

Abstract
Disruptions of endoplasmic reticulum (ER) Ca2+ homeostasis are heavily linked to neuronal pathology. Depletion of ER Ca2+ stores can result in cellular dysfunction and potentially cell death, although adaptive processes exist to aid in survival. We examined the age and region dependence of one postulated, adaptive response to ER store-depletion (SD), hyperpolarization-activated cation-nonspecific (h)-channel plasticity in neurons of the dorsal and ventral hippocampus (DHC and VHC, respectively) from adolescent and adult rats. With the use of whole-cell patch-clamp recordings from the soma and dendrites of CA1 pyramidal neurons, we observed a change in h-sensitive measurements in response to SD, induced by treatment with cyclopiazonic acid, a sarcoplasmic reticulum/ER Ca2+-ATPase blocker. We found that whereas DHC and VHC neurons in adolescent animals respond to SD with a perisomatic expression of SD h plasticity, adult animals express SD h plasticity with a dendritic and somatodendritic locus of plasticity in DHC and VHC neurons, respectively. Furthermore, SD h plasticity in adults was dependent on membrane potential and on the activation of L-type voltage-gated Ca2+ channels. These results suggest that cellular responses to the impairment of ER function, or ER stress, are dependent on brain region and age and that the differential expression of SD h plasticity could provide a neural basis for region- and age-dependent disease vulnerabilities.
Keywords: calcium stores, h channel, plasticity, hippocampus
the endoplasmic reticulum (ER) is central to the maintenance of Ca2+ homeostasis, protein handling, and regulation of biochemical signaling processes in mammalian cells (Alberts 2008; Berridge 2002). Depletion of the requisite high Ca2+ levels from the ER results in cellular dysfunction and potentially cell death. Several adaptive mechanisms, such as the unfolded protein response (UPR) and store-operated Ca2+ (SOC) entry, are initiated by ER store depletion (SD). The evolutionarily conserved UPR reduces the protein load on the ER by enhancing protein folding, inhibiting translation, and promoting degradation of misfolded proteins (Ron and Walter 2007; Xu 2005). SOC entry pathways are initiated to facilitate the refilling of the stores (Cahalan 2009). Although these SD-initiated events have been examined mainly in nonexcitable cells, it has been shown that these and other responses to ER SD occur in neurons (Klejman et al. 2009; Koss et al. 2013; Park et al. 2010; Verkhratsky 2005).
Recently, we found that ER Ca2+ depletion induces a form of intrinsic plasticity that is dependent on Ca2+ release through inositol 1,4,5 trisphosphate receptors (InsP3Rs), SOC entry, and activation of PKA (Narayanan et al. 2010). This form of plasticity is mediated by an increase in the functional density and a depolarizing shift in the activation range of hyperpolarization-activated cation-nonspecific (HCN), or h, channels. By reducing neuronal excitability, SD h plasticity might be an adaptive response to ER SD.
Two main lines of evidence led us to examine SD h plasticity further in other age groups and hippocampal regions. First, SD h plasticity has, thus far, only been examined in neurons from the middle hippocampus. In addition to functional differences across the longitudinal axis of the hippocampus (Fanselow and Dong 2010; Moser et al. 1995), the dorsal and ventral (posterior and anterior in humans) hippocampus (DHC and VHC, respectively) differ in susceptibility to epilepsy and stress (Bannerman et al. 2004; Henke 1990; Kesslak et al. 1995; Kjelstrup et al. 2002), which are also associated with changes in intracellular Ca2+ and ER function (Harte et al. 2007; Naidoo et al. 2008; Pal et al. 2001; Pascual et al. 2007). Recent work has also shown that the intrinsic excitability and integrative and resonant properties of dorsal and ventral neurons differ, which appears to be, at least partially, mediated by a differential subunit composition of h channels (Dougherty et al. 2012, 2013; Marcelin et al. 2012). Changes in h channels and intrinsic excitability have been demonstrated in numerous models of stress and epilepsy (Arnsten 2009; Fan et al. 2008; Jung et al. 2007; McDermott et al. 2003). Together, these observations suggest that there may be regional differences in the plasticity of h channels in response to cellular stress caused by ER SD. Second, another SD-initiated process—the UPR—undergoes age-dependent modification (Brown and Naidoo 2012; Naidoo 2009; Naidoo et al. 2008; Paz Gavilán et al. 2006). ER dysfunction is also associated with numerous events that are known to affect the brain differentially in adolescence and adulthood, including epilepsy, sleep deprivation, stress, and alcohol exposure (Badawy et al. 2013; Brenhouse and Andersen 2011; Holmes 1997; Markwiese et al. 1998; Novati et al. 2011; Varlinskaya and Spear 2008; Verkhratsky 2005), raising the question of whether the SD h plasticity in adolescent animals is mature in form or if the adult form of SD h plasticity is different.
To test the hypothesis that SD h plasticity is age and region dependent, we examined SD h plasticity in CA1 DHC and VHC neurons from adolescent and adult rats. With age, we found a loss of plasticity at the somata of DHC neurons and a spatial shift in the locus of plasticity from adolescence to adulthood. Adolescents show a perisomatic expression of plasticity, and adults show somatodendritic plasticity in VHC neurons and dendritic plasticity in DHC neurons. Furthermore, we found that plasticity in adult animals was dependent on membrane potential and activation of L-type voltage-gated Ca2+ channels (VGCCs). We suggest that observed changes in SD h plasticity could underlie region- and age-dependent vulnerabilities to cellular stress and disease.
