

Abstract
Autophagy is an essential eukaryotic pathway required for cellular homeostasis. Numerous key autophagy effectors and regulators have been identified, but the mechanism by which they carry out their function in autophagy is not fully understood. Our rigorous bioinformatic analysis shows that the majority of key human autophagy proteins include intrinsically disordered regions (IDRs), which are sequences lacking stable secondary and tertiary structure; suggesting that IDRs play an important, yet hitherto uninvestigated, role in autophagy. Available crystal structures corroborate the absence of structure in some of these predicted IDRs. Regions of orthologs equivalent to the IDRs predicted in the human autophagy proteins are poorly conserved, indicating that these regions may have diverse functions in different homologs. We also show that IDRs predicted in human proteins contain several regions predicted to facilitate protein-protein interactions, and delineate the network of proteins that interact with each predicted IDR-containing autophagy protein, suggesting that many of these interactions may involve IDRs. Lastly, we experimentally show that a BCL2 homology 3 domain (BH3D), within the key autophagy effector BECN1 is an IDR. This BH3D undergoes a dramatic conformational change from coil to α-helix upon binding to BCL2s, with the C-terminal half of this BH3D constituting a binding motif, which serves to anchor the interaction of the BH3D to BCL2s. The information presented here will help inform future in-depth investigations of the biological role and mechanism of IDRs in autophagy proteins.
Keywords: autophagy, natively unstructured proteins, protein interactions, sequence analysis, BECN1, BH3 domain, BCL2
Introduction
Autophagy is a catabolic cellular pathway responsible for sequestering intracellular macromolecular assemblies, such as obsolete, damaged or harmful proteins, organelles and pathogens, into multi-layered vesicles called autophagosomes, followed by lysosomal degradation of the sequestered molecules and recycling of nutrients.1, 2 Hence, autophagy is essential for cellular homeostasis as well as survival during cellular and organismal stress. The disruption or dysfunction of autophagy is implicated in numerous diseases such as neurodegenerative disorders, muscular diseases, cardiomyopathy, cancer and infectious diseases.3–5
Proteins that execute the process of autophagy are conserved in all eukaryotes and many of these conserved autophagy-related effectors are called ATG proteins.2, 6 Known autophagy effectors (Table S1) may be divided into four groups according to their function and association with other effectors, and at least three of these four groups function via formation of multi-protein complexes.1, 2, 7 The four groups implicated in different functional stages of autophagy are: (i) the ULK1/ATG1 kinase signaling complex that initiates autophagy; (ii) the class III phosphatidylinositol-3-kinase (PIK3C3) – BECN1/Beclin 1/ATG6 complex implicated in autophagosome nucleation; (iii) the ubiquitin-like ATG12 and ATG8 conjugation complexes implicated in autophagosome expansion; and (iv) proteins involved in autophagosome maturation and lysosomal fusion. In addition to these autophagy-related effectors, two other groups of proteins are important for autophagy: (v) proteins that select and target cytoplasmic macromolecular assemblies to the autophagosome and (vi) numerous autophagy regulators that modulate levels of autophagy within a cell via direct interactions with the different autophagy-related effectors included in the previous groups. The biological role of many of these proteins is known and preliminary sequence analyses has helped delineate their domain architecture; but a detailed, mechanistic understanding of the function of these proteins in autophagy is only now beginning to emerge.
Intrinsically disordered regions (IDRs), also called natively unstructured regions, are protein regions that have negligible folded tertiary structure or stable secondary structure elements like α-helices and β-sheets.8 The amino acid composition of these domains dictates an inherent structural flexibility, preventing the formation of a well-ordered, central hydrophobic core for packing. Thus, these regions appear to be exceptions to canonical paradigms that postulate that the three-dimensional structure of a protein dictates its function;9 and that three-dimensional structure is conserved more strongly than primary sequence. The importance of IDRs in cellular processes is often overlooked, as biological roles and mechanisms of most IDRs are poorly understood.
Here we perform a rigorous primary structure analysis using multiple programs to predict consensus IDRs in key human autophagy effectors and regulators, and find that the majority of proteins with established roles in autophagy contain potential IDRs. Available crystal structures provide experimental proof for the lack of structure in a few of these predicted IDRs. Further, we find that regions analogous to the IDRs predicted in the human proteins are substantially less well conserved in autophagy effector homologs from diverse eukaryotes, compared to regions that lie outside the predicted IDRs. An investigation of the potential functions of the predicted IDRs demonstrates that many of them may serve as interaction sites for other proteins and may adopt more ordered conformations upon binding to other proteins. For each predicted IDR-containing protein, we also mine available biological databases to identify potential protein partners that might interact via the IDRs. Lastly, we show that a functional domain responsible for protein interactions, the BCL2-homology 3 domain (BH3D) within the key autophagy effector BECN1, is part of the BECN1 IDR. The BH3D contains a region predicted to stabilize structure upon binding, and this region constitutes the most critical component for the interaction of BECN1 with BCL2 homologs.
The results reported here provide important information needed to inform future cellular, biochemical and structural research that will be needed to completely understand the function and mechanism of IDRs in autophagy. A better understanding of the role and mechanism of these IDRs will not only improve our understanding of autophagy and other related cellular pathways, but also help identify new and unique therapeutic targets to manipulate cellular levels of autophagy for health benefits.
Materials and Methods
Identifying IDRs in autophagy proteins
The amino acid sequences of 59 key autophagy-related effectors and regulators were downloaded from the NCBI human genome RefSeq Protein database, cross-checked with the autophagy database (http://autophagy.info/autophagy/) and analyzed using the four different programs below to predict IDRs based on different properties. The disorder predictor, DISOPRED (http://bioinf.cs.ucl.ac.uk/disopred) calculates the probability of a residue being disordered based on similarity to regions with missing electron density in known crystal structures.10 The protein disorder prediction system, PrDOS (http://prdos.hgc.jp), provides a disorder probability for each residue based on a combination of the conservation of intrinsic disorder in protein families and on local amino acid sequence information, such as a higher frequency of hydrophilic or charged residues, or low sequence complexity.11 The various short and long 2B algorithm, VSL2B algorithm, in the Predictors of Natural Disordered Regions, PONDR, program suite (http://www.dabi.temple.edu/disprot/predictor.php),12, 13 optimized for short (<=30 residues) and long (>30 residues) disordered regions, estimates a disorder probability by assessing amino acid frequencies, sequence complexity, ratio of net charge / hydrophobicity from averaged PSI-BLAST14 profiles based on a training data set comprising 1,335 non-redundant protein sequences. The intrinsically unstructured protein predictor, IUPred (http://iupred.enzim.hu/), calculates a disorder probability by estimating pairwise energy content from amino acid composition, to assess formation of favorable interactions and identify regions that do not adopt a stable structure because of the lack of such interactions.15 In each program, residues with a disorder probability greater than 0.5, are marked as disordered. Further, contiguous stretches of at least 15 disordered residues in DISOPRED (Table S2; only regions ≥ 25 residues are listed) and 30 disordered residues in IUPred, are listed as potential IDRs by these programs (Table S2). For the PrDOS and PONDR analysis (Table S2), we listed contiguous stretches of at least 25 disordered residues predicted by each program as potential IDRs.
Identification of IDR-containing autophagy protein orthologs
Sequences of the closest identifiable homologs from Drosophila melanogaster, Arabidopsis thaliana and Saccharomyces cerevisiae were identified by a combination of methods: from the autophagy database (http://autophagy.info/autophagy/); GeneCards (http://www.genecards.org/); by BLASTP searches of Genomic RefSeq Protein databases (http://blast.ncbi.nlm.nih.gov/) for each organism; and in selected cases, by the examination of relevant literature.
Sequence alignment of IDR-containing autophagy protein orthologs
Multiple sequence alignment of each set of orthologs was done using CLUSTALW16 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The overall % identity and similarity, reported in Table I for each set of orthologs, was calculated from each alignment, simply as the ratio of the total number of invariant residues, or conservative substitutions, to the length of the shortest homolog. The % identity or similarity between regions analogous to each human IDR was calculated as the ratio of the number of invariant or conserved residues to the length of the human IDR regions. Gaps and insertions in the alignment, if any, were not penalized for either calculation in the calculation of % identity or similarity.
Table I.
