

Abstract
Cardioprotection may be genome dependent. One example is the increased tolerance to cardiac ischemia-reperfusion (IR) in Brown Norway (BN) compared with Dahl salt-sensitive (SS) rats. By narrowing the genetic difference to chromosome 6 only, we found the consomic SS6BN to be similarly IR tolerant as BN. We hypothesized that better preserved mitochondrial structure and function are genetically determined and therefore critically linked to myocardial IR tolerance associated with BN chromosome 6. Langendorff-prepared BN, SS, and SS6BN rat hearts were subjected to IR, while corresponding controls were continuously perfused. Though largely equal in nonischemic controls, assessment of functional data and ventricular infarct size in IR experiments confirmed that BN and SS6BN have an equally higher tolerance to IR than SS hearts. This was complemented by equally better preserved mitochondrial structure, oxidative phosphorylation, and calcium retention capacity in BN and SS6BN vs. SS hearts. For the first time, our data indicate that SS6BN are as resistant to IR injury as BN hearts in mitochondrial and myocardial function and viability compared with SS hearts. These findings not only link myocardial and mitochondrial protection in a genetic model but also suggest that genetic information on rat chromosome 6 is critical for mitochondrial preservation and IR tolerance.
Keywords: Brown Norway, Dahl salt sensitive, SS6BN, myocardium
cardiovascular disease continues to be the most common cause of death, accounting for nearly 40% of the deaths in the developed world (13). In the United States alone, one in every six deaths is caused by coronary artery disease (CAD) (25). Various behavioral and metabolic risk factors can be modified by treatment or prevention. Although each of these factors may be in part genetically determined, family history is an independent predisposing factor suggesting that additional susceptibility genes exist (6). Greater knowledge of the relationship between genetic determinants and outcome after myocardial ischemia-reperfusion (IR) injury would help to identify target genes and pathways that convey higher tolerance to IR.
Interestingly, profound differences in individual IR tolerance have been found among (27) and even within different species. For example, Brown Norway (BN) rats exhibit more resistance to myocardial IR injury than does the Dahl salt-sensitive (SS) strain (3). Moreover, introgression of BN chromosome 6 into an SS background (SS6BN) conferred significant cardioprotection (17). In other words, one or more genes on chromosome 6 with or without linkage to other chromosomes may be responsible for higher IR tolerance. Furthermore, since human studies have implicated genes on chromosome 14 to increase the risk for myocardial infarction (MI) (6) and human chromosome 14 is homologous to rat chromosome 6, the consomic SS6BN strain constitutes a unique and promising model to study the genetic basis of cardioprotection.
To date, studies have focused primarily on differences in the cardiac functional phenotype between BN, SS6BN, and SS strains undergoing IR. The impact of BN chromosome 6 substitution on mitochondrial function is unknown. Altered mitochondrial function has been demonstrated to be both a trigger and effector of cardioprotection (7, 8, 22). Therefore, the objective of our study was to link mitochondrial preservation with genome-dependent endogenous cardioprotection in BN, SS6BN, and SS rats.
MATERIALS AND METHODS
Our methods have been described in detail previously (1, 12, 21–23). Unless otherwise indicated, all drugs were purchased from Sigma (St. Louis, MO). All indicated concentrations are final.
Animals.
All investigations conformed to the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research, National Academy of Sciences, 8th edition, 2011) and were approved by the Institutional Animal Care and Use Committee (AUA 1147, Medical College of Wisconsin, and ACORP 7435-1, VA Medical Center, Milwaukee, WI). The use of BN and SS rat strains as animal models to study cardiovascular disease processes is well established (10, 17, 28). The SS6BN panel was constructed by crossing parental BN and SS to generate a heterozygous Fl population; a male F1 is then backcrossed with a female SS to generate N2 rats that are subsequently backcrossed for up to 10 generations to yield offspring with an isogenic SS background for all but chromosome 6 (10). Because the N2 is generated from an SS mother, the mitochondrial genome is derived from the SS strain. To avoid the confounding influence of sex (19) or increasing phenotypical baseline differences developed over time, we used only 8 wk old normoxic male BN/NHsdMcwi, SS6BN, and SS/JrHsdMcwi rats on a 0.4% NaCl diet.
Langendorff heart preparation.
