ribose, fructose, methylglyoxal, glyoxal) | Murine RAW-cells | AGE–RAGE mediated Jak–Stat-pathway | Glyoxal/MG-BSA: 10 µM/mg BSA, fructose-, glucose, ribose-BSA: 25 µM/mg BSA | →Increased expression of immunoproteasomal subunits (LMP7, LMP2, MECL-1) | [30] |
| Phosphorylation: Jak-2, Stat-1 | →Decreased expression of proteasome subunits (β1, β2, β5) | ||||
| →Expression of MHC-1 complexes | |||||
| CML-BSA, AGE-BSA, human-serum-AGE (HsA) | Rat mesanglia cells | Angiotensin II-TGFβ-SMAD signaling | AGE-BSA: 10–100 µg/ml | →Increase of Angiotensin II induced TGFβ signaling | [220] |
| Angiotensin II, TGF-β, Smad2, p27Kip1 | CML-BSA: 10–50 µg/ml | →Smad2 phosphorylation | |||
| HsA: 500 µg/ml | →Upregulation of cyclin-dependent kinase inhibitor 1B (p27Kip1) leading to cell hypertrophy and increased fibronectin | ||||
| →increased RAGE expression | |||||
| → ROS generation by interaction with RAGE | |||||
| CML-collagen | Dermal fibroblasts | Induction of apoptosisviaFOXO1 | 200 µg/ml | →Activation of caspase-3-activity and apoptosis via induction of ROS, ceramide and NO, leading to activation MAPK p38 and JNK pathways and FOXO1 followed by TNF-mediated pro-apoptotic gene expression | [221] |
| p38 and JNK, FOXO1, TNFs | |||||
| BSA–AGE (methylglyoxal) | Bovine retinal pericytes | Induction of apoptosisviadiaglycerol (DAG) and ceramides | 3 µM (1–20 µM) | →Phosphatidylcholine-specific phospholipase C mediated DAG production | [222] |
| →Activation acidic sphingomyelinase, producing ceramides→apoptose | |||||
| BSA–AGE (glucose) | Rat vascular smooth muscle cells (A7R5) | Induction of autophagyviaAkt-mTOR | 100 µg/ml | →AGE–RAGE interaction increased autophagy via ERK | [223] |
| Phosphorylation of ERK, MAPK p38, JNK Inhibited phosphorylation Akt | →Increased autophagic vacuoles | ||||
| →Increased LC3-II to LC3-I ratio | |||||
| →Increased proliferation of VSMCs | |||||
| →Activation of Akt via IGF-1 decreased AGE-mediated autophagy | |||||
| BSA–AGE | Human chondrocyte from osteoarthritis patients | Augment inflammatory responses | Up to 800 µg/ml | →AGE–RAGE mediated activation of PGE2 and NO via MAPKs, NF-κB | [224] |
| COX-2, COX-1, mPGES-1 and INOS, JNK, MAPK p38, ERK, NF-κB | →COX-2, COX-1, mPGES-1, INOS expression | ||||
| BSA–AGE (glucose) | Human aortic smooth muscle cells | Calcification of vascular smooth muscle cells Phosphorylation of MAPK 38 | 1–100 µg/ml | →Activation of alkaline phosphatase promote mineralization and vascular calcification | [162] |
| →Calcification is due to AGE–RAGE activated p38 MAPK pathway | |||||
| BSA–AGE (glucose) | Rat vascular smooth muscle cell | AGE-mediated activation of inducible nitric oxidase (iNOS) | 1–500 µg/ml | →AGE-mediated induction of ROS via NADPH oxidase | [154] |
| NF-κB, Nox1 | →Upregulation of NF-κB followed by increased expression of iNOS via Nox1 leading to induced superoxide production | ||||
| →Induced nitrotyrosin formation | |||||
| BSA–AGE | Murine mesangial cells | AGE turnoverviaAGE-recptor 1 | 100 µg/ml | →Increased AGE turnover and reduction of NF-κB in AGE-R1 overexpressing mesangial cells | [83] |
| MAPK p42/44, NF-κB | →Suppressed RAGE-signaling and MCP-1 synthesis by AGE-R1 | ||||
| →silencing AGE-R1 led to enhanced p44/42 phosphorylation | |||||
| BSA–AGE | Rat heart myoblast cell line H9c2 | MEK-ERK pathway | 50–200 µg/ml | →Increased ROS production | [225] |
| Phosphorylation MEK, ERK, JNK, Akt | →Increase cell size and total protein | ||||
| →AGE meditated cell hypertrophy via MEK/ERK pathway | |||||
| Glycerol-, glycol-AGE | Human endothelia cells | Angiogenesis | 100 µg/ml | →Increased expression vascular endothelial growth factor (VEGF) and angiopoietin-2 | [226] |
| →Activation of NF-κB and activation protein-1 (AP-1) | |||||
| AGE | Rat cardiac myofibroblasts H9C2 | Upregulation of inflammatory cytokines | 100 µg/ml | →Increase ROS production and RAGE and NOX-p47 phox protein expression | [227] |
| →Increased cytokine expression: iNOS, TNF-α, TGF-β | |||||
| →Inhibition of cytokine expression with gallic acid | |||||
| BSA–AGE (glucose, ribose) | Rat pulmonary artery smooth muscle cells | p21ras-dependent mitogen-activated protein kinase pathway | Up to 100 µg/ml | →Activation of endogenous p21rasvia AGE–RAGE interaction, ERK1/2 and NF-κB | [228] |
| →Inhibition of p21ras blocked activation of NF-κB | |||||
AGE–RAGE interaction in oxidative and nitrosative stress: a vicious circle
As described above, AGE formation depends on the concentration and reactivity of glucose, amount of AGE-precursors and the availability of free amino groups. AGE formation is facilitated by ROS. Furthermore, the effect of reactive nitrogen species (RNS) on AGE formation has also been investigated. Reactive nitrogen species are reaction products of nitric oxide (NO•) produced by nitric oxide synthase (NOS) and superoxide anion (O2•−), produced by NAPDH-oxidases [7], [151]. While NOS is inducible by cytokines (iNOS) [152], NADPH-oxidase can be activated through AGE–RAGE interactions. The increase of NO•− and O2•− promotes the formation of peroxynitrite (ONOO−), a nitrating and oxidizing agent, leading to inactivation of functional proteins. Furthermore, the increase of ROS leads to an activation of transcription factor nuclear factor kappa-light-chain-enhancer of activated B (Nf-κB), resulting in upregulation of iNOS [153] and additional induction of nitrosative stress and the formation of elevated levels of peroxynitrite [154]. Summarizing, an AGE–RAGE interaction promotes NADPH-oxidase activity, resulting in activation of Nf-κB, followed by expression of iNOS and increased formation of ONOO−. Additionally, Liu et al. determined that RAGE is involved in nitrotyrosine production [155]. The authors were able to show a possible link between activation of RAGE by AGEs and an increase of nitrated thioredoxin in mice after diabetic myocardial ischemia–reperfusion (MI/R) injury. They have shown that increased RAGE levels led to the inactivation of thioredoxin by an increased nitration, followed by its loss in anti-apoptotic, anti-oxidative function and its functionality in cardiac protection. While RAGE modulates the nitration of thioredoxin through induced peroxynitrite generation, reduced non-nitrated thioredoxin inhibits RAGE expression, suggesting a cross-talk between both [155]. Furthermore, Nagai et al. showed that RNS are involved in AGE generation [156]. The incubation of glycated human serum albumin (HsA) with peroxynitrite results in an increase of CML formation. Glucose and peroxynitrite were shown to react to glucosone and glyoxal, which further promote CML generation [145]. Summarizing, the cross-talk between AGEs and nitrosative stress is mainly due to the amount of reactive oxygen and nitrogen species as well as the antioxidant property of the cells. An imbalance between antioxidative enzymes and ROS-meditated AGE production promotes the development of further ROS and RNS, such as peroxynitrite and leads to a vicious cycle of nitrosative stress and further protein damage.
AGE–RAGE-mediated signaling via Janus kinase (Jak) signal transducers and activators of transcription (Stat)
Jak/Stat pathway activation can be driven by various substances and are connected to many different pathways. Today, four Jaks (TYK2, Jak-1, Jak-2, and Jak-3) and seven Stats (Stat-1, -2, -3, -4, -5a, -5b and -6) are known. Interferon γ (INF-γ) is an excellent example for triggering the signaling cascade of Jak/Stat. Binding of INF-γ to its cell-surface receptor INF-γR activates the receptor-associated Janus kinase 1 (Jak-1) und Jak-2 via phosphorylation, followed by the activation of Stat-1 via phosphorylation of its tyrosin residue 701 [30], [157]. After activation two Stat-1 molecules dimerize and further translocate together with interferon regulatory factor 1 (IRF-1) into the nucleus. There, the complex binds on interferon stimulated response element (ISRE) sequences, inducing cytokine production, such as INF-γ and an increased expression of the immunoform of the proteasom (Fig. 4) [145]. Grimm et al. have shown a similar mechanism for AGE-mediated Jak–Stat activation. After treatment of macrophages with different AGE-precursors for 72 h they observed an increase in Jak-2 and Stat-1 phosphorylation. Furthermore, they demonstrated an increased expression of immunoproteasomal subunits. Inhibition of Stat-1 and Jak-2 reduced AGE induced expression of proteolytic subunits of the immunoproteasome. Silencing of RAGE confirmed that the receptor is involved in AGE mediated induction of immunoproteasomal subunits. Thus, AGEs are able to activate Jak-2 and Stat-1 via RAGE and thereby affect the composition and activity of proteasomal subunits [30]. Besides the regulation of the proteasome, AGEs are also involved in the reduction of collagen type II and chondrial cartilage via Jak–Stat signaling. Therefore, AGE–RAGE interaction activates Jak-2/Jak-3 resulting in activation of Stat-3 and the expression of matrix-metallopeptidase 13 (MMP-13, collagen III) and ADAMST 4/5 (cleavage of proteoglycan aggrecan). At high levels both enzymes are responsible for rapid degradation of collagen type II, resulting in osteoarthritis. Inhibition of Stat-3 shows a reduction of AGE-induced loss of collagen II [158]. In contrast, Huang et al. showed an AGE-mediated increase of collagen IV production in rat kidney fibroblasts via Jak-2/Stat-1/Stat-3 activity. Due to mutations and increasing levels of collagen IV, the glomerular membrane gets thinner until it dissolves [159], [160], [161]. This shows that the impact of the modification on collagen is type and tissue specific. In conclusion, AGE–RAGE interaction can regulate different transcription factors via activation of Jak/Stat signaling. Some of them were shown to block by inhibition of Jak/Stat activity as well as RAGE inhibition [30], [159].
Fig. 4.
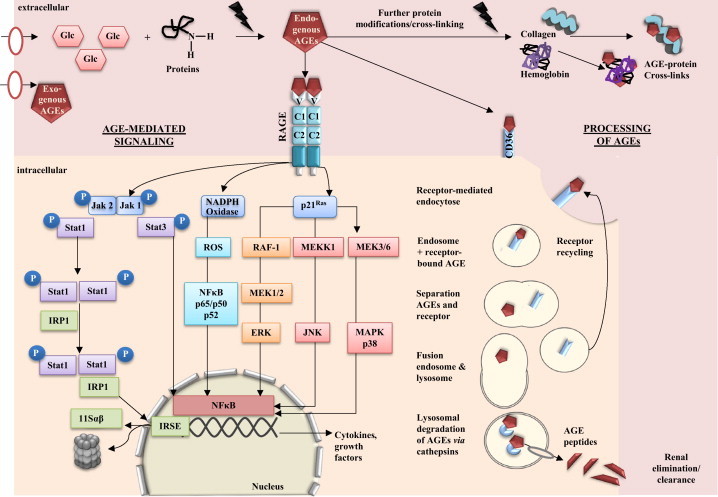
AGE-mediated signaling and detoxification of AGEs via lysosomal system. AGE–RAGE interaction stimulates a various number of signaling cascades, including Jak/Stat, NADPH oxidase, mitogen activated protein kinasen (MAPK), such as p38, extracellular regulated (ERK)-1/2 and c-Jun N-terminal kinase (JNK). AGE-mediated signaling via RAGE leads to the activation of transcription factors, such as nuclear factor (NF-kB) or IFN-stimulated response elements (ISRE) followed by an increased expression of cytokines, growth factors or i.e., immunoproteasomal subunits. While RAGE–AGE interactions are believed to activate inflammatory pathways, other receptors i.e., the family of scavenger receptors play an important role in receptor-mediated endocytosis, leading to intracellular uptake and (parial) degradation of AGEs by fusion with lysosomes. Furthermore, AGE peptides can be transferred to the renal system, while the receptors will be recycled and available for endocytosis processes (modified from [30], [54], [83], [86], [139], [215]).
