

Abstract
The urokinase receptor (uPAR) is a clinically relevant target for novel biological therapies. We have previously rescued oncolytic measles viruses fully retargeted against human (MV-h-uPA) or murine (MV-m-uPA) uPAR. Here, we investigated the in vivo effects of systemic administration of MV-m-uPA in immunocompetent cancer models. MV-m-uPA induced in vitro cytotoxicity and replicated in a receptor dependent manner in murine mammary (4T1), and colon (MC-38 and CT-26) cancer cells. Intravenous administration of MV-m-uPA to 4T1 tumor bearing mice was not associated with significant clinical or laboratory toxicity. Higher MV-N RNA copy numbers were detected in primary tumors, and viable viral particles were recovered from tumor bearing tissues only. Non-tumor bearing organs did not show histological signs of viral induced toxicity. Serum anti-MV antibodies were detected at day 14 of treatment. Immunohistochemistry and immunofluorescence studies confirmed successful tumor targeting and demonstrated enhanced MV-m-uPA induced tumor cell apoptosis in treated, compared to control mice. Significant antitumor effects and prolonged survival were observed after systemic administration of MV-m-uPA in colon (CT-26) and mammary (4T1) cancer models. The above results demonstrate safety and feasibility of uPAR targeting by an oncolytic virus, and confirm significant antitumor effects in highly aggressive syngeneic immunocompetent cancer models.
Keywords: Urokinase receptor, measles virus, tumor targeting, syngeneic models
Introduction
The oncolytic virotherapy field has significantly expanded in the last decade, and recently, several novel viral vectors have reached late phase clinical evaluation (1). Among the novel oncolytic viruses under development, the Edmonston vaccine strain of measles virus (MV-Edm) is a promising one, whose safety and efficacy are well established (2, 3). In ovarian cancer, MV-CEA has demonstrated clinical safety after intraperitoneal administration (4). Recombinant, non-targeted oncolytic MVs are currently being evaluated in brain tumors, multiple myeloma and mesothelioma (1).
Redirecting MV-Edm's target specificity provides potential advantages over non-targeted viral agents, by enhancing the selectivity, and therefore, safety and efficacy of the oncolytic virus (5-8). However, the majority of published recombinant MV vectors are targeted against receptors that are present in human, but not host (rodent) tissues. This limits the ability to fully characterize issues such as safety, selectivity and antitumor efficacy of targeted oncolytic MVs in syngeneic models with an intact immune system. In order to obtain preclinical data more predictive of the potential virus-tumor-host interactions that may occur in humans, it is critical to develop viral agents directed at targets that are expressed in human and murine tumors and tissues in a similar manner.
The urokinase receptor (uPAR) is a glycosylphosphoinositol (GPI) anchored cell surface receptor whose role in tumor progression and angiogenesis is well recognized (9). It is overexpressed in a variety of human and murine cancers, and its presence has been associated with metastatic potential and poor prognosis (10-17). During normal conditions, tissue expression of uPAR is very limited, and restricted to during active tissue remodeling, injury and inflammation (9, 17-23). Genetic and biological anti-uPAR strategies have shown to induce potent antitumor effects (24-27), and agents like anti-uPAR antibodies or uPAR targeted nanoparticles successfully target tumors and micrometastases in vivo, without affecting non-tumor bearing organs (28, 29). This makes uPAR as a promising target for oncolytic viral therapies.
MV-uPA is a novel oncolytic measles virus fully retargeted against urokinase receptor (30). MV-uPA was engineered by displaying the aminoterminal fragment (ATF) of either human (MV-h-uPA) or mouse (MV-m-uPA) urokinase in the C-terminus of a CD46 and SLAM “blind” MV-H glycoprotein (HAALS) (30). The fully retargeted viruses were shown to bind to human or murine uPAR expressing cells in a receptor and species specific manner. In our previous report, we demonstrated antitumor effects of MV-h-uPA against human breast cancer xenografts (30). MV-m-uPA (which targets murine uPAR) offers the unique opportunity for in vivo characterization of the safety and antitumor effects of a fully retargeted oncolytic MV in syngeneic models of cancer, where the target is naturally expressed by tumors and tissues, similar to the human situation.
In this study, the safety, biodistribution, organ toxicity, targeting, and antitumor effects of MV-m-uPA in syngeneic, immunocompetent cancer models were investigated. As uPAR is a highly relevant human and murine tumor target, results from our in vivo studies will be useful to predict safety and efficacy during preclinical and clinical development of uPAR targeted oncolytic viral therapies.
Results
uPAR dependent in vitro cytotoxicity and viral replication in murine cancer cells
To assess differences in MV-m-uPA induced cytotoxicity in murine cancer cells with different levels of uPAR expression, receptor levels were determined in murine colon cancer (MC-38 and CT-26), murine mammary cancer (4T1) and melanoma (B16F10) cells. 4T1, MC-38, and CT-26 had increased uPAR expression compared to B16F10 cells, which had markedly less expression (Fig 1. A and Fig S. 2. A, for quantitative analysis). This was correlated with successful infection, syncytia formation (Fig. 1. B, C, and Fig. S. 1), and significantly increased (p < 0.001, compared to controls) viral induced cytotoxicity in uPAR overexpressing cells (CT-26, MC-38 and 4T1), as opposed to B16F10 cells, where the levels of infection were markedly decreased (Fig 1. D, and Fig. S. 2. B). MV-m-uPA successfully replicated in uPAR overexpressing murine cancer cells (viral titers -TCID50- at 48 and 72 hours: MC-38= 26600/6300; CT-26= 6309/199000; 4T1: 3548/11220). We observed that MV-m-uPA replicated at significantly higher levels in CT-26 cells (p <0.001), compared to 4T1 cells at 72 hours (Fig. 1.E) .
Figure 1. In vitro viral infection, cytotoxicity and replication by MV-m-uPA in murine cancer cells.