A total of 90 animals were anesthetized by intraperitoneal injection of 30 mg ketamine along with 1,000 U heparin and euthanized by decapitation after a negative response to a noxious stimulus. After thoracotomy, the aorta was cannulated distal to the aortic valve, and the heart perfused retrograde with 4°C cold oxygenated Krebs solution of the following composition (in mM): 148 Na+, 4.7 K+, 1.2 Mg2+, 1.6 Ca2+, 127 Cl−, 27.8 HCO3−, 1.2 H2PO4−, 1.2 SO42−, 5.5 glucose, 2 pyruvate, 0.026 EDTA, and 5 U/l insulin. The latter has been shown to have no effect on myocardial injury in isolated hearts (14). The venae cavae were ligated, and the heart was rapidly placed into the support system and perfused at 70 mmHg at 37°C. The perfusate was equilibrated with ∼95% O2 and ∼5% CO2 to maintain a constant pH of 7.40 (carbon dioxide partial pressure Pco2 40 mmHg; oxygen partial pressure Po2 570 mmHg). The perfusate was filtered (5 μm pore size) in-line. Left ventricular pressure (LVP) was measured isovolumetrically with a saline-filled latex balloon (Radnoti, Monrovia, CA) inserted into the left ventricle. Its volume was initially adjusted to achieve a diastolic LVP of 10 mmHg so that any subsequent increase reflected diastolic contracture. LVP-derived data were: systolic, diastolic, and developed (systolic-diastolic) LVP, and its maximal and minimal first derivatives (dLVP/dtmax and dLVP/dtmin) as indexes of ventricular contractility and relaxation, respectively. Spontaneous heart rate (HR) was monitored with electrodes attached to the right atrial and ventricular walls. An ultrasonic flow-meter (model T106X; Transonic Systems, Ithaca, NY) measured coronary inflow. All analog signals were digitized (PowerLab/16 SP; AD Instruments, Castle Hill, Australia) and recorded at 200 Hz (Chart & Scope version 5.6.6, AD Instruments) for later analysis.
Protocols.
Experimental protocols are illustrated in Fig. 1. Hearts from all three strains were allowed to stabilize for 20 min followed by baseline readings. They were then subject to 35 min of perfusion and 30 min of global no-flow ischemia. Hearts were reperfused for 120 min if functional data and ventricular infarct size were measured (n = 6 per group); during experiments in which isolated mitochondria (n = 6 per group) were harvested or Western blotting (n = 3 per group) was performed, hearts were reperfused for 30 min. Time matched nonischemic controls were perfused for 185 or 95 min, respectively. The longer duration of reperfusion was chosen to allow full development of infarction, the shorter duration to test for disturbance in Ca2+ homeostasis without loss of nonviable mitochondria.
Fig. 1.

Experimental protocols. Isolated, perfused hearts (n = 90) from 8 wk old male Brown Norway (BN), Dahl salt-sensitive (SS), and consomic (SS6BN) rats were stabilized for 20 min. After baseline (bl) readings nonischemic control hearts were perfused for 185 min, whereas ischemia-reperfusion (IR) hearts were perfused for 35 min before they underwent 30 min global no-flow ischemia followed by 120 min reperfusion (RP). Ventricular infarct size (IS) was determined by TTC staining and cumulative planimetry (n = 6 per group). When Western blots (n = 3 per group) or functional experiments with isolated mitochondria (n = 6 per group) were performed, hearts were perfused for only 95 min or harvested after only 30 min reperfusion.
Infarct size measurement.
Hearts were removed and atria discarded; ventricles were cut into 2 mm transverse sections with a heart matrix and incubated in 1% 2,3,5-triphenyltetrazolium chloride (TTC) in 0.1 M KH2PO4 buffer (pH 7.4, 38°C) for 10 min (23). TTC stains viable tissue red, indicating the presence of a formazan precipitate that results from TTC reduction by dehydrogenase enzymes present in viable tissue. All slices were digitally imaged on green background by a photoscanner, and the infarcted areas of each slice were measured automatically by planimetry using Image J 1.44i software (National Institutes of Health, Bethesda, MD), its ColorThreshold plug-in, and a recently developed and calibrated macro ensuring fast and operator-independent measurements (29). Infarcted areas of individual slices were averaged on the basis of their weight to calculate the total infarction of both ventricles.
Mitochondrial isolation.
Hearts were immediately removed from the setup and immersed in 4°C cold isolation buffer containing (in mM) 200 mannitol, 50 sucrose, 5 KH2PO4, 5 3-(N-morpholino)propanesulfonic acid (MOPS), and 1 EGTA, with 0.1% bovine serum albumin (BSA), pH 7.15 (adjusted with KOH) (1, 21). The atria were discarded, and the ventricles were minced into 1 mm3 pieces. The suspension was initially homogenized for 15 s in 2.5 ml of isolation buffer containing 5 U/ml protease and for another 15 s after addition of 17 ml of isolation buffer. Mitochondria were then isolated by differential centrifugation. First, the suspension was centrifuged at 8,000 g for 10 min; the resulting pellet was then resuspended in 25 ml of isolation buffer and centrifuged again at 750 g for 10 min to remove cellular debris. Next, the supernatant containing the mitochondrial fraction was further centrifuged at 8,000 g for 10 min, and the final pellet was suspended in 0.5 ml of isolation buffer and kept on ice. Total protein concentration was determined by the Bradford method with BSA as a standard (4). All isolation procedures were conducted at 4°C; subsequent isolated mitochondrial experiments were conducted at 25°C.