AGE–RAGE interaction during vascular calcification and ossification
During the aging process, vascular calcification is a common feature in vessel walls. It correlates with plaque instability and inflammation during atherogenesis. Due to the response on inflammatory stimuli, vascular cells produce matrix vesicles in which calcium precipitates and forms calcified nuclei. Subsequently, the induction of necrosis and apoptosis and the activation of matrix degrading processes, results in plaque rupture [8]. Thus, vascular calcification ends in a decrease of elasticity and stability of the vessel walls. In diabetes mellitus arterial calcification is often associated with an elevated level of mortality, morbidity and stroke. Due to the fact that AGEs can cause inflammation, they have the ability to promote vascular calcification. Moreover, Wei et al. demonstrated that AGEs increase vascular calcification by inducing osteoblast-like differentiation via RAGE pathway [7]. They investigated elevated levels of aorta AGEs, RAGE expression and arterial calcification. Furthermore, they have demonstrated that an inhibition of RAGE by an anti-RAGE blocking antibody is able to reduce calcification in rat vascular smooth muscle cells [162], suggesting that the AGE–RAGE–ROS pathway plays an important role in vascular calcification [7] and bone metabolism [146], [147], [148]. In contrast, Li et al. showed that RAGE expression impaired osteoblast proliferation. They demonstrated that the negative regulation of RAGE goes back to the repression of the Wnt, phosphoinositol 3 kinase (PI3K) and extracellular-signal regulated kinase (ERK) pathway. Wnt/β-catenin signaling is known for regulating osteoblast proliferation and differentiation, leading to bone mass formation. An overexpression of RAGE was shown to reduce the expression of Wnt-related genes, which control cell proliferation. In contrast to vascular calcification, RAGE is also associated with bone loss-associated diseases [163].
Cross-talk of RAGE, AGE-R1, AGE-R3/galectin3
As mentioned in the section above, RAGE plays an important role in AGE-mediated signaling, inflammatory response and ROS-related NF-κB activation. In contrast, AGE-R1 and AGE-R3/galectin3 showed opposite effects. Moreover, several studies describe a negative regulation of RAGE via the other AGE-receptors [83], [86], [103], [164], [165]. Cai et al. demonstrated that AGE-R1 reduced pro-inflammatory response by suppressing phosphorylation of protein p66Shc. P66Shc signaling pathway is induced by AGE-mediated ROS and regulated via epidermal growth factor receptor (EGFR), mitogen-activated protein/ERK kinase kinases (MEKK) and ERK, leading to inactivation of forkhead transcription factor (FKHRL1) and impaired manganese superoxide dismutase (MnSOD) activity. Cells overexpressing AGE-R1 were resistant against oxidative damage and cell injury, suggesting that AGE-R1 controls inflammatory signals by inhibiting p66Shc signaling. Hence it was demonstrated that the pathway can be blocked by AGE-R1 which represents an alternative path against AGE-induced ROS formation [85]. Additionally, increased expression of AGE-R1 impaired AGE-induced NF-κB activity and MAPK1/2 phosphorylation and lead to an elevated degradation of AGEs [84]. Accordingly, Lu et al. investigated the involvement of RAGE in downregulation of NF-κB activity. They demonstrated that AGE-induced ROS formation activates MAPK/p42/44 and NF-κB, resulting in enhanced expression of RAGE. In a model with overexpressed AGE-R1 they have shown a higher binding of AGEs by AGE-R1, resulting in decreased RAGE activity and intracellular AGE-mediated signaling. Inhibition of AGE-R1 by siRNA leads to an elevated activity of MAPK/p44/42. Besides AGE-R1 the interaction of AGE-R3/galectin3 has also been studied. AGE-R3/galectin3 belongs to the lactose-binding lectin families as component of 80K-H [166]. As an AGE-receptor, it is directly involved in the modulation of Wnt/β-catenin signaling, responsible for osteogenic differentiation [167].
Although AGE-R1 and AGE-R3/galectin3 have the ability to reduce AGE-mediated ROS and RAGE activity, their expression and function decreases during aging and under higher levels of oxidative stress. Besides the antioxidative enzymes, the activity of AGE-R1 and AGE-R3/galectin3 is also impaired. It was demonstrated that accumulation of AGEs/ALEs and the reduced function of AGE-R3/galectin3 promote dysregulation of vascular calcification induced by AGE–RAGE interaction. Cai et al. conclude that decreasing AGE-R1 during diabetes and aging reduced the antioxidant defense and led to increasing levels of oxidative stress [85]. Besides the downregulation of AGE-R1, NAD+-dependent deacetylase and survival factor 1 (SIRT1) levels were also decreased under elevated state of oxidative stress. SIRT1 is furthermore responsible for repression of inflammatory signals and insulin efficiency. Increased ROS via AGE–RAGE interaction can cause the decrease in AGE-R1 and SIRT1, leading to increased inflammation processes and impaired insulin-signaling pathways [165]. Due to the fact that elevated ROS levels already play an important role in the young generation, the decrease of AGE-R1 and SIRT1 is not only limited to the aging population. Levels of AGE-R1 depend mainly on circulating AGEs, AGE metabolism, state of oxidative stress and elimination via kidney. Elevated dietary intake of AGEs can promote the abnormal ROS formation, followed by a decrease expression and function of AGE-R1, AGE-R3/galectin3 and SIRT1, resulting in an increasing development of chronic diseases [86].
In summary, it has been shown that AGEs influence many different signaling pathways. Thereby, reactive oxygen and nitrogen species as well as the intake of glucose and dietary AGEs play an important role in the formation of endogenous AGEs. The prevention of AGE formation by reducing dietary glucose intake, inhibiting cellular AGE–RAGE interaction and promoting AGE clearance may provide primary approaches against increasing AGE-levels [168].
Physiological role of RAGE
The question of the functional relevance of RAGE has become more and more popular. Besides the potential role of RAGE in embryogenesis and aging, the physiological function is largely undefined. As RAGE is highly expressed in the lung, the lung is a desired organ for investigation. Thus, most data exist from lung, followed by its role in the immune system and less is known about its involvement in bone metabolism or neuronal processes (Fig. 5).
Fig. 5.
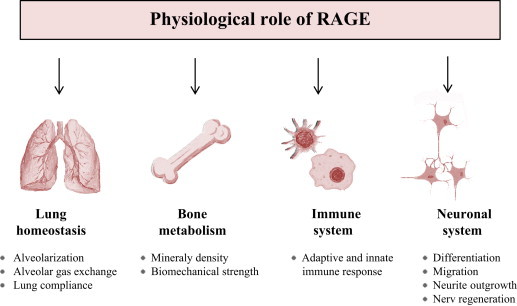
Physiological role of RAGE. AGE–RAGE mediated changes in physiology of lung homeostasis, bone metabolism, immune system and neuronal system.
RAGE expression: from fetal to adult
The first detectable RAGE mRNA-expression goes back to the early blastocyst stage at day 4 in the rabbit embryo. The expression increases in the gastrulating blastocyst on day 6 (stage 0–2, Fig. 6A). During the blastocyst formation RAGE expression is mainly localized in the embryoblast and barely in trophoblast cells (Fig. 6B). Immunohistochemical staining reveals a dominant staining of the membrane. The role of RAGE in the early blastocyst is unclear. In the human Konishi et al. confirmed the presence of RAGE in trophoblast cell in chorionic villi of the first trimester, but just with its association to induce oxidative stress [169]. There are barely data about RAGE during embryogenesis in the literature. Reynolds et al. described RAGE expression in endothelial cells of embryonic vessels and alveolar capillaries in the rat [170]. Hori et al. found RAGE mRNA and protein expression in (E17) embryonic cortical neurons from rats and, for the first time, with a functional relevance for neurite outgrowth [171]. However, most data dealing with RAGE during embryogenesis suggest RAGE to be crucial for organogenesis of the lung. Western blot analysis revealed a steady increase in RAGE expression from fetal (E19), term, 4 day, 8 day to adult lungs in rats [172], with 2-fold increase of mRAGE expression in the adult lung compared to the neonate. This gradual increase of RAGE from fetal to birth and adult life is discussed to be due to an increase in type I epithelial cells during alveolarization. Therefore, RAGE expression in the fetal lung is associated with lung development. Reynolds et al. assessed the RAGE expression in the mouse lung from embryonic day (E) 13.5 onwards. RAGE is expressed initially in undifferentiated parenchymal cells in the saccular stage (E17.5-PN5). A persistent RAGE expression is detected just before birth at E18.5. A remarkable upregulation of RAGE is observed in type I alveolar cells during the alveolar (PN5–PN20) stage of lung development, demonstrating once again its association to alveolarization [170]. High RAGE expression prior to the alveolarization period has deleterious effects including the development of pulmonary dysplasia [173], [174]. Although RAGE is highly expressed in lung its depletion does neither mediate serious pulmonary changes nor reduces the lifespan [175], [176]. During aging RAGE is constantly high expressed in the lung [177]. Interestingly, under pathophysiological condition RAGE is downregulated in malignant tumor of the lung, what is a striking paradox, since in many other tumor entities, the pattern of RAGE expression follows the opposite direction [178], [179]. In other tissues the moderate expression of RAGE can change during aging. In aged rat hearts Gao et al. found high levels of RAGE and equally high levels of AGEs, with a NF-kB activation [180]. The increase in RAGE during aging is possibly due to the accumulation of RAGE ligands, which in turn upregulates receptor expressions in a positive feedback loop. Simm et al. also found an age-dependent upregulation in the protein expression of RAGE in the human heart tissue with an unaltered level of mRNA suggesting a reduced turnover of the receptor during aging [181]. In contrast sRAGE, a splice variant of RAGE, is downregulated in the plasma during aging [182]. High levels of plasma sRAGE are even assumed to have positive impact on human longevity [183].
Fig. 6.
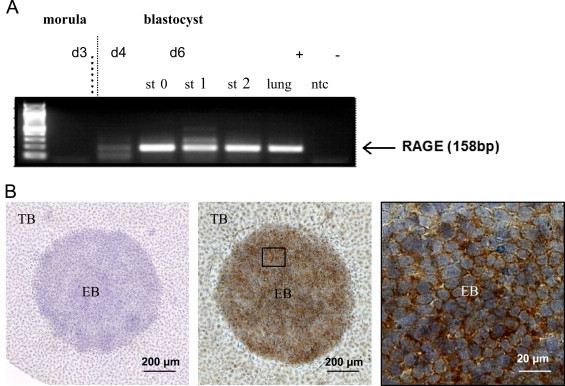
Expression of RAGE in the early rabbit embryo. Transcripts of RAGE were detected in day 3, 4, and 6 p.c. embryos and blastocysts in gastrulation stages 0 (st 0), 1 (st 1), and 2 (st 2). A probe without cDNA was used as negative control (ntc), cDNA from lung tissue was used as positive control. Internal control was the expression of GAPDH in all probes (A). Immunohistochemical detection of RAGE in 6 day old rabbit blastocysts. RAGE localization was visualized by peroxidase-diaminobenzidine reaction (brown color). The nucleus was counterstained with hemalum (blue color). The negative control is the control reaction of the HRP-conjugated secondary goat-anti mouse-IgG. RAGE is mainly localized in the membrane of embryoblast cells (EB). In trophoblast cells (TB) RAGE is barely present (B) (own unpublished data from EH and BF).
RAGE expression and lung homeostasis
RAGE shows histological presence in pulmonary endothelium, bronchial and vascular smooth muscle, as well as in alveolar macrophages and leiomyocytes and on the visceral pleural surface in bovine tissues [184]. Although it is widely accepted that RAGE is strongly expressed in the alveolar epithelium cells there are contradictory results concerning the exact differentiation state of these cells. However, there is more evidence that RAGE is a marker for type I alveolar cells rather than type II [185], [186], [187]. Within type I cell, RAGE has been shown to be specifically localized towards the basolateral membrane and to be co-localized with markers specific to type I alveolar epithelial cells [186], [188]
The strong expression of RAGE in the lung suggests a specific role of this receptor in maintaining lung homeostasis. Its potential role during lung development was described earlier. Although mice lacking RAGE do not show serious pulmonary alterations [175] several studies indicate adverse effects of RAGE in development fibrosis and acute lung injury. Al-Robaiy et al. elucidate the importance of the high RAGE expression in supporting the respiratory mechanics of the lung. Lungs from RAGE knockout (RAGE−/−) mice show a significant increase in dynamic lung compliance and a decrease in the maximal expiratory air flow [176]. It is discussed that the biomechanically interaction of cells and their matrix, as well as the mechanically coupling of cells are affected. Bartling et al. were the first group describing an preferential localization of RAGE at cell–cell contacts [178]. Based on these data RAGE was discussed to have an adhesive role. Later it was found that RAGE indeed enhances the adherence of cells to matrix proteins and, most remarkably, has an exceptional capacity to induce cell spreading [186]. The degree of adherence could be decreased by blocking RAGE. These studies give its apparently important role in modulating adhesion of alveolar epithelial cells to the basement membrane. This is important for pathologies like fibrosis or cancer/metastasis where this is impaired. Beside the interaction of RAGE with the extracellular matrix, the trans interaction of RAGE can promote tissue stiffness. This assumption is supported by the finding in RAGE−/−mice that showed lower elastin mRNA and protein levels [176].
RAGE expression and bone metabolism
Recent studies provide evidence for a contribution of RAGE to bone remodeling. RAGE is expressed in both osteoclasts and osteoblasts [146], [148]. In primary cultured bone marrow macrophages (BMM) RAGE appears to be up-regulated during osteoclast differentiation [148]. BMMs differentiated from RAGE−/− bone marrow cultures appear smaller in size compared with cells from WT cultures, that suggest a morphologic defect in in vitro-differentiated RAGE−/− osteoclasts [148]. Zhou and coworkers also found a reduced integrin-mediated cell adhesion signaling in BMMs. In RAGE−/− mice both bone mineral density and bone biomechanical strength are increased associated with a decreased number of osteoclasts [147], [148]. Ding et al. suggest that the lack of RAGE mainly decreases the osteoclast function in RAGE−/− mice and may not physiologically affect osteoblast function [147] Contrary Philip et al. show an attenuated gene expression associated with osteoblast differentiation in RAGE deficiency mice [189]. However, there are more data describing the role of RAGE in osteoclast maturation and function that generally implicates a role of RAGE in the maturation of monocyte/macrophage, representing cell lineages of the immune system.