MATERIALS AND METHODS
Animals
Adolescent (6.75 ± 0.19 wk of age, n = 22) and adult (42.15 ± 3.12 wk of age, n = 39) male Sprague-Dawley rats were used in this study. Animals were given food and water ad libitum, and only group-housed animals were used. All procedures were approved and performed in accordance with the rules and regulations set forth by the University of Texas at Austin Institutional Animal Care and Use Committee.
Hippocampal Slice Preparation
Transverse slices (350 μm thick) were obtained from the DHC and VHC regions using isolation methods described previously (Dougherty et al. 2012). Animals were anesthetized with an intraperitoneal injection of a combination of ketamine and xylazine and perfused transcardially with ice-cold cutting saline composed of (in mM) 210 sucrose, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 7 dextrose, 1.3 ascorbic acid, and 3 sodium pyruvate, bubbled with 95% O2/5% CO2. Slices were transferred to a warmed (∼35°C) holding chamber containing (in mM) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2, 7 dextrose, 1.3 ascorbic acid, and 3 sodium pyruvate; continuously bubbled with 95% O2/5% CO2; and allowed to incubate for 15 min. Slices were then removed from incubation and allowed to rest at room temperature for at least 45 min before recording. For most animals, one hemisphere was used to obtain dorsal slices and the other for ventral slices. The hemisphere (left or right) used for dorsal or ventral slices was alternated daily.
Electrophysiology
Whole-cell patch-clamp recordings in current-clamp mode were made from the soma or the dendrites of CA1 pyramidal neurons using a BVC-700 amplifier (Dagan, Minneapolis, MN). Signals were low-pass filtered at 5 kHz and sampled at 10–30 kHz. Recordings were made at ∼34°C, and slices were perfused constantly with external recording solution (in mM): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, and 12.5 dextrose, continuously bubbled with 95% O2/5% CO2. A 5- to 20-ms hyperpolarizing current injection (Iinj; 300–500 pA) was used to monitor changes in series resistance. Experiments were discarded if the series resistance exceeded 35 MΩ. Data acquisition and analysis were performed with custom-written software in IGOR Pro (WaveMetrics, Lake Oswego, OR), which controlled an InstruTECH ITC-18 (Heka Instruments, Bellmore, NY) data-acquisition module. All experiments were performed in the presence of the synaptic blockers: 20 μM 6,7-dinitroquinoxaline-2,3-dione and 2 μM gabazine. Electrodes were filled with an internal solution of the following composition (in mM): 120 K-gluconate 20 KCl, 10 HEPES, 4 NaCl, 4 Mg-ATP, 0.3 Na-GTP, 7 K2-phosphocreatine, pH = 7.37, adjusted with potassium hydroxide. Voltages were not corrected for the liquid junction potential (∼8 mV).
Measurements
Input resistance (Rin) was measured as the slope of a linear fit to the linear portion of the average steady-state voltage-current (V-I) plot obtained by injecting subthreshold, 500 ms current pulses of amplitudes spanning −50 pA to 50 pA in steps of 10 pA. The average steady-state V-I plot consisted of two or three iterations of the V-I step family. The stimulus used for characterizing impedance parameters was a sinusoidal current of constant amplitude, a chirp stimulus, with its frequency linearly spanning 0–15 Hz in 15 s. The impedance amplitude was calculated from the ratio of the fast Fourier transforms of the voltage responses (two to three traces averaged) and current stimuli. The resonance frequency (fR) was defined as the frequency associated with the peak impedance value.
Neuron Inclusion Criteria and Histological Processing
Before electrophysiological recording, neurons were visualized with differential interference contrast microscopy using an Axioskop 2 microscope fitted with a 40× or 63× (Zeiss, Goettingen, Germany) water-immersion objective. Cells located in the center of the CA1 cell-body layer (equidistant from the CA2 and subiculum in the transverse axis) were targeted for patching.
Neurobiotin (Vector Laboratories, Burlingame, CA) was included in the patch pipette solution (0.1–0.2%) to allow for post hoc visualization of neuronal location and morphology. Slices were fixed in a 3% glutaraldehyde solution (prepared in 0.1 M phosphate buffer, pH 7.4) and stored at 4°C before undergoing histological processing, which was performed using an avidin-horseradish peroxidase system, activated by diaminobenzidine (Vector Laboratories). Processed slices were mounted in glycerol and visualized with a compound (Leitz Diaplan) microscope. Inclusion criteria for neuronal morphology and location are similar to those used in Dougherty et al. (2012).