Western blotting.
Mitochondrial protein (50 μg) was solubilized in Laemmli buffer and resolved by SDS-PAGE as described by Laemmli (18) and transferred onto polyvinylidene difluoride membranes using Transblot System (Bio-Rad, Carlsbad, CA) in 50 mM tricine and 7.5 mM imidazole transfer buffer. Membranes were blocked with 10% nonfat dry milk in Tris-buffered saline-TBSt (25 mM Tris·HCl at pH 7.5, 50 mM NaCl, and 0.1% Tween 20) by incubating for 1 h followed by incubation in anti-NDUFA9 antibody (9, 11, 12) (Invitrogen, Carlsbad, CA) solution overnight at 4°C. After three washes with TBSt the membrane was incubated with an appropriate secondary antibody conjugated to alkaline phosphatase for 3 h. After five washes with TBSt the membrane was incubated in alkaline phosphatase detection kit (Invitrogen). The membrane was then stripped using stripping buffer (100 mM 2-mercaptoethanol, 2% SDS, 62.5 mM Tris·HCl pH 6.7) at 50°C, followed by three washes in TBSt. Equivalence of protein loading was confirmed with an antibody against cytochrome c oxidase (complex IV) subunit 1 (Invitrogen) since complex IV is relatively more resistant to IR injury than complex I (12). Densitometry was performed with Image J software.
Mitochondrial respiration.
Mitochondria (0.25 mg protein/ml) were suspended in 500 μl respiration buffer containing (in mM): 130 KCl, 5 K2HPO4, 20 MOPS, and 2.5 EGTA, with 1 μM Na4P2O7, and 0.1% BSA, pH 7.15 adjusted with KOH (21). Chamber [O2] was measured polargraphically with a Clark-type O2 electrode (model 1302; Strathkelvin Instruments, Glasgow, Scotland) in a water-jacketed 500 μl chamber (model MT200A, Strathkelvin Instruments) equipped with a Teflon-coated magnetic stirring bar and monitored by an oxygen meter (model 782, Strathkelvin Instruments). The O2 electrode was calibrated with air-saturated water (Po2 150 mmHg) and Na2SO3 solution to achieve near-zero Po2. Chamber [O2] in μM was monitored for 5 min or until it approached zero. Respiratory control index (RCI) was determined as the ratio of the maximum [O2] decrease rate (state 3) after addition of pyruvate/malate (10 mM each final concentration) and 60 s later adenosine diphosphate (ADP, 250 μM final concentration) to the rate during state 4 respiration after complete phosphorylation of ADP to ATP. Data were stored online on a computer with the manufacturer's software. Results from three replicates per heart were averaged.
Mitochondrial calcium retention capacity.
Mitochondria were suspended in respiration buffer (composition see above) in the presence of 10 mM succinate and 50 nM of the fluorescent dye rhodamine 123 (50 nM, Invitrogen) to measure mitochondrial membrane potential (ΔΨm) in a cuvette-based spectrophotometer (model QM-8; Photon Technology International, Birmingham, NJ) (1). ΔΨm was monitored by rhodamine fluorescence at 503 nm excitation and 527 nm emission. After stabilization, pulses of CaCl2 (to yield 25 μM increases in calcium) were added every 60 s until ΔΨm depolarized as indicated by a final plateau phase. Following calcium-induced depolarization, carbonyl cyanide-m-chlorophenylhydrazenone (4 μM), a mitochondrial uncoupler, was added to maximally depolarize the membrane. Duration until calcium-induced depolarization was measured as an indicator of mitochondrial calcium retention capacity. Results from three replicates per heart were averaged.
Statistical analysis.
All data are expressed as means ± SE. Composite baseline data were compared by one-way analysis of variance (SigmaStat 3.5; Systat Software, San Jose, CA); all other data were compared by two-way analysis of variance with strain and IR as two independent factors. If F values were significant, Student-Newman-Keuls post hoc tests were conducted. Tests were considered statistically significant at P < 0.05 (2-tailed).
RESULTS
Baseline data.
In none of the three strains differences were found between the control and the IR groups at baseline. BN rats and their hearts were smaller than SS6BN and SS, as were their heart-to-body weight ratios (Table 1). Baseline diastolic LVP and HR were identical among all three strains. Other functional baseline data were similar only between SS6BN and SS; both strains were different compared with BN (Table 1).