RAGE and immune response
The upregulation of RAGE in inflammatory lesions of various diseases (rheumatoid arthritis, inflammatory kidney disease, arteriosclerosis, inflammatory bowel disease) and the capacity of RAGE to bind many proinflammatory ligands (amyloid-β fibrils, S100 proteins, and HMGB1) indicating that RAGE is an important player in the propagation of immune/inflammatory responses. As the majority of components in the innate immune system, the encoding gene for RAGE (Ager) is localized within the major histocompatibility (MHC) class III [190]. RAGE is known to be expressed on numerous immune cells including neutrophils, monocytes/macrophages, lymphocytes and dentritic cells [184]. Liliensiek and coworkers identified RAGE−/− mice are protected from the lethal effects of septic shock, which is largely based on the innate immune response. RAGE−/− mice show reduced local inflammation and decreased NF-κB activation in target organs of septic shock [191]. Blockage of RAGE causes a retarded inflammatory response by arresting central inflammation signaling pathways [192], [193]. RAGE activation on endothelium, mononuclear phagocytes, and lymphocytes triggers cellular activation, with releasing of key proinflammatory mediators [192]. Chavaki et al. found RAGE to be a counter-receptor for the leukocyte integrin Mac-1, thus being directly involved in leukocyte recruitment. The interaction of RAGE on leukocytes results in activation of sustained NF-κB-dependent gene expression [56]. Recently, RAGE has been identified to form a complex with Toll-like receptor 9 (TLR9), the principal DNA-recognizing receptor. This RAGE–TLR9 complex is able to detect pathogen DNA [65]. Regarding to these findings, Sirosis et al. show that the RAGE lowers the immune recognition threshold for the activation of TLR9 and promotes DNA uptake into endosomes. In vivo experiments show that RAGE−/− mice are unable to mount a typical inflammatory response to DNA in the lung [194]. The data supporting the role of RAGE in adaptive immune response are much weaker than those focused on the innate immunity. However, several findings have been recently reported. The sRAGE administration in naive mice leads to an influx of lymphocytes into the spleen and a reducing B-cell number in the bone marrow. Furthermore, sRAGE causes the activation and maturation of B cells resulting in a higher secretion of IgM and IgG and leads to the activation of T cells, as confirmed by the increased production of cytokines [195]. The absence of RAGE is manifest by reduced activation of T cells to alloantigens in vivo and in vitro. RAGE is involved in the differentiation of T cells along a Th1 phenotype and RAGE mRNA is more abundant in Th1 compared with Th2 cells [61].
RAGE expression and neuronal differentiation
RAGE expression in cell bodies and axonal processes was first observed in the bovine nervous system in motor neurons and in certain populations of cortical neurons [184]. Ligands for RAGE in the central nervous system are HMGB1, S100B, AGEs, and amyloid-β fibrils. The interaction of RAGE with AGEs or amyloid-β fibrils leads primarily to inflammation and oxidative stress and is involved in the pathogenesis of some neurological disorders i.e., Alzheimer's disease [196], [197], [198], [199], whereas HMGB1 and S100B are known to be involved in numerous neuronal functions, including neurite outgrowth, migration, differentiation and survival. However, the concentration of ligands seems be an important factor in predicting the effect of RAGE on cellular homeostasis. Low concentrations lead to more beneficial events (neurite outgrowth) and high concentrations, inducing more deleterious events (inflammatory signaling) [200]. Besides the concentration, also the cell type seems to be important. In neurons, the binding of HMGB1 results in neurite outgrowth, while in microglia HMGB1 induces potentially deleterious inflammatory signaling [201]. The multiple effects of RAGE involved it in nerve regeneration through recruitment of both inflammatory, in part mediated through macrophage activation, and axonal outgrowth pathways [202]. Blocking of RAGE suppresses peripheral nerve regeneration, reduces the functional recovery of the nerve and decreases the number of infiltrating phagocytes [203].
Prevention of AGE formation
As already mentioned, the AGE pool in cells, tissues and body fluids is a result from the interaction of dietary AGEs, formation of endogenous AGE and the renal clearance. While intake of dietary AGEs and AGEs from cigarettes plays also a role in cellular AGE levels, a reduction in AGE-uptake would decrease plasma levels. Otherwise Somoza et al. have demonstrated that bread crust, pronyl-BSA and dietary malt are able to improve the activity of chemopreventive enzymes and antioxidant capacity. They suggest, that the positive effects rely on compounds known as melanoidine, which are responsible for the activation of Phase II enzymes, such as glutathione-S-transferase [204]. Moreover, several studies have shown that melanoidine have the ability to increase the proliferation of bifidobacteria [205], [206]. The bioavailability of dietary AGEs can only be assumed. How and which structures of melanoidine are responsible for the induction of Phase II enzymes is not known yet and needs to be further investigated. According to the first analysis of Pötzsch et al. a separation of AGEs into high molecular weight (HMW>1 kDa) and low molecular weight (LMW<1 kDa) compounds shows that the LMW fraction leads to an inhibition of intracellular oxidation but differs from the cell lines that were used [207]. However, for the AGE-toxicity the endogenously formed AGEs are of more importance and their elimination is imperative to avoid the development of AGE-mediated diseases. For early detoxification proteins, such as defensins, lactoferrin and lysozyme circulate in blood and body fluids. They all depend to the innate defense system and are able to bind AGEs before cellular uptake or cross-linking to other molecules [83], [208], [209]. Therefore, Zheng et al. describe an acceleration of renal AGE-clearance and a suppression of intracellular AGE-mediated signaling by lysozymes, suggesting that lysozymes sequestered AGEs and protect proteins for further damage [210]. In conclusion, AGEs are metabolized by innate defense and/or intracellular degradation after receptor-dependent uptake [209]. For the AGE-uptake different receptors have been investigated which have been described in detail above. Two main systems are responsible for the intracellular degradation of damaged and modified proteins, lipids and nucleic acids, the proteasomal and the lysosomal system. Therefore, Grimm et al. found out that AGEs can alter the assembly of the proteasome [30]. Normally, the 20S core of the proteasome consists of two outer α- and inner β-rings, where the β subunits, β5 (chymotrypsin-like activities), β1 (peptidyl-glutamyl-peptide-hydrolyzing or caspase-like activity) and β2 (trypsin-like activity) are responsible for the proteolytical activity of the proteasome [31]. After treatment of RAW 264.7 cells with AGEs, a decrease of the constitutive proteasomal β-subunits and an increase in the inducible immunoproteasomal β-subunits β5i, β1i and β2i have been shown. The induction of immunoproteasomal subunits demonstrates the involvement of AGEs in immune response. Thereby, the immunoproteasome has the ability to process proteins to peptides which are later presented on the surface of MHC class I molecules [211]. It is also discussed whether the immunoproteasome is able to remove oxidized proteins which accumulate after oxidative stress [212]. Due to the fact that the proteasome is unable to degrade the complex structures of the AGEs [138], [213], the lysosomal system plays the main part in detoxification. Further, Grimm et al. have demonstrated that cathepsins, especially cathepsin D plays a major role in intracellular degradation of modified AGEs [139].
Enzymatic degradation of AGE-precursors, such as α-oxalaldehydes is also important in prevention of AGE formation [22], [209]. The glyoxalase system as well as aldehyde dehydrogenase are described to detoxified early sugar adducts and reactive carbonyl compounds, such as glyoxal and methylglyoxal [214], [215]. The cytosolic glyoxalase system, consisting of glyoxalase I and II, is responsible for converting reactive acyclic α-oxalaldehydes into to α-hydoxyacids. Under normal conditions α-oxalaldehydes, such as methylglyoxal spontaneously form a hemiacetal with glutathione. Then glyoxalase I catalyzes the hemiacetal further to S-2-hydroxyacylglutathione. Subsequently, glyoxalase II converted S-2-hydroxyacylglutathione to α-hydoxyacids and glutathione. Thereby, the glyoxalase system prevents the accumulation of reactive α-oxalaldehydes and suppresses further rearrangements of these reactive compounds. The reactions are proportional to the glutathione level in the cytosol [216]. When glutathione levels are low due to increased oxidative stress, the detoxification via the glyoxalase system is impaired [37]. Furthermore, it was suggested that hyperglycemia; methylglyoxal and other RAGE ligands significantly reduce glyoxalase I activity in endothelia and neuronal cells as well as in cultured fibroblasts. An inhibition of RAGE restores the activity of glyoxalase I [143], [217].
Another enzyme for metabolizing further glycation adducts is fructosamie 3-phosphokinase (F3NK). It is responsible for the phosphorylation of free fructosamine into the AGE-precursor 3-deoxyglucosone. This reactive carbonyl compound needs to be detoxified also to interrupt the vicious cycle of AGE formation [168]. For hemoglobin, it was demonstrated that glycation increased during incubation with high glucose concentrations and the parallel inhibition of F3NK in human erythrocytes. Thus, it is assumed that F3NK plays an important role in prevention of further glycation processes [218].
Due to the involvement of oxidants and metals, such as iron and cooper in the formation of AGEs, the search for antioxidative and chelating nutrients is of increasing interest [42]. The use of desferoxamine as iron chelator and anti-inflammatory compounds, such as acetylsalicylic acid have been described to inhibit further glycation processes [219]. The final elimination of degraded AGEs is carried out by the kidney; however these processes are also not well understood.