Drugs and Chemicals
Most extracellular solution salts were procured from Thermo Fisher Scientific (Waltham, MA). CaCl2, K-gluconate, Mg-ATP, Na-GTP, sucrose, ascorbic acid, and sodium pyruvate were from Sigma-Aldrich (St. Louis, MO). HEPES and K2-phosphocreatine were from EMD Millipore (Billerica, MA). ZD7288 (Abcam Biochemicals, Cambridge, UK) was dissolved in water; nimodipine (Tocris Bioscience, Bristol, UK) and cyclopiazonic acid (CPA; Abcam Biochemicals) were dissolved in DMSO. To control for possible DMSO vehicle effects, DMSO was applied for 10 min, and Rin, fR, and membrane potential (Vrest) were monitored for 40 min. Significant changes in Rin, fR, or Vrest were not observed over time. Possible changes in Rin, fR, and Vrest over time were also measured in the presence of ZD7288. We found that applying ZD7288 in the pipette, rather in the bath solution, gave us the most stable, long-term recordings. Furthermore, we sought to avoid nonspecific effects that have been reported when ZD7288 is applied externally (Sánchez-Alonso et al. 2008). After obtaining a whole-cell patch-clamp configuration, recordings were allowed to stabilize for 5–7 min so that sufficient ZD7288 could diffuse from the pipette into the cell. Experiments in the presence of 10 μM nimodipine were performed in the dark, and precautions were taken to avoid inactivation of this light-sensitive drug. Nimodipine was applied in the extracellular, artificial cerebrospinal fluid at least 5 min before recording and remained present for the entirety of the experiment. The application of nimodipine alone had no effect on Rin, fR, or Vrest over time.
Statistical Analysis
Group data are expressed as the mean ± SE. Statistical significance was calculated using unpaired or paired Student's t-tests. Wilcoxon signed-rank test was applied to data when n < 7. Two-way ANOVA was used to test for interactions of age (adolescent × adult) and location (DHC × VHC; soma × dendrite). Hypothesis testing was performed with α = 0.05. Statistical analyses were performed using Prism (GraphPad Software, La Jolla, CA) or IGOR Pro (WaveMetrics).
RESULTS
SD h plasticity was first characterized in neurons from the middle hippocampus from adolescent (4–9 wk old) animals (Narayanan et al. 2010). Given the significant differences in h-channel composition, intrinsic properties, and synaptic plasticity across the dorsoventral axis of the hippocampus (Dougherty et al. 2012, 2013; Grigoryan et al. 2012; Marcelin et al. 2012), we examined whether DHC and VHC neurons from adolescents differ in their response to SD.
Experimental Protocol for SD h Plasticity
The experimental protocol used for induction of SD h plasticity was identical to that used in Narayanan et al. (2010). Data from a representative adolescent VHC cell are shown for illustrative purposes in Fig. 1. After obtaining a whole-cell current-clamp configuration, the recording was allowed to stabilize for a few minutes, and the resting membrane potential (RMP) of the cell was noted (Fig. 1A). Following the stabilization period, current was injected to hold the cell at −65 mV to take baseline measurements of Rin and fR. Next, the cell was returned to its RMP (Iinj = 0 pA), and a current stimulus, consisting of a short, −50 pA current step and a chirp stimulus (Fig. 1A), was given once every 30–60 s to obtain estimates of Rin and fR. Following a baseline period of 5 min, 20 μM CPA was applied for 10 min. In some cases, cells depolarized from their RMP. In these cases, a negative holding current was applied to maintain the initial RMP. After application of CPA, the Rin decreased gradually, and fR increased. After monitoring the change in Rin and fR for 30–40 min, current was again applied to hold the cell at −65 mV for after-CPA measurement of Rin and fR. A comparison of the before- and after-CPA measurements at −65 mV is shown in Fig. 1, B and C.
Fig. 1.

Experimental protocol for induction of store-depletion (SD) hyperpolarization-activated cation-nonspecific (h-channel) plasticity. A: after obtaining the whole-cell recording configuration (top left), the neuron was allowed to stabilize, and the resting membrane potential [Vm; RMP; with 0 current injection (Iinj)] was noted. Current was then injected (Iinj) to hold the cell at −65 mV and before cyclopiazonic acid (CPA) measurements of input resistance (Rin) and resonance frequency (fR) were made. The cell was then returned to its RMP, and the chirp stimulus (top right) was applied to measure Rin and fR every 30–60 s. After a baseline period of 5 min, CPA was applied for 10 min. As depicted in this example, current had to be injected to maintain the membrane potential at the initial RMP. Following 40–60 min of recording after the baseline measurements, current was injected again to hold cells at −65 mV for the after-CPA measurement of Rin and fR. Representative time courses for the change in normalized Rin and normalized fR are shown. B: current steps from −50 pA to +50 pA in 10-pA steps were applied to measure Rin during the before- and after-CPA periods. Voltage deflections for the current steps are shown before and after for this cell. The slope in the steady-state voltage deflection vs. amplitude of Iinj was taken in each case, giving the Rin. C: a chirp stimulus was given before- and after-CPA measurement periods to estimate fR at −65 mV. Voltage responses before and after CPA are depicted. The fR was obtained from the transformation of these voltage responses to the frequency domain (bottom). The frequency at which the largest impedance was seen (fR) increased in this representative example.
SD h Plasticity is Observed at the Somata of Dorsal and Ventral Neurons from Adolescent Animals
Each neuron was maintained at its initial RMP for the induction of SD plasticity, whereas Rin and fR were measured once every 30–60 s. The average, initial RMPs for DHC and VHC neurons are displayed in Fig. 2A (DHC: −64.50 ± 0.87 mV; VHC: −62.88 ± 1.01 mV). Rin and fR were also measured before and 30–40 min after CPA at −65 mV.
Fig. 2.