Consensus human IDRs and their conservation in orthologs.
| Protein | Homolog length (residues) | Full protein (%) | Human consensus IDRs | IDR (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Human | Insect | Plant | Yeast | Identity | Similarity | Identity | Similarity | ||
| Autophagy initiation signaling complex | |||||||||
| ULK1 | 1050 | 855 | 733 | 897 | 10.8 | 32.3 | 291–833 | 0.2 | 7.9 |
| ULK2 | 1036 | 855 | 733 | 897 | 11.6 | 31.9 | 285–354; 386–819 |
0; 0.7 |
4.3; 8.8 |
| ATG13 | 517 | 523 | 603 | 738 | 3.1 | 21.7 | 302–363; 401–441 |
0; 0 |
14.5; 19.5 |
| RB1CC1 | 1594 | 1357 | --- | 417 | 6.5nP | 40.3nP | 224–294; 630–756; 1271–1324; 1350–1449 |
0nP; 0nPY; 0nPY; 0nPY |
18.3nP; 0nPY; 0nPY; 0nPY |
| Autophagosome nucleation complex | |||||||||
| PIK3C3 | 887 | 949 | 814 | 875 | 24.2 | 56.3 | 414–468 | 1.8 | 7.3 |
| PIK3R4 | 1358 | 1342 | 1494 | 1454 | 12.6 | 39.5 | 834–978 | 0 | 12.4 |
| BECN1 | 450 | 422 | 517 | 557 | 12.1 | 42.2 | 42–115 | 0 | 20.3 |
| ATG14 | 492 | 503 | --- | 344 | 6.7nP | 38.1nP | 442–472 | 0 | 22.6nP |
| AMBRA1 | 1298 | --- | 793 | 1201 | 4.9nIY | 26.4nIY | 249–282; 319–497; 515–564; 584–692; 739–805; 1093–1148; 1187–1264 |
0nIPY; 3.9nI; 0nI; 0nI; 0nI; 1.8nI; 0nIP |
0nIPY; 19.0nI; 18.0nI; 10.1nI; 20.9nI; 3.6nI; 0nIP |
| UVRAG | 699 | 696 | --- | 439 | 4.1nP | 27.1nP | 1–30; 267–299; 468–619 |
0nPY; 6.1nP; 1.3nP |
0nPY; 27.3nP; 11.2nP |
| VMP1 | 406 | 530 | 416 | -- | 27.3nY | 54.7nY | 1–36 | 5.6nY | 25.0nY |
| Autophagosome expansion complex | |||||||||
| ATG16L1 | 607 | 604 | 509 | 150 | 5.3 | 30.7 | 59–84; 212–262 |
0; 0nY |
11.5; 0nY |
| ATG12 | 140 | 111 | 96 | 186 | 16.7 | 56.3 | 1–49 | 0 | 6.1 |
| ATG3 | 314 | 330 | 313 | 310 | 21.9 | 49.0 | 129–169 | 2.4 | 36.6 |
| ATG4D | 474 | 411 | 477 | 494 | 9.2 | 30.2 | 1–61 | 0 | 3.3 |
| Autophagosome maturation complex | |||||||||
| ATG9A | 839 | 845 | 866 | 997 | 7.5 | 31.2 | 606–700; 715–839 |
1.0; 0 |
7.3; 9.6 |
| WIPI1 | 446 | 377 | 312 | 500 | 16.0 | 47.8 | 362–410 | 0 | 4.1 |
| ATG2A | 1938 | 1906 | 1892 | 1592 | 3.3 | 23.7 | 108–153; 337–424; 748–788; 1241–1276; 1318–1362; 1438–1477; 1606–1658 |
0; 0; 0; 0; 0; 0; 0nY |
10.9; 14.8; 7.3; 8.3; 15.6; 17.5; 0nY |
| SNX18 | 628 | 565 | -- | -- | 31.3 | 67.4 | 63–224 | 12.4 | 41.4 |
| Autophagy targeting proteins | ||| | ||||||||
| NBR1 | 966 | -- | 704 | -- | 18.0nIY | 54.5nIY | 80–145; 485–531; 598–643; 661–881 |
12.1nIY; 0nIPY; 2.2nIY; 7.7nIY |
48.5 IY; 0nIPY; 17.4nIY; 29.9nIY |
| SQSTM1 | 440 | 599 | -- | -- | 24.3nPY | 60.7nPY | 249–390 | 23.2nPY | 66.9nPY |
| SMURF1 | 757 | 1061 | --- | --- | 58.1nPY | 82.4nPY | 158–234; 268–307; 343–370 |
27.3nPY; 30.0nPY; 14.3nPY |
61.0nPY; 70.0nPY; 57.1nPY |
| Autophagy Regulation | || | ||||||||
| BNIP3L | 219 | 201 | -- | -- | 16.9nPY | 62.7nPY | 1–32; 77–129 |
9.4nPY; 13.2nPY |
53.1nPY; 66.0nPY |
| BNIP3 | 194 | 201 | -- | -- | 24.7nPY | 58.2nPY | 48–101 | 24.1nPY | 61.1nPY |
| BCL2 | 239 | 325 | -- | -- | 23.4nPY | 61.5nPY | 32–88 | 22.8nPY | 59.6nPY |
| BCL2L1 | 233 | 313 | -- | -- | 19.3nPY | 64.8nPY | 27–77 | 9.8nPY | 64.7nPY |
| KIAA0226 | 972 | 247 | 938 | -- | 3.6nY | 33.2nY | 214–458; 515–586 |
2.8nY; 0nY |
18.0nY; 23.6nY |
| CLN3 | 438 | 422 | -- | 408 | 23.5 | 49.8 | 237–268 | 0 | 12.5 |
| GOPC | 462 | -- | -- | -- | -- | -- | 255–279; 422–462 |
-- | -- |
| TMEM74 | 305 | -- | -- | -- | -- | -- | 6–96; 121–170 |
-- | -- |
| PINK1 | 581 | 706 | --- | --- | 32.0nPY | 66.8nPY | 179–210 | 9.4nPY | 56.2nPY |
| HMGB1 | 215 | 385 | 446 | 552 | 3.2 | 27.4 | 163–215 | 1.9 | 28.3 |
| EXOC8 | 725 | 671 | 814 | 753 | 1.2 | 17.3 | 116–158 284–332 |
0; 0 |
0; 10.2 |
| HDAC 6 | 1215 | 1138 | 564 | 706 | 14.9 | 34.2 | 1–72; 842–1092 |
0nPY; 0 |
0nPY; 3.2 |
Superscript text denotes human protein consensus IDRs not found in orthologs from: Insect, nI; Plant, nP; Yeast, nY; Insect and Plant, nIP; Plant and Yeast, nPY.
Predicting autophagy protein IDR regions that bind to other proteins
ELM (http://elm.eu.org),17 a searchable database of experimentally validated interaction motifs was used to predict linear interaction motifs. ANCHOR (http://anchor.enzim.hu)18 was used to analyze protein sequences to predict parts of IDRs, or regions flanking IDRs, likely to be structurally stabilized by interaction with a globular protein partner. Each residue is assigned a “binding probability” based on energetic gain upon interaction; and contiguous stretches of at least five residues with a binding probability higher than 0.5, which are also predicted to be disordered by IUPred, are identified as potential “Anchors”. Binding-associated energies calculated by ANCHOR approximate the corresponding energies calculated from known structures of globular proteins, providing evidence that disordered regions can be discriminated from ordered proteins by unfavorable estimated energies.
Interactome of IDR-containing autophagy proteins
The Biological General Repository for Interaction Datasets,19 BioGRID3.1 (http://thebiogrid.org/) online database - version 3.1.89, was mined to identify known interactions involving IDR-containing autophagy proteins, as well as the original research publications reporting each interaction. Prior to data mining, protein aliases were verified using GeneCards (http://www.genecards.org/). Duplicate interactions were removed from the output. Published research reporting each interaction was then manually examined to determine whether the techniques used unambiguously demonstrate direct protein-protein interactions rather than just participation in the same complex.
Production of IDR constructs
Peptides corresponding to the human BECN1 BH3D (105DGGTMENLSRRLKVTGDLFDIMSGQT130); as well as various BH3D-derived peptides; were chemically synthesized, HPLC purified to > 95% purity, and their purity confirmed by electrospray mass spectrometry (Protein Chem. Tech. Core, UTSW or EZBioLabs). For each construct, a 1 mM peptide stock solution in 10 mM potassium phosphate pH 6.0 or 5.7, and 50 mM NaCl was prepared. Expression plasmids for BECN1 constructs containing two other domains: the helical domain (HD) and coiled-coil domain (CCD) were obtained by cloning DNA constructs corresponding to the HD+CCD (BECN1 residues 141–265) and BH3D+HD+CCD (BECN1 residues 105–265) into the pMBP-parellel-1 (AF097412) expression vector. Protein from these plasmids was expressed at 20°C overnight in BL21(DE5) pLysS cells. Soluble protein in the cell lysate was first purified by Amylose Affinity Chromatography, followed by TEV protease-cleavage of the MBP tag. Subsequently, each protein construct was purified to homogeneity by Ion-Exchange Chromatography, (MonoQ 5/50GL, GE), and size exclusion chromatography (16/60 Superdex 200, GE).