Conclusion
The review has shown the various impacts of AGEs and their ability to directly alter protein function, forming involved in many chronic diseases. The heterogeneous group of AGEs and the variety of receptor-mediated uptake into cells lead to the activation of many signaling cascades, gene expression as well as to the processing and elimination of AGEs and differs in the various cell types. For the cellular uptake and response a number of AGE receptors have been identified but their internalization and physiological roles are not yet fully understood. From all the evidence presented in this review, it is obvious that RAGE plays a vital role in normal physiology. On a closer look to its involvement in physiological processes it is noticeable that RAGE is most often implicated in the immune response of the respective organ: osteoclast function in the bone, activation of microglia in the nervous system and activation of literal immune cells for immune response. However, beside these roles, RAGE is identified to have various specific effects in different organs. As RAGE is most often discussed to induce deleterious effects and many animal studies showed beneficial effects in clinical settings by blocking RAGE, RAGE is suggested as pharmaco-therapeutic target. Changes in receptor activity and the reciprocal effects of receptor-mediated AGE signaling and detoxification during the aging process is yet to be established. Impaired detoxification and elimination of AGEs is associated with cytotoxicity, apoptosis and increased inflammatory response. Besides the fact that AGEs are inducible under increased ROS levels, they are able to induce ROS, leading to an in vicious cycle. In addition to the negative effects of AGEs, positive effects have also been described. However, there are still critical issues that remain to be resolved before a RAGE-directed therapeutic strategy can be established. Future investigations are definitely required to develop a greater understanding of the impact of RAGE on physiological processes.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Vaupel J.W., Kistowski K.G.V. The remarkable rise in life expectancy and how it will affect medicine. Bundesgesundheitsblatt, Gesundheitsforschung, Gesundheitsschutz. 2005;48:586–592. doi: 10.1007/s00103-005-1043-4. [DOI] [PubMed] [Google Scholar]
- 2.Oeppen J., Vaupel J.W. Demography. Broken limits to life expectancy. Science. 2002;296:1029–1031. doi: 10.1126/science.1069675. [DOI] [PubMed] [Google Scholar]
- 3.Singh R., Barden A., Mori T., Beilin L. Advanced glycation end-products: a review. Diabetologia. 2001;44:129–146. doi: 10.1007/s001250051591. [DOI] [PubMed] [Google Scholar]
- 4.Farouque H.M., O’Brien R.C., Meredith I.T. Diabetes mellitus and coronary heart disease—from prevention to intervention: part I. Aust. N. Z. J. Med. 2000;30:351–359. doi: 10.1111/j.1445-5994.2000.tb00837.x. [DOI] [PubMed] [Google Scholar]
- 5.Cerami C., Founds H., Nicholl I., Mitsuhashi T., Giordano D., Vanpatten S., Lee A., Al-Abed Y., Vlassara H., Bucala R., Cerami A. Tobacco smoke is a source of toxic reactive glycation products. Proc. Natl. Acad. Sci. USA. 1997;94:13915–13920. doi: 10.1073/pnas.94.25.13915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamagishi S., Matsui T., Nakamura K. Possible involvement of tobacco-derived advanced glycation end products (AGEs) in an increased risk for developing cancers and cardiovascular disease in former smokers. Med. Hypotheses. 2008;71:259–261. doi: 10.1016/j.mehy.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 7.Wei Q., Ren X., Jiang Y., Jin H., Liu N., Li J. Advanced glycation end products accelerate rat vascular calcification through RAGE/oxidative stress. BMC Cardiovasc. Disord. 2013;13:13. doi: 10.1186/1471-2261-13-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansson G.K., Robertson A.K., Soderberg-Naucler C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- 9.Passos J.F., Saretzki G., Ahmed S., Nelson G., Richter T., Peters H., Wappler I., Birket M.J., Harold G., Schaeuble K., Birch-Machin M.A., Kirkwood T.B., von Zglinicki T. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q., Fischer A., Reagan J.D., Yan L.J., Ames B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. USA. 1995;92:4337–4341. doi: 10.1073/pnas.92.10.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sitte N., Merker K., von Zglinicki T., Grune T. Protein oxidation and degradation during proliferative senescence of human MRC-5 fibroblasts. Free Radic. Biol. Med. 2000;28:701–708. doi: 10.1016/s0891-5849(99)00279-8. [DOI] [PubMed] [Google Scholar]
- 12.Sohal R.S. Hydrogen peroxide production by mitochondria may be a biomarker of aging. Mech. Ageing Dev. 1991;60:189–198. doi: 10.1016/0047-6374(91)90130-r. [DOI] [PubMed] [Google Scholar]
- 13.Kirkwood T.B. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 14.Jakus V., Rietbrock N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiol. Res./Acad. Sci. Bohemoslov. 2004;53:131–142. [PubMed] [Google Scholar]
- 15.Schleicher E.D., Wagner E., Nerlich A.G. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J. Clin. Investig. 1997;99:457–468. doi: 10.1172/JCI119180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grillo M.A., Colombatto S. Advanced glycation end-products (AGEs): involvement in aging and in neurodegenerative diseases. Amino Acids. 2008;35:29–36. doi: 10.1007/s00726-007-0606-0. [DOI] [PubMed] [Google Scholar]
- 17.Bierhaus A., Hofmann M.A., Ziegler R., Nawroth P.P. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovasc. Res. 1998;37:586–600. doi: 10.1016/s0008-6363(97)00233-2. [DOI] [PubMed] [Google Scholar]
- 18.Simm A. Protein glycation during aging and in cardiovascular disease. J. Proteomics. 2013;92:248–259. doi: 10.1016/j.jprot.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 19.Sebastiani P., Solovieff N., Dewan A.T., Walsh K.M., Puca A., Hartley S.W., Melista E., Andersen S., Dworkis D.A., Wilk J.B., Myers R.H., Steinberg M.H., Montano M., Baldwin C.T., Hoh J., Perls T.T. Genetic signatures of exceptional longevity in humans. PLoS One. 2012;7:e29848. doi: 10.1371/journal.pone.0029848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.John W.G., Lamb E.J. The Maillard or browning reaction in diabetes. Eye. 1993;7(Pt 2):230–237. doi: 10.1038/eye.1993.55. [DOI] [PubMed] [Google Scholar]
- 21.O’Brien J., Morrissey P.A. Nutritional and toxicological aspects of the Maillard browning reaction in foods. Crit. Rev. Food Sci. Nutr. 1989;28:211–248. doi: 10.1080/10408398909527499. [DOI] [PubMed] [Google Scholar]
- 22.Nass N., Bartling B., Navarrete Santos A., Scheubel R.J., Borgermann J., Silber R.E., Simm A. Advanced glycation end products, diabetes and ageing. Z. Gerontol. Geriatr. 2007;40:349–356. doi: 10.1007/s00391-007-0484-9. [DOI] [PubMed] [Google Scholar]
- 23.Kasper M., Funk R.H. Age-related changes in cells and tissues due to advanced glycation end products (AGEs) Arch. Gerontol. Geriatr. 2001;32:233–243. doi: 10.1016/s0167-4943(01)00103-0. [DOI] [PubMed] [Google Scholar]
- 24.Badenhorst D., Maseko M., Tsotetsi O.J., Naidoo A., Brooksbank R., Norton G.R., Woodiwiss A.J. Cross-linking influences the impact of quantitative changes in myocardial collagen on cardiac stiffness and remodelling in hypertension in rats. Cardiovasc. Res. 2003;57:632–641. doi: 10.1016/s0008-6363(02)00733-2. [DOI] [PubMed] [Google Scholar]
- 25.Minois N. Longevity and aging: beneficial effects of exposure to mild stress. Biogerontology. 2000;1:15–29. doi: 10.1023/a:1010085823990. [DOI] [PubMed] [Google Scholar]
- 26.Radak Z., Chung H.Y., Goto S. Exercise and hormesis: oxidative stress-related adaptation for successful aging. Biogerontology. 2005;6:71–75. doi: 10.1007/s10522-004-7386-7. [DOI] [PubMed] [Google Scholar]
- 27.Schmeisser S., Priebe S., Groth M., Monajembashi S., Hemmerich P., Guthke R., Platzer M., Ristow M. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol. Metab. 2013;2:92–102. doi: 10.1016/j.molmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dupre-Crochet S., Erard M., Nubetae O. ROS production in phagocytes: why, when, and where? J. Leukoc. Biol. 2013;94:657–670. doi: 10.1189/jlb.1012544. [DOI] [PubMed] [Google Scholar]
- 29.Vitek M.P., Bhattacharya K., Glendening J.M., Stopa E., Vlassara H., Bucala R., Manogue K., Cerami A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1994;91:4766–4770. doi: 10.1073/pnas.91.11.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimm S., Ott C., Horlacher M., Weber D., Hohn A., Grune T. Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochem. J. 2012;448:127–139. doi: 10.1042/BJ20120298. [DOI] [PubMed] [Google Scholar]
- 31.Jung T., Catalgol B., Grune T. The proteasomal system. Mol. Asp. Med. 2009;30:191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Ahmed N., Argirov O.K., Minhas H.S., Cordeiro C.A., Thornalley P.J. Assay of advanced glycation endproducts (AGEs): surveying AGEs by chromatographic assay with derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to Nepsilon-carboxymethyl-lysine- and Nepsilon-(1-carboxyethyl)lysine-modified albumin. Biochem. J. 2002;364:1–14. doi: 10.1042/bj3640001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lapolla A., Fedele D., Martano L., Arico N.C., Garbeglio M., Traldi P., Seraglia R., Favretto D. Advanced glycation end products: a highly complex set of biologically relevant compounds detected by mass spectrometry. J. Mass Spectrom.: JMS. 2001;36:370–378. doi: 10.1002/jms.137. [DOI] [PubMed] [Google Scholar]
- 34.Nicholl I.D., Bucala R. Advanced glycation endproducts and cigarette smoking. Cell. Mol. Biol. 1998;44:1025–1033. [PubMed] [Google Scholar]
- 35.Valencia J.V., Weldon S.C., Quinn D., Kiers G.H., DeGroot J., TeKoppele J.M., Hughes T.E. Advanced glycation end product ligands for the receptor for advanced glycation end products: biochemical characterization and formation kinetics. Anal. Biochem. 2004;324:68–78. doi: 10.1016/j.ab.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 36.Dyer D.G., Dunn J.A., Thorpe S.R., Lyons T.J., McCance D.R., Baynes J.W. Accumulation of Maillard reaction products in skin collagen in diabetes and aging. Ann. N. Y. Acad. Sci. 1992;663:421–422. doi: 10.1111/j.1749-6632.1992.tb38687.x. [DOI] [PubMed] [Google Scholar]
- 37.Thornalley P.J. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification—a role in pathogenesis and antiproliferative chemotherapy. Gen. Pharmacol. 1996;27:565–573. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- 38.Thornalley P.J., Langborg A., Minhas H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999;344(Pt 1):109–116. [PMC free article] [PubMed] [Google Scholar]
- 39.Ferreira A.E., Ponces Freire A.M., Voit E.O. A quantitative model of the generation of N(epsilon)-(carboxymethyl)lysine in the Maillard reaction between collagen and glucose. Biochem. J. 2003;376:109–121. doi: 10.1042/BJ20030496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glomb M.A., Monnier V.M. Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J. Biol. Chem. 1995;270:10017–10026. doi: 10.1074/jbc.270.17.10017. [DOI] [PubMed] [Google Scholar]
- 41.Aldini G., Vistoli G., Stefek M., Chondrogianni N., Grune T., Sereikaite J., Sadowska-Bartosz I., Bartosz G. Molecular strategies to prevent, inhibit, and degrade advanced glycoxidation and advanced lipoxidation end products. Free. Radic. Res. 2013;47(Suppl. 1):93–137. doi: 10.3109/10715762.2013.792926. [DOI] [PubMed] [Google Scholar]
- 42.Price D.L., Rhett P.M., Thorpe S.R., Baynes J.W. Chelating activity of advanced glycation end-product inhibitors. J. Biol. Chem. 2001;276:48967–48972. doi: 10.1074/jbc.M108196200. [DOI] [PubMed] [Google Scholar]
- 43.Gkogkolou P., Bohm M. Advanced glycation end products: key players in skin aging? Derm.-Endocrinol. 2012;4:259–270. doi: 10.4161/derm.22028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmitt A., Schmitt J., Munch G., Gasic-Milencovic J. Characterization of advanced glycation end products for biochemical studies: side chain modifications and fluorescence characteristics. Anal. Biochem. 2005;338:201–215. doi: 10.1016/j.ab.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Kume S., Takeya M., Mori T., Araki N., Suzuki H., Horiuchi S., Kodama T., Miyauchi Y., Takahashi K. Immunohistochemical and ultrastructural detection of advanced glycation end products in atherosclerotic lesions of human aorta with a novel specific monoclonal antibody. Am. J. Pathol. 1995;147:654–667. [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y.M., Mitsuhashi T., Wojciechowicz D., Shimizu N., Li J., Stitt A., He C., Banerjee D., Vlassara H. Molecular identity and cellular distribution of advanced glycation endproduct receptors: relationship of p60 to OST-48 and p90 to 80K-H membrane proteins. Proc. Natl. Acad. Sci. USA. 1996;93:11047–11052. doi: 10.1073/pnas.93.20.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vlassara H., Li Y.M., Imani F., Wojciechowicz D., Yang Z., Liu F.T., Cerami A. Identification of galectin-3 as a high-affinity binding protein for advanced glycation end products (AGE): a new member of the AGE-receptor complex. Mol. Med. (Cambridge, MA) 1995;1:634–646. [PMC free article] [PubMed] [Google Scholar]