SD h plasticity in dorsal and ventral hippocampal (DHC and VHC, respectively) neurons from adolescent animals. A: current was injected to maintain the cell at the voltages used in each portion of the plasticity experiment. Before-CPA measures (Rin and fR) were made at −65 mV. Neurons were then returned to their RMP and maintained there for the subsequent 40- to 60-min recording period. Black and blue lines indicate average, initial RMP values for VHC and DHC neurons, respectively. Neurons were again held at −65 mV at the end of the experiment to collect after-CPA measurements. B: the time course of change in normalized Rin and fR for DHC and VHC neurons. C: percent change in normalized Rin vs. normalized fR. D: representative before- and after-CPA traces for the measurement of Rin. Voltage traces shown are responses to a −50 pA Iinj. E: before- and after-CPA measurements of Rin (at −65 mV) for all cells. In this and subsequent figures: blue (open circles) indicates data from DHC neurons, and black (closed circles) indicates data from VHC neurons; horizontal bars indicate average values; and asterisks indicate statistical significance with a paired statistical test (P < 0.05). F: representative before- and after-CPA impedance traces (performed at −65 mV). G: before- and after-CPA measurements of fR (at −65 mV). H: average percent changes after CPA (for measures made at −65 mV) for reduction in Rin and increase in fR. In this and subsequent figures of this type: blue bars indicate data from DHC neurons, and black bars indicate data from VHC neurons; asterisks indicate statistical significance (P < 0.05) when a paired test was performed on the ordinate data before and after CPA; and error bars are SE.
The change in Rin and fR over time for DHC and VHC neurons is shown in Fig. 2B, and the percent change in Rin vs. percent change in fR over time is shown in Fig. 2C. For DHC neurons, Rin was reduced significantly when measured at −65 mV after CPA (Fig. 2, D and E; P < 0.05; n = 4), and fR was significantly higher (Fig. 2, F and G; P < 0.05; n = 4). In VHC neurons, we also saw a significant reduction in Rin (Fig. 2, D and E; P < 0.05; n = 4) and a significant increase in fR (Fig. 2, F and G; P < 0.05; n = 4). In Fig. 2H, the percentage change in Rin and fR is shown for neurons from the adolescent DHC and VHC. No significant interaction was found for Rin (P > 0.25, two-way ANOVA, adolescent DHC × adolescent VHC) or fR (P > 0.25, two-way ANOVA, adolescent DHC × adolescent VHC) before and after CPA. Thus DHC and VHC neurons from adolescent animals show similar responses to SD.
SD h Plasticity is Observed at the Somata of Ventral but Not Dorsal Neurons from Adult Animals
To address the possible age dependence of SD h plasticity, we performed experiments in DHC and VHC neurons from adult animals. SD h plasticity was performed at the initial resting potential for adult DHC and VHC neurons (Fig. 3A; DHC: −65.90 ± 1.07 mV; VHC: −63.83 ± 0.69 mV). The observed measures of plasticity for DHC neurons showed a significant interaction across age for Rin (P < 0.01, two-way ANOVA, adolescent DHC × adult DHC) and fR (P < 0.05, two-way ANOVA, adolescent DHC × adult DHC) before and after CPA. For VHC neurons, however, no age-dependent interaction was found (Rin P > 0.1; fR P > 0.1; two-way ANOVA, adolescent VHC × adult VHC). The change in Rin and fR over time for DHC and VHC neurons is shown in Fig. 3B, and the percent change in Rin vs. percent change in fR over time is shown in Fig. 3C. In DHC neurons, Rin was not changed significantly when measured at −65 mV (Fig. 3, D and E; P > 0.05; n = 5) nor was fR (Fig. 3, F and G; P > 0.05; n = 5). In VHC neurons, however, we saw a significant reduction in Rin (Fig. 3, D and E; P < 0.05; n = 6) and a significant increase in fR (Fig. 3, F and G; P < 0.05; n = 6). In Fig. 3H, the percentage change in Rin and fR is shown for neurons from the adult DHC and VHC. A significant interaction was found for Rin (P < 0.01, two-way ANOVA, adult DHC × adult VHC) but not fR (P > 0.25, two-way ANOVA, adult DHC × adult VHC) before and after CPA when compared across region (DHC vs. VHC). Thus the response of DHC and VHC neurons from adult animals to SD h plasticity differs across region.
Fig. 3.

SD h plasticity in DHC and VHC neurons from adult animals. A: voltages used in each portion of the plasticity experiment. Before-CPA measures (Rin and fR) were made at −65 mV. Neurons were then returned to their RMP and maintained there for the subsequent 40- to 60-min recording period. Black and blue lines indicate average, initial RMP values for VHC and DHC neurons, respectively. Neurons were held at −65 mV at the end of the experiment to collect after-CPA measurements. B: the time course of change in normalized Rin and fR for DHC and VHC neurons. C: percent change in normalized Rin vs. normalized fR. D: representative before- and after-CPA traces for the measurement of Rin. Voltage deflections are responses to a −50 pA Iinj. E: before- and after-CPA measurements of Rin (at −65 mV) for all cells. Horizontal bars indicate average values. F: representative before- and after-CPA impedance traces (measured at −65 mV). G: before- and after-CPA measurements of fR (at −65 mV). H: average percent changes after CPA (for measures made at −65 mV) for reduction in Rin and increase in fR.
