

Abstract
Erythropoiesis may be limited by absolute or functional iron deficiency or when chronic inflammatory conditions lead to iron sequestration. Intravenous iron may be indicated when oral iron cannot address the deficiency. Ferric carboxymaltose (FCM) is a nondextran iron preparation recently approved in the United States for intravenous treatment of iron deficiency anemia (IDA) in adult patients with intolerance or unsatisfactory response to oral iron or with nondialysis-dependent chronic kidney disease. The full dose is two administrations of up to 750 mg separated by at least 7 days (up to 1500 mg total). FCM can be injected in 7–8 min or diluted in saline for slower infusion. The efficacy and safety of this dose was established in two prospective trials that randomized over 3500 subjects, 1775 of whom received FCM. One trial showed similar efficacy of FCM to an approved intravenous iron regimen (1000 mg of iron sucrose) in 2500 subjects with chronic kidney disease and additional cardiovascular risk factors. The other trial showed superior efficacy of FCM to oral iron in subjects with IDA due to various etiologies (e.g. gastrointestinal or uterine bleeding). In these trials, there was no significant difference between FCM and comparator with respect to an independently adjudicated composite safety endpoint, including death, myocardial infarction, or stroke. A database of 5799 subjects exposed to FCM provided a safety profile acceptable for regulatory approval. Mechanistic studies demonstrated that the transient, asymptomatic reduction in serum phosphate observed following FCM administration results from induction of fibroblast growth factor 23, which in turn induces renal phosphate excretion. An elevated hepcidin level may identify patients with IDA who will not respond to oral iron but will respond to FCM. The ability to administer FCM in two rapid injections or infusions will likely be viewed favorably by patients and healthcare providers.
Keywords: intravenous iron, iron deficiency anemia, hepcidin
Introduction
Erythropoiesis may be limited by absolute iron deficiency (e.g. inadequate dietary intake) or functional iron deficiency (e.g. intake inadequate to accommodate an increased erythropoietic rate due to circumstances such as blood loss or treatment with an erythropoiesis-stimulating agent). Erythropoiesis may also be inhibited in the presence of adequate iron stores when chronic inflammatory conditions lead to iron sequestration [Goodnough, 2012]. Oral iron (in addition to a thorough evaluation of the cause) is usually the appropriate initial treatment for anemia due to absolute iron deficiency, but a significant proportion of patients will not respond adequately [Killip et al. 2007]. This may be due to poor compliance secondary to gastrointestinal side effects which include nausea, vomiting, and cramping. In addition, poor absorption of iron in the gastrointestinal tract occurs in conditions of chronic inflammation [which may accompany disease states contributing to iron deficiency anemia (IDA)]. Chronic inflammatory conditions lead to elevated hepcidin levels which in turn lead to the intracellular degradation of ferroportin, the transport molecule present on intestinal epithelial cells required for the transport of iron from the gut lumen into plasma [Goodnough et al. 2010]. Lastly, oral iron may be inappropriate in conditions of severe IDA in which oral iron may not be able to replenish iron stores rapidly enough to avoid blood transfusion.
For these circumstances a variety of intravenous iron preparations have been developed. All are composed of elemental iron combined with a carbohydrate that tends to reduce the biological reactivity of iron, which may cause undesired adverse effects (sometimes referred to as nontransferrin-bound iron reactions). The earliest intravenous irons utilized dextran; while effective at reducing nontransferrin-bound iron reactions they have been associated with allergic/anaphylactic reactions, which may be fatal [Faich and Strobos, 1999; Qunibi, 2010]. Subsequent iron preparations utilized nondextran carbohydrates; iron sucrose injection (Venofer, Luitpold Pharmaceuticals, Shirley, NY, USA) and ferric gluconate injection (Ferrlecit, Sanofi-Aventis, Bridgewater, NJ, USA) were designed to result in fewer hypersensitivity reactions. However, a major limitation of the second-generation intravenous iron agents is that, due to a less stable iron–carbohydrate complex, there may be toxic reactions to nontransferrin-bound iron. They therefore cannot be administered in large doses and the typical 1000 mg therapy requires several clinic visits [Qunibi, 2010]. Several retrospective studies have provided evidence that low molecular weight iron dextran may be used to treat IDA with a similar safety profile to that of iron sucrose or ferric gluconate [Auerbach and Al, 2008; Sinha et al. 2009].
Two newer iron formulations permit much higher single doses of intravenous iron to be administered over shorter periods of time. Ferumoxytol, an iron oxide with a carbohydrate coating, has demonstrated superiority over oral iron supplementation in adult patients with chronic kidney disease (CKD), and was approved by the US Food and Drug Administration (FDA) for use in CKD in 2009 [Lu et al. 2010]. The recommended dose is two 510 mg injections, 3–8 days apart.
Ferric carboxymaltose injection (FCM) (Injectafer, Luitpold Pharmaceuticals, Inc., Shirley, NY, USA; Ferinject, Vifor Pharma Ltd, St Gallen, Switzerland), the subject of this review, is a nondextran preparation which can be administered at single doses of up to 750 mg (USA) to up to 1000 mg (outside USA) over short intervals [Lyseng-Williamson and Keating, 2009; FDA, 2013c]. FCM is approved in over 50 countries, including several in Europe, Australia, and South America since its initial approval in Europe in 2007. Since its first approval in Europe more than 1 million patients have been treated with FCM. FCM (Injectafer) was approved in July 2013 by the FDA for the intravenous treatment of IDA in adult patients intolerant to oral iron or who have had unsatisfactory response to oral iron as well as in adults who have nondialysis-dependent (NDD) CKD [FDA 2013a]. The usual total dose is two administrations of up to 750 mg separated by at least 7 days (up to 1500 mg total), depending on weight. It may be administered either as an undiluted slow intravenous push at the rate of approximately 100 mg per min (i.e. over 7–8 min) or by infusion, in which case 750 mg of FCM is diluted in up to 250 ml of 0.9% sodium chloride solution and administered over at least 15 min.
The approval of FCM was delayed in the US because at the time of its initial review by the FDA in 2006–2008 there were safety concerns, including a potential imbalance of deaths in randomized controlled trials of 5:1 (FCM to comparators). Although none of the deaths were considered to be drug related, after a meeting of the Drug Safety and Risk Management Advisory Committee in February 2008 [FDA, 2013d], the FDA did not approve FCM. Subsequent to the Committee meeting, the FDA recommended that Luitpold conduct further safety and efficacy trials, preferably at a reduced dose. This review summarizes the efficacy and safety database that was compiled in response to the FDA’s recommendations, focusing on two large efficacy and safety trials.