- 48.Araki N., Higashi T., Mori T., Shibayama R., Kawabe Y., Kodama T., Takahashi K., Shichiri M., Horiuchi S. Macrophage scavenger receptor mediates the endocytic uptake and degradation of advanced glycation end products of the Maillard reaction. Eur. J. Biochem. 1995;230:408–415. doi: 10.1111/j.1432-1033.1995.0408h.x. [DOI] [PubMed] [Google Scholar]
- 49.Ohgami N., Nagai R., Ikemoto M., Arai H., Kuniyasu A., Horiuchi S., Nakayama H. CD36, a member of class B scavenger receptor family, is a receptor for advanced glycation end products. Ann. N. Y. Acad. Sci. 2001;947:350–355. doi: 10.1111/j.1749-6632.2001.tb03961.x. [DOI] [PubMed] [Google Scholar]
- 50.Ohgami N., Nagai R., Ikemoto M., Arai H., Miyazaki A., Hakamata H., Horiuchi S., Nakayama H. CD36, serves as a receptor for advanced glycation endproducts (AGE) J. Diabetes Complicat. 2002;16:56–59. doi: 10.1016/s1056-8727(01)00208-2. [DOI] [PubMed] [Google Scholar]
- 51.Ohgami N., Nagai R., Miyazaki A., Ikemoto M., Arai H., Horiuchi S., Nakayama H. Scavenger receptor class B type I-mediated reverse cholesterol transport is inhibited by advanced glycation end products. J. Biol. Chem. 2001;276:13348–13355. doi: 10.1074/jbc.M011613200. [DOI] [PubMed] [Google Scholar]
- 52.Jono T., Miyazaki A., Nagai R., Sawamura T., Kitamura T., Horiuchi S. Lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) serves as an endothelial receptor for advanced glycation end products (AGE) FEBS Lett. 2002;511:170–174. doi: 10.1016/s0014-5793(01)03325-7. [DOI] [PubMed] [Google Scholar]
- 53.Tamura Y., Adachi H., Osuga J.-I., Ohashi K., Yahagi N., Sekiya M., Okazaki H., Tomita S., Iizuka Y., Shimano H., Nagai R., Kimura S., Tsujimoto M., Ishibashi S. FEEL-1 and FEEL-2 are endocytic receptors for advanced glycation end products. J. Biol. Chem. 2003;278:12613–12617. doi: 10.1074/jbc.M210211200. [DOI] [PubMed] [Google Scholar]
- 54.Vlassara H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes/Metab. Res. Rev. 2001;17:436–443. doi: 10.1002/dmrr.233. [DOI] [PubMed] [Google Scholar]
- 55.Neeper M., Schmidt A.M., Brett J., Yan S.D., Wang F., Pan Y.C., Elliston K., Stern D., Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 56.Chavakis T., Bierhaus A., Al-Fakhri N., Schneider D., Witte S., Linn T., Nagashima M., Morser J., Arnold B., Preissner K.T., Nawroth P.P. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003;198:1507–1515. doi: 10.1084/jem.20030800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohashi K., Takahashi H.K., Mori S., Liu K., Wake H., Sadamori H., Matsuda H., Yagi T., Yoshino T., Nishibori M., Tanaka N. Advanced glycation end products enhance monocyte activation during human mixed lymphocyte reaction. Clin. Immunol. (Orlando, FL) 2010;134:345–353. doi: 10.1016/j.clim.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y., Wang H., Piper M.G., McMaken S., Mo X., Opalek J., Schmidt A.M., Marsh C.B. sRAGE induces human monocyte survival and differentiation. J. Immunol. (Baltimore, MD) 2010;185:1822–1835. doi: 10.4049/jimmunol.0903398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang F., Kent K.C., Yamanouchi D., Zhang Y., Kato K., Tsai S., Nowygrod R., Schmidt A.M., Liu B. Anti-receptor for advanced glycation end products therapies as novel treatment for abdominal aortic aneurysm. Ann. Surg. 2009;250:416–423. doi: 10.1097/SLA.0b013e3181b41a18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akirav E.M., Preston-Hurlburt P., Garyu J., Henegariu O., Clynes R., Schmidt A.M., Herold K.C. RAGE expression in human T cells: a link between environmental factors and adaptive immune responses. PloS One. 2012;7:e34698. doi: 10.1371/journal.pone.0034698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Y., Akirav E.M., Chen W., Henegariu O., Moser B., Desai D., Shen J.M., Webster J.C., Andrews R.C., Mjalli A.M., Rothlein R., Schmidt A.M., Clynes R., Herold K.C. RAGE ligation affects T cell activation and controls T cell differentiation. J. Immunol. (Baltimore, MD) 2008;181:4272–4278. doi: 10.4049/jimmunol.181.6.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moser B., Desai D.D., Downie M.P., Chen Y., Yan S.F., Herold K., Schmidt A.M., Clynes R. Receptor for advanced glycation end products expression on T cells contributes to antigen-specific cellular expansion in vivo. J. Immunol. (Baltimore, MD) 2007;179:8051–8058. doi: 10.4049/jimmunol.179.12.8051. [DOI] [PubMed] [Google Scholar]
- 63.Pollreisz A., Hudson B.I., Chang J.S., Qu W., Cheng B., Papapanou P.N., Schmidt A.M., Lalla E. Receptor for advanced glycation endproducts mediates pro-atherogenic responses to periodontal infection in vascular endothelial cells. Atherosclerosis. 2010;212:451–456. doi: 10.1016/j.atherosclerosis.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dumitriu I.E., Baruah P., Valentinis B., Voll R.E., Herrmann M., Nawroth P.P., Arnold B., Bianchi M.E., Manfredi A.A., Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. (Baltimore, MD) 2005;174:7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 65.Tian J., Avalos A.M., Mao S.-Y., Chen B., Senthil K., Wu H., Parroche P., Drabic S., Golenbock D., Sirois C., Hua J., An L.L., Audoly L., La Rosa G., Bierhaus A., Naworth P., Marshak-Rothstein A., Crow M.K., Fitzgerald K.A., Latz E., Kiener P.A., Coyle A.J. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y., Liang C., Liu X., Liao B., Pan X., Ren Y., Fan M., Li M., He Z., Wu J., Wu Z. AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE, MAPK and NF-kappaB pathways. Atherosclerosis. 2010;208:34–42. doi: 10.1016/j.atherosclerosis.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 67.Kamioka M., Ishibashi T., Ohkawara H., Nagai R., Sugimoto K., Uekita H., Matsui T., Yamagishi S.-I., Ando K., Sakamoto T., Sakamoto N., Takuwa Y., Wada I., Shiomi M., Maruyama Y., Takeishi Y. Involvement of membrane type 1-matrix metalloproteinase (MT1-MMP) in RAGE activation signaling pathways. J. Cell. Physiol. 2011;226:1554–1563. doi: 10.1002/jcp.22492. [DOI] [PubMed] [Google Scholar]
- 68.Nah S.-S., Choi I.-Y., Yoo B., Kim Y.G., Moon H.-B., Lee C.-K. Advanced glycation end products increases matrix metalloproteinase-1, -3, and -13, and TNF-alpha in human osteoarthritic chondrocytes. FEBS Lett. 2007;581:1928–1932. doi: 10.1016/j.febslet.2007.03.090. [DOI] [PubMed] [Google Scholar]
- 69.Zhu P., Ren M., Yang C., Hu Y.-X., Ran J.-M., Yan L. Involvement of RAGE, MAPK and NF-κB pathways in AGEs-induced MMP-9 activation in HaCaT keratinocytes. Exp. Dermatol. 2012;21:123–129. doi: 10.1111/j.1600-0625.2011.01408.x. [DOI] [PubMed] [Google Scholar]
- 70.Kierdorf K., Fritz G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013;94:55–68. doi: 10.1189/jlb.1012519. [DOI] [PubMed] [Google Scholar]
- 71.Huttunen H.J., Fages C., Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999;274:19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 72.Dattilo B.M., Fritz G., Leclerc E., Kooi C.W.V., Heizmann C.W., Chazin W.J. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry. 2007;46:6957–6970. doi: 10.1021/bi7003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matsumoto S., Yoshida T., Murata H., Harada S., Fujita N., Nakamura S., Yamamoto Y., Watanabe T., Yonekura H., Yamamoto H., Ohkubo T., Kobayashi Y. Solution structure of the variable-type domain of the receptor for advanced glycation end products: new insight into AGE–RAGE interaction. Biochemistry. 2008;47:12299–12311. doi: 10.1021/bi800910v. [DOI] [PubMed] [Google Scholar]
- 74.Falcone C., Bozzini S., D’Angelo A., Matrone B., Colonna A., Benzi A., Paganini E.M., Falcone R., Pelissero G. Plasma levels of soluble receptor for advanced glycation end products and coronary atherosclerosis: possible correlation with clinical presentation. Dis. Markers. 2013;35:135–140. doi: 10.1155/2013/129360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yonekura H., Yamamoto Y., Sakurai S., Petrova R.G., Abedin M.J., Li H., Yasui K., Takeuchi M., Makita Z., Takasawa S., Okamoto H., Watanabe T., Yamamoto H. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003;370:1097–1109. doi: 10.1042/BJ20021371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang L., Bukulin M., Kojro E., Roth A., Metz V.V., Fahrenholz F., Nawroth P.P., Bierhaus A., Postina R. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J. Biol. Chem. 2008;283:35507–35516. doi: 10.1074/jbc.M806948200. [DOI] [PubMed] [Google Scholar]
- 77.Bierhaus A., Humpert P.M., Morcos M., Wendt T., Chavakis T., Arnold B., Stern D.M., Nawroth P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005;83:876–886. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 78.Yang Z., Makita Z., Horii Y., Brunelle S., Cerami A., Sehajpal P., Suthanthiran M., Vlassara H. Two novel rat liver membrane proteins that bind advanced glycosylation endproducts: relationship to macrophage receptor for glucose-modified proteins. J. Exp. Med. 1991;174:515–524. doi: 10.1084/jem.174.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Silberstein S., Kelleher D.J., Gilmore R. The 48-kDa subunit of the mammalian oligosaccharyltransferase complex is homologous to the essential yeast protein WBP1. J. Biol. Chem. 1992;267:23658–23663. [PubMed] [Google Scholar]
- 80.Kelleher D.J., Kreibich G., Gilmore R. Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kDa protein. Cell. 1992;69:55–65. doi: 10.1016/0092-8674(92)90118-v. [DOI] [PubMed] [Google Scholar]
- 81.Kelleher D.J., Gilmore R. DAD1, the defender against apoptotic cell death, is a subunit of the mammalian oligosaccharyltransferase. Proc. Natl. Acad. Sci. USA. 1997;94:4994–4999. doi: 10.1073/pnas.94.10.4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roboti P., High S. The oligosaccharyltransferase subunits OST48, DAD1 and KCP2 function as ubiquitous and selective modulators of mammalian N-glycosylation. J. Cell Sci. 2012;125:3474–3484. doi: 10.1242/jcs.103952. [DOI] [PubMed] [Google Scholar]
- 83.Lu C., He J.C., Cai W., Liu H., Zhu L., Vlassara H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc. Natl. Acad. Sci. USA. 2004;101:11767–11772. doi: 10.1073/pnas.0401588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cai W., He J.C., Zhu L., Lu C., Vlassara H. Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc. Natl. Acad. Sci. USA. 2006;103:13801–13806. doi: 10.1073/pnas.0600362103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cai W., He J.C., Zhu L., Chen X., Striker G.E., Vlassara H. AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am. J. Physiol. Cell. Physiol. 2008;294:C145–C152. doi: 10.1152/ajpcell.00350.2007. [DOI] [PubMed] [Google Scholar]
- 86.Vlassara H., Cai W., Goodman S., Pyzik R., Yong A., Chen X., Zhu L., Neade T., Beeri M., Silverman J.M., Ferrucci L., Tansman L., Striker G.E., Uribarri J. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: role of the antiinflammatory AGE receptor-1. J. Clin. Endocrinol. Metab. 2009;94:4483–4491. doi: 10.1210/jc.2009-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sakai K., Hirai M., Minoshima S., Kudoh J., Fukuyama R., Shimizu N. Isolation of cDNAs encoding a substrate for protein kinase C: nucleotide sequence and chromosomal mapping of the gene for a human 80 K protein. Genomics. 1989;5:309–315. doi: 10.1016/0888-7543(89)90063-3. [DOI] [PubMed] [Google Scholar]
- 88.Hirai M., Shimizu N. Purification of two distinct proteins of approximate Mr 80,000 from human epithelial cells and identification as proper substrates for protein kinase C. Biochem. J. 1990;270:583–589. doi: 10.1042/bj2700583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Radoff S., Vlassara H., Cerami A. Characterization of a solubilized cell surface binding protein on macrophages specific for proteins modified nonenzymatically by advanced glycosylated end products. Arch. Biochem. Biophys. 1988;263:418–423. doi: 10.1016/0003-9861(88)90654-6. [DOI] [PubMed] [Google Scholar]
- 90.Goh K.C., Lim Y.P., Ong S.H., Siak C.B., Cao X., Tan Y.H., Guy G.R. Identification of p90, a prominent tyrosine-phosphorylated protein in fibroblast growth factor-stimulated cells, as 80K-H. J. Biol. Chem. 1996;271:5832–5838. doi: 10.1074/jbc.271.10.5832. [DOI] [PubMed] [Google Scholar]