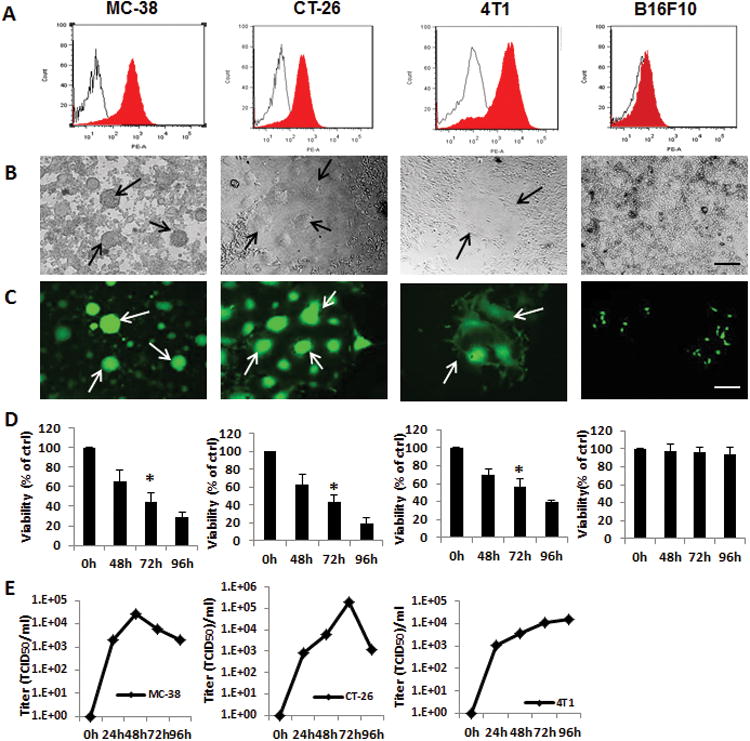
(A) uPAR expression in mouse cancer cells MC-38, CT-26, 4T1 and B16F10 was assessed by FACS, using murine anti-uPAR monoclonal antibodies (filled histograms) or isotype controls (open histograms). (B, C) Mouse cancer cells were infected with MV-m-uPA as indicated at an MOI= 1 and photographed 48 h after infection. Representative pictures of infected cells (B: light; C: fluorescence). Scale bar = 500 μm. Arrows indicate areas of virus induced syncytia. (D) In vitro cytopathic effects of MV-m-uPA. Murine cancer cells were infected with MV-m-uPA at an MOI=1 and viability was determined at different time points (48h, 72h, and 96h) by trypan blue exclusion and presented as percentage of controls. Bars represent averages +/- SD of triplicate experiments, p < 0.001. (E) MC-38, CT-26, and 4T1 cells were infected with MV-m-uPA (MOI = 3) and titers of virus were determined at different time points by the one-step growth curve.
In vivo safety and biodistribution of MV-m-uPA after intravenous administration
The orthotopic 4T1 tumor model was established in immunocompetent female Balb/C mice. Tumor bearing mice were treated with 2 doses of MV-m-uPA (1.5×106 TCID50, total dose: 3×106 TCID50) intravenously, and were sacrificed at 2, 5 and 28 days after treatment. No significant toxicity or treatment related deaths were observed throughout the study. No changes in feeding behavior or activity were observed, nor were signs of physical distress or neurotoxicity observed in treated mice.
Tumors and organs were harvested for viral biodistribution and toxicity studies. Total RNA was extracted from frozen specimens and qRT-PCR for MV-N mRNA was performed. Significantly more viral RNA was detected in tumors, compared to other organs at days 2 and 5 after treatment (Fig. 2). There was a sizeable increase in viral copy numbers in tumor tissues at day 5 compared to day 2 (p=0.0622, Fig. 2.A), strongly suggesting viral replication in the tumor. Conversely, levels of viral RNA decreased in the majority of non-tumor bearing organs from day 2 to day 5 after treatment (Figs. 2.B-F). At day 5 of treatment, tumor viral RNA copy numbers were significantly higher (p<0.003) than that for all other organs. Of note, viral RNA in the livers of one of the treated animals reached levels observed in primary tumors (6.5 ×105 TCID50) at day 2, but the levels decreased by day 5 (Fig. 2. C). By day 28 after virus administration, compared with day 2, there were significantly fewer copies of the viral RNA detectable in the lungs (p=0.0014), liver (p=0.0060), heart (p<0.001) and kidney (p<0.0001) of the MV-m-uPA treated animals.
Figure 2. In vivo biodistribution after systemic administration of MV-m-uPA.
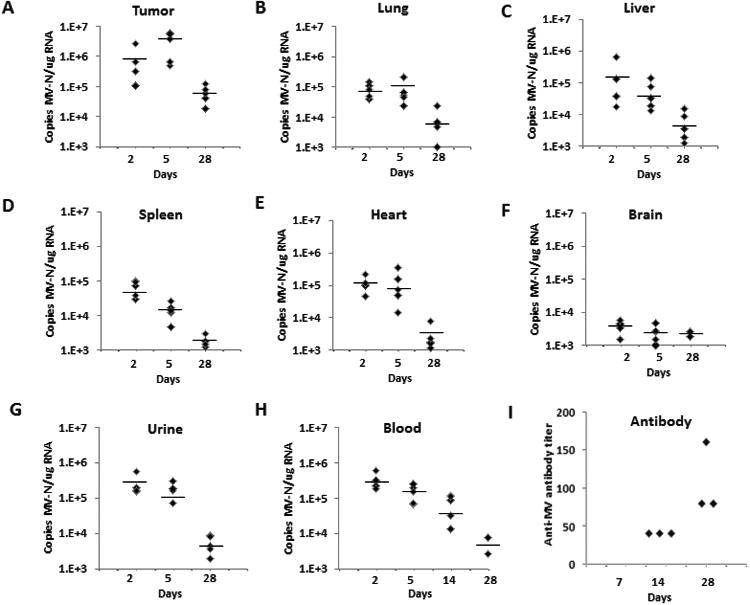
The orthotopic 4T1 tumor model was established in immunocompetent female Balb/c mice. Animals were treated and tissues processed as described in the methods section (n=5 mice per time point). At days 2, 5 and 28 days post-treatment, total RNA was extracted from frozen tumors (A), organs (B-F) and urine (G) for MV-N mRNA quantification by qRT-PCR. H. Blood samples were obtained for MV-N RNA quantification at days 2, 5, 14 and 28 after treatment (n=5 per time point). Results were expressed as copies of MV-RNA/μg of total RNA in each organ/tissue, and horizontal bars represent the mean value of the replicates. (I) Determination of serum anti-MV antibody. Serum was obtained from treated mice at 7 (no antibody detected), 14 and 28 days after treatment (n=3 per time point) for antibody determination (see methods section for details).
Viremia and antibody production
MV-m-uPA RNA was detected in the blood of treated animals at day 2, and levels significantly decreased at days 5, 14 and 28 after treatment (Fig. 2. H). At day 28, viral RNA was detected in 2/5 mice in the blood at low levels. Viral RNA was detected in the urine of treated animals at day 2, but significantly decreased at days 5 and 28 of treatment (Fig. 2. G). Serum from mice was obtained at days 7, 14 and 28 after treatment for determination of serum anti-MV antibody. While no anti-MV antibodies were detected at day 7, increasing titers were found in all mice (n=3 per group), from day 14 to day 28 (Fig 2. I).
Histologic analysis of tumors and organs of mice treated with MV-m-uPA
Five days after virus treatment, tumors and major organs were removed from treated and control (untreated) tumor bearing mice, for histological analysis (H&E). As shown in figure 3. A, significant necrosis and inflammatory changes were observed in tumors from treated animals. Lung, kidney, brain, spleen and heart did not show histological signs of viral induced toxicity. Of note, livers from some tumor bearing mice treated with MV-m-uPA had few microscopic tumor foci, and areas of focal inflammation. To further investigate these findings, and rule out the possibility of viral induced liver inflammation, we conducted additional experiments in MV-m-uPA treated and untreated tumor bearing mice, as well as in tumor free mice treated with virus. Mice were sacrificed 5 days after treatment, and livers were analyzed. No signs of inflammation or other signs of organ toxicity were observed in the livers of tumor free mice treated with MV-m-uPA (Fig. 3. B, center). In tumor bearing, untreated mice, obvious tumor foci (5/5 mice), and inflammatory changes were observed (Fig. 3. B, left). On the other hand, the prevalence of tumor foci was markedly decreased (2/5 mice) in tumor bearing, treated mice, and inflammation was not different compared to untreated mice (Fig. 3. B, right). No significant staining of viral (MV-N) protein was detected in the livers of treated mice (data no shown).