Pivotal 750 mg FCM studies
Two trials were designed and conducted in collaboration with the Duke Clinical Research Institute and an academic steering committee to demonstrate the efficacy and safety of FCM (two doses at 15 mg/kg to a maximum of 750 mg per dose), evaluating a cross section of subjects representative of the patient population for which FCM was intended. The inclusion criteria included subjects with IDA in the setting of CKD (trial 1) or IDA in any clinical setting (trial 2). The inclusion criteria for both trials were designed to be broader than typical phase III trials of intravenous iron formulations with no exclusions for high blood pressure, asthma, human immunodeficiency virus/acquired immunodeficiency syndrome, history of exposure to a hepatitis virus in the absence of active disease, or most drug allergies (except for known hypersensitivity reactions to FCM or iron sucrose). Both trials included an independently adjudicated primary composite cardiovascular safety endpoint composed of death due to any cause, nonfatal myocardial infarction, nonfatal stroke, unstable angina requiring hospitalization, congestive heart failure requiring hospitalization or medical intervention, arrhythmias, or protocol defined hypertension or hypotension events during the 120 days following randomization. A predefined secondary composite excluded protocol-defined hypertension or hypotension events. A predefined third composite was limited to the major adverse cardiac events of death, myocardial infarction, or stroke. The components of the composite safety endpoint, which was the same for both trials, are detailed in a manuscript describing the design of trial 1 [Szczech et al. 2010].
Additional study-specific design elements and efficacy results are provided below. Pooled safety data are provided subsequently.
Trial 1: IDA in high-risk NDD CKD (REPAIR-IDA)
Trial 1 (REPAIR-IDA) was conducted in over 2500 adults with CKD and other risk factors for cardiovascular disease. Its primary objective was to evaluate the cardiovascular safety and efficacy of FCM compared with iron sucrose (1000 mg administered as five infusions of 200 mg) in subjects with IDA and an elevated risk for cardiovascular events. It was a multicenter, randomized, active-controlled, open-label study in subjects with NDD CKD. Eligible patients had IDA defined as hemoglobin (Hgb) up to 11.5 g/dl as well as ferritin up to 100 ng/ml or ferritin up to 300 ng/ml if transferrin saturation (TSAT) was 30% or less; and high-risk NDD CKD defined as either glomerular filtration rate (GFR) less than 60 ml/min/1.73 m2 or GFR less than 90 ml/min/1.73 m2 and either evidence of renal injury by urinalysis or elevated Framingham cardiovascular risk score. Subjects were randomized to FCM or iron sucrose in a 1:1 ratio. The primary efficacy endpoint was mean change in Hgb from baseline to the highest observed Hgb between baseline and day 56 or time of intervention [transfusion, use of intravenous iron outside the protocol, or increased dose of erythropoiesis-stimulating agent (ESA)]. The primary safety endpoint was the proportion of subjects experiencing at least one of the following events: death due to any cause, nonfatal myocardial infarction, nonfatal stroke, unstable angina requiring hospitalization, congestive heart failure requiring hospitalization or medical intervention, arrhythmias, hypertension or hypotension during the 120 days following randomization. Additional efficacy endpoints included the proportion of subjects achieving an increase in Hgb of at least 1 g/dl any time between baseline and day 56 or time of intervention and the mean changes from baseline to the highest observed ferritin and TSAT values any time between baseline and day 56 or time of intervention [Szczech et al. 2010].
Efficacy results
The mean age of study patients was 67 years (range 19–96); 64% were women; 54% were white people, 26% were African American, 18% Hispanics, and 2% were other races.
With regard to the primary efficacy endpoint (change in Hgb from baseline to day 56 or time of intervention), the FCM regimen was shown to be noninferior to the iron sucrose regimen. Subjects randomized to FCM in the modified intention-to-treat (mITT) population (defined as subjects who received at least one dose of randomized study medication, had at least one post baseline hemoglobin assessment) (N = 1249) had a baseline hemoglobin of 10.3 g/dl with a change to highest level at day 56 or time of intervention of 1.1 g/dl and subjects randomized to iron sucrose in the mITT population (N = 1244) had the same baseline of 10.3 g/dl with a change to highest level at day 56 or time of intervention of 0.9 g/dl. Key efficacy endpoints (Hgb, ferritin, and TSAT) are shown in Table 1. The efficacy and safety results for REPAIR IDA have been published [Onken et al. 2013b].
Table 1.
Summary of mean change in iron indices from baseline to the highest value between baseline and day 56 or time of intervention in trial 1 (REPAIR IDA; modified intent-to-treat population).
| Variable | Group A: FCM |
Group B: iron sucrose |
||||
|---|---|---|---|---|---|---|
| Baseline | Highest | Change to highest | Baseline | Highest | Change to highest | |
| Hemoglobin (g/dl) | ||||||
| N | 1249 | 1249 | 1249 | 1244 | 1244 | 1244 |
| Mean (SD) | 10.31 | 11.44 | 1.13 | 10.33 | 11.25 | 0.92 |
| Treatment difference | 0.21 | |||||
| 95% CI* | 0.13–0.28 | |||||
| Ferritin (ng/ml) | ||||||
| N | 1249 | 1249 | 1249 | 1244 | 1244 | 1244 |
| Mean (SD) | 72.67 | 807.39 | 734.73 | 75.20 | 364.09 | 288.89 |
| Treatment difference | 445.84 | |||||
| 95% CI* | 424.83–466.85 | |||||
| TSAT (%) | ||||||
| N | 1247 | 1247 | 1247 | 1243 | 1243 | 1243 |
| Mean | 19.75 | 49.22 | 29.46 | 19.60 | 35.87 | 16.27 |
| Treatment difference | 13.19 | |||||
| 95% CI* | 11.96–14.42 | |||||
Source: Onken et al. [2013b].
CI based on normal distribution, assuming equal variances.
CI, confidence interval; FCM, ferric carboxymaltose; SD, standard deviation; TSAT, transferrin saturation.