- 91.Stitt A.W., Bucala R., Vlassara H. Atherogenesis and advanced glycation: promotion, progression, and prevention. Ann. N. Y. Acad. Sci. 1997;811:115–127. doi: 10.1111/j.1749-6632.1997.tb51994.x. (discussion 127-119) [DOI] [PubMed] [Google Scholar]
- 92.Kanai M., Göke M., Tsunekawa S., Podolsky D.K. Signal transduction pathway of human fibroblast growth factor receptor 3. Identification of a novel 66-kDa phosphoprotein. J. Biol. Chem. 1997;272:6621–6628. doi: 10.1074/jbc.272.10.6621. [DOI] [PubMed] [Google Scholar]
- 93.Brûlé S., Rabahi F., Faure R., Beckers J.F., Silversides D.W., Lussier J.G. Vacuolar system-associated protein-60: a protein characterized from bovine granulosa and luteal cells that is associated with intracellular vesicles and related to human 80K-H and murine beta-glucosidase II. Biol. Reprod. 2000;62:642–654. doi: 10.1095/biolreprod62.3.642. [DOI] [PubMed] [Google Scholar]
- 94.Hodgkinson C.P., Mander A., Sale G.J. Identification of 80K-H as a protein involved in GLUT4 vesicle trafficking. Biochem. J. 2005;388:785–793. doi: 10.1042/BJ20041845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ho M.K., Springer T.A. Mac-2, a novel 32,000 Mr mouse macrophage subpopulation-specific antigen defined by monoclonal antibodies. J. Immunol. (Baltimore, MD) 1982;128:1221–1228. [PubMed] [Google Scholar]
- 96.Barondes S.H., Cooper D.N., Gitt M.A., Leffler H. Galectins. Structure and function of a large family of animal lectins. J. Biol. Chem. 1994;269:20807–20810. [PubMed] [Google Scholar]
- 97.Barondes S.H., Castronovo V., Cooper D.N., Cummings R.D., Drickamer K., Feizi T., Gitt M.A., Hirabayashi J., Hughes C., Kasai K. Galectins: a family of animal beta-galactoside-binding lectins. Cell. 1994;76:597–598. doi: 10.1016/0092-8674(94)90498-7. [DOI] [PubMed] [Google Scholar]
- 98.Cowles E.A., Agrwal N., Anderson R.L., Wang J.L. Carbohydrate-binding protein 35. Isoelectric points of the polypeptide and a phosphorylated derivative. J. Biol. Chem. 1990;265:17706–17712. [PubMed] [Google Scholar]
- 99.Huflejt M.E., Turck C.W., Lindstedt R., Barondes S.H., Leffler H. L-29, a soluble lactose-binding lectin, is phosphorylated on serine 6 and serine 12 in vivo and by casein kinase I. J. Biol. Chem. 1993;268:26712–26718. [PubMed] [Google Scholar]
- 100.Mazurek N., Conklin J., Byrd J.C., Raz A., Bresalier R.S. Phosphorylation of the beta-galactoside-binding protein galectin-3 modulates binding to its ligands. J. Biol. Chem. 2000;275:36311–36315. doi: 10.1074/jbc.M003831200. [DOI] [PubMed] [Google Scholar]
- 101.Yoshii T., Fukumori T., Honjo Y., Inohara H., Kim H.-R.C., Raz A. Galectin-3 phosphorylation is required for its anti-apoptotic function and cell cycle arrest. J. Biol. Chem. 2002;277:6852–6857. doi: 10.1074/jbc.M107668200. [DOI] [PubMed] [Google Scholar]
- 102.Pricci F., Leto G., Amadio L., Iacobini C., Romeo G., Cordone S., Gradini R., Barsotti P., Liu F.T., Di Mario U., Pugliese G. Role of galectin-3 as a receptor for advanced glycosylation end products. Kidney Int. Suppl. 2000;77:S31–39. doi: 10.1046/j.1523-1755.2000.07706.x. [DOI] [PubMed] [Google Scholar]
- 103.Menini S., Iacobini C., Ricci C., Blasetti Fantauzzi C., Salvi L., Pesce C.M., Relucenti M., Familiari G., Taurino M., Pugliese G. The galectin-3/RAGE dyad modulates vascular osteogenesis in atherosclerosis. Cardiovasc. Res. 2013;100:472–480. doi: 10.1093/cvr/cvt206. [DOI] [PubMed] [Google Scholar]
- 104.Goldstein J.L., Ho Y.K., Basu S.K., Brown M.S. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA. 1979;76:333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Henriksen T., Mahoney E.M., Steinberg D. Enhanced macrophage degradation of low density lipoprotein previously incubated with cultured endothelial cells: recognition by receptors for acetylated low density lipoproteins. Proc. Natl. Acad. Sci. USA. 1981;78:6499–6503. doi: 10.1073/pnas.78.10.6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Steinbrecher U.P., Parthasarathy S., Leake D.S., Witztum J.L., Steinberg D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc. Natl. Acad. Sci. USA. 1984;81:3883–3887. doi: 10.1073/pnas.81.12.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parthasarathy S. Oxidation of low-density lipoprotein by thiol compounds leads to its recognition by the acetyl LDL receptor. Biochim. Biophys. Acta. 1987;917:337–340. doi: 10.1016/0005-2760(87)90139-1. [DOI] [PubMed] [Google Scholar]
- 108.Parthasarathy S., Printz D.J., Boyd D., Joy L., Steinberg D. Macrophage oxidation of low density lipoprotein generates a modified form recognized by the scavenger receptor. Arteriosclerosis (Dallas, TX) 1986;6:505–510. doi: 10.1161/01.atv.6.5.505. [DOI] [PubMed] [Google Scholar]
- 109.Palinski W., Rosenfeld M.E., Ylä-Herttuala S., Gurtner G.C., Socher S.S., Butler S.W., Parthasarathy S., Carew T.E., Steinberg D., Witztum J.L. Low density lipoprotein undergoes oxidative modification in vivo. Proc. Natl. Acad. Sci. USA. 1989;86:1372–1376. doi: 10.1073/pnas.86.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kodama T., Freeman M., Rohrer L., Zabrecky J., Matsudaira P., Krieger M. Type I macrophage scavenger receptor contains alpha-helical and collagen-like coiled coils. Nature. 1990;343:531–535. doi: 10.1038/343531a0. [DOI] [PubMed] [Google Scholar]
- 111.Emi M., Asaoka H., Matsumoto A., Itakura H., Kurihara Y., Wada Y., Kanamori H., Yazaki Y., Takahashi E., Lepert M. Structure, organization, and chromosomal mapping of the human macrophage scavenger receptor gene. J. Biol. Chem. 1993;268:2120–2125. [PubMed] [Google Scholar]
- 112.Freeman M., Ekkel Y., Rohrer L., Penman M., Freedman N.J., Chisolm G.M., Krieger M. Expression of type I and type II bovine scavenger receptors in Chinese hamster ovary cells: lipid droplet accumulation and nonreciprocal cross competition by acetylated and oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA. 1991;88:4931–4935. doi: 10.1073/pnas.88.11.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Peiser L., Gordon S. The function of scavenger receptors expressed by macrophages and their role in the regulation of inflammation. Microbes Infect./Inst. Pasteur. 2001;3:149–159. doi: 10.1016/s1286-4579(00)01362-9. [DOI] [PubMed] [Google Scholar]
- 114.Tao N., Wagner S.J., Lublin D.M. CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J. Biol. Chem. 1996;271:22315–22320. doi: 10.1074/jbc.271.37.22315. [DOI] [PubMed] [Google Scholar]
- 115.Gu X., Trigatti B., Xu S., Acton S., Babitt J., Krieger M. The efficient cellular uptake of high density lipoprotein lipids via scavenger receptor class B type I requires not only receptor-mediated surface binding but also receptor-specific lipid transfer mediated by its extracellular domain. J. Biol. Chem. 1998;273:26338–26348. doi: 10.1074/jbc.273.41.26338. [DOI] [PubMed] [Google Scholar]
- 116.Acton S., Rigotti A., Landschulz K.T., Xu S., Hobbs H.H., Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science (New York, NY) 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 117.Nakata A., Nakagawa Y., Nishida M., Nozaki S., Miyagawa J., Nakagawa T., Tamura R., Matsumoto K., Kameda-Takemura K., Yamashita S., Matsuzawa Y. CD36, a novel receptor for oxidized low-density lipoproteins, is highly expressed on lipid-laden macrophages in human atherosclerotic aorta. Arterioscler. Thromb. Vasc. Biol. 1999;19:1333–1339. doi: 10.1161/01.atv.19.5.1333. [DOI] [PubMed] [Google Scholar]
- 118.Kuniyasu A., Ohgami N., Hayashi S., Miyazaki A., Horiuchi S., Nakayama H. CD36-mediated endocytic uptake of advanced glycation end products (AGE) in mouse 3T3-L1 and human subcutaneous adipocytes. FEBS Lett. 2003;537:85–90. doi: 10.1016/s0014-5793(03)00096-6. [DOI] [PubMed] [Google Scholar]
- 119.Horiuchi S., Unno Y., Usui H., Shikata K., Takaki K., Koito W., Sakamoto Y.-I., Nagai R., Makino K., Sasao A., Wada J., Makino H. Pathological roles of advanced glycation end product receptors SR-A and CD36. Ann. N. Y. Acad. Sci. 2005;1043:671–675. doi: 10.1196/annals.1333.076. [DOI] [PubMed] [Google Scholar]
- 120.Sawamura T., Kume N., Aoyama T., Moriwaki H., Hoshikawa H., Aiba Y., Tanaka T., Miwa S., Katsura Y., Kita T., Masaki T. An endothelial receptor for oxidized low-density lipoprotein. Nature. 1997;386:73–77. doi: 10.1038/386073a0. [DOI] [PubMed] [Google Scholar]
- 121.Yamanaka S., Zhang X.Y., Miura K., Kim S., Iwao H. The human gene encoding the lectin-type oxidized LDL receptor (OLR1) is a novel member of the natural killer gene complex with a unique expression profile. Genomics. 1998;54:191–199. doi: 10.1006/geno.1998.5561. [DOI] [PubMed] [Google Scholar]
- 122.Biocca S., Arcangeli T., Tagliaferri E., Testa B., Vindigni G., Oteri F., Giorgi A., Iacovelli F., Novelli G., Desideri A., Falconi M. Simulative and experimental investigation on the cleavage site that generates the soluble human LOX-1. Arch. Biochem. Biophys. 2013;540:9–18. doi: 10.1016/j.abb.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 123.Li S., Guo Y., Zhu P., Yang T. Role of Ox-LDL/LOX-1/NF-κB signaling pathway in regulation of atherosclerotic plaque growth by testosterone in male rabbits. Vasc. Pharmacol. 2012 doi: 10.1016/j.vph.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 124.Shiu S.W.M., Wong Y., Tan K.C.B. Effect of advanced glycation end products on lectin-like oxidized low density lipoprotein receptor-1 expression in endothelial cells. J. Atheroscler. Thromb. 2012;19:1083–1092. doi: 10.5551/jat.11742. [DOI] [PubMed] [Google Scholar]
- 125.Mueller M.A., Beutner F., Teupser D., Ceglarek U., Thiery J. Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR−/− mice despite severe hypercholesterolemia. Atherosclerosis. 2008;198:39–48. doi: 10.1016/j.atherosclerosis.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 126.Adachi H., Tsujimoto M. FEEL-1, a novel scavenger receptor with in vitro bacteria-binding and angiogenesis-modulating activities. J. Biol. Chem. 2002;277:34264–34270. doi: 10.1074/jbc.M204277200. [DOI] [PubMed] [Google Scholar]
- 127.Irjala H., Elima K., Johansson E.-L., Merinen M., Kontula K., Alanen K., Grenman R., Salmi M., Jalkanen S. The same endothelial receptor controls lymphocyte traffic both in vascular and lymphatic vessels. Eur. J. Immunol. 2003;33:815–824. doi: 10.1002/eji.200323859. [DOI] [PubMed] [Google Scholar]
- 128.Adachi H., Tsujimoto M. Adaptor protein sorting nexin 17 interacts with the scavenger receptor FEEL-1/stabilin-1 and modulates its expression on the cell surface. Biochim. Biophys. Acta. 2010;1803:553–563. doi: 10.1016/j.bbamcr.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 129.Li R., McCourt P., Schledzewski K., Goerdt S., Moldenhauer G., Liu X., Smedsrød B., Sørensen K.K. Endocytosis of advanced glycation end-products in bovine choriocapillaris endothelial cells. Microcirculation (New York, NY) 2009;16:640–655. doi: 10.1080/10739680903133185. [DOI] [PubMed] [Google Scholar]
- 130.Miyazaki A., Nakayama H., Horiuchi S. Scavenger receptors that recognize advanced glycation end products. Trends Cardiovasc. Med. 2002;12:258–262. doi: 10.1016/s1050-1738(02)00171-8. [DOI] [PubMed] [Google Scholar]
- 131.Vlassara H., Palace M.R. Diabetes and advanced glycation endproducts. J. Intern. Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 132.Horiuchi S., Sakamoto Y., Sakai M. Scavenger receptors for oxidized and glycated proteins. Amino Acids. 2003;25:283–292. doi: 10.1007/s00726-003-0029-5. [DOI] [PubMed] [Google Scholar]
- 133.Mukherjee S., Ghosh R.N., Maxfield F.R. Endocytosis. Physiol. Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- 134.Zastrow M.V., Sorkin A. Signaling on the endocytic pathway. Curr. Opin. Cell Biol. 2007;19:436–445. doi: 10.1016/j.ceb.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Platta H.W., Stenmark H. Endocytosis and signaling. Curr. Opin. Cell Biol. 2011;23:393–403. doi: 10.1016/j.ceb.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 136.Sorkin A., Zastrow M.V. Endocytosis and signalling: intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Frommhold D., Kamphues A., Hepper I., Pruenster M., Lukic I.K., Socher I., Zablotskaya V., Buschmann K., Lange-Sperandio B., Schymeinsky J., Ryschich E., Poeschl J., Kupatt C., Nawroth P.P., Moser M., Walzog B., Bierhaus A., Sperandio M. RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo. Blood. 2010;116:841–849. doi: 10.1182/blood-2009-09-244293. [DOI] [PubMed] [Google Scholar]
- 138.Grimm S., Horlacher M., Catalgol B., Hoehn A., Reinheckel T., Grune T. Cathepsins D and L reduce the toxicity of advanced glycation end products. Free Radic. Biol. Med. 2012;52:1011–1023. doi: 10.1016/j.freeradbiomed.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 139.Grimm S., Ernst L., Grotzinger N., Hohn A., Breusing N., Reinheckel T., Grune T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE-modified proteins. Free Radic. Res. 2010;44:1013–1026. doi: 10.3109/10715762.2010.495127. [DOI] [PubMed] [Google Scholar]