BCL2 production
Soluble human BCL2 (residues 1–218), lacking the C-terminal transmembrane helix, was expressed and purified as previously described.20
Circular Dichroism spectroscopy (CD)
BECN1 constructs were diluted to 10–100 μM in a “CD buffer” comprising 10 mM potassium phosphate, pH 7.6, with 100 mM (NH4)2SO4. For experiments involving complexes of BCL2 and BECN1 BH3D-derived peptides, the BCL2 and peptides were mixed in 1:1, 1:10 or 2:1 molar ratios, then dialyzed against and diluted in the CD buffer. A Jasco J-815 spectrometer equipped with a PFD-425S peltier cell holder was used to measure CD spectra at 4°C between 180–250 nm. The spectra were smoothened and buffer-subtracted, and secondary structure content of various constructs calculated using K2D221 (http://www.ogic.ca/projects/k2d2/); and the CONTIN, CDSTTR and SELCON programs provided by the CDPro program package22 obtained with Jasco software.
NMR spectroscopy
The 1D-1H NMR spectrum were recorded at 25°C using a Varian Inova 500-MHz spectrometer equipped with a swpfg 2 channel probe, after the addition of 10% v/v D2O to 500 mL of a 1 mM peptide solution prepared as above. NMR data were analyzed with the program MestreNova 5.3.1 and the chemical peak dispersion in the amide and methyl regions qualitatively assessed for the presence of secondary structure and folded tertiary structure content.23, 24 The region between 4.5 ppm – 7 ppm was removed from the spectra due to poor water peak suppression. Additionally, since there were no signals less than −1.0 ppm and beyond 10.5 ppm, these regions were also removed from the displayed spectra.
Isothermal Titration Calorimetry (ITC)
To ensure buffer match, BCL2 and the various BECN1 BH3D-derived peptides were loaded into separate dialysis cassettes, and simultaneously dialyzed against 2 L of 50 mM HEPES, pH 7.5, 150 mM NaCl, and 2 mM β-mercaptoethanol buffer. ITC experiments were performed using a TA Instruments Low Volume Gold Nano ITC (TA Instruments), at 25°C with 25 injections of 2 μL each. BCL2 was placed in the ITC cell into which different BH3D-derived peptides were titrated. Data were plotted and analyzed with NanoAnalyze software, using an independent binding model.
Results
Identification of consensus IDRs
IDRs can be reliably identified solely from protein sequence,12 based on distinctive features of amino acid composition such as low propensity to form well-packed hydrophobic cores or secondary structure, high charge fraction, low complexity sequences, and sequence similarity to disordered loops in crystal structures. Here we have identified consensus IDRs based on predictions from four programs that use different subsets of these criteria: DISOPRED;10 PrDOS;11 IUPred;15 and the VSL2B algorithm in PONDR.12, 13 Each program was used to analyze and predict IDRs within 59 human autophagy effectors, regulators and targeting proteins (Table S1). Not surprisingly, we find that there is significant overlap in disordered regions predicted by the various programs (Table S2).
There is disagreement regarding the sequence length that should be used to define IDRs; with the programs we used defining stretches of either 15 (DISOPRED)10 or 30 (IUPred)15 predicted disordered residues as IDRs. We selected a 25-residue length criteria, as this is comparable to the sizes of the smallest structured domains and corresponds to the size of a previously established functional domain that we show is disordered in this study; yet allows us to distinguish IDRs from smaller disordered sequences that may be classified as loops. The combined predictions from the four programs listed above were used to identify consensus IDRs (Table I), which we define as regions comprising at least 25 contiguous residues that were predicted to be disordered by at least three of the four programs. Further, disordered regions separated by short stretches of fifteen or less residues that were predicted to be ordered by only one or two programs, were defined as a single, contiguous IDR. In summary, a consensus IDR was defined as an unbroken stretch of at least 25 residues, within which every residue was predicted to be disordered by at least two of the four programs; constituting a criteria more stringent than that used by each individual program.
Based on these criteria, 34 of 59 proteins analyzed were found to contain consensus IDRs, and these are listed and grouped according to function in Table I. It is likely that these consensus IDRs represent a conservative minimal boundary for each IDR and that actual IDR boundaries extend beyond the residues listed. For four additional proteins, IDRs were predicted by only one or two programs, or the consensus length was a little shorter than our 25-residue definition, and consequently, while these fail our definition of consensus IDRs, it is possible that these proteins do contain IDRs. Further, some of the consensus IDR-containing proteins listed may also have additional IDRs that did not meet our stringent criteria for a consensus IDR. Lastly, we note that of the consensus IDRs identified in Table I, only three: ATG16L1 (59–84), SMURF1 (343–370) and GOPC (255–279) are shorter than 30 residues, the length previously used to describe long disordered domains;25 but in each case the protein bearing the IDR also had additional larger IDRs. Therefore, the use of a 30-residue definition would not change any of our general conclusions; but would wrongly prevent inclusion of shorter, previously-identified, functional domains such as the BECN1 BH3D in this study.
Human autophagy proteins that contain IDRs
The signaling network that triggers autophagy comprises of at least four proteins: either one of the ULK1/ATG1 S/T kinase homologs, ULK1 or ULK2; the ULK1/ATG1-activating protein, ATG13; the protein interaction hub, RB1CC1; and the ATG13-interacting protein, C12orf44/ATG101. Of these, only C12orf44 does not appear to contain an IDR. Each of the other four proteins contains a consensus IDR larger than sixty residues (Table I). Thus, four out of five proteins important in signaling autophagy initiation contain consensus IDRs.
All key autophagy effectors that participate in the PIK3C3-BECN1 complex contain large consensus IDRs (Table I). The core components of this complex are the VPS34 homolog, PIK3C3, that catalyzes the synthesis of phosphatidylinositol 3-phosphate; PIK3R4/p150, a S/T kinase VPS15 homolog; and the ATG6 homolog, BECN1/Beclin 1, a protein interaction hub. Other important effectors such as ATG14/Barkor, AMBRA1, VMP1 and UVRAG, differentially associate with BECN1, modulating the function of the core complex, although the mechanism of these effectors in autophagy is unclear. Thus, consensus IDRs are present in all the key proteins of the PIK3C-BECN1 complex.
Autophagosome expansion is carried out by two ubiquitin-like conjugation complexes: the ATG12-ATG5 complex and the ATG8-phosphatidylethanolamine complex. Both ATG12 and the various GABARAPL2/ATG8 homologs have ubiquitin-like folds. The ATG12 ubiquitin-like fold is preceded by a consensus IDR at its N-terminus. In contrast, none of the ATG8 paralogs, MAP1LC3A/ATG8E, MAP1LC3B/ATG8F, MAP1LC3C/ATG8J, GABARAP/ATG8A, GABARAPL2/ATG8C or GABARAPL1/ATG8L, contain CONSENSUS IDRs. Humans have four paralogs of ATG4, a protease that cleaves GABARAPL2/ATG8 homologs to release the C-terminal glycine required for conjugation. Of these paralogs, only ATG4D bears a consensus IDR, located at its N-terminus (Table I); while ATG4A, ATG4B and ATG4C do not contain IDRs. ATG7, the E1 activating enzyme shared by both conjugation systems; and ATG10, the E2 conjugating enzyme for the ATG12 system; do not contain IDRs. However, ATG3, the E2 enzyme for the GABARAPL2/ATG8 system, has a 41-residue consensus IDR (Table I). ATG12 is conjugated to ATG5, which does not contain an IDR; but the ATG12-ATG5 conjugate forms a complex with another effector, ATG16L1, which contains two consensus IDRs. Thus, although IDRs are not a common feature of human proteins involved in autophagosome expansion, four key effectors contain consensus IDRs.