Table 1.
Baseline data
| BN | SS6BN | SS | |
|---|---|---|---|
| Heart weight, g | 1.00 ± 0.02 | 1.41 ± 0.04* | 1.46 ± 0.03* |
| Body weight, g | 230 ± 7 | 304 ± 4* | 313 ± 4* |
| Heart-to-body weight ratio, % | 0.43 ± 0.00 | 0.47 ± 0.01* | 0.47 ± 0.01* |
| Systolic LVP, mmHg | 79.8 ± 1.1 | 92.1 ± 2.1* | 89.1 ± 2.0* |
| Diastolic LVP, mmHg | 9.8 ± 0.2 | 9.8 ± 0.1 | 10.1 ± 0.2 |
| Developed LVP, mmHg | 70.0 ± 1.1 | 82.4 ± 2.2* | 79.1 ± 2.0* |
| dLVP/dtmax, mmHg/s | 1,473 ± 18 | 1,552 ± 24* | 1,582 ± 31* |
| dLVP/dtmin, mmHg/s | −1,055 ± 7 | −1,294 ± 18* | −1,253 ± 15* |
| HR, beats/min | 272 ± 9 | 291 ± 12 | 280 ± 17 |
| Coronary flow, ml·min−1·g−1 | 12.2 ± 0.1 | 9.9 ± 0.1* | 9.7 ± 0.2* |
Shown are baseline data for the 3 rat strains Brown Norway (BN), Dahl salt-sensitive (SS), and consomic SS6BN (n = 30 per strain). Systolic, diastolic, and developed, i.e., systolic–diastolic, left ventricular pressure (LVP), its positive and negative first derivatives dLVP/dtmax and dLVP/dtmin as indexes of contractility and relaxation, respectively, spontaneous heart rate (HR) in beats/min, and coronary flow (in ml per min per g heart wt) were measured at baseline, i.e., after 20 min stabilization. None of the baseline parameters showed a difference between the control and its respective ischemia-reperfusion (IR) group in any of the 3 strains (data not shown). All values are means ± SE;
vs. BN.
Myocardial and coronary function.
Functional parameters at the end of the experiments are displayed in Fig. 2. Systolic LVP (Fig. 2A) was not significantly different among the strains in control or IR hearts and not different between control and IR hearts in any strain; the nonsignificance in the upward trend from BN- to SS-IR hearts may be secondary to a lack of power. In contrast, diastolic LVP (Fig. 2B) was not only increased in all three strains after IR, but significantly more so in SS than in BN and SS6BN hearts. There was no difference in developed LVP (Fig. 2C) among the strains in control hearts, but IR led to significantly lower developed LVP in all three strains, in SS more than BN or SS6BN hearts. HR (Fig. 2D) was not different among the strains in control or IR hearts and not different between control and IR hearts in any strain. Both contractility (Fig. 2E) and relaxation (Fig. 2F) were decreased by IR in all three strains, but markedly more so in SS compared with BN and SS6BN hearts. Coronary flow (Fig. 2G), at constant perfusion pressure inversely proportional to coronary resistance, was also decreased by IR in all three strains, but more so in SS compared with BN and SS6BN hearts.
Fig. 2.

Functional data expressed as % of their baseline (bl) values (unless otherwise indicated) and ventricular infarct size in % after 35 min perfusion, 30 min ischemia, and 120 min reperfusion (IR, right half of each panel), or 185 min of perfusion (Con, left half of each panel) following stabilization. A: systolic left ventricular pressures (LVP); B: diastolic LVP (in mmHg); C: developed, i.e., systolic-diastolic, LVP; D: heart rate (HR); E and F: dLVP/dtmax and dLVP/dtmin, as indexes of contractility and relaxation, respectively; G: coronary flow; H: ventricular infarct size in the 3 strains BN (white), SS (black), and SS6BN (striped). All values are means ± SE; *vs. Con, †vs. BN, ‡vs. SS6BN; n = 6 per group.
Ventricular infarct size.
There was no significant difference in MI among the three strains in control hearts after 205 min perfusion (Fig. 2H). The fact that even nonischemic hearts suffered a certain degree of dysfunction and MI, likely caused by a combination of surgery and extended Krebs perfusion, underlines the importance of comparing IR data to appropriately time-matched nonischemic controls to be able to distinguish treatment from time effects. IR increased MI in all three strains, but significantly more so in SS than in BN or SS6BN hearts.
Mitochondrial complex I.
Western blots of complex I subunit NDUFA9 showed significantly lower protein levels in the SS strain following IR (Fig. 3). There was no difference among the strains in control hearts.
Fig. 3.