Trial 2: IDA in a broad range of patients
Trial 2 was a randomized, open-label, multicenter study of subjects with IDA in a broad range of clinical settings, including heavy uterine bleeding, gastrointestinal disorders, the postpartum state, and subjects with IDA and a history of bariatric surgery. Inclusion criteria included Hgb up to 11.0 g/dl, ferritin up to 100 ng/ml or up to 300 ng/ml if TSAT was 30% or less. Subjects who satisfied all of the inclusion and exclusion criteria [Onken et al. 2013a] entered cohort 1 or 2.
Cohort 1 was designed to assess subjects who did not respond adequately to an initial course of oral iron. The primary objective was to demonstrate the efficacy and safety of FCM compared with oral iron in subjects in cohort 1. Subjects underwent a 14-day (run-in) course of oral iron (ferrous sulfate, 325 mg, three times daily). The 14-day run-in criteria were selected based on a review of phase III studies of subjects with IDA in the postpartum period or IDA and heavy uterine bleeding or inflammatory bowel disease. It was observed that subjects who achieved an increase of less than 1 g/dl in 14 days were less likely to achieve satisfactory responses to oral iron at the end of the respective studies than those who achieved an increase of at least 1 g/dl in 14 days. Thus, subjects with an increase in Hgb of at least 1 g/dl despite compliance of 67% or more based on pill count were not randomized. All other subjects were randomized to group A, FCM (two injections of 750 mg of iron injected at a rate of 100 mg/min on days 0 and 7) or group B, oral iron for an additional 14 days.
Cohort 2 - Subjects with either a history of severe intolerance to oral iron, considered too anemic for oral iron, or who were intolerant of oral iron during the 14-day run-in period for cohort 1 were assigned to cohort 2 and randomized to group C, FCM (two injections of 750 mg injected at a rate of 100 mg/min on days 0 and 7, the same as group A) or group D, another intravenous iron [intravenous standard of care (SC)]. Intravenous SC was iron sucrose for 90% of subjects. For both cohorts, the primary efficacy endpoint was the change in Hgb from baseline to highest value between baseline and day 35 or time of intervention (transfusion, use of oral or intravenous iron outside the protocol, or increased dose of ESA) in the mITT population.
Efficacy results
In this trial, the mean Hgb increase was significantly greater in group A FCM than group B oral iron: 1.57 versus 0.80 g/dl (p = 0.001), satisfying the primary efficacy objective. A post hoc comparison of group C (FCM) and group D (intravenous SC) also demonstrated significant mean increase in Hgb from baseline to highest value by day 35 or time of intervention in group C versus group D: 2.90 versus 2.16 g/dl (p = 0.001). Changes in Hgb are summarized in table 2. The efficacy and safety results for this trial have been published [Onken et al. 2013a].
Table 2.
Summary of mean change in hemoglobin from baseline to the highest value between day 35 or time of intervention in trial 2 (modified intent-to-treat population).
| Cohort 1 |
Cohort 2 |
|||
|---|---|---|---|---|
| Group A: FCM (N = 244) | Group B: oral iron (N = 251) | Group C: FCM (N = 245) | Group D: intravenous SC* (N = 237) | |
| Baseline | ||||
| Mean (SD) | 10.59 (1.008) | 10.62 (1.033) | 9.12 (1.598) | 9.02 (1.465) |
| Highest value | ||||
| Mean (SD) | 12.16 (1.112) | 11.42 (1.181) | 12.02 (1.222) | 11.17 (1.256) |
| Change to highest value | ||||
| Mean (SD) | 1.57 (1.194) | 0.80 (0.799) | 2.90 (1.640) | 2.16 (1.252) |
| Adjusted mean (SE)* | 1.52 (0.068) | 0.76 (0.067) | 2.93 (0.072) | 2.12 (0.073) |
| p value* | 0.001 | 0.001 | ||
Source: Onken et al. [2013a].
Group A versus group B and group C versus group D from ANCOVA with treatment and etiology of IDA as fixed factors and baseline hemoglobin as a continuous covariate.
ANCOVA, analysis of covariance; FCM, ferric carboxymaltose; IDA, iron deficiency anemia; SC, standard of care; SD, standard deviation; SE, standard error.
Pooled safety results for trials 1 and 2
In trials 1 and 2 collectively, a total of 192 subjects (10.82%) in the FCM group and 172 subjects (9.65%) in the pooled comparators group met the primary composite safety endpoint. The 95% confidence interval (CI) for the difference between FCM and pooled comparators was −0.88% to 3.22%. The inclusion of zero difference in the CI provides evidence for equivalence in cardiovascular risk for FCM and pooled comparators. The pooled comparators group included subjects without CKD who received oral iron in trial 2 and were at lower cardiovascular risk than the subjects with CKD who received FCM in trial 1. The most common component of the primary composite safety endpoint was protocol-defined hypertensive events in the FCM, iron sucrose, and pooled comparator groups (5.97%, 4.13%, and 3.48% respectively). These events were transient elevations in systolic blood pressure, sometimes occurring with facial flushing, dizziness, or nausea. These elevations generally occurred immediately after dosing and resolved within 30 min. No significant clinical sequelae were associated with these elevations [Onken et al. 2013a, 2013b; FDA, 2013b].
More subjects in the pooled comparator group (47, 2.64%) experienced an event of protocol-defined hypotension, defined as a transient decrease in blood pressure, than in the FCM group (27, 1.52%). The prespecified secondary composite endpoint, which excluded protocol-defined hypertensive or hypotensive events, was more nearly equal with 72 (4.06%) subjects in the FCM group and 74 (4.15%) subjects in the pooled comparator group experiencing such events. In addition, the number of major adverse cardiac events (death, myocardial infarction, or stroke) was trending lower in the FCM group, 26 subjects (1.46%), compared with the pooled comparator group, 39 subjects (2.19%), although the difference did not reach statistical significance. Importantly, the number of deaths due to any cause was noticeably higher in the pooled comparator group (21, 1.18%) compared with the FCM group (16, 0.90%), strongly indicating that the death imbalance observed during the initial FDA review was the play of chance and that the cardiovascular safety profile of FCM is favorable. A summary of the primary composite safety endpoint for trials 1 and 2 is presented by treatment group in Table 3.
Table 3.