SD h Plasticity in Adult VHC Neurons is Blocked by ZD7288
To test the hypothesis that the modified form of plasticity observed in VHC neurons is due to a change in a current other than the h current, plasticity experiments were performed in the presence of the h-channel blocker, ZD7288 (20 μM in the pipette). As expected from a blockade of h channels, ZD7288 produced a significant hyperpolarization of the RMP (VHC without ZD7288: −63.83 ± 0.69 mV; VHC with ZD7288: −68.20 ± 1.24 mV). For consistency in the experimental protocol, neurons in the presence of ZD7288 were maintained at the average, initial RMP for a VHC neuron without ZD7288 (−63 mV). In the presence of ZD7288, no changes were observed in Rin (Fig. 4, A and C; P > 0.05; n = 5) or fR (Fig. 4, B and C; P > 0.1; n = 5) after CPA application, suggesting that the plasticity observed in VHC neurons from adult animals was mediated by a change in h channels.
Fig. 4.

SD h plasticity in adult animals is blocked by ZD7288. Whole-cell current-clamp recordings were obtained from somata of adult VHC neurons with 20 μM ZD7288 in the pipette. A: representative before- and after-CPA traces in the presence of ZD7288 for the measurement of Rin. Voltage deflections are responses to a −50 pA Iinj. B: representative before- and after-CPA impedance traces in the presence of ZD7288 (performed at −65 mV). C: before- and after-CPA measurements of Rin and fR (at −65 mV). Horizontal bars indicate average values. D: average percent changes after CPA (for measures made at −65 mV) for reduction in Rin and increase in fR for adult VHC somata with and without ZD7288.
SD h Plasticity is Observed in the Dendrites of Adult, Not Adolescent Animals
Although Narayanan et al. (2010) showed that plasticity in adolescent animals was expressed perisomatically in neurons from the middle hippocampus, it was not known whether plasticity was perisomatic in DHC and VHC neurons from adolescents. Dendrites of neurons from adolescent DHC and VHC were patched at distances 150–250 μm from the soma (DHC: 174.29 ± 13.56 μm; VHC: 155 ± 2.89 μm), and experiments were performed with the standard plasticity protocol. No changes were observed in Rin (Fig. 5, A and C; DHC P > 0.5, n = 7; VHC P > 0.1, n = 4) or fR (Fig. 5, B and C; DHC P > 0.5, n = 7; VHC P > 0.1, n = 4) of dendrites in adolescent DHC or VHC neurons. The percent change in Rin and fR was plotted with the somatic data (Fig. 5D), revealing that plasticity in DHC and VHC neurons from adolescents showed a dramatic reduction in the dendrites. A direct comparison of SD plasticity at the soma vs. dendrite revealed that these two locations have a significantly different response to SD for both measures of plasticity (Rin: DHC P < 0.0001; VHC P < 0.01; fR: DHC P < 0.05; VHC P < 0.01; two-way ANOVA, adolescent soma × adolescent dendrite). These results, together with those from Narayanan et al. (2010), indicate that SD plasticity is perisomatic in expression for CA1 pyramidal neurons across the dorsoventral axis of the hippocampus in adolescent animals.
Fig. 5.
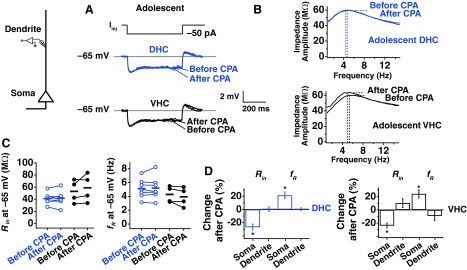
SD h plasticity is not observed in the dendrites of adolescent animals. Whole-cell current-clamp recordings were obtained from dendrites of DHC and VHC neurons. A: representative traces from measurements of Rin before and after CPA for DHC and VHC dendrites. All measurements were made at −65 mV, and Iinj is −50 pA. B: representative impedance profiles before and after CPA for DHC and VHC dendrites for adolescents. C: before- and after-CPA measurements of Rin and fR (at −65 mV). Horizontal bars indicate average values. D: percent change in Rin and fR after CPA. Somatic values included here display the somatodendritic relationship.
Our results, however, suggest significant differences in the nature of SD h plasticity between adolescent and adult animals. Previous descriptions of h-channel plasticity in neurons have demonstrated that the subcellular distribution of h plasticity can vary greatly (Campanac et al. 2008; Narayanan et al. 2010; Narayanan and Johnston 2007, 2008; Wang et al. 2003), giving us reason to investigate whether there might be a different locus to plasticity in adult animals. Dendritic recordings were made from DHC and VHC neurons at dendritic distances, approximately 150–250 μm from the soma (DHC 178.75 ± 24.53 μm; VHC 185.83 ± 22.00 μm). Rin was reduced after CPA in adult DHC and VHC neurons (Fig. 6, A and C; DHC P < 0.05, n = 5; VHC P < 0.05, n = 6), and fR increased for VHC but not DHC neurons (Fig. 6, B and C; DHC P > 0.1, n = 5; VHC P < 0.005, n = 6), indicating that SD plasticity is somatodendritic in adult VHC neurons and dendritic in adult DHC neurons (Fig. 6D).
Fig. 6.