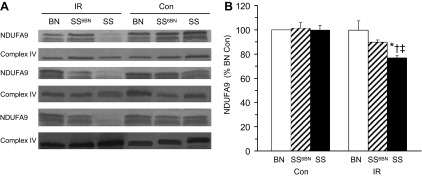
Original Western blot data for complex I subunit NDUFA9 and complex IV subunit 1 as a loading control are shown in A. The summary of NDUFA9 levels in % BN Con is shown in B for BN (white), SS (black), and consomic SS6BN rats (striped) after 35 min perfusion, 30 min ischemia, and 120 min reperfusion (IR, right half of B) or 185 min of perfusion (Con, left half of B) following stabilization. Complex IV subunit 1 was not different among the 6 groups (data not shown). All values are means ± SE; *vs. Con, †vs. BN, ‡vs. SS6BN; n = 3 per group.
Mitochondrial respiration.
RCIs were not different among the three strains in control hearts. Following IR, SS mitochondria had a lower RCI than SS6BN and BN mitochondria, indicating that coupling of oxidative phosphorylation was better preserved in BN and SS6BN than in SS mitochondria (Fig. 4).
Fig. 4.

Mitochondrial respiration and respiratory control index (RCI). A–C: representative traces of polarographically measured chamber [O2] concentrations (in μM) of mitochondria isolated from a nonischemic control (Con) vs. mitochondria isolated after IR from BN (A), SS6BN (B), and SS hearts (C). Following 1 min stabilization, the complex I substrates pyruvate and malate (10 mM each) were added to initiate state 2 respiration. State 3 respiration was determined after addition of adenosine diphosphate (ADP, 250 μM) 60 s later, and state 4 respiration after complete ADP phosphorylation to ATP. RCI was calculated as the state 3-to-state 4 ratio. Results from 3 replicates per heart were averaged. D: summary of RCIs in Con (left) and IR mitochondria (right) from BN (white), SS (black), and the consomic SS6BN strain (striped). All values are means ± SE; *vs. Con, †vs. BN, ‡vs. SS6BN; n = 6 per group.
Mitochondrial calcium retention capacity.
Experiments in isolated mitochondria following IR revealed a significant difference among the strains in their ability to maintain ΔΨm during calcium-induced stress (Fig. 5). Whereas there were no differences among the strains in controls, the duration to depolarization, i.e., calcium-handling capacity, in SS was lower than in BN and SS6BN mitochondria after IR and lower than in SS controls. In contrast, there was no difference between control and IR in BN or SS6BN mitochondria.
Fig. 5.

Mitochondrial calcium retention capacity. A: representative traces of rhodamine 123 fluorescence (in arbitrary fluorescence units, afu), indicative of membrane potential (ΔΨm), in mitochondria isolated from BN, SS6BN, and SS hearts following IR in the presence of 10 mM succinate. Arrows indicate when CaCl2 pulses (to yield 25 μM increases) were added every 60 s until ΔΨm depolarization. After CaCl2-induced depolarization, 4 μM carbonyl cyanide-m-chlorophenylhydrazenone (CCCP), a mitochondrial uncoupler, was added for maximal depolarization. B: summary of the time until depolarization (in s) in Con (left) and IR mitochondria (right) from BN (white), SS (black), and the consomic SS6BN strain (striped). All values are means ± SE; *vs. Con, †vs. BN, ‡vs. SS6BN; n = 6 per group.
DISCUSSION
Our study reveals novel findings at several different levels: 1) genome-dependent differences in myocardial function and viability are linked to mitochondrial preservation in this consomic rat model; 2) these differences become most visible after IR, while baseline phenotypical differences appear to play no role; and 3) myocardial and mitochondrial protection are linked to genetic information on BN chromosome 6.
Genetic predisposition may be an important confounding factor when one is trying to utilize cardioprotective strategies in clinical practice. For basic research in this area to be successful it is critical to sufficiently narrow the genetic differences between study groups, but to maintain adequately large and measurable phenotypical differences. Baker et al. (3) showed strain-dependent cardioprotection in BN rats as evidenced by a reduction in infarct size, decreased postischemic enzyme leakage, and increased function compared with SS and laid the foundation for further genetic studies of myocardial IR tolerance. While 20% of the traits differ in any one strain, the BN has the highest number of differences because it is the most genetically distinct laboratory rat strain (30). There are 281 traits related to diseases of the heart, lung, and blood that have been studied in BN rats compared with the same traits measured across multiple genetic backgrounds, both sexes, and differing environments (17).