Components of the primary composite safety endpoint (pivotal 750 mg FCM infusion studies; trials 1 and 2).
| Composite safety endpoint | FCM (N = 1775) (n/N, %) | Pooled comparators* (N = 1783) (n/N, %) | Oral iron (N = 253) (n/N, %) | Venofer (N = 1503) (n/N, %) |
|---|---|---|---|---|
| Any | 192 (10.82) | 172 (9.65) | 4 (1.58) | 167 (11.11) |
| Components of composite endpoint$ | ||||
| Death due to any cause | 16 (0.90) | 21 (1.18) | 2 (0.79) | 19 (1.26) |
| Nonfatal myocardial infarction | 9 (0.51) | 14 (0.79) | 0 | 14 (0.93) |
| Nonfatal stroke | 3 (0.17) | 4 (0.22) | 1 (0.40) | 3 (0.20) |
| Unstable angina requiring hospitalization | 11 (0.62) | 4 (0.22) | 1 (0.40) | 3 (0.20) |
| Congestive heart failure | 38 (2.14) | 34 (1.91) | 0 | 34 (2.26) |
| Arrhythmias | 18 (1.01) | 14 (0.79) | 1 (0.40) | 13 (0.86) |
| Protocol-defined hypertensive events | 106 (5.97) | 62 (3.48) | 0 | 62 (4.13) |
| Protocol-defined hypotensive events | 27 (1.52) | 47 (2.64) | 1 (0.40) | 45 (2.99) |
| Composite endpoint, excluding protocol-defined hypotensive and hypertensive events | 72 (4.06) | 74 (4.15) | 4 (1.58) | 70 (4.66) |
| Death, myocardial infarction, or stroke | 26 (1.46) | 39 (2.19) | 3 (1.19) | 36 (2.40) |
Source: FDA [2013b].
Includes oral iron, Venofer, and other forms of intravenous iron.
Subjects may have experienced more than one component of the composite safety endpoint.
FCM, ferric carboxymaltose.
Other adverse events
Drug-related adverse reactions reported by at least 1% of treated patients in the two primary maximum 750 mg FCM infusion studies are shown in Table 4. Other adverse reactions reported by at least 0.5% of patients treated with FCM include abdominal pain, diarrhea, γ glutamyl transferase increased, injection site pain/irritation, rash, paraesthesia, and sneezing. Transient decreases in laboratory blood phosphate levels (<2 mg/dl) were observed in 27% of patients in trials 1 and 2 [FDA, 2013c]. The change in blood phosphate is described in detail below.
Table 4.
Drug-related adverse reactions reported in at least 1% of study patients in any treatment group in trials 1 and 2.
| Preferred term | FCM (N = 1775) | Pooled comparators* (N = 1783) | Oral iron (N = 253) |
|---|---|---|---|
| Nausea | 7.2 | 1.8 | 1.2 |
| Hypertension | 3.8 | 1.9 | 0.4 |
| Flushing/hot flush | 3.6 | 0.2 | 0 |
| Blood phosphorus decrease | 2.1 | 0.1 | 0 |
| Dizziness | 2.0 | 1.2 | 0 |
| Vomiting | 1.7 | 0.5 | 0.4 |
| Injection site discoloration | 1.4 | 0.3 | 0 |
| Headache | 1.2 | 0.9 | 0 |
| Alanine aminotransferase increased | 1.1 | 0.2 | 0 |
| Dysgeusia | 1.1 | 2.1 | 0 |
| Hypotension | 1.0 | 1.9 | 0 |
| Constipation | 0.5 | 0.9 | 3.2 |
Source: FDA [2013c].
Includes oral iron and all formulations of intravenous iron other than FCM.
FCM, ferric carboxymaltose.
Other trials of the 750 mg dose
In addition to trials 1 and 2, three other trials were conducted to evaluate the safety of the 750 mg dose (i.e. 15 mg/kg up to a maximum of 750 mg); two of them also evaluated efficacy. Two of these three trials compared FCM with equivalent numbers of subjects randomized to SC iron regimens (which could include any approved intravenous or oral iron regimen, at the investigator’s discretion) [Barish et al. 2012]. These two trials exposed a total of 709 subjects to FCM [as either single (N = 366) or multiple (N = 343) doses]. The third trial, with a safety database of 82 subjects exposed to FCM, compared the efficacy and safety of FCM, 750 mg to 78 subjects exposed to iron dextran injection [Hussain et al. 2013]. Collectively, the safety and efficacy findings of these trials were entirely consistent with those of trials 1 and 2, although there was no composite cardiovascular endpoint. Not surprisingly, there were more hypersensitivity-related reactions in subjects randomized to iron dextran injection (10.3%) compared with subjects randomized to FCM (0%) [Hussain et al. 2013].
Pooled phase II/III database
In addition to the five trials that evaluated the 750 mg dose, 15 additional short-term phase II or phase III trials were conducted, several of which are published. These trials included subjects with IDA in association with CKD [Charytan et al. 2013; Qunibi et al. 2011], heavy uterine bleeding [Van Wyck et al. 2009], gastrointestinal disease [Evstatiev et al. 2011; Kulnigg et al. 2008], and the postpartum state [Seid et al. 2008; Van Wyck et al. 2007], as well as subjects with iron deficiency and chronic heart failure (CHF) [Anker et al. 2009]. In some of these trials, subjects were administered up to 1000 mg in a single dose. The total short-term phase II/III database of 20 trials, including the five trials with the 750 mg dose, evaluated a pooled safety database of 5799 subjects exposed to FCM, a larger database than for any other intravenous iron formulation reported to date. Drug-related adverse reactions reported by at least 1% of treated patients are shown in Table 5. In this larger database the event rates are compared between subjects exposed to FCM and subjects exposed to oral iron, any intravenous iron, or pooled comparators (includes any intravenous iron formulation available at the time the trials were conducted, i.e. iron sucrose, ferric gluconate, or low or high molecular weight iron dextran, or oral iron as well as placebo). As described above, intravenous iron is used in clinical situations when oral iron is inadequate or inappropriate, and its adverse event profile is known to be different than that of oral iron. When comparing FCM to other intravenous iron formulations, the rates of nausea, injection site reactions (i.e. discoloration, extravasation, or pain), headache, hypertension, dizziness, vomiting and diarrhea are fairly similar. The rates of dysgeusia and hypotension are lower in the FCM group compared with the other intravenous iron group. However, the rates of decreased blood phosphate, flushing, and increased alanine aminotransferase are higher in the FCM group than the other intravenous iron group.