SD h plasticity is observed in the dendrites of adult animals. Whole-cell current-clamp recordings were obtained from dendrites of DHC and VHC neurons. A: representative traces from measurements of Rin before and after CPA for DHC and VHC dendrites. All measurements were made at −65 mV, and Iinj is −50 pA. B: representative impedance profiles before and after CPA for DHC and VHC dendrites for adolescents. C: before- and after-CPA measurements of Rin and fR (at −65 mV). Horizontal bars indicate average values. D: percent change in Rin and fR after CPA. Somatic values included here display the somatodendritic relationship. E: representative before- and after-CPA traces from DHC and VHC dendrites in the presence of ZD7288 for the measurement of Rin. Voltage deflections are all responses to −50 pA Iinj. F: representative before- and after-CPA impedance traces from DHC and VHC dendrites in the presence of ZD7288 (performed at −65 mV). G: before- and after-CPA measurements of Rin and fR (at −65 mV) from dendritic recordings in the presence of ZD7288. Horizontal bars indicate average values. H: average percent change after CPA (for measures made at −65 mV) for reduction in Rin vs. initial RMP.
To test whether the observed changes in DHC and VHC dendrites were mediated by a change in h channels, we performed SD h-plasticity experiments with ZD7288 in the patch pipette. As expected from a blockade of HCN channels, ZD7288 produced a significant hyperpolarization of the RMP. For consistency in the experimental protocol, dendrites in the presence of ZD7288 were maintained near the average, initial RMP for a VHC and DHC dendrite without ZD7288 (−63 mV). In the presence of ZD7288, no changes were observed in Rin (Fig. 6, E and G; DHC P > 0.5, n = 5; VHC P > 0.5, n = 5) or fR (Fig. 6, F and G; DHC P > 0.5, n = 5; VHC P > 0.5, n = 5) after CPA, suggesting that the plasticity observed in VHC neurons from mature, adult animals was mediated by a change in h channels.
Induction of SD h Plasticity in Adult Animals is Dependent on Membrane Potential
We explored the relationship between the magnitude of plasticity expression and several basic intrinsic properties to understand further the unique pattern of SD h-plasticity expression in adult animals. The most striking relationship was found when examining the percent reduction in Rin after CPA vs. the initial RMP. The percent reduction in Rin after CPA was greater with more depolarized membrane potentials (Fig. 6H), which could explain the absence of effect at the DHC soma in adults (Fig. 3). This raised the question of whether the expression of plasticity is dependent on induction potential.
To test whether the membrane potential during plasticity induction influenced the expression of plasticity, the experimental protocol was modified so that adult DHC somata were maintained at more a more depolarized potential (−63 mV) during plasticity induction, and adult VHC somata were maintained at a more hyperpolarized potential (−66 mV; Fig. 7A). Surprisingly, these small deviations from the initial RMP were able to reverse plasticity expression for adult DHC and VHC neurons. The effect of depolarization on plasticity in DHC neurons was observed when Rin (Fig. 7C; P < 0.05; n = 5) and fR (Fig. 7D; P < 0.05; n = 5) were measured at −65 mV before and after CPA application. Hyperpolarization of VHC neurons blocked the changes in Rin (Fig. 7C; P > 0.05; n = 5) and fR (Fig. 7D; P > 0.1; n = 5) before and after CPA. When we put these results in the context of what we found when induction was performed at each cell's initial RMP (Fig. 7E), this suggested that a voltage-dependent change in the range of a few millivolts is capable of determining whether SD plasticity occurs in adult DHC and VHC neurons.
Fig. 7.
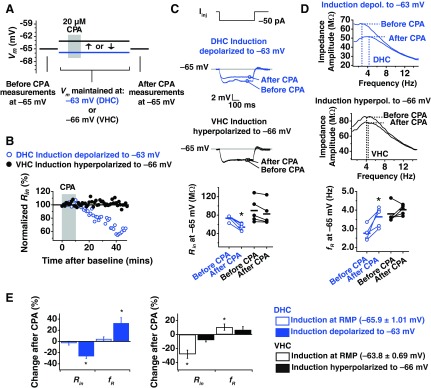
Induction of SD h plasticity in somata from adult animals is dependent on membrane voltage. A: voltages used in each portion of the plasticity experiment. Before-CPA measurements (Rin and fR) were made at −65 mV. Instead of maintaining neurons at their initial RMP, DHC neurons were maintained at the average, initial RMP for a VHC neuron (−63 mV). Neurons were again held at −65 mV at the end of the experiment to collect after-CPA measurements. B: change in normalized Rin (%) over time. C: representative traces from measurements of Rin before and after CPA for DHC neurons depolarized to −63 mV during induction and VHC neurons hyperpolarized to −66 mV during induction (top). Before and after measurements were made at −65 mV, and Iinj is −50 pA (bottom). D: example of impedance profiles from measurement of fR before and after CPA for DHC neurons depolarized (depol.) to −63 mV during induction and VHC neurons hyperpolarized (hyperpol.) to −66 mV during induction (top). Before and after measurements of fR were made at −65 mV (bottom). E: percent change in Rin and fR after CPA when induction was performed at RMP, when induction was depolarized to −63 mV (DHC), or when induction was hyperpolarized to −66 mV (VHC).
We next asked whether a voltage-dependent mechanism determined the expression of plasticity in the dendrites of adult animals. The dendrites of DHC and VHC showed a reduced Rin after CPA and rested more depolarized than the somata of DHC neurons (DHC dendrite: −62.75 ± 1.11 mV; VHC dendrite: −62.83 ± 0.70 mV). When DHC and VHC dendrites were hyperpolarized for the induction of SD h plasticity, we found no change in Rin (Fig. 8, A and C; DHC P > 0.5, n = 5; VHC P > 0.5, n = 6) or fR (Fig. 8, B and D; DHC P > 0.5, n = 5; VHC P > 0.1, n = 6) following SD. This suggested that SD plasticity in dendrites of DHC and VHC dendrites is also determined by small differences in membrane potential.