Through marker-assisted selection the genetic differences between BN and SS could be made even smaller (10) in so-called “consomic” animals where all chromosomes are derived from one strain except for one pair that is introgressed from a different strain. Using a unique consomic rat panel at the Medical College of Wisconsin, Kwitek et al. (17) have shown that a significant degree of cardioprotection was conferred when BN chromosome 6 was introgressed into SS. Cardioprotection was evidenced by a decrease in ischemic diastolic contracture and ventricular infarct size, while all other parameters remained inconclusive (http://pga.mcw.edu). More recently, myonecrosis and left ventricular contractile dysfunction were found to be ameliorated in BN and SS6BN compared with SS hearts in a model of cardioplegic and deep hypothermic circulatory arrest (5).
Despite these efforts, we still have very limited knowledge about the intracellular, let alone genetic mechanisms responsible for the observed cardioprotective phenotype in BN and SS6BN vs. SS rats. Although Shi et al. (28) have suggested that increased association of heat shock protein 90 with endothelial nitric oxide synthase (eNOS) plays an important role in increased IR tolerance of BN vs SS, they did not include SS6BN in their study nor did they examine mitochondrial function. Furthermore, the gene coding for eNOS is located on rat chromosome 4 and therefore cannot directly explain the cardioprotected phenotype associated with BN chromosome 6.
It is well known that mitochondria play an important role in IR injury and that mitochondria are triggers and effectors of cardioprotection (7, 8, 22). However, to the best of our knowledge, this is the first study to show that cardioprotection in BN and SS6BN rats is closely linked to mitochondrial protection and preservation. Our results confirm, but more importantly extend, existing findings that SS6BN hearts are as protected as BN hearts compared with SS. This is evidenced not only by decreased diastolic contracture and infarct size, but also by increased contractility, relaxation, and coronary flow. Moreover, key mitochondrial functions such as coupling of oxidative phosphorylation and resistance against calcium-induced depolarization are significantly less preserved in SS compared with BN and SS6BN hearts. Activity of complex I has been shown to be decreased as a result of IR injury (26). In addition, the Western blot analyses indicate that complex I integrity as assessed by subunit NDUFA9 protein levels is diminished as well in SS compared with BN and SS6BN hearts after IR. Impaired intracellular calcium handling, impaired mitochondrial tolerance to intracellular calcium overload, and subsequently impaired oxidative phosphorylation may be the common denominator of these findings.
Interestingly, all of these findings are apparent only after IR, indicating that cardiac injury exposes the genetic differences. No significant differences in mitochondrial function between BN, SS6BN, and SS were observed under control conditions. Indeed, none of the obvious phenotypical baseline differences between BN and SS such as heart and body weight, systolic and developed pressure, contractility and relaxation, or coronary flow can account for the decreased IR injury because SS6BN share these baseline traits with SS but are as protected as BN, so that there is no association between baseline differences and outcome. The fact that there are only differences in mitochondrial function (RCI and calcium retention capacity) between SS and BN/SS6BN hearts following IR, but no difference between control and IR hearts of BN or SS6BN rats is further evidence that preservation of mitochondrial function is necessary but not sufficient by itself to protect against IR injury. The same is true for the relation of preserved coronary flow and myocardial function (15).
Given the high prevalence of CAD and MI (25), the genome-dependent association of mitochondrial preservation and cardioprotection makes mitochondria a promising target for pharmacogenomic interventions (8). If ischemia occurs, more resistant individuals may suffer a smaller MI given the same ischemic insult, or, for the same outcome, they may tolerate ischemia longer (16). Only by unraveling the genetic basis of endogenous and exogenous cardioprotection will we ultimately be able to develop genetic tests to identify and distinguish patients at high risk for adverse cardiac events from those at low risk and to be able to adapt treatment to the individual's personal risk profile.
