

Abstract
Intratumoral heterogeneity including genetic and nongenetic mechanisms refers to biological differences amongst malignant cells originated within the same tumor. Both, cell differentiation hierarchy and stochasticity in gene expression and signaling pathways may result in phenotypic differences of cancer cells. Since a tumor consists of cancer cell clones that display distinct behaviours, changes in clonal proliferative behavior may also contribute to the phenotypic variability of tumor cells. There is a need to reveal molecular actions driving chemotherapeutic resistance in colon cancer cells. In general, it is widely hypothesized that therapeutic resistance in colorectal cancer is a consequence of the preferential survival of cancer stem cells. However, recent data regarding colorectal cancer suggest that resistance to anticancer therapy and post-therapeutic tumor reappearence could be related to variations of clonal dynamics. Understanding the interaction of genetic and nongenetic determinants influencing the functional diversity and therapy response of tumors should be a future direction for cancer research.
Keywords: Colorectal cancer, Clonal dynamics, Functional heterogeneity, Chemotherapy, Xenograft, Oxaliplatin
Core tip: Beyond genetic diversity, a complex level of nongenetic mechanisms exists and drives the intratumoral inherent functional heterogeneity of tumor cells. Recent data suggest that changes in clonal dynamics of colorectal cancer cells can lead to drug resistance and tumor reappearance.
COMMENTARY ON HOT TOPICS
Cancer is a major worldwide health problem. It exists and grows due to uncontrolled proliferation of aberrant cells that are characterized by several different morphological and pathophysiological properties. This intratumoral cellular diversity remains a major challenge in our understanding both, the cancerous process and therapeutic resistance. Cellular heterogeneity can be resulted due to genetic and nongenetic mechanisms. However, the degree of interplay between these processes and their relative involvement in cancer propagation needs to be clarified.
Intratumoral heterogeneity, in part, arises through accumulated genetic changes that, within single tumors, result in several cellular subclones with significant biological differences[1-3]. On the basis of genetic changes, cell differentiation hierarchies can also contribute to cancer cell diversity[4-6]. Similarly, resistance to antitumoral therapies can arise on the basis of genetic mutations[7,8]. Nevertheless, the results of Kreso et al[9] indicate that biological differences amongst colorectal cancer cells can be provoked by the involvement of additional, nongenetic mechanisms.
INTRATUMORAL GENETIC HETEROGENEITY
Cancer is the final result of successive genetic changes, disturbing regulatory processes and investing tumor cells with survival and growth advantages[10]. Genetic mutations lead to the selection of affected cells and their progeny[11]. Since intratumoral genetic heterogeneity is generally accompanied by variation in malignant behaviors[12], clonal genetic diversity of tumor cells has been correlated with poor prognosis[13]. In several types of cancers[3,14], heterogeneity in sequence mutations has been identified by exome sequencing of different regions of primary and metastatic tumors. These findings are of important clinical merit, given the current focus on using drugs that target specific mutant proteins and downstream signaling molecules.
INTRATUMORAL NONGENETIC HETEROGENEITY
Besides genetic changes, some nongenetic (e.g., epigenetic changes, posttranslational modifications, cell differentiation hierarchy) factors may also influence cell-cell variability within a tumor.
To explore functional equivalence of cancer cells within single genetic clones Kreso et al[9] combined deoxyribonucleic acid (DNA) copy number alteration (CNA) profiling, sequencing, and lentiviral lineage tracking, followed the repopulation dynamics of 150 single lentivirus-marked lineages from 10 human colorectal cancers over multiple serial transplantations in mouse xenografts.
DNA CNA profiling and mutational hotspot deep sequencing in 42 cancer-related genes indicated that a number of tumor xenografts preserved the genomic profile of the primary tumor, whilst in some cases substantial genetic differences were observed between the first transplant and the parental tumor, indicating the presence of clonal selection during xenograft growth. However, the latter cancer cells also remained genetically stable in subsequent transplants. Furthermore, the results of deep sequencing proved high concurrence amongst distinct single cell-derived clones. Based on these results, clonal stability of colorectal cancer cells seems to be maintained through serial tumor transplantations. Consistent with earlier results[15], Kreso et al[9] showed that xenografting itself did not select for a significantly different tumor cell population in relation of multiple recipients at each stage of serial transplantation.
In spite of the observed genetic homogeneity, the different, lentiviral marked cancer cell clones displayed different biological behaviours during serial transplantations. Persistent cell clones were present in all serial transplants. Short-term clones did not persist, but exhausted before reaching the final passage. Transient clones were only detected in the first recipient, and were not detected in the second and subsequent recipients, therefore these clones lacked tumor-propagating ability. The presence of these clones suggests that there is a substantial functional diversity with respect to clonal longevity in the course of successive tumor transplantations. Additionally, cell clones with different dynamic behaviour were also observed. Resting clones were likely produced by cancer cells that were initially dormant (or slowly proliferating), but became activated during later transplants, finally resulting in a measurable clone. Fluctuating clones consisted of cells whose progeny appeared early, but they became undetectable in a subsequent transplantation, and only to reappear later. These clones displayed intermittently extensive proliferative capacity. These results indicate that not all cancer cells having the potential for tumor propagation actually function and contribute to tumor growth at a given time. There are cells that can become activated at a later point of tumor progression. The distinct clonal proliferative kinetics observed by Kreso et al[9] underscore the functional variability of individual cells. Taking into account the absence of changes in CNAs and single nucleotide variants with serial transplantations, these data provide evidence for functional heterogeneity amongst individual tumor-propagating cells with a shared common genetic lineage. This phenotypic diversity results from the integration of both genetic and nongenetic influences. Nongenetic factors include stochasticity in gene expression, epigenetic regulation, and microenvironmental variability[16,17].