Autophagosome maturation and lysosomal fusion involves some of the GABARAPL2/ATG8 paralogs listed above, and other proteins with membrane-associating domains. These include four WD40 domain-containing WIPI1 paralogs. Of these, WIPI1 contains a large consensus IDR (Table I); but WIPI2, WIPI3 and WIPI4 do not contain IDRs. In contrast, seven consensus IDRs were identified in ATG2A, which binds to WIPI1. Lastly, the first transmembrane protein implicated in autophagy, ATG9, also contains two consensus IDRs, each over 90 residues long; and the PX-BAR domain-containing SNX18, shown to play a critical role in autophagosome tubulation, also contains a very large, 162-residue consensus IDR (Table I). Thus, although a couple of proteins like GOSR1 and RAB24 do not contain IDRs, most of the key proteins responsible for autophagosome maturation bear consensus IDRs.
Amongst proteins involved in selective targeting of macromolecular assemblies to the autophagosome, NBR1, SQSTM1 and SMURF1 contain consensus IDRs. SQSTM1 contains one large, 142-residue consensus IDR, while NBR1 and SMURF1 contain multiple IDRs, including a very large 221-residue consensus IDR in NBR1 (Table I). NBR2 is the only targeting protein lacking a consensus IDR, although it bears an 18-residue, consensus disordered region which is predicted to be longer by a subset of programs. Thus, IDRs appear to be common in proteins responsible for targeting macromolecular assemblies for autophagy.
Several proteins modulate cellular autophagy levels via direct interactions with autophagy effectors. Some regulators such as HIF1α, TRAF2, DDIT3, PDPK2, IRGM and RALB, do not contain consensus IDRs. However, many others, especially those that regulate the PIK3C3-BECN1 complex, such as BCL2, BCL2L1, BNIP3, BNIP3L, AMBRA1, KIAA0226, CLN3, GOPC, PINK1, HMGB1, EXOC8 and TMEM74 contain consensus IDRs (Table I). HDAC6, shown to selectively regulate autophagosome maturation in response to the presence of defective macromolecular assemblies, also contains a large consensus IDR.
Ortholog sequences analogous to human autophagy protein IDRs are poorly conserved
We identified the closest ortholog of each consensus IDR-containing human autophagy protein in Drosophila melanogaster (insect), Arabidopsis thaliana (plant) and Saccharomyces cerevisiae (yeast), ensuring a wide diversity of eukaryotes for our analysis (Table I). Each species had orthologs of all consensus IDR-containing autophagy effectors, emphasizing the conservation of the autophagy pathway in eukaryotes. Interestingly however, each species did not all have orthologs of the autophagy targeting proteins, NBR1 and SQSTM1, or of many human autophagy regulators, including BNIP3, BNIP3L, BCL2, BCL2L1, KIAA0226, CLN3, GOPC, TMEM74 and PINK1.
Sequence alignments of each set of orthologs revealed that ortholog sequences that align with human protein consensus IDRs are poorly conserved, but sequences that align with protein regions outside the human protein consensus IDRs identified are better conserved (Table I and Figure S1; alignments of ATG13 and PIK3C3 are shown as examples). Strikingly, the consensus IDR-equivalent regions in orthologs consistently share low sequence similarity, regardless of the overall conservation of the autophagy-related effector (Table I). For instance, although PIK3C3 orthologs are highly conserved overall, sharing ~24% identity and ~56% similarity (Table I); the PIK3C3 consensus IDR shares less than 2% sequence identity and 8% sequence similarity. Other effectors such as ATG13 appear to be less well conserved, sharing less than 3.1% identity and 21.7% similarity respectively; yet, ATG13 consensus IDRs share ~0% identity and 14–20% similarity; which is comparable to the conservation of the PIK3C3 consensus IDR. This low level of sequence conservation, and lack of correlation with the conservation of the rest of the protein, is a feature of all consensus IDRs in the autophagy proteins analyzed (Table I). A corollary to this finding is that, although structured regions of consensus-IDR containing proteins may be significantly conserved, the presence of IDRs reduces the overall identity and similarity of ortholog sequences.
IDRs in autophagy effectors may be responsible for protein interactions
A potential IDR function is to facilitate protein-protein interactions; therefore, we investigated whether these were likely functions of IDRs within human autophagy effectors. We first searched the sequences of consensus IDR-containing effectors for Eukaryotic Linear Motifs (ELMs), using the ELM server17. ELMs are short, evolutionarily-plastic, linear sequence motifs experimentally shown to be key for various protein-protein interactions. ELMs are found in each effector, but have a variable distribution between IDRs and ordered regions, with 5–70% of the ELMs found in each effector mapping to IDRs (Table II). The distribution of ELMs correlated somewhat imperfectly with the fraction of total protein residues comprising the IDR (Tables I and II).
Table II.
Binding motifs associated with consensus IDRs in human autophagy effectors.
| Protein | Total no. of | No. associated with consensus IDRs | ||
|---|---|---|---|---|
| ELMs | Anchors | ELMs | Anchors | |
| ULK1 | 60 | 16 | 40 | 15 |
| ULK2 | 44 | 18 | 31 | 17 |
| ATG13 | 37 | 5 | 8 | 4 |
| RB1CC1 | 58 | 9 | 14 | 7 |
| PIK3C3 | 18 | 0 | 4 | 0 |
| PIK3R4 | 53 | 7 | 6 | 3 |
| BECN1 | 40 | 5 | 11 | 4 |
| ATG14 | 37 | 4 | 5 | 2 |
| AMBRA1 | 76 | 20 | 40 | 18 |
| UVRAG | 53 | 8 | 13 | 7 |
| VMP1 | 19 | 0 | 1 | 0 |
| ATG16L1 | 17 | 11 | 6 | 5 |
| ATG12 | 15 | 2 | 2 | 2 |
| ATG3 | 1 | 1 | 0 | 1 |
| ATG4D | 36 | 2 | 2 | 2 |
| ATG9A | 48 | 5 | 10 | 5 |
| WIPI1 | 34 | 1 | 4 | 3 |
| ATG2A | 131 | 22 | 35 | 11 |
| SNX18 | 19 | 6 | 13 | 4 |
Next, we used the program ANCHOR18 to identify sequences flanking or overlapping IDRs that could be stabilized as secondary structures upon binding to a globular protein partner26. Here we call such sequences “Anchors”. We find that most autophagy effector IDRs contain or are adjacent to Anchors. Perhaps not surprisingly, the number of Anchors associated with each protein IDR is somewhat correlated to the length of the consensus IDR. Interestingly, in ATG13, AMBRA1 and ATG9A, short Anchors bridge two consensus IDRs, suggesting that in the absence of an appropriate binding partner, the Anchor sequence is part of the IDR. In ULK1 and AMBRA1, ELMs are predicted within Anchors, suggesting that these ELMs might nucleate interactions to stabilize secondary structure.
Lastly, we mined BioGRID3.1;19 to identify potential binding partners for each consensus IDR-containing autophagy-related effector (Figure 1). Most IDR-containing effectors appear to be involved in multiple protein-protein interactions (Figure 1). We supplemented and overlaid this data with published information regarding the nature of these interactions. Many of the interactions included in BioGRID3.1 were identified based on cellular pull-down experiments followed by protein identification, rather than by experiments showing direct interactions between proteins (Figure 1, dashed lines). Hence, some of these interactions likely represent participation in the same cellular protein complex inside cells, rather than direct interactions between proteins. For interactions that have been shown to represent direct interactions between proteins, we further distinguish between interactions involving coiled-coil, ubiquitin-like, WD40 or other structured domains (Figure 1, black lines); those suspected to involve IDRs (Figure 1, red lines) and those that involve only consensus IDRs (Figure 1, sinusoidal red lines). Notably, proteins with consensus IDRs that did not contain either ELMs or Anchors, such as PI3KC3, also have fewer interaction partners. It is possible that these IDRs have roles other than in binding.
Figure 1.

Interactomes of IDR-containing autophagy effectors. Labeled ovals represent proteins, with autophagy-related effectors colored-coded according to function: autophagy signaling (yellow), autophagosome nucleation (pink), enlargement (green) and maturation (blue). Effectors that neither contain IDRs themselves, nor are implicated in interactions with other IDR-containing effectors are not shown. Effectors that contain consensus IDRs are outlined in red. Dotted lines indicate protein interactions not yet shown to be direct. Solid lines indicate demonstrated direct interactions; with black lines denoting interactions involving structured domains; red lines denoting interactions involving IDRs; and sinusoidal lines denoting interactions involving only the IDR.