Figure 3. Histologic analysis of tumors and organs of mice treated with MV-m-uPA.
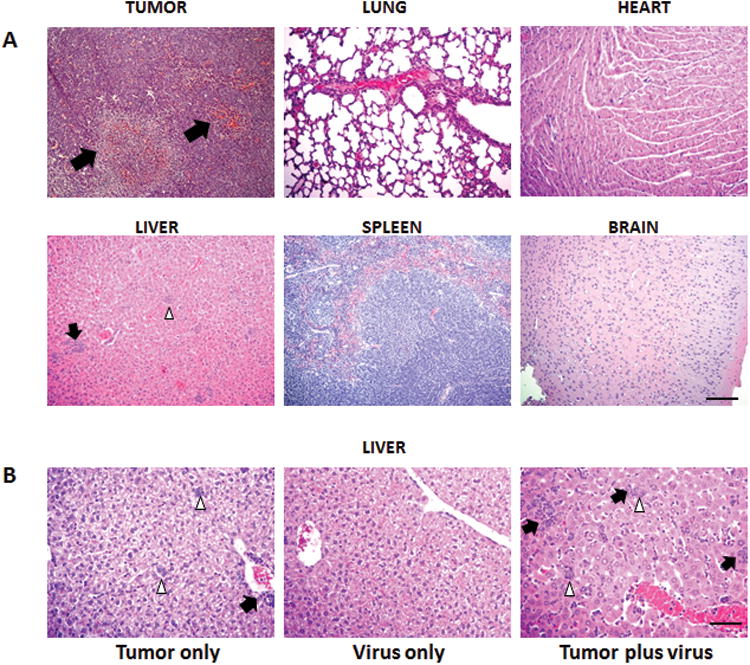
(A). 4T1 tumor bearing Balb/c mice (n = 5) were given 2 doses of 1.5×106 TCID50 of MV-m-uPA or PBS via tail vein. Mice were sacrificed 5 days after virus treatment and primary tumors and major organs (lung, heart, liver, spleen and brain) were removed for histological analysis (H&E). Arrows indicate the necrotic and inflammatory areas. White arrowheads (liver) represent tumor foci. Scale bar = 400 μm. (B). Effects of MV-m-uPA in the liver of tumor bearing and tumor free mice (n=5 per group). Virus treatment and tissue processing was performed as in methods section. Representative pictures of livers in the 3 groups. Left: Tumor bearing mice treated with PBS (micrometastases detected in 5/5 mice). Center: Tumor free mice treated with virus. Right: Tumor bearing mice treated with virus (micrometastases detected in 2/5 mice, picture is shown from a mouse with positive micrometastases). Note micrometastatic foci (white arrowheads) and areas of inflammation (black arrows). Scale bar = 200 μm.
Virus rescue, tumor targeting and induction of apoptosis
To correlate viral RNA copy numbers with presence of viable virus in tissues, virus recovery assays were performed from tumors and selected tissues at day 5 and day 28 after treatment. At day 5, no virus was rescued from lung, heart, spleen, brain samples, while virus was rescued in 4 of 5 tumor samples, and 1 of 5 liver samples in mice treated with MV-m-uPA (Fig 4. A). At day 28, MV-m-uPA was rescued from 2 of 5 tumors samples, while none of the organs tested had viable virus (Fig 4. B).
Figure 4. Tumor Targeting, antibody production, and induction of apoptosis in vivo.
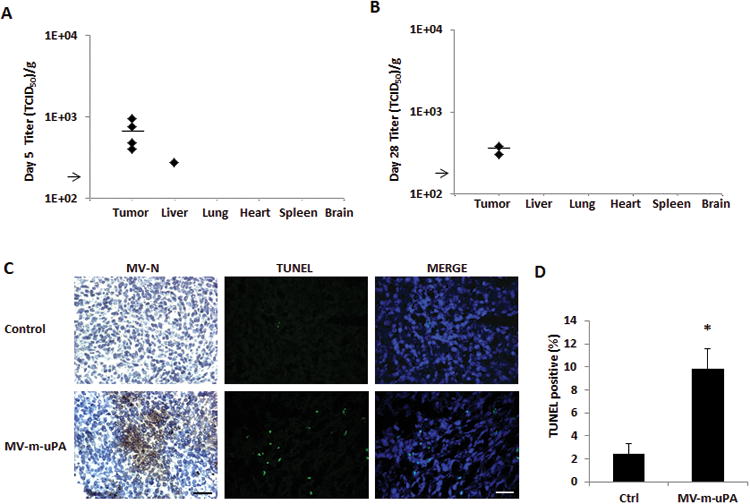
(A, B) Recovery of viable viral particles from tumor, liver, lung, heart, spleen and brain from mice at day 5 (A) and Day 28 (B) after virus treatment (n=5 mice per group). Tissues were processed and assays were preformed as in methods. Viral titers are displayed as TCID50/gram of tissue . Arrow represents the assay's limit of detection-LOD (1.26×102 TCID50/gram of tissue). (C) Immunocompetent (Balb/c) mice (n = 3 per group) bearing 4T1 tumors received two intravenous injections of either PBS or MV-m-uPA (1.5×106 TCID50). Tumors were harvested 3 days later and frozen tumor sections were used for immunostaining for measles N protein. Viral protein was detected in tumors after intravenous administration of the virus. TUNEL assay of tumors from mice treated with MV-m-uPA or PBS was performed as in materials and methods (n=3 per group). Scale bar = 200 μm. (D) Quantitative analysis of TUNEL–positive nuclei in 4 microscopic fields per section per sample (displayed as percentage of positive/total nuclei; n = 3 per group). *, P < 0.01, MV-m-uPA versus ctrl.
To further evaluate tumor targeting of the recombinant viruses after systemic administration, 4T1 tumor bearing mice were treated with MV-m-uPA, and tumors were resected 72 hours after the last injection, for IHC determination of viral proein (MV-N) in treated tumors. Viral protein was detected in the tumors of treated animals, but not in the controls (Fig. 4. C).
Next, we evaluated the effects of systemic administration of MV-m-uPA on tumor cell apoptosis (TUNEL) and proliferation (Ki67). Tumors from mice treated with MV-m-uPA had higher frequency of TUNEL positive areas, compared to untreated controls (MV-m-uPA (9.79%±1.77) vs Ctrl (2.45%±0.9), p < 0.01) (Fig. 4. C, D). No differences in tumor cell proliferation were observed between treated and control mice (not shown).