Table 5.
Drug-related treatment-emergent adverse events (≥1% in the FCM group) (pooled phase II/III database).
| Preferred term | FCM (N = 5799) | Pooled comparators (N = 5272) | Oral iron (N = 2497) | Any intravenous iron (N = 2439) |
|---|---|---|---|---|
| Nausea | 3.1 | 2.8 | 2.5 | 2.2 |
| Blood phosphorus decreased | 1.9 | 2 | 0 | 0 |
| Injection site reaction* | 1.6 | 0.7 | 0 | 1.8 |
| Headache | 1.4 | 1.1 | 1.1 | 1.3 |
| Hypertension | 1.3 | 0.7 | 0.1 | 1.4 |
| Dizziness | 1.2 | 0.8 | 0.3 | 1.3 |
| Flushing | 1.0 | 0.1 | 0 | 0.2 |
| Alanine aminotransferase increased | 1.0 | 0.5 | 0.8 | 0.2 |
| Dysgeusia | 0.9 | 1.0 | 0.2 | 2.0 |
| Constipation | 0.8 | 4.1 | 8.0 | 0.4 |
| Vomiting | 0.7 | 0.9 | 1.0 | 0.8 |
| Diarrhea | 0.5 | 1.1 | 1.6 | 0.7 |
| Hypotension | 0.5 | 0.8 | 0 | 1.7 |
Source: FDA [2013b].
Injection site reaction includes injection site discoloration, injection site extravasation, or injection site pain.
FCM, ferric carboxymaltose.
Postmarketing experience
FCM is currently marketed in approximately 40 countries, most prevalently in Europe, and has approval in over 50 countries. The following serious adverse reactions have been most frequently reported from the postmarketing spontaneous reports with FCM: urticaria, dyspnea, pruritis, tachycardia, erythema, pyrexia, chest discomfort, chills, angioedema, back pain, arthralgia, and syncope [FDA, 2013c].
Hypersensitivity reactions
Hypersensitivity reactions, including some which are fatal, are adverse events that occur to some extent for all intravenous iron formulations [European Medicines Agency, 2013; Wysowski et al. 2010]. So far, a single case of fatality has been associated with FCM [FDA, 2013b]. Standard warning text is required by the FDA to be included in all the prescribing information of all intravenous irons marketed in the USA, advising that patients may present with shock, clinically significant hypotension, loss of consciousness, or collapse. The text states it is necessary to monitor patients for signs and symptoms of hypersensitivity during and after intravenous iron administration for at least 30 min and until clinically stable following completion of the infusion. In addition, it is stated to only administer such agents when personnel and therapies are immediately available for the treatment of serious hypersensitivity reactions. Similarly, the European Medicines Agency recently completed a review of postmarketing safety data for intravenous irons (including FCM) and concluded that all intravenous irons have a small risk of causing allergic reactions which can be life threatening if not treated promptly [European Medicines Agency, 2013].
In the two primary FCM 750 mg trials, serious anaphylactic/anaphylactoid reactions were reported in 0.1% (2/1775) of subjects receiving FCM and 0.1% (1/1783) of subjects receiving a comparator. Other serious or severe adverse reactions potentially associated with hypersensitivity included, but were not limited to, pruritus, rash, urticaria, wheezing, or hypotension and were reported in 1.5% (26/1775) of subjects; the same rate of 1.5% (26/1783) was observed in the pooled comparator group. In the larger pooled phase II/III database the rate for FCM was 0.9% (51/5799) compared with 0.8% (42/5272) for all comparators combined, and 1.5% (37/2439) for the other intravenous iron comparators [FDA, 2013b].
Hypertension
As described above, protocol-defined hypertensive events were the most frequent events in the adjudicated composite endpoint of the primary 750 mg infusion trials, and were found to be significantly higher in the FCM group compared with comparator groups (Table 3). These events included elevated blood pressure readings taken during the observation period immediately following study drug administration and adverse events of elevated blood pressure (i.e. necessitating an unscheduled healthcare visit) that could be reported any time after the subject left the study site. Most of the events in the FCM group occurred during the observation period immediately following study drug administration and most resolved within 30 min. The adverse event of hypertension was also reported more frequently in the FCM group than the pooled comparator group in trials 1 and 2 (3.8% versus 1.9%, Table 4). It was also greater for FCM than pooled comparators in the pooled phase II/III database (1.3% versus 0.7%, Table 5), although the rate was similar to that of FCM in the pooled IV iron group (1.4%, Table 5). The FCM prescribing information recommends monitoring patients for signs and symptoms of hypertension following each FCM administration [FDA, 2013c].
Injection site reactions
Injection site reactions (including injection site discoloration, injection site extravasation, or injection site pain) may occur with any intravenous iron. Such reactions were reported in 1.6% of subjects receiving FCM in the pooled phase II/III database compared with 1.8% of subjects receiving any other intravenous iron (Table 5). These discolored regions may be long lasting (several months) and can be a cosmetic concern. It has been found that the incidence of injection site discoloration after FCM administration may be greatly reduced by the practice of flushing the infusion catheter with saline before withdrawing the needle to avoid dribbling of FCM into the subcutaneous tissue.
Transient reductions in blood phosphate levels
A consistent finding in clinical trials of ferric carboxymaltose has been reductions in serum phosphate levels. The serum phosphate level normally ranges from 2.5 to 4.5 mg/dl (0.81–1.45 mmol/liter) in adults. Clinical sequelae of low serum phosphate are rarely observed unless the phosphate level is approximately 1 mg/dl (0.3 mmol/liter) or lower. The complications associated with phosphate levels lower than 1 mg/dl for 2 or more days may include rhabdomyolysis, respiratory failure, acute hemolytic anemia, arrhythmias, and death [Liamis et al. 2010]. In the two primary 750 mg studies, 26.9% of subjects exposed to FCM experienced reductions in serum phosphate from normal levels to levels less than 2 mg/dl compared with 0.7% in those exposed to iron sucrose or pooled comparators. The median number of days to first value below the normal range was 14 days in the FCM, iron sucrose, and pooled comparators groups. Additionally, among subjects whose phosphate value returned to within reference range, number of median days to recovery was 21 days in the FCM group and 14 days in the iron sucrose and pooled comparators groups. None of the low phosphate values in either trial were associated with serious adverse events [FDA, 2013b]. In nearly all cases there was no clinical sign except for the low laboratory value. In a single literature report, a 55-year-old women with chronic anemia who received six injections of FCM (500–1000 mg/dose) over 9 months was diagnosed with hypophosphatemic osteomalacia which resolved after discontinuation of FCM [Moore et al. 2013].