Evidence of IDRs from autophagy protein structures
Next, we investigated the correlation between the consensus IDRs predicted here and relevant autophagy protein structures previously determined by other groups that are available from the protein structure database (http://www.rcsb.org/pdb/home/home.do). Almost all the available autophagy protein structures correspond to either proteins that we predict lack consensus IDRs, such as ATG4A, ATG4B, ATG5, various ATG8 homologs, ATG7, and ATG10; or for protein constructs in which regions corresponding to the consensus IDRs were excluded during expression, purification and structure determination, such as ATG3, ATG12, ATG13, NBR1, SQSTM1, SMURF1, BNIP3 and HDAC6. This is not surprising, as the lack of structure often prevents structure determination, and may also make IDRs difficult to express and purify.
We found three protein structures wherein the entire consensus IDRs were included in the constructs crystallized: human BCL2L1,27 human BCL2,20 and Drosophila VPS34.28 Each of these structures provides strong support for our consensus IDR predictions (Table I). The BCL2L1 structure27 lacks a model for consensus IDR residues 27–77 because electron density corresponding to these residues was missing, indicative of a disordered region; whereas in the BCL2 structure,20 one-third of consensus IDR residues 32–88 are modeled in a coil conformation and the remaining two-thirds lack a model indicating a disordered region. Residues analogous to the human PIK3C3 consensus IDR residues 414–468, residues 437–526 of the Drosophila VPS34, are not modeled in the recently determined X-ray crystal structure28, confirming this region of the protein is disordered.
Another set of structures, that of the BECN1 BH3D (residues 105–130) crystallized in complex with different BCL2 homologs,29–32 overlaps the BECN1 consensus IDR predicted by us (Table I). These structures indicate that the BCL2 IDRs discussed above are not implicated in this interaction, as they are located on the BCL2 face opposite that of the BH3D-binding groove. Conversely however, these structures and associated biochemical results demonstrate that the BECN1 BH3D constitutes a functional domain required and sufficient for binding to BCL2s; wherein the bound BH3D is an amphipathic α-helix, bearing a conserved sequence motif: -φ-xxx-φ-K-xx-G-D-x-φ- (with φ representing hydrophobic residues),33 with hydrophobic residues buried in a hydrophobic groove on the surface of the BCL2 homolog(Figure 2). Therefore, we decided to further investigate BH3D structure in the absence of BCL2 binding.
Figure 2.
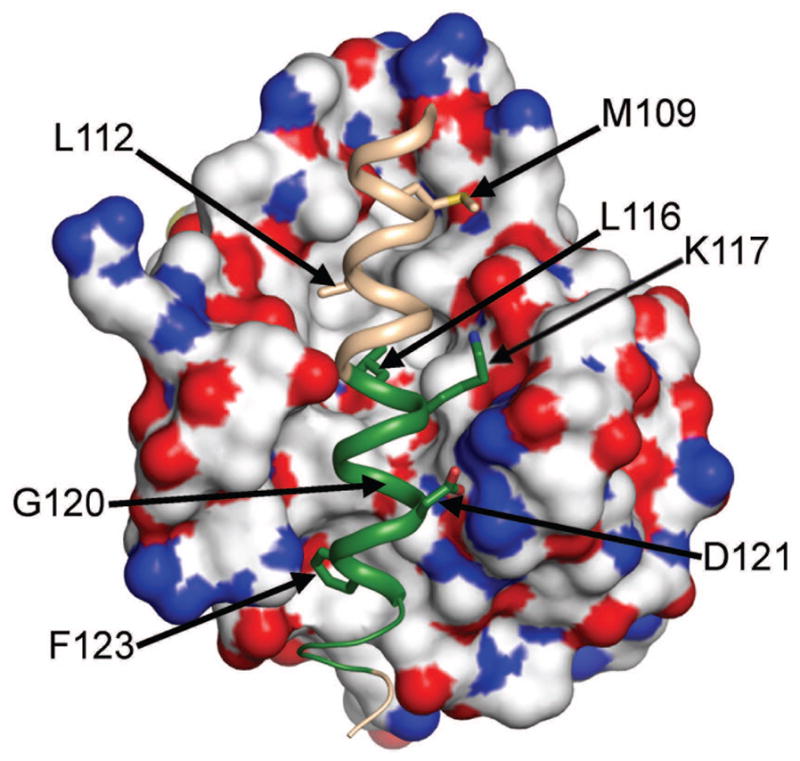
The BECN 1 BH3D structure when bound to BCL2. The BCL2 molecular surface is colored by atom type. The BH3D is shown as a salmon ribbon, with the Anchor region highlighted in green. BH3D residues involved in binding are shown in atomic detail and are labeled. This figure was made with PyMol.66
The BECN1 BH3D is an IDR in the absence of other interactions
Despite extensive efforts by our and other groups, to date, it has not been possible to purify full-length BECN1, or non-aggregated preparations of the BECN1 IDR that either include the BH3D (residues 1–135) or exclude the BH3D (residues 1–104). Therefore, we evaluated the structure of the isolated BH3D using 1D NMR and CD. Further, we used CD to evaluate BH3D structure in the context of the two BECN1 domains that follow it: the helical domain (HD) comprising residues 141–170, and the coiled-coil domain (CCD) comprising residues 175–265.
1D-1H NMR spectroscopy is now routinely used to assess the folded state of proteins.23, 24, 34, 35 Importantly, 1D-1H NMR spectra reflect peptide backbone flexibility, not backbone conformations or secondary structure content. As expected for disordered peptides,23, 24 the 1D-1H NMR spectra (Figure 3A) measured for the BH3D lacked peaks upfield of the strong methyl peaks at 0.8 ppm; contained broad peaks at about 8.3 ppm, the region characteristic for amide groups in random-coil conformation; and had little signal dispersion visible downfield of ~8.5 ppm, with the few peaks visible below 8.5 ppm being attributable to Trp sidechains.
Figure 3.
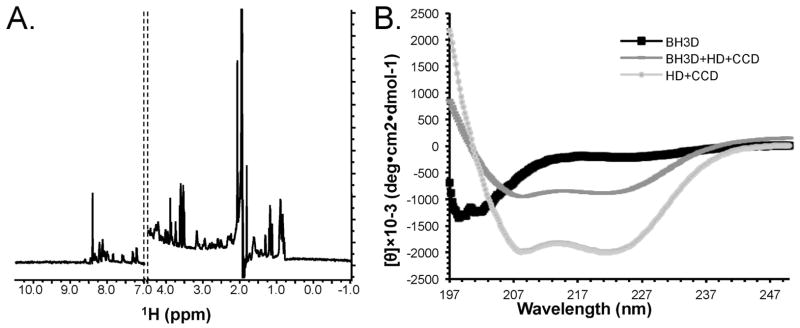
Structure of the BECN1 BH3D: (A) 1D-1H NMR spectrum. Only 1H chemical shifts between −1.0 ppm to 4.5 ppm (methyl region) and 7.0 ppm to 10.5 ppm (amide) are shown. (B) CD spectra of the BECN1 BH3D (black squares), HD+CCD (light gray circles), and BH3D+HD+CCD (dark gray dashes).
The BECN1 BH3D CD spectra showed a large single negative transition at ~195 nm, indicative of a random coil conformation (Figure 3B); compared to the CD spectra of α-helical polypeptides which are expected to display a positive band at ~192 nm and a large negative split transition at 208 nm and 222 nm; and of β-strand polypeptides which would show a positive band at 195 nm and a negative band at 218 nm.36 In contrast to the isolated BH3D, the molar ellipticity at 222 nm indicated that a BECN1 BH3D+HD+CCD construct has a much higher helical content, although not as high as that observed for a HD+CCD construct. This reduction in helical content suggests that the BH3D is disordered even when attached to the helical domains that follow it.
Secondary structure content was quantified by analyzing these CD spectra using four different programs: K2D2,21 CONTIN,37 CDSSTR38 and SELCON3.39 Even though each program has previously been shown36 to underestimate coil conformations in proteins, the analysis from each program indicated that most of the BH3D residues had a coil conformation. SELCON3, which was trained on a reference data set that included denatured proteins, is expected to provide the most accurate estimation of unordered structure.40 The secondary structure content estimated using SELCON3 (Table III) indicates that 83% of the BH3D residues have a coil or turn conformation; while 9% have a helical conformation and 10% have a β-strand conformation. This number of residues estimated to be in either α-helix or β-strand conformation is insufficient to form stable secondary structures; confirming that the BH3D is disordered. In contrast, the 125-residue HD+CCD has a ~81% helical content (Table III) consistent with available crystal structures of the HD (our unpublished data) and the CCD41 (and our unpublished data) demonstrating that these are helical domains. Strikingly, the 161-residue BH3D+HD+CCD is only ~64% helical, consistent with the BH3D being disordered even when attached to these helical domains.