Laboratory parameters
At day 5, blood samples were obtained for complete blood count and clinical chemistry (liver and renal function) analysis. Hematology studies showed that both treated and untreated tumor bearing mice had elevated white blood cell counts, with the treated mice having higher white blood cell counts than the controls (Table 1). In addition, lymphocyte percentages were decreased in both treated and control groups, albeit the degree of relative lymphopenia was greater in virus treated mice. Calculated absolute lymphocyte counts, however were not markedly different between the two groups (5,651 ×103/microliter in treated mice, vs. 5,348 × 103/microliter in untreated mice). Otherwise, no clinically significant differences were observed in other hematological or chemistry values in treated vs. control mice. To determine if the increase in white blood cell counts was directly related to the virus treatment, a separate group of tumor free mice were treated with MV-m-uPA as described above, and complete blood counts were performed No changes in the white blood cells or lymphocytes were observed in tumor free mice treated with the virus (Supplementary table 1).
Table 1. Hematology and chemistry values in 4T1 bearing mice treated with MV-m-uPA*.
| TEST | Treatment | Normal range | Units of Measure | |
|---|---|---|---|---|
| Ctrl (vehicle) | MV-m-uPA | |||
| White Blood Cell Count | 16.37±3.27 | 24.57 ±7.40 | 4.5-9.1 | ×103/ul |
| Red Blood Cell Count | 9.93±0.37 | 9.95±0.23 | 7.51-9.66 | ×106/ul |
| Hemoglobin | 14.53±0.25 | 14.77±0.50 | 12.8-16.1 | g/dL |
| Hematocrit | 46.67.±1.15 | 46±2.65 | 34-50 | % |
| MCV | 46.67±0.58 | 46±1.73 | 41-60 | fL |
| MCH | 14.67±0.58 | 14.67±0.58 | 13-19 | pg |
| MCHC | 31.33±0.58 | 32.3±1.15 | 30-39 | % |
| Segmented Neutrophis | 64±2.65 | 71.33±2.31 | 21-57 | % |
| Band Neutrophils | 0±0 | 0±0 | 0-1 | % |
| Lymphocytes | 32.67±3.06 | 23±3.61 | 49-82 | % |
| Monocytes | 0.67±0.58 | 1.67±0.58 | 2-8 | % |
| Eosinophils | 2.33±0.58 | 4±1 | 0-3 | % |
| Basophils | 0.33±0.58 | 0 | 0-3 | % |
| NRBC | 0±0 | 0 | ||
| RBC Morphology | Normal | Normal | ||
| Platelet Morphology | Normal | Normal | ||
| WBC Morphology | Normal | Normal | ||
| Hemolysis | 2 ±0 | 1.67±0.58 | 0 | |
| Lipemia Index | 0±0 | 0±0 | 0 | |
| Glucose | 161.67±54.24 | 145±55.82 | 90-193 | mg/dL |
| BUN | 15.33±1.15 | 13.67.±1.52 | 18-29 | mg/dL |
| CREA | 0.1±0 | 0.13±0.06 | 0.1-0.4 | mg/dL |
| Calcium | 8.6±0.6 | 9.33±0.42 | 8.7-10.1 | mg/dL |
| Phosphorus | 9.37±0.25 | 8.63±0.47 | 5.4-9.3 | mg/dL |
| Total Protein | 5.2±0.1 | 5.4±0.53 | 4.6-6.9 | g/dL |
| ALT | 99.67±10.01 | 54.67±13.2 | 29-191 | U/L |
4T1 tumor bearing Balb/c mice (n = 5) were given 2 doses of 1.5×106 MV-m-uPA or PBS via tail vein as described in the methods section At day 5, blood samples were obtained for complete blood count and clinical chemistry (liver and renal function) analysis using a Hemavet 1700 multispecies hematology analyzer and an Ortho Vitros 250 analyzer, respectively.
In vivo antitumor effects
Next, the in vivo antitumor effects of systemic administration of MV-m-uPA were investigated. First, immunocompetent Balb/C mice bearing orthotropic 4T1 tumors (approximately 0.5 cm) were treated (n=8 per group) with either vehicle, or escalating doses of MV-m-uPA, as follows: A) 1×105,(total dose, divided in three individual doses of 3.33 × 104); B) 1×106 (3 doses of 3.33 ×105); C) 1×107 (3 doses of 3.33 ×106); D) 5×107 (3 doses of 1.66 ×106) TCID50, as in methods. As shown in figure 5. A, treatment with MV-m-uPA (total) doses of 1×107 and 5×107 TCID50 was associated with significant delay in tumor growth (p< 0.0001) compared to control mice. This was associated with significant prolongation of survival in mice treated with 1×107 (p =0.0001) or 5×107 (p =0.0006) TCID50 of MV-m-uPA, compared to controls (Fig. 5. B). Importantly, systemic administration of escalating doses of MV-m-uPA was not associated with acute and subacute toxicity in tumor bearing mice during or after intravenous administration.
Figure 5. In vivo antitumor effects and tumor targeting in mammary and colon cancer models.
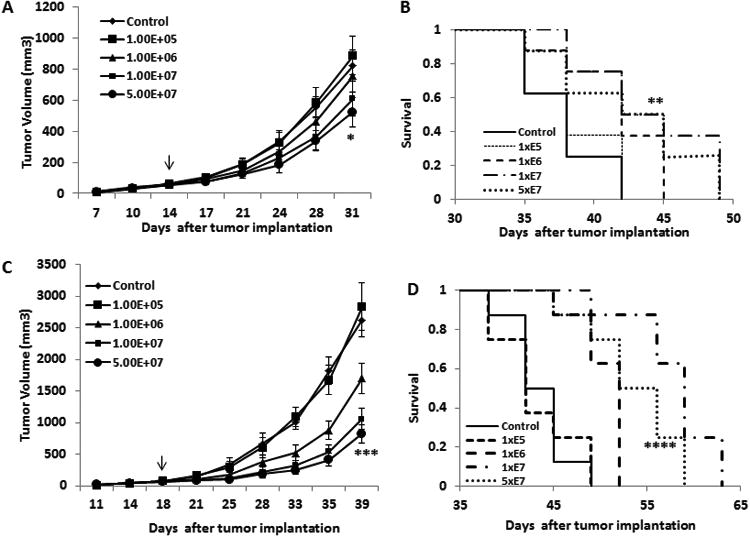
(A). 4T1 cells were implanted into the mammary fat pad of female Balb/c mice. When tumors reached a mean diameter of 0.4-0.5 cm, the animals (8 per group) were treated with total doses of 1×105, 1×106, 1×107, 5×107 TCID50 of MV-m-uPA, or equal volumes of PBS (control group). Total doses were split in three separate administrations, given via tail vein every other day × 3. Tumor growth was monitored as in methods. * p < 0.0001, day 17, MV-m-uPA (5×107) vs. control. Arrow indicates the time (day) of MV treatment, relative to time of tumor cell implantation. (B). Kaplan-Mier analysis of survival of tumor bearing mice treated with vehicle control or MV-m-uPA. Mice were monitored until they reached sacrifice criteria (see materials and methods). MV-m-uPA 5×107 vs. control ** p=0.0006. (C). Murine colon cancer (CT-26) cells were implanted into the right flank of female Balb/c mice. When the tumors reached a mean diameter of 0.4-0.5 cm, the animals (8 per group) were treated and followed as in the 4T1 model. *** p < 0.0001: day 21, MV-m-uPA (5×107) vs. control. Arrow indicates the time (day) of MV treatment, relative to time of tumor cell implantation. (D). Kaplan-Mier analysis of survival of tumor bearing mice treated with vehicle control or MV-m-uPA. There was a significant prolongation of survival in the MV-m-uPA treatment group compared with control. **** p=0.0297, MV-m-uPA 5×107 vs. control.