The cause of low phosphate following FCM has been speculated to possibly result from increased cellular uptake of phosphate by hematopoietic cells during accelerated erythropoiesis [Van Wyck et al. 2007]. However, a randomized clinical trial was conducted by the sponsor in which women with IDA due to heavy uterine bleeding were treated with 1000 mg of either FCM or iron dextran. Hemoglobin levels were increased to similar degrees in both groups. However, only FCM-treated subjects experienced decreases in phosphate, indicating that the phenomenon is due to something unique to FCM, possibly the carbohydrate moiety of the molecule [Wolf et al. 2013]. The study showed that levels of fibroblast growth factor 23 (FGF23), an osteocyte-derived hormone that regulates phosphate and vitamin D homeostasis, became transiently increased in the FCM group alone, which was followed by increased urinary fractional excretion of phosphate, decreased calcitriol levels, and increased parathyroid hormone levels, as well as a transient, asymptomatic reduction in serum phosphate to levels less than 2.0 mg/dl. These data appear to confirm the hypothesis suggested by others that certain iron preparations induce levels of FGF23 which induces transient renal excretion of phosphate [Schouten et al. 2009].
The trial by Wolf and colleagues also revealed that in situations of IDA (i.e. before treatment with intravenous iron when the average baseline Hgb level was 9.5–9.8 mg/dl) subjects had remarkably elevated levels of FGF23, but only when measured with an antibody that recognized the C-terminal moiety of the molecule (cFGF23). Levels of FGF23 measured with an antibody that recognizes only full-length, intact FGF23 molecule (iFGF23), demonstrated normal levels at baseline. The levels of iFGF23 dropped transiently only after treatment with FCM. These data support a model, suggested by others [Imel et al. 2011], that IDA leads to an increase in the secretion of FGF23 by osteocytes, possibly by inducing its transcription. Levels of cFGF23 accumulate but levels of iFGF23 remain the same, possibly due to a counterbalancing post-translational cleavage of the FGF23 protein. Treatment with intravenous iron may inhibit transcription of the fgf23 gene so cFGF23 protein levels decrease, but only certain intravenous iron preparations, such as FCM, appear to also inhibit the post-translational cleavage of FGF23 protein so that full-length FGF23 (iFGF23) transiently accumulates. This model is illustrated in Figure 1.
Figure 1.

Hypothesis for differential effects of iron deficiency and its correction with iron dextran or FCM on regulation of iFGF23 and cFGF23 levels. (a) fgf23 transcription in osteocytes is upregulated by iron deficiency, but a counterbalancing increase in post-translational FGF23 cleavage maintains normal net production of intact protein. Increased fgf23 transcription accompanied by increased intracellular FGF23 cleavage results in markedly elevated levels of FGF23 fragments that are detectable by the C-terminal assay. (b) Correction of iron deficiency with iron dextran restores normal fgf23 transcription, thereby decreasing production of FGF23 fragments while maintaining normal production of intact protein. (c) Correction of iron deficiency with FCM restores normal fgf23 transcription, but production of intact FGF23 protein increases nevertheless, perhaps because of a greater magnitude of concomitant inhibition of FGF23 cleavage by the carbohydrate moiety of FCM. Reproduced from original source [Wolf et al. 2013]. cFGF23, C-terminal moiety of fibroblast growth factor 23; FCM, ferric carboxymaltose; FGF, fibroblast growth factor 23; iFGF23, intact fibroblast growth factor 23.
Hepcidin levels may identify patients who will not respond to oral iron
As described above, IDA may be treated with oral iron, which is inexpensive and not associated with the risk of serious hypersensitivity reactions. However, many patients may not respond adequately to restore their hemoglobin levels to the normal range or avoid a blood transfusion, either because oral iron could not restore the iron levels fast enough or because the absorption of iron from the gastrointestinal tract is inadequate [Weiss and Goodnough, 2005]. Transport of orally ingested iron from the gut lumen to the circulation requires the transmembrane molecule, ferroportin. Intracellular levels of this transporter are downregulated when hepcidin levels become elevated, which occurs when iron stores are adequate or high (i.e. negative feedback) or in response to inflammatory cytokines such as interleukin 6 and C-reactive protein [Nemeth, 2010]. Inflammatory cytokines similarly downregulate ferroportin in reticuloendothelial cells responsible for the recycling of stored iron [Goodnough, 2012]. Intravenously administered iron may still be clinically useful under such circumstances because it may be transported directly to erythropoietic cells bound to molecules such as transferrin without the need to enter or exit reticuloendothelial cells.
Trial 2 was designed to evaluate FCM in subjects with IDA of any etiology who did not demonstrate a 1 g/dl increase in Hgb after the 14-day oral iron run-in period (with documented oral iron compliance ≥67%) [Onken et al. 2013a]. It also provided the opportunity to test whether baseline hepcidin level could be used to prospectively identify subjects unlikely to respond adequately to oral iron [Bregman et al. 2013]. Based on hepcidin values recorded from 240 subjects, it was shown that a baseline hepcidin level of over 20 ng/ml, predicting unresponsiveness to oral iron with a sensitivity of 41.3%, a specificity of 84.4%, and a positive predictive value (PPV) of 81.6%. PPVs for baseline ferritin greater than 30 ng/ml or baseline TSAT greater than 15% were 59.2% and 55% respectively. Negative predictive values for hepcidin, ferritin, and TSAT were 46.3%, 22.7%, and 19.7% respectively. A receiver-operator analysis demonstrated that hepcidin was a superior predictor of unresponsiveness to oral iron than ferritin or TSAT [i.e. only the hepcidin curve was above the line defined by X (1 − specificity) =Y (sensitivity); Figure 2] [Bregman et al. 2013]. Nonresponse to oral iron therapy does not rule out responsiveness to intravenous iron: subjects who did not respond adequately to the 14-day oral iron run in who were randomized to intravenous iron still demonstrated a robust response to FCM, with Hgb increases of 1.7 ± 1.3, compared with a Hgb increase of 0.6 ± 0.9 g/dl for subjects randomized to an additional 14 days of oral iron treatment (p = 0.0025).