Based on the presence of functionally heterogenous cancer cell clones, Kreso et al[9] further investigated the response of these cells to oxaliplatin chemotherapy. Although the authors have found that chemotherapy reduced tumor mass, no apparent change in the absolute number of marked clones, or the proportions of clone types were observed. Tumor propagation capability of cancer cell clones was also found to be altered after oxaliplatin therapy. Despite eradication of some lentiviral marked (mainly persistent cell) clones, resting or slowly proliferating cancer cells endured oxaliplatin therapy and reinitiated tumor growth in a slower manner. The results of CNAs, single nucleotide variant and methylation pattern analyses indicated that oxaliplatin-treated cells genetically closely matched to the control recipients. These data suggest that therapeutic tolerance is not always caused by the acquisition of new driver mutations. Behalf, alterations of tumor propagation behaviour of individual cancer cells can act as a nongenetic determinant of tumor response to chemotherapy. Similarly, in human lung cancer a small subpopulation of “anti-epidermal growth factor receptor therapy-tolerant” cells were found[18]. This subpopulation of cells demonstrated a highly reduced drug sensitivity and maintained viability via engagement of insulin-like growth factor 1 receptor (IGF1R) signaling and an altered chromatin state that required histone demethylase activity. However, the drug-tolerant subpopulation could be selectively ablated by treatment with IGF1R-inhibitors or chromatin-modifying agents, potentially yielding a therapeutic opportunity.
Although Kreso et al[9] did not discover mechanisms responsible for the variability in clonal behavior, their study has several important values. It emphasizes the need of adequate understanding of nongenetic processes that underlie phenotypic heterogeneity of cancer cells.
Upon technical procedures, a shift from classical assaying techniques using bulk cell populations (and masking single-cell level heterogeneity) to newly developed/advanced methods (e.g., combining laser pressure catapulting techniques with bisulfite-based arrays, next-genome sequencing arrays or whole genome gene expression arrays) is expected. Moreover, their findings highlight on demand of efforts to reveal molecular actions driving chemotherapeutic resistance in colorectal cancer cells. In general, it is widely hypothesized that therapeutic resistance in cancer is a consequence of the preferential survival of cancer stem cells. However, the results of Kreso et al[9] suggest that cellular drug resistance and post-therapeutic tumor reappearence could not only be related to the stem-cell characteristics, but also to variations of clonal dynamics (Figure 1).
Figure 1.
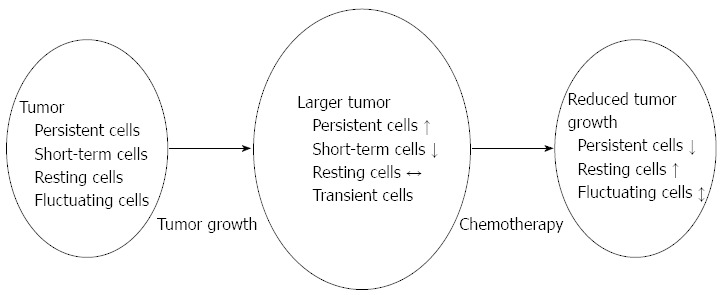
Connection between clonal behaviour and cellular constitution of the tumor. Differences in clonal behavior result in changes of the cellular constitution of colorectal cancer during normal tumor growth and after chemotherapy as well. In the case of unperturbed tumor growth the proportion of the persistent cell clone increases, short-term cells fade away, while the number of resting cells remains unchanged. Transient cells may appear but later they also fade away. After oxaliplatin therapy the number of the highly proliferative (hence chemotherapy sensitive) persistent cells significantly decreases, while the drug resistant resting cell clone contributes to tumor reappearance.
Recent findings of Kreso et al[9] reveal that, beyond genetic diversity, a complex level of nongenetic mechanisms exists and drives the intratumoral inherent functional heterogeneity of tumor cells. Thus, understanding the interaction of genetic and nongenetic determinants influencing the functional diversity and therapy response for cancers should be a prominent future direction for cancer research.
Footnotes
P- Reviewers: Chen JX, Hoensch HP, Libra M, Mickevicius A, Ma H, Pazienza V, Zhang L S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–361. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 5.Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H, Croul S, Bouffet E, Fults DW, Eberhart CG, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482:529–533. doi: 10.1038/nature10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 8.Roche-Lestienne C, Laï JL, Darré S, Facon T, Preudhomme C. A mutation conferring resistance to imatinib at the time of diagnosis of chronic myelogenous leukemia. N Engl J Med. 2003;348:2265–2266. doi: 10.1056/NEJMc035089. [DOI] [PubMed] [Google Scholar]
- 9.Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AM, Ng K, Ma J, Wienholds E, Dunant C, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–548. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138–141. doi: 10.1016/0168-9525(93)90209-z. [DOI] [PubMed] [Google Scholar]
- 11.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 12.Anaka M, Hudson C, Lo PH, Do H, Caballero OL, Davis ID, Dobrovic A, Cebon J, Behren A. Intratumoral genetic heterogeneity in metastatic melanoma is accompanied by variation in malignant behaviors. BMC Med Genomics. 2013;6:40. doi: 10.1186/1755-8794-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques RA, Rabinovitch PS, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 14.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, Traulsen A, Nowak MA, Siegel C, Velculescu VE, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci USA. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 17.Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- 18.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]