- 140.Sano H., Higashi T., Matsumoto K., Melkko J., Jinnouchi Y., Ikeda K., Ebina Y., Makino H., Smedsrod B., Horiuchi S. Insulin enhances macrophage scavenger receptor-mediated endocytic uptake of advanced glycation end products. J. Biol. Chem. 1998;273:8630–8637. doi: 10.1074/jbc.273.15.8630. [DOI] [PubMed] [Google Scholar]
- 141.Sevillano N., Girón M.D., Salido M., Vargas A.M., Vilches J., Salto R. Internalization of the receptor for advanced glycation end products (RAGE) is required to mediate intracellular responses. J. Biochem. 2009;145:21–30. doi: 10.1093/jb/mvn137. [DOI] [PubMed] [Google Scholar]
- 142.Nicholl I.D., Stitt A.W., Moore J.E., Ritchie A.J., Archer D.B., Bucala R. Increased levels of advanced glycation endproducts in the lenses and blood vessels of cigarette smokers. Mol. Med. 1998;4:594–601. [PMC free article] [PubMed] [Google Scholar]
- 143.Vlassara H., Cai W., Crandall J., Goldberg T., Oberstein R., Dardaine V., Peppa M., Rayfield E.J. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc. Natl. Acad. Sci. USA. 2002;99:15596–15601. doi: 10.1073/pnas.242407999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Uribarri J., Peppa M., Cai W., Goldberg T., Lu M., Baliga S., Vassalotti J.A., Vlassara H. Dietary glycotoxins correlate with circulating advanced glycation end product levels in renal failure patients. Am. J. Kidney Dis.: Off. J. Natl. Kidney Found. 2003;42:532–538. doi: 10.1016/s0272-6386(03)00779-0. [DOI] [PubMed] [Google Scholar]
- 145.Hofmann S.M., Dong H.J., Li Z., Cai W., Altomonte J., Thung S.N., Zeng F., Fisher E.A., Vlassara H. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes. 2002;51:2082–2089. doi: 10.2337/diabetes.51.7.2082. [DOI] [PubMed] [Google Scholar]
- 146.Cecil D.L., Johnson K., Rediske J., Lotz M., Schmidt A.M., Terkeltaub R. Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J. Immunol. 2005;175:8296–8302. doi: 10.4049/jimmunol.175.12.8296. [DOI] [PubMed] [Google Scholar]
- 147.Ding K.-H., Wang Z.-Z., Hamrick M.W., Deng Z.-B., Zhou L., Kang B., Yan S.-L., She J.-X., Stern D.M., Isales C.M., Mi Q.-S. Disordered osteoclast formation in RAGE-deficient mouse establishes an essential role for RAGE in diabetes related bone loss. Biochem. Biophys. Res. Commun. 2006;340:1091–1097. doi: 10.1016/j.bbrc.2005.12.107. [DOI] [PubMed] [Google Scholar]
- 148.Zhou Z., Immel D., Xi C.-X., Bierhaus A., Feng X., Mei L., Nawroth P., Stern D.M., Xiong W.-C. Regulation of osteoclast function and bone mass by RAGE. J. Exp. Med. 2006;203:1067–1080. doi: 10.1084/jem.20051947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Geissler S., Hellwig M., Zwarg M., Markwardt F., Henle T., Brandsch M. Transport of the advanced glycation end products alanylpyrraline and pyrralylalanine by the human proton-coupled peptide transporter hPEPT1. J. Agric. Food Chem. 2010;58:2543–2547. doi: 10.1021/jf903791u. [DOI] [PubMed] [Google Scholar]
- 150.Forster A., Kuhne Y., Henle T. Studies on absorption and elimination of dietary maillard reaction products. Ann. N. Y. Acad. Sci. 2005;1043:474–481. doi: 10.1196/annals.1333.054. [DOI] [PubMed] [Google Scholar]
- 151.Zhang M., Kho A.L., Anilkumar N., Chibber R., Pagano P.J., Shah A.M., Cave A.C. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation. 2006;113:1235–1243. doi: 10.1161/CIRCULATIONAHA.105.581397. [DOI] [PubMed] [Google Scholar]
- 152.Mizutani K., Ikeda K., Ito T., Tamaki K., Nara Y., Yamori Y. Protective effect of inducible type nitric oxide synthase against intracellular oxidative stress caused by advanced glycation end-products in vascular smooth muscle cells from stroke-prone spontaneously hypertensive rats. J. Hypertens. 2000;18:1071–1079. doi: 10.1097/00004872-200018080-00012. [DOI] [PubMed] [Google Scholar]
- 153.Wong A., Dukic-Stefanovic S., Gasic-Milenkovic J., Schinzel R., Wiesinger H., Riederer P., Munch G. Anti-inflammatory antioxidants attenuate the expression of inducible nitric oxide synthase mediated by advanced glycation endproducts in murine microglia. Eur. J. Neurosci. 2001;14:1961–1967. doi: 10.1046/j.0953-816x.2001.01820.x. [DOI] [PubMed] [Google Scholar]
- 154.San Martin A., Foncea R., Laurindo F.R., Ebensperger R., Griendling K.K., Leighton F. Nox1-based NADPH oxidase-derived superoxide is required for VSMC activation by advanced glycation end-products. Free Radic. Biol. Med. 2007;42:1671–1679. doi: 10.1016/j.freeradbiomed.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 155.Liu Y., Qu Y., Wang R., Ma Y., Xia C., Gao C., Liu J., Lian K., Xu A., Lu X., Sun L., Yang L., Lau W.B., Gao E., Koch W., Wang H., Tao L. The alternative crosstalk between RAGE and nitrative thioredoxin inactivation during diabetic myocardial ischemia-reperfusion injury. Am. J. Physiol. Endocrinol. Metab. 2012;303:E841–852. doi: 10.1152/ajpendo.00075.2012. [DOI] [PubMed] [Google Scholar]
- 156.Nagai R., Mera K., Nakajou K., Fujiwara Y., Iwao Y., Imai H., Murata T., Otagiri M. The ligand activity of AGE-proteins to scavenger receptors is dependent on their rate of modification by AGEs. Biochim. Biophys. Acta. 2007;1772:1192–1198. doi: 10.1016/j.bbadis.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 157.Chung E.Y., Kim B.H., Hong J.T., Lee C.K., Ahn B., Nam S.Y., Han S.B., Kim Y. Resveratrol down-regulates interferon-gamma-inducible inflammatory genes in macrophages: molecular mechanism via decreased STAT-1 activation. J. Nutr. Biochem. 2011;22:902–909. doi: 10.1016/j.jnutbio.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 158.Huang C.Y., Lai K.Y., Hung L.F., Wu W.L., Liu F.C., Ho L.J. Advanced glycation end products cause collagen II reduction by activating Janus kinase/signal transducer and activator of transcription 3 pathway in porcine chondrocytes. Rheumatology. 2011;50:1379–1389. doi: 10.1093/rheumatology/ker134. [DOI] [PubMed] [Google Scholar]
- 159.Huang J.S., Guh J.Y., Chen H.C., Hung W.C., Lai Y.H., Chuang L.Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001;81:102–113. doi: 10.1002/1097-4644(20010401)81:1<102::aid-jcb1027>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 160.Yagame M., Kim Y., Zhu D., Suzuki D., Eguchi K., Nomoto Y., Sakai H., Groppoli T., Steffes M.W., Mauer S.M. Differential distribution of type IV collagen chains in patients with diabetic nephropathy in non-insulin-dependent diabetes mellitus. Nephron. 1995;70:42–48. doi: 10.1159/000188542. [DOI] [PubMed] [Google Scholar]
- 161.Khoshnoodi J., Pedchenko V., Hudson B.G. Mammalian collagen IV. Microsc. Res. Tech. 2008;71:357–370. doi: 10.1002/jemt.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tanikawa T., Okada Y., Tanikawa R., Tanaka Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J. Vasc. Res. 2009;46:572–580. doi: 10.1159/000226225. [DOI] [PubMed] [Google Scholar]
- 163.Li G., Xu J., Li Z. Receptor for advanced glycation end products inhibits proliferation in osteoblast through suppression of Wnt, PI3K and ERK signaling. Biochem. Biophys. Res. Commun. 2012;423:684–689. doi: 10.1016/j.bbrc.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 164.Pugliese G., Pricci F., Iacobini C., Leto G., Amadio L., Barsotti P., Frigeri L., Hsu D.K., Vlassara H., Liu F.T., Di Mario U. Accelerated diabetic glomerulopathy in galectin-3/AGE receptor 3 knockout mice. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2001;15:2471–2479. doi: 10.1096/fj.01-0006com. [DOI] [PubMed] [Google Scholar]
- 165.Cai W., Ramdas M., Zhu L., Chen X., Striker G.E., Vlassara H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. USA. 2012;109:15888–15893. doi: 10.1073/pnas.1205847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nakano N., Fukuhara-Takaki K., Jono T., Nakajou K., Eto N., Horiuchi S., Takeya M., Nagai R. Association of advanced glycation end products with A549 cells, a human pulmonary epithelial cell line, is mediated by a receptor distinct from the scavenger receptor family and RAGE. J. Biochem. 2006;139:821–829. doi: 10.1093/jb/mvj092. [DOI] [PubMed] [Google Scholar]
- 167.Kubota T., Michigami T., Ozono K. Wnt signaling in bone metabolism. J. Bone. Miner. Metab. 2009;27:265–271. doi: 10.1007/s00774-009-0064-8. [DOI] [PubMed] [Google Scholar]
- 168.Veiga da-Cunha M., Jacquemin P., Delpierre G., Godfraind C., Theate I., Vertommen D., Clotman F., Lemaigre F., Devuyst O., Van Schaftingen E. Increased protein glycation in fructosamine 3-kinase-deficient mice. Biochem. J. 2006;399:257–264. doi: 10.1042/BJ20060684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Konishi H., Nakatsuka M., Chekir C., Noguchi S., Kamada Y., Sasaki A., Hiramatsu Y. Advanced glycation end products induce secretion of chemokines and apoptosis in human first trimester trophoblasts. Hum. Reprod. 2004;19:2156–2162. doi: 10.1093/humrep/deh389. [DOI] [PubMed] [Google Scholar]
- 170.Reynolds P.R., Kasteler S.D., Cosio M.G., Sturrock A., Huecksteadt T., Hoidal J.R. RAGE: developmental expression and positive feedback regulation by Egr-1 during cigarette smoke exposure in pulmonary epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2008;294:L1094–1101. doi: 10.1152/ajplung.00318.2007. [DOI] [PubMed] [Google Scholar]
- 171.Hori O., Brett J., Slattery T., Cao R., Zhang J., Chen J.X., Nagashima M., Lundh E.R., Vijay S., Nitecki D., Morser J., Stern D., Schmidt A.M. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin mediation of neurite outgrowth and co-expression of RAGE and amphoterin in the developing nervous system. J. Biol. Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 172.Lizotte P.-P., Hanford L.E., Enghild J.J., Nozik-Grayck E., Giles B.-L., Oury T.D. Developmental expression of the receptor for advanced glycation end-products (RAGE) and its response to hyperoxia in the neonatal rat lung. BMC Dev. Biol. 2007;7:15. doi: 10.1186/1471-213X-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Reynolds P.R., Stogsdill J.A., Stogsdill M.P., Heimann N.B. Up-regulation of receptors for advanced glycation end-products by alveolar epithelium influences cytodifferentiation and causes severe lung hypoplasia. Am. J. Respir. Cell Mol. Biol. 2011;45:1195–1202. doi: 10.1165/rcmb.2011-0170OC. [DOI] [PubMed] [Google Scholar]
- 174.Fineschi S., De Cunto G., Facchinetti F., Civelli M., Imbimbo B.P., Carnini C., Villetti G., Lunghi B., Stochino S., Gibbons D.L., Hayday A., Lungarella G., Cavarra E. Receptor for advanced glycation end products contributes to postnatal pulmonary development and adult lung maintenance program in mice. Am. J. Respir. Cell Mol. Biol. 2013;48:164–171. doi: 10.1165/rcmb.2012-0111OC. [DOI] [PubMed] [Google Scholar]
- 175.Buckley S.T., Ehrhardt C. The receptor for advanced glycation end products (RAGE) and the lung. J. Biomed. Biotechnol. 2010;2010 doi: 10.1155/2010/917108. , Article ID 917108, 11 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Al-Robaiy S., Weber B., Simm A., Diez C., Rolewska P., Silber R.-E., Bartling B. The receptor for advanced glycation end-products supports lung tissue biomechanics. Am. J. Physiol. Lung Cell Mol. Physiol. 2013;305:L491–L500. doi: 10.1152/ajplung.00090.2013. [DOI] [PubMed] [Google Scholar]
- 177.Bartling B., Fuchs C., Somoza V., Niemann B., Silber R.-E., Simm A. Lung level of HMBG1 is elevated in response to advanced glycation end product-enriched food in vivo. Mol. Nutr. Food Res. 2007;51:479–487. doi: 10.1002/mnfr.200600223. [DOI] [PubMed] [Google Scholar]
- 178.Bartling B., Hofmann H.-S., Weigle B., Silber R.-E., Simm A. Down-regulation of the receptor for advanced glycation end-products (RAGE) supports non-small cell lung carcinoma. Carcinogenesis. 2005;26:293–301. doi: 10.1093/carcin/bgh333. [DOI] [PubMed] [Google Scholar]
- 179.Sparvero L.J., Asafu-Adjei D., Kang R., Tang D., Amin N., Im J., Rutledge R., Lin B., Amoscato A.A., Zeh H.J., Lotze M.T. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009;7:17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gao Z.Q., Yang C., Wang Y.Y., Wang P., Chen H.L., Zhang X.D., Liu R., Li W.L., Qin X.J., Liang X., Hai C.X. RAGE upregulation and nuclear factor-kappaB activation associated with ageing rat cardiomyocyte dysfunction. Gen. Physiol. Biophys. 2008;27:152–158. [PubMed] [Google Scholar]
- 181.Simm A., Casselmann C., Schubert A., Hofmann S., Reimann A., Silber R.E. Age associated changes of AGE-receptor expression: RAGE upregulation is associated with human heart dysfunction. Exp. Gerontol. 2004;39:407–413. doi: 10.1016/j.exger.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 182.Fujii E.Y., Nakayama M. The measurements of RAGE, VEGF, and AGEs in the plasma and follicular fluid of reproductive women: the influence of aging. Fertil. Steril. 2010;94:694–700. doi: 10.1016/j.fertnstert.2009.03.029. [DOI] [PubMed] [Google Scholar]