Fig. 8.

Induction of SD h plasticity in dendrites from adult animals is dependent on membrane voltage. A: representative traces from measurements of Rin before and after CPA for DHC and VHC dendrites hyperpolarized to −66 mV during induction and then returned to −65 mV for measurements. B: example of impedance profiles from measurement of fR before and after CPA for DHC and VHC dendrites hyperpolarized to −66 mV during induction. C: before and after measurements of Rin were made at −65 mV (left). Percent change in Rin when induction was performed at RMP or if neurons were hyperpolarized during induction (right). D: before and after measurements of fR were made at −65 mV (left). Percent change in fR when induction was performed at RMP or if neurons were hyperpolarized during induction (right).
Voltage Dependence of SD h Plasticity in Adult Animals Involves Activation of L-Type Channels
Thus far, we have demonstrated that SD h plasticity is expressed at the somata of DHC, VHC, and middle hippocampal neurons (Narayanan et al. 2010) from adolescent animals. Even though DHC and VHC neurons from adolescents rest at different potentials, the observed changes in Rin and fR after CPA were not different between the two regions. In contrast with the results in adolescent animals, we found that SD h plasticity in adults is highly dependent on the membrane potential during induction of plasticity. We identified L-type Ca2+ channels as a potential mechanism for voltage-dependent induction in adults, because L-type channels are highly expressed in hippocampal neurons and are active near resting potential (Avery and Johnston 1996; Magee et al. 1996), and age-dependent increases in L-type channels have been described (Gant et al. 2006; Thibault and Landfield 1996). To test the hypothesis that L-type channels may contribute to the voltage-dependent induction of SD h plasticity in adult animals, we performed experiments in the presence of the L-type channel blocker nimodipine (10 μM), which did not significantly change the initial RMP of VHC neurons (VHC with nimodipine, −63.50 ± 0.65 mV), so induction was performed at each cell's initial RMP as before. Experiments on adult VHC neurons showed that Rin was not reduced significantly by SD in the presence of nimodipine (Fig. 9, B, D, and E; P > 0.1; n = 6). We also found no change in fR after CPA (Fig. 9, C, D, and E; P > 0.5; n = 6). When adult DHC neurons were depolarized in the presence of nimodipine, no significant change in Rin was seen (Fig. 9, G, I, and J; P > 0.1; n = 8) nor was a change seen for fR (Fig. 9, H, I, and J; P > 0.1; n = 8). Thus the voltage dependence of SD h-plasticity induction in adult animals appears to require activation of L-type channels in DHC and VHC neurons.
Fig. 9.

SD h plasticity in somata from adult animals involves activation of L-type channels. A: voltages used in each portion of the plasticity experiment. Before-CPA measurements (Rin and fR) were made at −65 mV. Neurons were then returned to their RMP and maintained there for the subsequent 40- to 60-min recording period. Solid black and blue lines indicate average, initial RMP values for VHC and DHC neurons, respectively. Neurons were again held at −65 mV at the end of the experiment to collect after-CPA measurements. All measurements are made in the presence of 10 μM nimodipine. B: representative traces for measurement of Rin before and after CPA in the presence of nimodipine for VHC neurons. C: representative impedance profiles before and after CPA in the presence of nimodipine for adult VHC neurons. D: values of Rin and fR before and after CPA in the presence of nimodipine. Horizontal bars are average values. E: percent change in Rin and fR after CPA when experiments were performed in normal artificial cerebrospinal fluid (ACSF) or in ACSF plus nimodipine. F–J: plasticity in adult DHC neurons is not restored with depolarization in the presence of nimodipine. F: DHC neurons were maintained at −63 mV (similar to experiments in Fig. 5), although this time in the presence of nimodipine. G: representative traces for measurement of Rin before and after CPA in the presence of nimodipine for DHC neurons from adult animals. H: representative impedance profiles before and after CPA in the presence of nimodipine for adult VHC neurons. I: values for Rin and fR before and after CPA in the presence of nimodipine. Horizontal bars are average values. J: percent change in Rin and fR after CPA when neurons were depolarized in normal ACSF or depolarized in ACSF plus nimodipine.
Lastly, we performed dendritic SD h-plasticity experiments in the presence of nimodipine to test whether plasticity induction in adult dendrites also involves activation of L-type channels. Measurement of Rin at −65 mV, before and after CPA, showed that Rin was not reduced significantly by SD in the presence of nimodipine (Fig. 10, A and C; DHC P > 0.5, n = 4; VHC P > 0.5, n = 5). We also found no change in fR after CPA (Fig. 10, B and D; DHC P > 0.5, n = 4; VHC P > 0.5, n = 5). Together, this shows that the voltage-dependent mechanism of plasticity induction at the soma and dendrites of adult animals requires activation of L-type channels.
Fig. 10.