To further validate MV-m-uPA as a therapeutic viral vector in other immunocompetent models, we assessed the antitumor effects of MV-m-uPA in the CT-26 murine colon cancer model. CT-26 cells were implanted into the right flank of female Balb/c mice. When the tumors reached 0.5 cm, mice were treated with either vehicle, or escalating doses of MV-m-uPA, as follows: A) 1×105 (total dose, divided in three individual doses of 3.33 × 104); B) 1×106 (3 doses of 3.33 ×105); C) 1×107 (3 doses of 3.33 ×106); D) 5×107 (3 doses of 1.66 ×106) TCID50. Intravenous administration of MV-m-uPA at doses of 1×106 TCID50 and above resulted in significant (p < 0.0001) inhibition of tumor progression (Fig.5. C), and significant prolongation of survival (1×106 vs. ctrl: p =0.0256, 1×107 vs ctrl: p =0.0089, 5×107 vs ctrl: p =0.0297, Fig.5. D). No acute of subacute toxicity was observed in animals treated with escalating doses of MV-m-uPA, compared to controls.
Discussion
An important requisite for the development of a retargeted oncolytic virus from bench to bedside is the characterization of its safety, biodistribution and feasibility of systemic administration in syngeneic, immunocompetent models of cancer that resemble human malignancies. Preclinical development efforts have been made with measles viruses targeted or retargeted against CD38 (7), CD20 (31), CD133 (32), EGFRvIII (6), IL-13 (1), carcinoembrionic antigen (33), Prostate stem cell antigen (34), among others; however, characterization of preclinical safety or virus-host interactions was limited by the use of either immunodeficient xenograft models, models where the target was not expressed by host tissues, or immunocompetent models where targets were artificially expressed in murine cancer cells. Ideally, the retargeted oncolytic virus should be directed against targets that are biologically relevant and naturally expressed in murine cancers and tissues in a similar way as in humans. Under these conditions, data on safety, tissue distribution and tumor targeting may be more predictive of the human situation.
The urokinase receptor is a clinically validated and biologically important target that is overexpressed in many human as well as murine cancers (11, 13, 14, 28). Non-oncolytic viral strategies that target the uPAR have been shown to be tumor specific and to be associated with antitumor effects in vitro and in vivo (24, 25, 28). We have previously shown that human uPAR retargeted viruses (MV-h-uPA) induce significant antitumor effects in a human breast cancer xenograft model in vivo, in immunodeficient mice (30). The effects of this viral vector in syngeneic, immunocompetent cancer models have not been previously described.
In the current study, we focused on characterizing the virus-tumor-host interactions in syngeneic cancer models using the murine uPAR retargeted MV (MV-m-uPA). We demonstrated uPAR dependent in vitro cytotoxicity induced by MV-m-uPA, as well as successful in vitro viral replication in murine cancer cells that overexpress uPAR (4T1, CT-26, MC-38), while no efficacy was observed in murine melanoma cell lines (B16F10), which express low levels of uPAR.
Systemic administration of MV-m-uPA in immunocompetent mice bearing syngeneic mammary tumors was found to be safe and feasible. This was demonstrated both in the biodistribution studies (4T1 model), as well as the dose escalation studies in the efficacy studies (4T1 and CT-26 models). MV-m-uPA preferentially accumulates in tumor tissues after IV administration, compared to other organs. The observation that viral RNA copy numbers increased in tumor tissues from day 2 to day 5, while they decreased in other organs, as well as viable virus recovery in tumor tissues only strongly suggests tumor selective viral replication. Tumor targeting by MV-m-uPA was also demonstrated by IHC analysis of MV-N in treated, but not in control tumors (Fig. 4, C).
Serum biochemistry studies did not show evidence of liver or renal toxicity (Table 1). The observation that white blood cell counts were elevated in treated and untreated tumor bearing mice (table 1), but not in treated non-tumor bearing mice (supplementary table 1) suggests that changes in WBCs may be secondary to tumor burden, and not directly due to the virus treatment. As expected in immunocompetent models, an antibody response was observed in treated mice at 14 days after systemic administration of MV-m-uPA.
Correlative histopathologic analysis of the organs where viral RNA was detected showed no signs of significant organ toxicity induced by the virus. The observation that the livers of some tumor bearing mice treated with MV-m-uPA had higher viral RNA titers, viable virus (in 1/5 treated mice), and focal inflammatory changes can be explained by probable targeting by MV-m-uPA to liver micrometastases –which spontaneously develop in orthotopic 4T1 tumors- and not due to virus induced hepatotoxicity. Lack of hepatotoxicity is supported by several lines of evidence, including lack of histological signs of liver injury in tumor free mice treated with the virus (Figure 3.B) and lack of abnormalities in serum chemistry (AST) in either treated tumor free or tumor bearing mice. MV-m-uPA targeting of liver micrometastases is supported by additional experiments showing that while all (5/5) tumor bearing untreated mice had micrometastatic liver nodules, only 2/5 treated mice had metastatic foci. Studies to further characterize the abilities of MV-m-uPA to target and treat metastases in vivo are underway.
The biological significance of the findings was demonstrated by significant antitumor effects and prolonged survival in the very aggressive murine models of mammary carcinoma (4T1) and murine colon carcinoma (CT-26). TUNEL assays showed increased in vivo apoptosis in tumors treated with the retargeted virus, compared to controls. This finding provides further in vivo validation of prior in vitro studies reporting that one mechanism of oncolytic measles virus cytotoxicity is induction of apoptosis (35-37). Further studies are required, however, to elucidate the cellular, molecular, and immunological mechanisms of MV-m-uPA induced cytotoxicity in vitro and in vivo.
The in vivo antitumor effects (both antitumor response and survival) were more marked in the colon cancer than the mammary carcinoma model. The in vivo differences were correlated with the in vitro results (Fig. 1), where CT-26 cells were found to be more sensitive for viral infection and permissive for viral replication than 4T1 cells, even though uPAR expression was similar between the two cell lines. These findings may reflect differences in cellular innate antiviral responses, or in oncogenic pathways , which may render some cancer cells less sensitive to the cytotoxic effects of oncolytic viruses (38, 39). The in vitro and in vivo differences in sensitivity to MV-m-uPA between the two models (which grow in the same mouse strains -(Balb/c-), offer the opportunity to investigate mechanisms of intrinsic resistance to this and other oncolytic measles viruses, and identify strategies to improve the virus' in vitro and in vivo antitumor effects.