Table III.
CD analysis of secondary structure content of BH3D-containing constructs and complexes.
| Protein | Helix | Strand | Turn + Coil | Total |
|---|---|---|---|---|
| Different BECN1 Constructs | ||||
| BH3D | 0.04 | 0.038 | 0.919 | 0.997 |
| BH3D+HD | 0.345 | 0.208 | 0.441 | 0.994 |
| BH3D+HD+CCD | 0.644 | 0.052 | 0.346 | 1.008 |
| HD+CCD | 0.806 | 0.002 | 0.2 | 1.008 |
| BCL2+BH3D complex | ||||
| Bcl2 | 0.81 | −0.003 | 0.199 | 1.006 |
| BH3D | 0.04 | 0.038 | 0.919 | 0.997 |
| Bcl2:BH3D 1:1 | 0.809 | −0.003 | 0.198 | 1.004 |
| Bcl2:BH3D 1:10 | 0.763 | 0.001 | 0.249 | 1.013 |
| Bcl2:BH3D 2:1 | 0.809 | 0 | 0.202 | 1.011 |
| BCL2-DS BH3D complex | ||||
| DS BH3D | 0.09 | 0.104 | 0.831 | 1.025 |
| Bcl2:DS BH3D 1:1 | 0.666 | −0.003 | 0.347 | 1.01 |
| Bcl2:DS BH3D 1:10 | 0.446 | 0.114 | 0.451 | 1.011 |
| Bcl2:DS BH3D 2:1 | 0.809 | −0.004 | 0.193 | 0.998 |
Thus, our combined CD and NMR experimental evidence unambiguously show that the entire BECN1 BH3D (residues 105–130) is unstructured in the absence of other interactions. As only part of the BH3D was predicted to be a consensus IDR (Table I), this indicates that the BECN1 IDR likely extends well beyond the consensus IDR we have listed (Table I).
The BECN1 BH3D undergoes a helical transition upon binding, which is nucleated by an Anchor
Previous experimental structures29–32 of the BECN1 BH3D bound to different BCL2 homologs show that the bound BH3D forms an amphipathic α-helix, with hydrophobic residues buried in the interaction interface (Figure 2). BH3D residues 116–128, which flank the BECN1 consensus IDR at the C-terminus, are predicted to be an Anchor. Although this Anchor comprises only half the BH3D, it bears most of the residues involved in binding to BCL2 (Figure 2), as well as four of five residues essential for binding (Table IV). ITC measurements indicate that the BECN1 BH3D binds to BCL2 with a Kd of 10.4 μM (Table IV). Binding is abolished by Ala substitutions of the single residues L112, L116, or F123, as well as the double G120E+D121A substitution (DS). Of these, only L112 lies outside the BH3D Anchor. It is likely that for binding of these BH3D-derived peptides, the lower ΔH due to diminished interactions is not compensated by increased ΔS arising from improved BH3D helicity from increased Ala content.43 The role of the BECN1 BH3D Anchor in binding is further emphasized by the finding that Kd, ΔH and ΔS are minimally affected for a M109A BH3D peptide, which lies outside the Anchor; while binding affinity halves for the K117Q BH3D peptide, which lies within the Anchor; as the reduced ΔH is offset by a decrease in ΔS (Table III). Thus, the BH3D anchor likely nucleates α-MoRF structure within the BH3D by triggering concomitant binding and folding.
Table IV.
Thermodynamics of Bcl-2 binding to Beclin 1 BH3D mutants.
| Mutant | Kd (μM) | ΔH (kJ/mol) | ΔG (kJ/mol) | ΔS (J/K•mol) |
|---|---|---|---|---|
| Wt | 10.4 ± 3.5 | −17.8 ± 6.5 | −28.2 ± 0.7 | 35.2 ± 24.1 |
| M109A | 14.5 ± 2.3 | −15.8 ± 2.9 | −27.6 ± 0.4 | 39.6 ± 11.2 |
| L112A | No binding | -- | -- | -- |
| L116A | No binding | -- | -- | -- |
| K117Q | 24.5 ± 6.0 | −21.4 ± 4.6 | −26.4 ± 0.6 | 16.7 ± 13.5 |
| G120E+D121A | No binding | -- | -- | -- |
| F123A | No binding | -- | -- | -- |
Lastly, using CD methods similar to those used previously to analyze coupled folding and binding of calmodulin binding targets to calmodulin;44–46 we directly confirmed the structural transition of the BH3D by assessing and comparing the secondary structure content (Figure 4 and Table III) of different molar mixtures of BCL2 and the BECN 1 BH3D, as well as of BCL2 and DS BH3D, which does not bind to BCL2 (Table IV) and is also disordered in solution (Figure 4 and Table III). Our CD data (Figure 4 and Table III) indicate that BCL2 is 80% helical, consistent with crystal structures of BCL2 that show that it comprises of eight α-helices and a large disordered region. This secondary structure content is unchanged upon addition of equimolar or 0.5-fold molar BECN1 BH3D, suggesting that the BH3D folds into a helix upon binding (Table III). However, addition of a 10-fold molar excess of the BH3D reduces the helical content of the sample to 76%, consistent with a significant population of BH3D molecules being in a random coil conformation. In contrast to the BH3D, addition of equimolar or 10-fold excess molar DS BH3D peptide dramatically reduces the helical content of the sample to ~67% and ~45% respectively, indicating that there is no change in the secondary structure of the DS BH3D peptide, consistent with lack of binding to BCL2s.
Figure 4.
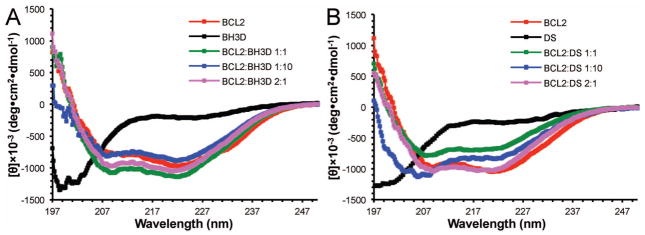
CD spectra of mixtures of different molar ratios of BCL2 and (A) the BECN1 BH3D or (B) BECN1 BH3D DS. The variously colored lines correspond to different molar ratios of BCL2:BH3D peptide: red, 0:1; black, 1:0; green, 1:1; blue, 1:10; and magenta, 2:1.
Thus, the combined information from IDR predictions, CD, NMR and co-complex crystal structures indicates that the BECN1 BH3D undergoes a disorder to αhelix transition upon binding. IDRs experimentally confirmed to undergo a disorder to secondary structure transition have been called molecular recognition features (MoRFs), that are further classified as α-, β- or σ-MoRFs, depending on whether they fold into αhelices, β-strands or restrained coils.42 Thus, the BECN1 BH3D appears to be an α-MoRF. Strikingly, in organisms that lack BCL2 orthologs, BECN1/ATG6 orthologs also appear to lack a BH3D, suggesting that the BECN1 IDR may serve to facilitate species-specific regulatory interactions.
Discussion
Although de novo prediction of structure from sequence is challenging, unique features of amino acid composition and sequence enable a more reliable prediction of unstructured regions within proteins. The application of multiple criteria further increases the robustness of these predictions. Despite the ease of IDR prediction, and their common occurrence in proteins, comparatively little research has been focused on understanding the mechanistic basis of the biological roles of such regions.47 Our approach, which focuses on delineating and investigating IDRs in proteins involved in a single, essential, cellular process provides a unique perspective on the potentially important role of IDRs in autophagy and related cellular processes.
IDRs have been predicted in ~30% of prokaryotic proteins and 45–50%, of eukaryotic proteins,48 although these ratios vary depending on species.49 Here we show that IDRs are present in at least 57% of human autophagy proteins identified to date, and most likely in additional proteins where identification was ambiguous. Twelve of the 25 proteins in which consensus IDRs were not identified are paralogs: six GABARAPL2/ATG8 paralogs, three ATG4 paralogs and three WIPI paralogs; hence, the proportion of unique autophagy protein homologs that contain IDRs appears to be over 70%. It is important to note that as our objective was to unambiguously delineate IDRs, we used the rather stringent protocol of identifying consensus IDRs based on predictions of four programs; hence it is likely that the consensus IDRs identified represent conservative boundaries, and that the additional disordered regions predicted beyond these boundaries by each individual program, also constitute parts of IDRs (Table S2). When present, consensus IDRs constitute between 6–60% of these proteins.