Figure 2.
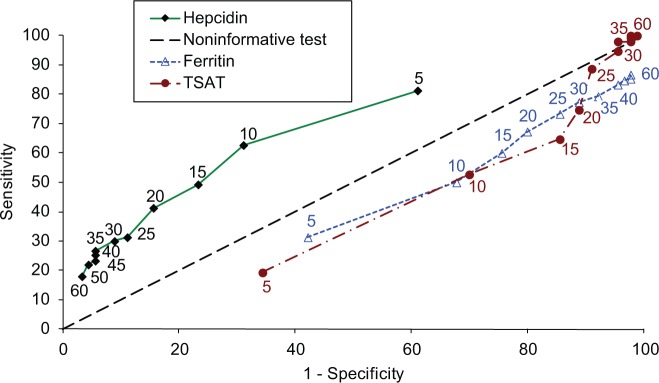
ROC curve for three potential predictors of response to oral iron in subjects with IDA (study 2). The ROC curves present plots of sensitivity versus (1 − specificity) for hepcidin (ng/ml), ferritin (ng/ml), and TSAT (%) utilizing data from study 2 at the various cutoff levels indicated near the respective curves in the same color as the respective curves. Reproduced from original source [Bregman et al. 2013]. IDA, iron deficiency anemia; ROC, receiver-operating characteristic; TSAT, transferrin saturation.
Conclusion
FCM has been available in Europe since its approval in 2007 and it is currently marketed in over 40 countries in Europe and elsewhere. Approval in the USA was delayed due to an imbalance in deaths that existed at the time of its initial review by the FDA in 2006–2008. Since that time several additional trials have been completed, including two trials exposing over 3500 subjects with IDA to FCM (two doses of 15 mg/kg up to a maximum single dose of 750 mg administered at least 7 days apart for a total of up to 1500 mg). These two trials, which included an independently adjudicated composite cardiovascular endpoint, confirmed the safety of FCM for the treatment of IDA in a variety of clinical settings. There was no imbalance of deaths in these two new trials or in the complete database, which included 5799 subjects exposed to FCM. Taken together, the clinical trial data for FCM support its ability to treat IDA in adult patients for whom oral iron is either not tolerated or is not effective and in adult patients with CKD. The management of IDA should focus on identifying and if possible treating the underlying cause. In many cases oral iron may be the appropriate adjunct for restoring iron and hemoglobin levels. Frequently, however, oral iron may not be able to restore iron levels rapidly enough to prevent the need for a blood transfusion, or oral iron may not be tolerated or may not be absorbed appropriately from the gastrointestinal tract. For example, when anemia is accompanied by chronic inflammation, elevated levels of hepcidin may induce the proteolytic degradation of ferroportin molecules which are necessary for transporting iron from the gastrointestinal tract to the circulation. Evidence exists that hepcidin may provide a useful clinical tool for identifying such patients who will never respond adequately to oral iron. Preliminary evidence also exists that FCM may be useful for treating iron deficiency in other clinical situations, including restless legs syndrome and CHF. The ability to treat IDA that is unresponsive to oral iron in a broad range of patients in two rapid administrations is likely to increase patient compliance and reduce the expenditure of healthcare resources (e.g. infusion chair time).
Footnotes
Funding: The clinical trials reviewed in this article were funded by Luitpold Pharmaceuticals.
Conflict of interest statement: David B. Bregman was an employee of Luitpold Pharmaceuticals, Inc. when trials 1 and 2 were conducted. Lawrence T. Goodnough is a consultant for Luitpold Pharmaceuticals, Inc.
Contributor Information
David B. Bregman, Department of Pathology, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA
Lawrence T. Goodnough, Departments of Pathology and Medicine, Stanford University School of Medicine, Stanford, CA, USA
References
- Anker S., Comin C., Filippatos G., Willenheimer R., Dickstein K., Drexler H., et al. (2009) Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med 361: 2436–2448 [DOI] [PubMed] [Google Scholar]
- Auerbach M., Al T. (2008) Low-molecular weight iron dextran and iron sucrose have similar comparative safety profiles in chronic kidney disease. Kidney Int 73: 528–530 [DOI] [PubMed] [Google Scholar]
- Barish C., Koch T., Butcher A., Morris D., Bregman D. (2012) Safety and efficacy of intravenous ferric carboxymaltose (750 mg) in the treatment of iron deficiency anemia: two randomized, controlled trials. Anemia 2012: 172104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregman D., Morris D., Koch T., He A., Goodnough L. (2013) Hepcidin levels predict nonresponsiveness to oral iron therapy in patients with iron deficiency anemia. Am J Hematol 88: 97–101 [DOI] [PubMed] [Google Scholar]
- Charytan C., Bernardo M., Koch T., Butcher A., Morris D., Bregman D. (2013) Intravenous ferric carboxymaltose versus standard medical care in the treatment of iron deficiency anemia in patients with chronic kidney disease: a randomized, active-controlled, multi-center study. Nephrol Dial Transplant 28: 953–964 [DOI] [PubMed] [Google Scholar]
- European Medicines Agency (2013) New recommendations to manage risk of allergic reactions with intravenous iron-containing medicines. http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2013/06/WC500144874.pdf (accessed 9 January 2013).
- Evstatiev R., Marteau P., Iqbal T., Khalif I., Stein J., Bokemeyer B., et al. (2011) FERGIcor, a randomized controlled trial on ferric carboxymaltose for iron deficiency anemia in inflammatory bowel disease. Gastroenterology 141: 846–853 [DOI] [PubMed] [Google Scholar]
- Faich G., Strobos J. (1999) Sodium ferric gluconate complex in sucrose: safer intravenous iron therapy than iron dextrans. Am J Kidney Dis 33: 464–470 [DOI] [PubMed] [Google Scholar]
- FDA (US Food and Drug Administration) (2013a) Injectafer (Ferric carboxymaltose) approval letter. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/203565Orig1s000Approv.pdf (accessed 9 January 2013).
- FDA (US Food and Drug Administration) (2013b) Injectafer (ferric carboxymaltose) medical review. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/203565Orig1s000MedR.pdf (accessed 9 January 2013).