The results presented above make this viral vector a very useful tool to better understand the virus-tumor-host interactions in vivo, for translational and clinical development of retargeted viral agents. The demonstration of safety and tumor targeting of MV-m-uPA in immunocompetent cancer will allow further studies that will exploit the abilities of this novel agent for preclinical development of uPAR targeted viral agents in general, and MV-uPA in particular. Current efforts are focused on characterization of the tumor-host-virus interactions in regards to changes in the innate and adaptive immunity induced by the virus, and development of strategies to improve the virus' antitumor efficacy in immune competent animals. As uPAR targeting is associated with important antitumor effects, our results will lead to development of other viral vectors retargeted against the uPAR.
In conclusion, this is the first report to our knowledge of safety, feasibility and antitumor efficacy of systemic administration of an oncolytic measles virus fully redirected to a naturally expressed, biologically relevant human and murine tumor target, in immunocompetent cancer models. The promising safety and antitumor activity of this novel viral agent warrant further studies and consideration for further clinical development of uPAR targeted oncolytic viral therapies.
Materials and Methods
Virus preparation and cell culture
Construction of MV-m-uPA, virus rescue, propagation, titration, infection and in vitro cytopathic effects were performed as previously reported (30, 40). 4T1 cells (murine mammary carcinoma), MC-38 cells (murine colon carcinoma), CT-26 cells (murine colon cancer), B16F10 cells (murine melanoma) were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin and streptomycin at 37 °C and 5% CO2. Vero-αHis cells (40) were grown in DMEM containing 10% FBS at 37 °C and 5% CO2.
Flow cytometry
Mouse uPA receptor expression was detected in cancer cell lines by flow cytometry, using a Phycoerythrin (PE)-conjugated rat monoclonal anti-mouse uPAR (R&D systems, Minneapolis, MN), as previously described (30, 41).
Animal studies
Animal studies were approved by Institutional Animal Care and Use Committee of University of Miami.
Mouse biodistribution and organ toxicity studies
8-10 week old female Balb/c mice (Jackson Laboratories, Bal Harbor, ME) were injected with 1×105 4T1 cells in 50 μl PBS into the 5th mammary fat pad. When tumors reached a diameter of 0.7 cm, mice (5 mice/group/time point) were treated with MV-m-uPA at a dose of 1.5×106 TCID50 (in 100 μl PBS) via tail vein every other day for two doses, or 100μl PBS as control. At days 2, 5, and 28 after treatment, mice (n=5 per group) were euthanized. Tumors and major organs (brain, lung, heart, liver, spleen, pancreas, kidney, and ovaries) were harvested for viral RNA determination and histopathology studies. At each time point, urine were obtained before euthanasia and frozen (− 80 °C). At days 2, 5, 14 and 28 after treatment, blood was collected by retro-orbital plexus puncture.
Additional studies were conducted to compare the effects of MV-m-uPA in the livers of tumor bearing vs. non-tumor bearing mice. Three groups of mice (n=5) were established: a)Tumor free Balb/c mice treated with MV-m-uPA, 1.5×106 TCID50 (in 100 μl PBS for two doses), b) 4T1 tumor bearing Balb/c mice treated as above, and c) 4T1 tumor bearing Balb/c mice treated with PBS. At days 5 after treatment, mice were euthanized and livers were removed and fixed in 10% neutral buffered formalin, paraffin embedded, sectioned at 5 microns and stained with hematoxylin and eosin for histological analysis by a veterinary pathologist. The presence of any lesions (inflammatory, necrotic, infectious, neoplastic, etc) was assessed.
Characterization of MV-m-uPA's in vivo oncolytic effects in immunocompetent cancer murine models
Eight to 10 week old female Balb/c mice were injected with 1×105 4T1 cells into the 5th mammary fat pad. CT-26 (2×105) cells were inoculated into the right flank of Balb/c mice. When tumors reached 0.4-0.5 cm, mice were treated with either PBS (control group) or escalating (total) doses of MV-m-uPA, given intravenously -via tail vein- every other day for three doses, as follows: 1×105 TCID50 (3.33×104 per dose × 3); 1×106 TCID50 (3.33 × 105 per dose × 3); 1×107 (3.33×106 per dose × 3); TCID50 , 5×107 TCID50 (1.66 ×107 per dose × 3).
Tumor volume was measured twice a week and calculated with the following formula (Width2 × Length × 0.5). Clinical signs of toxicity were closely monitored. Tumor bearing animals were followed until they reached sacrifice criteria (when tumor burden reached 10% of body weight, if tumor ulceration occurred or mice became moribund). To evaluate tumor targeting of the recombinant viruses after systemic administration, 4T1 mammary tumor model was established as above. Tumor bearing mice (n = 3) were treated with two intravenous injections of 1.5×106 TCID50 of MV-m-uPA, and tumors were resected after 72 hours of the last injection, for immunohistochemistry studies for MV-N protein.
Viral RNA quantification
Total RNA was extracted from frozen specimens using the RNeasy tissue mini kit (for tumor, brain, lung, liver, spleen, pancreas, kidney, and ovaries) or the RNeasy fibrous tissue mini kit (Qiagen, Valencia, CA) for heart. RNA from blood samples was isolated using the QIAamp RNA blood mini kit. RNA from urine was isolated using the QIAamp viral RNA minikit (Qiagen), following the manufacturer's recommendations. qRT-PCR for MV-N(ucleoprotein) mRNA was performed as previously reported (42).
Hematology and chemistry analysis
Whole blood and serum were harvested from mice at specific timepoints, and host toxicity was evaluated by assessment of changes in hematological and biochemical parameters using a Hemavet 1700 multispecies hematology analyzer and an Ortho Vitros 250 analyzer, respectively.
Anti-MV antibody assay
Mice bearing 4T1 tumors were treated as described above, sacrificed (n=3 per time point) at 7, 14 and 28 days after treatment and serum was collected for anti-MV antibody studies. The MV-specific IgG titer was measured by an indirect immunofluorescence test using MV antigen substrate slides (Bion Enterprises, Des Plaines, IL) according to the manufacturer's instructions, and performed as described (43, 44). The protocol was modified using goat anti-mouse IgG fluorescein isothiocyanate-conjugated antibodies (Invitrogen) for detection of murine antibodies.
Virus Recovery
Tissues were weighed and homogenized in three volumes (w/v) of Opti-MEM utilizing mechanical crushing and a single freeze thaw cycle. The supernatant was clarified by centrifugation and ten-fold serial dilutions of samples were prepared in Opti-MEM. Aliquots (50 μL) of each dilution were placed in 96 well plates containing Vero-his cells and TCID50 titrations were performed. TCID50 calculations were normalized per gram of tissue.