Consensus IDRs are especially prevalent in proteins participating in the ULK1/ATG1 signaling complex responsible for autophagy initiation; the PIK3C3-BECN1 complex implicated in autophagosome nucleation; autophagosome maturation and lysosomal fusion; and the targeting of macromolecular assemblies to the autophagosome; as between 75–100% of the proteins involved in these stages contain IDRs. Consensus IDRs are also present in key proteins involved in the two ubiquitin-like conjugation complexes implicated in autophagosome expansion and in many regulators that interact directly with autophagy effectors. Together these results suggest that unstructured regions play important roles in almost all stages of autophagy.
Our investigation of IDR conservation also highlights an important general feature of autophagy – i.e. while the basic autophagy machinery is conserved in all eukaryotes, proteins involved in selecting and targeting macromolecular assemblies for autophagy and those that regulate autophagy via direct interactions with the autophagy machinery are not as well conserved. This suggests that equivalent functions in diverse organisms are performed either by proteins that share very low, undetectable sequence identity to the human proteins or by alternate, completely different proteins. Consistent with previous analysis of IDRs,50–55 we find that IDRs appear to be the most variable regions of ATG proteins. An important corollary of this observation is that proteins with large IDRs show lower conservation across species, and consequently, homologs are difficult to identify based solely on sequences. As has been suggested for other IDR-containing proteins,51 the low sequence conservation of IDRs suggests that freedom from the sequence constraints implicit in maintaining a defined 3D structure, enables increased mutability of these regions, facilitating protein plasticity during evolution. This makes IDRs a powerful mechanism for evolution as it enables the involvement of IDRs in multiple, variable, species-specific, protein-protein interactions and / or signaling processes.
IDRs are unstructured because internal residue interactions do not provide sufficient enthalpic compensation for entropic loss due to folding. However, this energy may be provided by interactions of binding partners with smaller regions within an IDR,56, 57 either an ELM or an Anchor.18 Anchors were predicted to be associated with all but two consensus IDRs identified in autophagy proteins (Table II), suggesting that many of these IDRs may be MoRFs. IDRs not containing Anchors may be entropic chains or non-folders,58, 59 and their function is unclear. Potential protein interaction partners identified from the biological interaction database, BioGRID3.1, show that most consensus IDR-containing proteins have multiple partners, but proteins whose consensus IDRs have fewer binding motifs also have fewer partners. The importance of IDRs in macromolecular interactions may be three-fold: 1) The entropic cost of converting from a disordered or flexible state to an ordered or restrained state enforces specificity, even with low binding affinities; allowing an interaction to be specific, yet reversible; 2) their flexibility enables them to undergo conformational changes to form diverse interaction surfaces complementary to multiple proteins and; 3) different domains within an IDR may simultaneously bind to different partners. This functional elasticity is a valuable asset in signaling processes.
Without exception, all consensus IDRs for which experimental data are available have been shown to be actual IDRs. Experimental crystal structures previously determined by other groups provide additional experimental support for consensus IDRs predicted within human BCL2L1,27 human BCL2, and Drosophila PI3KC3/VPS3428. A fourth set of structures correspond to complexes of different BCL2 homologs bound to a BH3D from BECN1, an essential autophagy effector that serves as a protein interaction hub60, 61 (Figure 1). The BH3D is required and sufficient for binding to BCL2 homologs, and for autophagy inhibition by BCL2s. While the first half of the BECN1 BH3D overlaps the consensus IDR predicted in BECN1, the second half constitutes an Anchor. Our combined CD and NMR data experimentally demonstrates that the BECN1 BH3D is an IDR, even when attached to other structured BECN1 domains, indicating that the BECN1 IDR extends beyond the conservative consensus IDR boundaries listed in Table II. This suggests that our stringent predictions identify conservative or minimal IDR boundaries, and that actual IDRs may well extend beyond the boundaries listed in Table I. Further, this study demonstrates that the BECN1 BH3D is an α-MoRF, as was previously shown for the BIM BH3D.62
Lastly, we find that although the Anchor constitutes only half the BECN1 BH3D, it bears four of five residues critical for binding BCL2. Perhaps this Anchor transitions between helical and disordered conformations on time scales faster than CD and NMR measurements, with a helical conformation stabilized only upon binding to BCL2s. The concomitant binding and stabilization of the BH3D Anchor as a helix may then promote helicity in the remaining BH3D, which is stabilized further by BCL2 interactions. These results also draw a distinction between Anchors and MoRFs: Anchors may constitute the entire MoRF, or only a part of the MoRF that nucleates concomitant folding and binding of a larger IDR.
As the BECN1 IDR contains many ELMs and is associated with four other potential Anchors (Table II), we suggest that other parts of the BECN1 IDR are likely to also be involved in protein interactions. It is important to investigate the role of these IDR interaction motifs, and their potentially competing interactions to elucidate the function of BECN1 as a protein interaction hub. Strikingly, BH3Ds have only been found in BECN1/ATG6 orthologs from organisms that also have BCL2 orthologs, suggesting that IDRs might constitute sites of species-specific interactions rather than evolutionarily-conserved interactions. It is likely that in BECN1/ATG6 orthologs lacking BH3Ds, other proteins interact with the IDR of these orthologs to regulate autophagy. This provides a powerful example of how IDR sequence variability enables functional variability. A better understanding of the mechanism by which different BECN1/ATG6 IDR interactions regulate autophagy will further our understanding of the mechanism and regulation of ATG6/BECN1 orthologs in autophagy, as well as elucidate the similarities and differences of this mechanism in various organisms. Further, this will also improve our understanding of the general biological importance of IDRs.
Given the high preponderance of IDRs in autophagy proteins, it is likely that protein disorder plays a very important function in this cellular process, and a mechanistic understanding of autophagy will be incomplete without an understanding of the role and the mechanism of these IDRs. The importance of IDRs is often overlooked, as biological roles of most IDRs have not yet been investigated. Many IDRs investigated to date have been implicated in signaling processes.63 This function is probably facilitated by their widely varying sequences and enhanced ability to participate in diverse protein interactions and post-translational modifications. Hence, future research focused on identifying interaction partners of each IDR implicated in protein interactions followed by research investigating these complexes and potential competing interactions, is essential to understanding the mechanism of IDR-containing proteins. The results presented here constitute an essential step toward understanding IDRs in autophagy, by highlighting their prevalence, delineating boundaries and outlining potential functions and functional domains. Mutations implicated in important diseases like cancer, neurodegenerative and cardiac diseases often map to IDRs.64 Hence, IDRs are novel and important targets for treating such diseases65 and a better mechanistic understanding of IDRs in autophagy proteins will be essential to developing therapeutics targeting them.
Supplementary Material
Acknowledgments
This work was funded by the following grants to SCS and CLC: NIH grants P20-RR015566 and P30-GM103332-01 and NSF grant EPS-0814442, as well as NIH grants R21-AI078198 and NSF grant HRD-0811239 to SCS. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or NSF. The authors declare no conflict of interest.