Some limitations of our study need to be acknowledged. The use of a blood-free high-Po2 perfusate causes a greater use of coronary flow reserve and in animals with less radical scavenging capacity may contribute to some of the observed differences among species and strains. Even though in vivo experiments would provide a more physiological insight into the interplay of coronary, endothelial, and myocyte function, they lack the distinct advantage of isolated heart models of having fewer confounding factors such as neural, humoral, hematologic, and hemodynamic influences that would alter some of the outcome differences. Although several methodological differences may be responsible for the differences observed between our and other studies (3, 17), a key determinant of the degree of IR injury remains the chosen duration of ischemia (15), especially after the onset of diastolic contracture secondary to cytosolic calcium overload (2) and mitochondrial deterioration evidenced by calcium overload (24) and increased superoxide production (15). The duration of 30 min (20) was chosen over a shorter duration (3, 17) to produce an IR injury large enough to be potentially attenuated by cardioprotective strategies not only in IR-susceptible strains, i.e., SS, but also in -resistant strains, i.e., BN and SS6BN, to elucidate further strain-dependent cardioprotective mechanisms. Despite their clear association with BN chromosome 6, it cannot be ruled out that the observed differences are secondary to other traits mapping to chromosome 6, or that a BN allele on chromosome 6 interacts with other relevant SS genes; this, however, can be addressed in the future by using the SS6BN as a starting point for the generation of congenic strains with a chromosomal region small enough for positional cloning. Other potential targets on chromosome 6 known to affect mitochondrial function including the sodium calcium exchanger, VIP, HIF1alpha, G3P dehydrogenase, VDAC, TOM, TIM, AKT, and PKC epsilon may be worthwhile investigating in this context. The use of another available unprotected consomic strain, e.g., SS19BN, but with similar baseline characteristics as BN (http://rgd.mcw.edu) would not necessarily add to the delineation between baseline characteristics and mitochondrial and myocardial protection. A reverse consomic strain, i.e., BN6SS, however, could confirm the role of BN chromosome 6 for cardioprotection by reestablishing an unprotected SS phenotype in a BN background. Similarly, an evaluation of the other IR-resistant consomic, SS2BN, could provide an additional opportunity to clarify a possible causative link between chromosome 6 and mitochondrial IR sensitivity in this model.
In summary, we have successfully used an established and translationally relevant genetic rat model to demonstrate for the first time that SS6BN are as resistant as BN hearts in both mitochondrial and myocardial function and viability compared with SS hearts. Not only do our results link myocardial and mitochondrial protection in a genetic model but they also indicate that genetic information on rat chromosome 6 is, directly or indirectly, responsible for mitochondrial preservation and IR tolerance. Improved preservation of postischemic mitochondrial structure and function may become key in pharmacogenomic treatment of patients at risk for CAD and MI.
GRANTS
This work was supported in part by Department of Veterans Affairs Grant CARA-026-10F, National Heart, Lung, and Blood Institute Grant 5R01 HL-098490-03, and the inaugural Roizen Anesthesia Research Foundation New Investigator Grant from the Society of Cardiovascular Anesthesiologists (all to M. L. Riess).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: R.N., A.K.G., and M.L.R. performed experiments; R.N., A.K.G., and M.L.R. analyzed data; R.N., A.K.G., and M.L.R. interpreted results of experiments; R.N. and M.L.R. drafted manuscript; R.N., A.K.G., J.R.K., D.F.S., J.L., and M.L.R. edited and revised manuscript; R.N., A.K.G., J.R.K., D.F.S., J.L., and M.L.R. approved final version of manuscript; A.K.G. and M.L.R. prepared figures; J.R.K., D.F.S., J.L., and M.L.R. conception and design of research.
ACKNOWLEDGMENTS
The authors thank Mohammed Aldakkak, Amadou K. S. Camara, Qunli Cheng, James S. Heisner (all Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, WI), and Sushrut V. Shidham (Medical College of Wisconsin Affiliated Hospitals, Milwaukee, WI) for valuable contributions to this study.
REFERENCES
- 1.Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol Res 64: 381–392, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.An JZ, Varadarajan SG, Novalija E, Stowe DF. Ischemic and anesthetic preconditioning reduces cytosolic [Ca2+] and improves Ca2+ responses in intact hearts. Am J Physiol Heart Circ Physiol 281: H1508–H1523, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Baker JE, Konorev EA, Gross GJ, Chilian WM, Jacob HJ. Resistance to myocardial ischemia in five rat strains: is there a genetic component of cardioprotection? Am J Physiol Heart Circ Physiol 278: H1395–H1400, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254, 1976 [DOI] [PubMed] [Google Scholar]
- 5.Brar S, Ma Q, Dobra A, Zhang Z, Mackensen B, Podgoreanu MV. Susceptibility to global myocardial ischemia-reperfusion injury in the rat is genetically determined. Anesthesia Analgesia 112: SCA46, 2011 [Google Scholar]