Immunohistochemistry studies
Tissue samples were collected and frozen, and cryostat sections were fixed in cold acetone for 10 min and endogenous peroxidase activity were quenched with 0.3% H2O2 for 10 min. The slides were washed in PBS and incubated with biotinylated mouse anti-MV-nucleoprotein antibody (Chemicon International, Temecula, CA) for 30 min at 37°C. After washing in PBS, the slides were developed with VECTASTAIN ABC horseradish peroxidase (HRP) kit (Vector Laboratories) and 3, 3, 9-diaminobenzidine (DAB) HRP substrate (Vector Laboratories) according to the manufacturer's instructions.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Apoptosis was detected with an in situ cell death detection kit (Roche Applied Science, Indianapolis, IN), as previously described by us (45).
Statistical analysis
The data in this study fell into three types: 1) continuous measures where only one observation was made on the experimental unit; 2) continuous measures where there were repeated observations, usually over time, where each experimental unit had multiple observations under different conditions; and, 3) time to survival after tumor transplant. The data in type 1 were analyzed using the Analysis of Variance (ANOVA) and Multiple Linear and Non-Linear Regression. Sub-group comparisons were made after the overall analyses using the Student t-test in single degree of freedom contrasts. Analyses were performed using SAS PROC GLM and SAS PROC REG statistical programs. The data from type 2 observations were analyzed using Mixed Model ANOVAs suitable for repeated measures. Single degree of freedom contrasts were performed between and within conditions, at specific times, and across time using the methods proposed by (46). The survival data were analyzed using Kaplan-Meir stratified analyses in SAS PROC LIFETEST and the Cox Proportional Hazards model in SAS PROC PHREG. Statistical significance was set at p = 0.05 with adjustments for multiple comparisons when appropriate.
Supplementary Material
Acknowledgments
This work was supported by a research grant from the National Cancer Institute (1R01CA149659-01 to JRM, JZ, RD), and by the Sylvester Comprehensive Cancer Center (JRM).
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Supplementary information is available at GT's website.
References
- 1.Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Translational research : the journal of laboratory and clinical medicine. 2013;161:355–64. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura T, Russell SJ. Oncolytic measles viruses for cancer therapy. Expert Opin Biol Ther. 2004;4:1685–92. doi: 10.1517/14712598.4.10.1685. [DOI] [PubMed] [Google Scholar]
- 3.Russell SJ. RNA viruses as virotherapy agents. Cancer Gene Ther. 2002;9:961–6. doi: 10.1038/sj.cgt.7700535. [DOI] [PubMed] [Google Scholar]
- 4.Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–82. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen C, Paraskevakou G, Iankov I, Giannini C, Schroeder M, Sarkaria J, et al. Interleukin-13 displaying retargeted oncolytic measles virus strains have significant activity against gliomas with improved specificity. Mol Ther. 2008;16:1556–64. doi: 10.1038/mt.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen C, Vongpunsawad S, Nakamura T, James CD, Schroeder M, Cattaneo R, et al. Retargeted oncolytic measles strains entering via the EGFRvIII receptor maintain significant antitumor activity against gliomas with increased tumor specificity. Cancer Res. 2006;66:11840–50. doi: 10.1158/0008-5472.CAN-06-1200. [DOI] [PubMed] [Google Scholar]
- 7.Peng KW, Donovan KA, Schneider U, Cattaneo R, Lust JA, Russell SJ. Oncolytic measles viruses displaying a single-chain antibody against CD38, a myeloma cell marker. Blood. 2003;101:2557–62. doi: 10.1182/blood-2002-07-2195. [DOI] [PubMed] [Google Scholar]
- 8.Springfeld C, von Messling V, Frenzke M, Ungerechts G, Buchholz CJ, Cattaneo R. Oncolytic efficacy and enhanced safety of measles virus activated by tumor-secreted matrix metalloproteinases. Cancer Res. 2006;66:7694–700. doi: 10.1158/0008-5472.CAN-06-0538. [DOI] [PubMed] [Google Scholar]
- 9.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932–43. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 10.Boyd D, Florent G, Kim P, Brattain M. Determination of the levels of urokinase and its receptor in human colon carcinoma cell lines. Cancer Res. 1988;48:3112–6. [PubMed] [Google Scholar]
- 11.Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev. 2008;34:122–36. doi: 10.1016/j.ctrv.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 12.Ohba K, Miyata Y, Kanda S, Koga S, Hayashi T, Kanetake H. Expression of urokinase-type plasminogen activator, urokinase-type plasminogen activator receptor and plasminogen activator inhibitors in patients with renal cell carcinoma: correlation with tumor associated macrophage and prognosis. J Urol. 2005;174:461–5. doi: 10.1097/01.ju.0000165150.46006.92. [DOI] [PubMed] [Google Scholar]
- 13.Gutova M, Najbauer J, Gevorgyan A, Metz MZ, Weng Y, Shih CC, et al. Identification of uPAR-positive chemoresistant cells in small cell lung cancer. PLoS ONE. 2007;2:e243. doi: 10.1371/journal.pone.0000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rabbani SA, Xing RH. Role of urokinase (uPA) and its receptor (uPAR) in invasion and metastasis of hormone-dependent malignancies. Int J Oncol. 1998;12:911–20. doi: 10.3892/ijo.12.4.911. [DOI] [PubMed] [Google Scholar]
- 15.Li P, Gao Y, Ji Z, Zhang X, Xu Q, Li G, et al. Role of urokinase plasminogen activator and its receptor in metastasis and invasion of neuroblastoma. J Pediatr Surg. 2004;39:1512–9. doi: 10.1016/j.jpedsurg.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Lester RD, Jo M, Montel V, Takimoto S, Gonias SL. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol. 2007;178:425–36. doi: 10.1083/jcb.200701092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianchi E, Cohen RL, Thor AT, Todd RF, 3rd, Mizukami IF, Lawrence DA, et al. The urokinase receptor is expressed in invasive breast cancer but not in normal breast tissue. Cancer Res. 1994;54:861–6. [PubMed] [Google Scholar]
- 18.Plesner T, Ralfkiaer E, Wittrup M, Johnsen H, Pyke C, Pedersen TL, et al. Expression of the receptor for urokinase-type plasminogen activator in normal and neoplastic blood cells and hematopoietic tissue. Am J Clin Pathol. 1994;102:835–41. doi: 10.1093/ajcp/102.6.835. [DOI] [PubMed] [Google Scholar]
- 19.Romer J, Lund LR, Eriksen J, Pyke C, Kristensen P, Dano K. The receptor for urokinase-type plasminogen activator is expressed by keratinocytes at the leading edge during re-epithelialization of mouse skin wounds. J Invest Dermatol. 1994;102:519–22. doi: 10.1111/1523-1747.ep12373187. [DOI] [PubMed] [Google Scholar]