References
- 1.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B, Klionsky DJ. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 3.Shintani T, Klionsky DJ. Autophagy in health and disease: A double-edged sword. Science. 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N, Levine B, Cuervo A, Klionsky D. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Legakis JE, Klionski DJ. Overview of autophagy. In: Deretic V, editor. Autophagy in immunity and infection. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2006. pp. 3–17. [Google Scholar]
- 7.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 8.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 9.Creamer TP. Unfolded Proteins: From Denatured to Intrinsically Disordered. Nova Science Publishers; 2008. [Google Scholar]
- 10.Ward J, McGuffin L, Bryson K, Buxton B, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004 doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- 11.Ishida T, Kinoshita K. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007;35:W460–W4. doi: 10.1093/nar/gkm363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obradovic Z, Peng K, Vucetic S, Radivojac P, Dunker AK. Exploiting Heterogeneous Sequence Properties Improves Prediction of Protein Disorder. Proteins. 2005;61:176–82. doi: 10.1002/prot.20735. [DOI] [PubMed] [Google Scholar]
- 13.Peng K, Radivojac P, Vucetic SA, KD, Obradovic Z. Length-Dependent Prediction of Protein Intrinsic Disorder. BMC Bioinformatics. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dosztányi Z, Csizmok V, Tompa P, Simon In. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–4. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 16.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalities and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dinkel H, Michael S, Weatheritt RJ, Davey NE, Roey KV, Altenberg B, et al. ELM—the database of eukaryotic linear motifs. Nucleic Acids Res. 2012;40:D242–51. doi: 10.1093/nar/gkr1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dosztányi Z, Mészáros B, Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–6. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Research. 2006;34:D535–D9. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petros AM, Medek A, Nettesheim DG, Kim DH, Yoon HS, Swift K, et al. Solution structure of the antiapoptotic protein bcl-2. Proceedings of the National Academy of Sciences. 2001;98:3012–7. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez-Iratxeta C, Andrade-Navarro MA. K2D2: Estimation of protein secondary structure from circular dichroism spectra. BMC Structural Biology. 2008:8. doi: 10.1186/1472-6807-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sreerama N, Venyaminov SY, Woody RW. Analysis of protein circular dichroism spectra based on the tertiary structure classification. Analytical biochemistry. 2001;299:271–4. doi: 10.1006/abio.2001.5420. [DOI] [PubMed] [Google Scholar]
- 23.Rehm T, Huber R, Holak T. Application of NMR in structural proteomics: screening for proteins amenable to structural analysis. Structure. 2002;10:1613–8. doi: 10.1016/s0969-2126(02)00894-8. [DOI] [PubMed] [Google Scholar]
- 24.Hill JM. NMR Screening for Rapid Protein Characterization in Structural Proteomics. In: Kobe B, Guss M, Huber T, editors. Methods in Molecular Biology: Structural Proteomics: High-throughput Methods. Totowa, NJ: Humana Press; 2008. pp. 437–46. [DOI] [PubMed] [Google Scholar]
- 25.Dosztányi Z, Márk S, Tompa P, Simon I. Prediction of Protein Disorder at the Domain Level. Current Protein and Peptide Science. 2007;8:161–71. doi: 10.2174/138920307780363406. [DOI] [PubMed] [Google Scholar]
- 26.Dyson H, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 27.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, et al. X-ray and NMR structure of human Bcl-XL, an inhibitor of programmed cell death. Nature. 1996;381:335–41. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 28.Miller S, Tavshanjian B, Oleksy A, Perisic O, Houseman B, Shokat K, et al. Shaping Development of Autophagy Inhibitors with the Structure of the Lipid Kinase Vps34. Science. 2010;327:1638–42. doi: 10.1126/science.1184429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3 only protein. J Biol Chem. 2007;282:13123–32. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 30.Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl-XL’s target recognition versatility revealed by the structure of Bcl-XL in complex with the BH3 domain of Beclin-1. J Mol Biol. 2007;372:223–35. doi: 10.1016/j.jmb.2007.06.069. [DOI] [PubMed] [Google Scholar]
- 31.Ku B, Woo J-S, Liang C, Lee K-H, Hong H-S, Xiaofei E, et al. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral Bcl-2 of murine γ-Herpesvirus 68. PLoS Pathogens. 2008;4:e25. doi: 10.1371/journal.ppat.0040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha S, Colbert CL, Becker N, Wei Y, Levine B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the γ-herpesvirus 68 Bcl-2 homolog M11. Autophagy. 2008;4:989–97. doi: 10.4161/auto.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinha S, Levine B. The autophagy effector Beclin 1: A novel BH3-only protein. Oncogene. 2009;27:S137–S48. doi: 10.1038/onc.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yee A, Chang X, Pineda-Lucena A, Wu B, Semesi A, Le B, et al. An NMR approach to structural proteomics. Proceedings of the National Academy of Sciences. 2002;99:1825–30. doi: 10.1073/pnas.042684599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Page R, Peti W, Wilson IA, Stevens RC, Wüthrich K. NMR screening and crystal quality of bacterially expressed prokaryotic and eukaryotic proteins in a structural genomics pipeline. Proceedings National Academy of Science, USA. 2004;102:1901–5. doi: 10.1073/pnas.0408490102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenfield N. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–90. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Provencher SW, Glöckner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–7. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 38.Manavalan P, Johnson WCJ. Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal Biochem. 1987;167:76–85. doi: 10.1016/0003-2697(87)90135-7. [DOI] [PubMed] [Google Scholar]
- 39.Sreerama N, Woody RW. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal Biochem. 1993;209:32–44. doi: 10.1006/abio.1993.1079. [DOI] [PubMed] [Google Scholar]
- 40.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–60. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 41.Li X, He L, Che KH, Funderburk SF, Pan L, Pan N, et al. Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat Commun. 2012;3:1–11. doi: 10.1038/ncomms1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohan A, Oldfield C, Radivojac P, Vacic V, Cortese M, Dunker A, et al. Analysis of molecular recognition features (MoRFs) J Mol Biol. 2006;362:1043–59. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 43.Pace C, Scholtz JM. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J. 1998;75:422–7. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rumi-Masante J, Rusinga FI, Lester TE, Dunlap TB, Williams TD, Dunker AK, et al. Structural basis for activation of calcineurin by calmodulin. Journal of molecular biology. 2012;415:307–17. doi: 10.1016/j.jmb.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan T, Gomes AV, Barnes JA, Hunter HN, Vogel HJ. Spectroscopic characterization of the calmodulin-binding and autoinhibitory domains of calcium/calmodulin-dependent protein kinase I. Archives of biochemistry and biophysics. 2004;421:192–206. doi: 10.1016/j.abb.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 46.Dunlap TB, Kirk JM, Pena EA, Yoder MS, Creamer TP. Thermodynamics of binding by calmodulin correlates with target peptide α-helical propensity. Proteins. 2013;81:607–12. doi: 10.1002/prot.24215. [DOI] [PubMed] [Google Scholar]
- 47.Tompa P. Unstructural biology coming of age. Curr Opin Struct Biol. 2011;21:419–25. doi: 10.1016/j.sbi.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 48.Dunker A, Oldfield C, Meng J, Romero P, Yang J, Chen J, et al. The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics. 2008;9:S1. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pancsa R, Tompa P. Structural Disorder in Eukaryotes. PLoS ONE. 2012;7:e34687. doi: 10.1371/journal.pone.0034687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huntley M, Golding GB. Evolution of simple sequence in proteins. Journal of molecular evolution. 2000;51:131–40. doi: 10.1007/s002390010073. [DOI] [PubMed] [Google Scholar]
- 51.Romero P, Obradovic Z, Dunker AK. Intelligent Data Analysis for Protein Disorder Prediction. Artif Intell Rev. 2000;14:447–84. [Google Scholar]
- 52.Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW, Oldfield CJ, et al. Evolutionary rate heterogeneity in proteins with long disordered regions. Journal of molecular evolution. 2002;55:104–10. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 53.Mohan A, Uversky VN, Radivojac P. Influence of sequence changes and environment on intrinsically disordered proteins. PLoS computational biology. 2009;5:e1000497. doi: 10.1371/journal.pcbi.1000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown CJ, Johnson AK, Daughdrill GW. Comparing models of evolution for ordered and disordered proteins. Molecular biology and evolution. 2010;27:609–21. doi: 10.1093/molbev/msp277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeong CS, Kim D. Coevolved residues and the functional association for intrinsically disordered proteins. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2012:140–51. [PubMed] [Google Scholar]
- 56.Schulz GE. Molecular Mechanism of Biological Recognition. Elsevier/North-Holland Biomedical Press; 1977. Nucleotide Binding Proteins; pp. 79–94. [Google Scholar]
- 57.Dunker AK, Garner SE, Guilliot S. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac Symp Biocomput. 1998:473–84. [PubMed] [Google Scholar]
- 58.Tompa P. Structure and function of intrinsically disordered proteins. CRC Press/Taylor and Francis Group; Boca Raton, FL: 2009. [Google Scholar]
- 59.Rauscher S, Pomes R. Molecular simulations of protein disorder. Biochemistry and Cell Biology. 2010;88:269–90. doi: 10.1139/o09-169. [DOI] [PubMed] [Google Scholar]
- 60.He C, Levine B. The Beclin 1 interactome. Current Opinion in Cell Biology. 2010;22:140–9. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kang R, Zeh H, Lotze M, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–80. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu X, Dai SC, Zhu Y, Marrack P, Kappler JW. The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity. 2003;19:341–52. doi: 10.1016/s1074-7613(03)00234-6. [DOI] [PubMed] [Google Scholar]
- 63.Tantos A, Han K, Tompa P. Intrinsic disorder in cell signaling and gene transcription. Mol Cell Endocrinol. 2012;348:457–65. doi: 10.1016/j.mce.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 64.Uversky V, Oldfield C, Dunker A. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 65.Metallo S. Intrinsically disordered proteins are potential drug targets. Curr Opin Chem Biol. 2010;14:481–8. doi: 10.1016/j.cbpa.2010.06.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.