- FDA (US Food and Drug Administration) (2013c) Injectafer (ferric carboxymaltose) prescribing information. http://wwwaccessdatafdagov/drugsatfda_docs/nda/2013/203565Orig1s000Lblpdf (accessed 9 January 2013).
- FDA (US Food and Drug Administration) (2013d) Summary minutes of the Drug Safety and Risk Management Advisory Committee, 1 February 2008. http://www.fda.gov/ohrms/dockets/ac/08/minutes/2008–4337m1-Final.pdf ( accessed 18 January 2014).
- Goodnough L. (2012) Iron deficiency syndromes and iron-restricted erythropoiesis (CME). Transfusion 52: 1584–1592 [DOI] [PubMed] [Google Scholar]
- Goodnough L., Nemeth E., Ganz T. (2010) Detection, evaluation, and management of iron-restricted erythropoiesis. Blood 116: 4754–4761 [DOI] [PubMed] [Google Scholar]
- Hussain I., Bhoyroo J., Butcher A., Koch T., He A., Bregman D. (2013) Direct comparison of the safety and efficacy of ferric carboxymaltose versus iron dextran in patients with iron deficiency anemia. Anemia 2013: 169107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imel E., Peacock M., Gray A., Padgett L., Hui S., Econs M. (2011) Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab 96: 3541–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killip S., Bennett J., Chambers M. (2007) Iron deficiency anemia. Am Fam Physician 75: 671–678 [PubMed] [Google Scholar]
- Kulnigg S., Stoinov S., Simanenkov V., Dudar L., Karnafel W., Garcia L., et al. (2008) A novel intravenous iron formulation for treatment of anemia in inflammatory bowel disease: the ferric carboxymaltose (FERINJECT) randomized controlled trial. Am J Gastroenterol 103: 1182–1192 [DOI] [PubMed] [Google Scholar]
- Liamis G., Milionis H., Elisaf M. (2010) Medication-induced hypophosphatemia: a review. QJM 103: 449–459 [DOI] [PubMed] [Google Scholar]
- Lu M., Cohen M., Rieves D., Pazdur R. (2010) FDA report: ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am J Hematol 85: 351–319 [DOI] [PubMed] [Google Scholar]
- Lyseng-Williamson K., Keating G. (2009) Ferric carboxymaltose: a review of its use in iron-deficiency anaemia. Drugs 69: 739–756 [DOI] [PubMed] [Google Scholar]
- Moore K., Kildahl-Andersen O., Kildahl-Andersen R., Tjonnfjord G. (2013) Uncommon adverse effect of a common medication. Tidsskr Nor Laegeforen 133: 165. [DOI] [PubMed] [Google Scholar]
- Nemeth E. (2010) Targeting the hepcidin-ferroportin axis in the diagnosis and treatment of anemias. Adv Hematol 2010: 750643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken J., Bregman D., Harrington R., Morris D., Acs P., Akright B., et al. (2013a) A multicenter, randomized, active-controlled study to investigate the efficacy and safety of intravenous ferric carboxymaltose in patients with iron deficiency anemia. Transfusion 17 June (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- Onken J.E., Bregman D., Harrington R., Morris D., Buerkert J., Hamerski D., et al. (2013b) Ferric carboxymaltose in patients with iron-deficiency anemia and impaired renal function: the REPAIR-IDA trial. Nephrol Dial Transplant 20 August (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- Qunibi W. (2010) The efficacy and safety of current intravenous iron preparations for the management of iron-deficiency anaemia: a review. Arzneimittelforschung 60: 399–412 [DOI] [PubMed] [Google Scholar]
- Qunibi W., Martinez C., Smith M., Benjamin J., Mangione A., Roger S. (2011) A randomized controlled trial comparing intravenous ferric carboxymaltose with oral iron for treatment of iron deficiency anaemia of non-dialysis-dependent chronic kidney disease patients. Nephrol Dial Transplant 26: 1599–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten B., Hunt P., Livesey J., Frampton C., Soule S. (2009) FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: a prospective study. J Clin Endocrinol Metab 94: 2332–2337 [DOI] [PubMed] [Google Scholar]
- Seid M., Derman R., Baker J., Banach W., Goldberg C., Rogers R. (2008) Ferric carboxymaltose injection in the treatment of postpartum iron deficiency anemia: a randomized controlled clinical trial. Am J Obstet Gynecol 199: 435–437 [DOI] [PubMed] [Google Scholar]
- Sinha S., Chiu D., Peebles G., Kolakkat S., Lamerton E., Fenwick S., et al. (2009) Comparison of intravenous iron sucrose versus low-molecular-weight iron dextran in chronic kidney disease. J Ren Care 35: 67–73 [DOI] [PubMed] [Google Scholar]
- Szczech L., Bregman D., Harrington R., Morris D., Butcher A., Koch T., et al. (2010) Randomized evaluation of efficacy and safety of ferric carboxymaltose in patients with iron deficiency anaemia and impaired renal function (REPAIR-IDA): rationale and study design. Nephrol Dial Transplant 25: 2368–2375 [DOI] [PubMed] [Google Scholar]
- Van Wyck D., Mangione A., Morrison J., Hadley P., Jehle J., Goodnough L. (2009) Large-dose intravenous ferric carboxymaltose injection for iron deficiency anemia in heavy uterine bleeding: a randomized, controlled trial. Transfusion 49: 2719–2728 [DOI] [PubMed] [Google Scholar]
- Van Wyck D., Martens M., Seid M., Baker J., Mangione A. (2007) Intravenous ferric carboxymaltose compared with oral iron in the treatment of postpartum anemia: a randomized controlled trial. Obstet Gynecol 110: 267–278 [DOI] [PubMed] [Google Scholar]
- Weiss G., Goodnough L. (2005) Anemia of chronic disease. N Engl J Med 352: 1011–1023 [DOI] [PubMed] [Google Scholar]
- Wolf M., Koch T., Bregman D. (2013) Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res 28: 1793–1803 [DOI] [PubMed] [Google Scholar]
- Wysowski D., Swartz L., Borders-Hemphill B., Goulding M., Dormitzer C. (2010) Use of parenteral iron products and serious anaphylactic-type reactions. Am J Hematol 85: 650–654 [DOI] [PubMed] [Google Scholar]