- 183.Geroldi D., Falcone C., Minoretti P., Emanuele E., Arra M., D'Angelo A. High levels of soluble receptor for advanced glycation end products may be a marker of extreme longevity in humans. J. Am. Geriatr. Soc. 2006;54:1149–1150. doi: 10.1111/j.1532-5415.2006.00776.x. [DOI] [PubMed] [Google Scholar]
- 184.Brett J., Schmidt A.M., Yan S.D., Zou Y.S., Weidman E., Pinsky D., Nowygrod R., Neeper M., Przysiecki C., Shaw A. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 185.Shirasawa M., Fujiwara N., Hirabayashi S., Ohno H., Iida J., Makita K., Hata Y. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells. 2004;9:165–174. doi: 10.1111/j.1356-9597.2004.00712.x. [DOI] [PubMed] [Google Scholar]
- 186.Demling N., Ehrhardt C., Kasper M., Laue M., Knels L., Rieber E.P. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006;323:475–488. doi: 10.1007/s00441-005-0069-0. [DOI] [PubMed] [Google Scholar]
- 187.Dahlin K., Mager E.M., Allen L., Tigue Z., Goodglick L., Wadehra M., Dobbs L. Identification of genes differentially expressed in rat alveolar type I cells. Am. J. Respir. Cell Mol. Biol. 2004;31:309–316. doi: 10.1165/rcmb.2003-0423OC. [DOI] [PubMed] [Google Scholar]
- 188.Fehrenbach H., Kasper M., Tschernig T., Shearman M.S., Schuh D., Müller M. Receptor for advanced glycation endproducts (RAGE) exhibits highly differential cellular and subcellular localisation in rat and human lung. Cell. Mol. Biol. (Noisy-le-grand) 1998;44:1147–1157. [PubMed] [Google Scholar]
- 189.Philip B.K., Childress P.J., Robling A.G., Heller A., Nawroth P.P., Bierhaus A., Bidwell J.P. RAGE supports parathyroid hormone-induced gains in femoral trabecular bone. Am. J. Physiol. —Endocrinol. Metab. 2010;298:E714–E725. doi: 10.1152/ajpendo.00564.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sugaya K., Fukagawa T., Matsumoto K., Mita K., Takahashi E., Ando A., Inoko H., Ikemura T. Three genes in the human MHC class III region near the junction with the class II: gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics. 1994;23:408–419. doi: 10.1006/geno.1994.1517. [DOI] [PubMed] [Google Scholar]
- 191.Liliensiek B., Weigand M.A., Bierhaus A., Nicklas W., Kasper M., Hofer S., Plachky J., Gröne H.-J., Kurschus F.C., Schmidt A.M., Yan S.D., Martin E., Schleicher E., Stern D.M., Hämmerling G G.ü., Nawroth P.P., Arnold B. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004;113:1641–1650. doi: 10.1172/JCI18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hofmann M.A., Drury S., Fu C., Qu W., Taguchi A., Lu Y., Avila C., Kambham N., Bierhaus A., Nawroth P., Neurath M.F., Slattery T., Beach D., McClary J., Nagashima M., Morser J., Stern D., Schmidt A.M. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 193.Kislinger T., Tanji N., Wendt T., Qu W., Lu Y., Ferran L.J., Jr, Taguchi A., Olson K., Bucciarelli L., Goova M., Hofmann M.A., Cataldegirmen G., D'Agati V., Pischetsrieder M., Stern D.M., Schmidt A.M. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2001;21:905–910. doi: 10.1161/01.atv.21.6.905. [DOI] [PubMed] [Google Scholar]
- 194.Sirois C.M., Jin T., Miller A.L., Bertheloot D., Nakamura H., Horvath G.L., Mian A., Jiang J., Schrum J., Bossaller L., Pelka K., Garbi N., Brewah Y., Tian J., Chang C., Chowdhury P.S., Sims G.P., Kolbeck R., Coyle A.J., Humbles A.A., Xiao T.S., Latz E. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J. Exp. Med. 2013;210:2447–2463. doi: 10.1084/jem.20120201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Brisslert M., Amu S., Pullerits R. Intra-peritoneal sRAGE treatment induces alterations in cellular distribution of CD19(+), CD3 (+) and Mac-1 (+) cells in lymphoid organs and peritoneal cavity. Cell Tissue Res. 2013;351:139–148. doi: 10.1007/s00441-012-1508-3. [DOI] [PubMed] [Google Scholar]
- 196.Mattson M.P. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Deane R., Du Yan S., Submamaryan R.K., LaRue B., Jovanovic S., Hogg E., Welch D., Manness L., Lin C., Yu J., Zhu H., Ghiso J., Frangione B., Stern A., Schmidt A.M., Armstrong D.L., Arnold B., Liliensiek B., Nawroth P., Hofman F., Kindy M., Stern D., Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat. Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 198.Kim S.J., Ahn J.W., Kim H., Ha H.J., Lee S.W., Kim H.K., Lee S., Hong H.S., Kim Y.H., Choi C.Y. Two beta-strands of RAGE participate in the recognition and transport of amyloid-beta peptide across the blood brain barrier. Biochem. Biophys. Res. Commun. 2013;439:252–257. doi: 10.1016/j.bbrc.2013.08.047. [DOI] [PubMed] [Google Scholar]
- 199.Chen C., Li X.H., Tu Y., Sun H.T., Liang H.Q., Cheng S.X., Zhang S. Abeta-AGE aggravates cognitive deficit in rats via RAGE pathway. Neuroscience. 2013;257C:1–10. doi: 10.1016/j.neuroscience.2013.10.056. [DOI] [PubMed] [Google Scholar]
- 200.Huttunen H.J., Kuja-Panula J., Sorci G., Agneletti A.L., Donato R., Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J. Biol. Chem. 2000;275:40096–40105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- 201.Ding Q., Keller J.N. Evaluation of rage isoforms, ligands, and signaling in the brain. Biochim. Biophys. Acta. 2005;1746:18–27. doi: 10.1016/j.bbamcr.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 202.Rong L.L., Yan S.-F., Wendt T., Hans D., Pachydaki S., Bucciarelli L.G., Adebayo A., Qu W., Lu Y., Kostov K., Lalla E., Yan S.D., Gooch C., Szabolcs M., Trojaborg W., Hays A.P., Schmidt A.M. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004;18:1818–1825. doi: 10.1096/fj.04-1900com. [DOI] [PubMed] [Google Scholar]
- 203.Rong L.L., Trojaborg W., Qu W., Kostov K., Yan S.D., Gooch C., Szabolcs M., Hays A.P., Schmidt A.M. Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB J. 2004;18:1812–1817. doi: 10.1096/fj.04-1899com. [DOI] [PubMed] [Google Scholar]
- 204.Somoza V., Wenzel E., Lindenmeier M., Grothe D., Erbersdobler H.F., Hofmann T. Influence of feeding malt, bread crust, and a pronylated protein on the activity of chemopreventive enzymes and antioxidative defense parameters in vivo. J. Agric. Food Chem. 2005;53:8176–8182. doi: 10.1021/jf0512611. [DOI] [PubMed] [Google Scholar]
- 205.Borrelli R.C., Fogliano V. Bread crust melanoidins as potential prebiotic ingredients. Mol. Nutr. Food Res. 2005;49:673–678. doi: 10.1002/mnfr.200500011. [DOI] [PubMed] [Google Scholar]
- 206.Ames J.M., Wynne A., Hofmann A., Plos S., Gibson G.R. The effect of a model melanoidin mixture on faecal bacterial populations in vitro. Br. J. Nutr. 1999;82:489–495. [PubMed] [Google Scholar]
- 207.Pötzsch S., Blankenhorn A., Navarrete Santos A., Silber R.-E., Somoza V., Simm A. The effect of an AGE-rich dietary extract on the activation of NF-κB depends on the cell model used. Food Funct. 2013;4:1023–1031. doi: 10.1039/c3fo30349g. [DOI] [PubMed] [Google Scholar]
- 208.Liu H., Zheng F., Cao Q., Ren B., Zhu L., Striker G., Vlassara H. Amelioration of oxidant stress by the defensin lysozyme. Am. J. Physiol. Endocrinol. Metab. 2006;290:E824–832. doi: 10.1152/ajpendo.00349.2005. [DOI] [PubMed] [Google Scholar]
- 209.Vlassara H., Uribarri J., Cai W., Striker G. Advanced glycation end product homeostasis: exogenous oxidants and innate defenses. Ann. N. Y. Acad. Sci. 2008;1126:46–52. doi: 10.1196/annals.1433.055. [DOI] [PubMed] [Google Scholar]
- 210.Zheng F., Cai W., Mitsuhashi T., Vlassara H. Lysozyme enhances renal excretion of advanced glycation endproducts in vivo and suppresses adverse age-mediated cellular effects in vitro: a potential AGE sequestration therapy for diabetic nephropathy? Mol. Med. 2001;7:737–747. [PMC free article] [PubMed] [Google Scholar]
- 211.Sijts A., Sun Y., Janek K., Kral S., Paschen A., Schadendorf D., Kloetzel P.M. The role of the proteasome activator PA28 in MHC class I antigen processing. Mol. Immunol. 2002;39:165–169. doi: 10.1016/s0161-5890(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 212.Seifert U., Bialy L.P., Ebstein F., Bech-Otschir D., Voigt A., Schroter F., Prozorovski T., Lange N., Steffen J., Rieger M., Kuckelkorn U., Aktas O., Kloetzel P.M., Kruger E. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. 2010;142:613–624. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 213.Bulteau A.L., Verbeke P., Petropoulos I., Chaffotte A.F., Friguet B. Proteasome inhibition in glyoxal-treated fibroblasts and resistance of glycated glucose-6-phosphate dehydrogenase to 20 S proteasome degradation in vitro. J. Biol. Chem. 2001;276:45662–45668. doi: 10.1074/jbc.M105374200. [DOI] [PubMed] [Google Scholar]
- 214.Knecht K.J., Feather M.S., Baynes J.W. Detection of 3-deoxyfructose and 3-deoxyglucosone in human urine and plasma: evidence for intermediate stages of the Maillard reaction in vivo. Arch. Biochem. Biophys. 1992;294:130–137. doi: 10.1016/0003-9861(92)90146-n. [DOI] [PubMed] [Google Scholar]
- 215.Goldin A., Beckman J.A., Schmidt A.M., Creager M.A. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 216.Abordo E.A., Minhas H.S., Thornalley P.J. Accumulation of alpha-oxoaldehydes during oxidative stress: a role in cytotoxicity. Biochem. Pharmacol. 1999;58:641–648. doi: 10.1016/s0006-2952(99)00132-x. [DOI] [PubMed] [Google Scholar]
- 217.Uribarri J., Cai W., Sandu O., Peppa M., Goldberg T., Vlassara H. Diet-derived advanced glycation end products are major contributors to the body's AGE pool and induce inflammation in healthy subjects. Ann. N. Y. Acad. Sci. 2005;1043:461–466. doi: 10.1196/annals.1333.052. [DOI] [PubMed] [Google Scholar]
- 218.Delpierrre G., Vertommen D., Communi D., Rider M.H., Van Schaftingen E. Identification of fructosamine residues deglycated by fructosamine-3-kinase in human hemoglobin. J. Biol. Chem. 2004;279:27613–27620. doi: 10.1074/jbc.M402091200. [DOI] [PubMed] [Google Scholar]
- 219.Peppa M., Goldberg T., Cai W., Rayfield E., Vlassara H. Glycotoxins: a missing link in the “relationship of dietary fat and meat intake in relation to risk of type 2 diabetes in men”. Diabetes Care. 2002;25:1898–1899. doi: 10.2337/diacare.25.10.1898. [DOI] [PubMed] [Google Scholar]
- 220.Fukami K., Ueda S., Yamagishi S., Kato S., Inagaki Y., Takeuchi M., Motomiya Y., Bucala R., Iida S., Tamaki K., Imaizumi T., Cooper M.E., Okuda S. AGEs activate mesangial TGF-beta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004;66:2137–2147. doi: 10.1111/j.1523-1755.2004.66004.x. [DOI] [PubMed] [Google Scholar]
- 221.Alikhani M., Maclellan C.M., Raptis M., Vora S., Trackman P.C., Graves D.T. Advanced glycation end products induce apoptosis in fibroblasts through activation of ROS, MAP kinases, and the FOXO1 transcription factor. Am. J. Physiol. Cell. Physiol. 2007;292:C850–C856. doi: 10.1152/ajpcell.00356.2006. [DOI] [PubMed] [Google Scholar]
- 222.Denis U., Lecomte M., Paget C., Ruggiero D., Wiernsperger N., Lagarde M. Advanced glycation end-products induce apoptosis of bovine retinal pericytes in culture: involvement of diacylglycerol/ceramide production and oxidative stress induction. Free Radic. Biol. Med. 2002;33:236–247. doi: 10.1016/s0891-5849(02)00879-1. [DOI] [PubMed] [Google Scholar]
- 223.Hu P., Lai D., Lu P., Gao J., He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int. J. Mol. Med. 2012;29:613–618. doi: 10.3892/ijmm.2012.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Nah S.S., Choi I.Y., Lee C.K., Oh J.S., Kim Y.G., Moon H.B., Yoo B. Effects of advanced glycation end products on the expression of COX-2, PGE2 and NO in human osteoarthritic chondrocytes. Rheumatology. 2008;47:425–431. doi: 10.1093/rheumatology/kem376. [DOI] [PubMed] [Google Scholar]
- 225.Ko S.Y., Lin I.H., Shieh T.M., Ko H.A., Chen H.I., Chi T.C., Chang S.S., Hsu Y.C. Cell hypertrophy and MEK/ERK phosphorylation are regulated by glyceraldehyde-derived AGEs in cardiomyocyte H9c2 cells. Cell Biochem. Biophys. 2013;66:537–544. doi: 10.1007/s12013-012-9501-8. [DOI] [PubMed] [Google Scholar]
- 226.Okamoto T., Yamagishi S., Inagaki Y., Amano S., Koga K., Abe R., Takeuchi M., Ohno S., Yoshimura A., Makita Z. Angiogenesis induced by advanced glycation end products and its prevention by cerivastatin. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2002;16:1928–1930. doi: 10.1096/fj.02-0030fje. [DOI] [PubMed] [Google Scholar]
- 227.Umadevi S., Gopi V., Vellaichamy E. Inhibitory effect of gallic acid on advanced glycation end products induced up-regulation of inflammatory cytokines and matrix proteins in H9C2 (2-1) cells. Cardiovasc. Toxicol. 2013;13:396–405. doi: 10.1007/s12012-013-9222-2. [DOI] [PubMed] [Google Scholar]
- 228.Lander H.M., Tauras J.M., Ogiste J.S., Hori O., Moss R.A., Schmidt A.M. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 1997;272:17810–17814. doi: 10.1074/jbc.272.28.17810. [DOI] [PubMed] [Google Scholar]