- 20.Romer J, Nielsen BS, Ploug M. The urokinase receptor as a potential target in cancer therapy. Curr Pharm Des. 2004;10:2359–76. doi: 10.2174/1381612043383962. [DOI] [PubMed] [Google Scholar]
- 21.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 22.Solberg H, Ploug M, Hoyer-Hansen G, Nielsen BS, Lund LR. The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J Histochem Cytochem. 2001;49:237–46. doi: 10.1177/002215540104900211. [DOI] [PubMed] [Google Scholar]
- 23.Uszynski M, Perlik M, Uszynski W, Zekanowska E. Urokinase plasminogen activator (uPA) and its receptor (uPAR) in gestational tissues; Measurements and clinical implications. Eur J Obstet Gynecol Reprod Biol. 2004;114:54–8. doi: 10.1016/j.ejogrb.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 24.D'Alessio S, Margheri F, Pucci M, Del Rosso A, Monia BP, Bologna M, et al. Antisense oligodeoxynucleotides for urokinase-plasminogen activator receptor have anti-invasive and anti-proliferative effects in vitro and inhibit spontaneous metastases of human melanoma in mice. Int J Cancer. 2004;110:125–33. doi: 10.1002/ijc.20077. [DOI] [PubMed] [Google Scholar]
- 25.Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS. Downregulation of uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA) inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biol. 2004;1:165–76. doi: 10.1017/s1740925x04000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gondi CS, Lakka SS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, et al. Adenovirus-mediated expression of antisense urokinase plasminogen activator receptor and antisense cathepsin B inhibits tumor growth, invasion, and angiogenesis in gliomas. Cancer Res. 2004;64:4069–77. doi: 10.1158/0008-5472.CAN-04-1243. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Lu H, Griscelli F, Opolon P, Sun LQ, Ragot T, et al. Adenovirus-mediated delivery of a uPA/uPAR antagonist suppresses angiogenesis-dependent tumor growth and dissemination in mice. Gene Ther. 1998;5:1105–13. doi: 10.1038/sj.gt.3300742. [DOI] [PubMed] [Google Scholar]
- 28.Rabbani SA, Gladu J. Urokinase receptor antibody can reduce tumor volume and detect the presence of occult tumor metastases in vivo. Cancer Res. 2002;62:2390–7. [PubMed] [Google Scholar]
- 29.Yang L, Peng XH, Wang YA, Wang X, Cao Z, Ni C, et al. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin Cancer Res. 2009;15:4722–32. doi: 10.1158/1078-0432.CCR-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jing Y, Tong C, Zhang J, Nakamura T, Iankov I, Russell SJ, et al. Tumor and vascular targeting of a novel oncolytic measles virus retargeted against the urokinase receptor. Cancer Res. 2009;69:1459–68. doi: 10.1158/0008-5472.CAN-08-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bucheit AD, Kumar S, Grote DM, Lin Y, von Messling V, Cattaneo RB, et al. An oncolytic measles virus engineered to enter cells through the CD20 antigen. Molecular therapy : the journal of the American Society of Gene Therapy. 2003;7:62–72. doi: 10.1016/s1525-0016(02)00033-3. [DOI] [PubMed] [Google Scholar]
- 32.Bach P, Abel T, Hoffmann C, Gal Z, Braun G, Voelker I, et al. Specific elimination of CD133+ tumor cells with targeted oncolytic measles virus. Cancer research. 2013;73:865–74. doi: 10.1158/0008-5472.CAN-12-2221. [DOI] [PubMed] [Google Scholar]
- 33.Ungerechts G, Springfeld C, Frenzke ME, Lampe J, Parker WB, Sorscher EJ, et al. An Immunocompetent Murine Model for Oncolysis with an Armed and Targeted Measles Virus. Mol Ther. 2007 doi: 10.1038/sj.mt.6300291. [DOI] [PubMed] [Google Scholar]
- 34.Bossow S, Grossardt C, Temme A, Leber MF, Sawall S, Rieber EP, et al. Armed and targeted measles virus for chemovirotherapy of pancreatic cancer. Cancer gene therapy. 2011;18:598–608. doi: 10.1038/cgt.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhaskar A, Bala J, Varshney A, Yadava P. Expression of measles virus nucleoprotein induces apoptosis and modulates diverse functional proteins in cultured mammalian cells. PLoS ONE. 2011;6:e18765. doi: 10.1371/journal.pone.0018765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esolen LM, Park SW, Hardwick JM, Griffin DE. Apoptosis as a cause of death in measles virus-infected cells. J Virol. 1995;69:3955–8. doi: 10.1128/jvi.69.6.3955-3958.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laine D, Bourhis JM, Longhi S, Flacher M, Cassard L, Canard B, et al. Measles virus nucleoprotein induces cell-proliferation arrest and apoptosis through NTAIL-NR and NCORE-FcgammaRIIB1 interactions, respectively. The Journal of general virology. 2005;86:1771–84. doi: 10.1099/vir.0.80791-0. [DOI] [PubMed] [Google Scholar]
- 38.Borrego-Diaz E, Mathew R, Hawkinson D, Esfandyari T, Liu Z, Lee PW, et al. Pro-oncogenic cell signaling machinery as a target for oncolytic viruses. Curr Pharm Biotechnol. 2012;13:1742–9. doi: 10.2174/138920112800958788. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen TL, Abdelbary H, Arguello M, Breitbach C, Leveille S, Diallo JS, et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14981–6. doi: 10.1073/pnas.0803988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol. 2005;23:209–14. doi: 10.1038/nbt1060. [DOI] [PubMed] [Google Scholar]
- 41.Jing Y, Kovacs K, Kurisetty V, Jiang Z, Tsinoremas N, Merchan JR. Role of plasminogen activator inhibitor-1 in urokinase's paradoxical in vivo tumor suppressing or promoting effects. Molecular cancer research : MCR. 2012;10:1271–81. doi: 10.1158/1541-7786.MCR-12-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng KW, Frenzke M, Myers R, Soeffker D, Harvey M, Greiner S, et al. Biodistribution of oncolytic measles virus after intraperitoneal administration into Ifnar-CD46Ge transgenic mice. Hum Gene Ther. 2003;14:1565–77. doi: 10.1089/104303403322495070. [DOI] [PubMed] [Google Scholar]
- 43.Iankov ID, Pandey M, Harvey M, Griesmann GE, Federspiel MJ, Russell SJ. Immunoglobulin g antibody-mediated enhancement of measles virus infection can bypass the protective antiviral immune response. J Virol. 2006;80:8530–40. doi: 10.1128/JVI.00593-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Myers RM, Greiner SM, Harvey ME, Griesmann G, Kuffel MJ, Buhrow SA, et al. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther. 2007;82:700–10. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merchan JR, Kovacs K, Railsback JW, Kurtoglu M, Jing Y, Pina Y, et al. Antiangiogenic activity of 2-deoxy-D-glucose. PLoS ONE. 2010;5:e13699. doi: 10.1371/journal.pone.0013699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Littell RC, Henry PR, Ammerman CB. Statistical analysis of repeated measures data using SAS procedures. Journal of animal science. 1998;76:1216–31. doi: 10.2527/1998.7641216x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.