- 6.Broeckel U, Hengstenberg C, Mayer B, Holmer S, Martin LJ, Comuzzie AG, Blangero J, Nurnberg P, Reis A, Riegger GA, Jacob HJ, Schunkert H. A comprehensive linkage analysis for myocardial infarction and its related risk factors. Nat Genet 30: 210–214, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 287: C817–C833, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Camara AK, Lesnefsky EJ, Stowe DF. Potential therapeutic benefits of strategies directed to mitochondria. Antioxid Redox Signal 13: 279–347, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carroll J, Shannon RJ, Fearnley IM, Walker JE, Hirst J. Definition of the nuclear encoded protein composition of bovine heart mitochondrial complex I. Identification of two new subunits. J Biol Chem 277: 50311–50317, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Cowley AW, Jr, Liang M, Roman RJ, Greene AS, Jacob HJ. Consomic rat model systems for physiological genomics. Acta Physiol Scand 181: 585–592, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Fearnley IM, Walker JE. Conservation of sequences of subunits of mitochondrial complex I and their relationships with other proteins. Biochim Biophys Acta 1140: 105–134, 1992 [DOI] [PubMed] [Google Scholar]
- 12.Gadicherla AK, Stowe DF, Antholine WE, Yang M, Camara AK. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim Biophys Acta 1817: 419–429, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaziano TA, Bitton A, Anand S, Abrahams-Gessel S, Murphy A. Growing epidemic of coronary heart disease in low- and middle-income countries. Curr Probl Cardiol 35: 72–115, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jonassen AK, Sack MN, Mjos OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ Res 89: 1191–1198, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Kevin LG, Camara AK, Riess ML, Novalija E, Stowe DF. Ischemic preconditioning alters real-time measure of O2 radicals in intact hearts with ischemia and reperfusion. Am J Physiol Heart Circ Physiol 284: H566–H574, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Kevin LG, Katz P, Camara AK, Novalija E, Riess ML, Stowe DF. Anesthetic preconditioning: effects on latency to ischemic injury in isolated hearts. Anesthesiology 99: 385–391, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Kwitek AE, Jacob HJ, Baker JE, Dwinell MR, Forster HV, Greene AS, Kunert MP, Lombard JH, Mattson DL, Pritchard KA, Jr, Roman RJ, Tonellato PJ, Cowley AW., Jr. BN phenome: detailed characterization of the cardiovascular, renal, and pulmonary systems of the sequenced rat. Physiol Genomics 25: 303–313, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685, 1970 [DOI] [PubMed] [Google Scholar]
- 19.Murphy E, Steenbergen C. Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovasc Res 75: 478–486, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Nabbi R, Kersten J, Stowe D, Lazar J, Aldakkak M, Riess M. Characterizing the cardioprotective phenotype of Brown Norway rats: importance of optimal ischemia duration. FASEB J 24: 1037.–6., 2010 [Google Scholar]
- 21.Riess ML, Eells JT, Kevin LG, Camara AKS, Henry MM, Stowe DF. Attenuation of mitochondrial respiration by sevoflurane in isolated cardiac mitochondria is mediated in part by reactive oxygen species. Anesthesiology 100: 498–505, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Riess ML, Novalija E, Camara AK, Eells JT, Chen Q, Stowe DF. Preconditioning with sevoflurane reduces changes in nicotinamide adenine dinucleotide during ischemia-reperfusion in isolated hearts: reversal by 5-hydroxydecanoic acid. Anesthesiology 98: 387–395, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Riess ML, Rhodes SS, Stowe DF, Aldakkak M, Camara AK. Comparison of cumulative planimetry versus manual dissection to assess experimental infarct size in isolated hearts. J Pharmacol Toxicol Methods 60: 275–280, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riess ML, Camara AKS, Novalija E, Chen Q, Rhodes SS, Stowe DF. Anesthetic preconditioning attenuates mitochondrial Ca2+ overload during ischemia in guinea pig intact hearts: reversal by 5-hydroxydecanoic acid. Anesth Analg 95: 1540–1546, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation 125: e2-e220, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sadek HA, Szweda PA, Szweda LI. Modulation of mitochondrial complex I activity by reversible Ca2+ and NADH mediated superoxide anion dependent inhibition. Biochemistry (Mosc) 43: 8494–8502, 2004 [DOI] [PubMed] [Google Scholar]
- 27.Shen YT, Vatner SF. Differences in myocardial stunning following coronary artery occlusion in conscious dogs, pigs, and baboons. Am J Physiol Heart Circ Physiol 270: H1312–H1322, 1996 [DOI] [PubMed] [Google Scholar]
- 28.Shi Y, Hutchins W, Ogawa H, Chang CC, Pritchard KA, Jr, Zhang C, Khampang P, Lazar J, Jacob HJ, Rafiee P, Baker JE. Increased resistance to myocardial ischemia in the Brown Norway vs Dahl S rat: role of nitric oxide synthase and Hsp90. J Mol Cell Cardiol 38: 625–635, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Shidham SV, Nabbi R, Camara AKS, Riess ML. Development of automated infarct size measurement in TTC stained rat isolated hearts after global ischemia/reperfusion. FASEB J 25: 1130.–2., 2011 [Google Scholar]
- 30.Thomas MA, Chen CF, Jensen-Seaman MI, Tonellato PJ, Twigger SN. Phylogenetics of rat inbred strains. Mamm Genome 14: 61–64, 2003 [DOI] [PubMed] [Google Scholar]