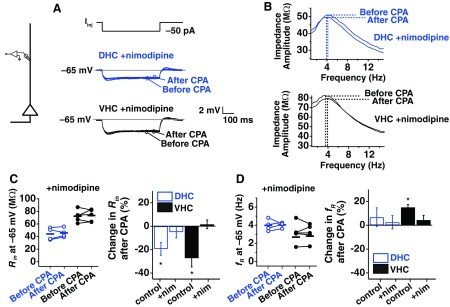
SD h plasticity in dendrites from adult animals involves activation of L-type channels. A: representative traces from measurements of Rin before and after CPA for DHC and VHC dendrites in the presence of nimodipine. B: example of impedance profiles from measurement of fR before and after CPA for DHC and VHC dendrites in the presence of nimodipine. C: before and after measurements of Rin were made at −65 mV (left). Percent change in Rin when experiments were performed with or without nimodipine (right). D: before and after measurements of fR were made at −65 mV (left). Percent change in fR when experiments were performed with or without nimodipine (right). nim, nimodipine.
DISCUSSION
We examined ER SD plasticity of h channels in DHC and VHC neurons from adolescent and adult animals. We found the following changes between these two ages: 1) there is a loss of plasticity at the somata of DHC neurons with age; 2) there is a spatial shift in the locus of plasticity from adolescence to adulthood, where adolescents show a perisomatic expression of plasticity, and adults show somatodendritic plasticity in VHC and dendritic plasticity in DHC neurons; 3) plasticity in adult animals is dependent on membrane voltage; and 4) the voltage dependence of plasticity is, at least partially, mediated by the activation of L-type VGCCs.
Age-Dependent Shift in the Somatodendritic Location of SD h Plasticity
Adolescent animals.
Although DHC and VHC neurons in adolescent animals have distinct baseline intrinsic properties (RMP, Rin, fR), the percent changes in Rin and fR following SD h plasticity were similar (Fig. 2). The percent reduction in Rin and the percent increase in fR were not different across DHC, VHC, and middle hippocampal neurons (Fig. 2) (Narayanan et al. 2010). Neuronal models, however, suggest a nonlinear relationship between fR and HCN channel density, implying that the number of h channels required to produce a change in fR from 3.39 to 4.11 Hz (observed for adolescent DHC neurons) was different than the number of channels required to produce a change from 4.51 to 5.80 Hz (observed for adolescent VHC neurons) (Hutcheon et al. 1996; Narayanan and Johnston 2007).
Adult animals.
With age, we saw significant changes in SD h plasticity. Although there was no plasticity at the soma of adult DHC neurons, we observed a reduction in Rin of adult DHC dendrites after SD h plasticity. In adult VHC neurons, we observed a comparable reduction in Rin with what was observed in adolescent VHC neurons; however, the increase in fR was not as large (Fig. 3). Frequency-dependent signals show greater distance-dependent attenuation compared with steady-state signals (Rall and Segev 1985; Spruston et al. 1993), suggesting that the location of plasticity in adult animals may have shifted to the dendrites with age. It is possible that a dendritic change in h channels, measured at the soma, could detect a change in Rin without a comparable change in fR, due to these features of attenuation or the differential influence of dendritic h channels on somatic Rin and fR (Rathour and Narayanan 2012).
Mechanisms.
We found that the spatial extent of SD h plasticity varied based on age and hippocampal region, even though the SD agent—CPA—was delivered by bath application, and the distribution of sarcoplasmic reticulum Ca2+ ATPase (SERCA) is thought to be uniform in hippocampal neurons (Jacob et al. 2005). Assuming that there are no dorsoventral differences or adolescent-to-adult changes in SERCA distribution, it is possible that another Ca2+ signaling component mediates the spatial extent of plasticity.
It was suggested that the perisomatic SD h plasticity observed by Narayanan et al. (2010) could be due to the InsP3R-interacting molecule chromogranin B (CGB) (Schmidt and Ehrlich 2010), which increases the open probability of the InsP3R and is expressed in the soma and proximal dendrites of hippocampal neurons (Jacob et al. 2005). If CGB becomes more highly expressed in dendrites with age, then this could explain why SD h plasticity is seen only in the dendrites of adult animals. Additionally, one study showed that at between 2 and 10 wk of age, InsP3R1 expression shifts from a high density in the proximal apical dendrites to a uniform dendritic distribution in CA1 pyramidal neurons (Hertle and Yeckel 2007). Thus an age-dependent shift in the InsP3R or its associated molecules could be responsible for the observed results. Other candidates of the neuronal Ca2+ signaling toolkit, such as calcium-activated PKA, SOC, and L-type channels, could also lend to the observed spatial shift in plasticity.
Implications for synaptic integration.
A perisomatic increase in h, as was observed for adolescent animals, should have a different impact on synaptic integration than the dendritic and somatodendritic increase in h observed for adult neurons. Because action potential initiation normally occurs near the soma, the observed perisomatic reduction in Rin should impact the integration of all dendrite-to-soma spreading events regardless of where they originate. The dendritic changes in Rin that were observed for adult animals, however, could be capable of preferentially influencing some inputs and not others. For example, if overexcitation is occurring at a select subset of synapses, then it might be advantageous for the neuron to reduce excitability in the dendritic region where the overexcitation is occurring and not for the entire cell. Subcellular localization of intrinsic plasticity has a proposed role in mnemonic memory processes, where the throughput of specific synapses may be affected (Zhang and Linden 2003). Our results demonstrate that the subcellular extent of intrinsic plasticity is highly age and region dependent; it would be interesting to determine whether similar trends exist with associative forms of intrinsic plasticity, such as theta-burst pairing plasticity.
SD h-Plasticity Induction Depends on Membrane Potential and Activation of L-type Channels in Adult Neurons
We found that the expression of SD h plasticity, measured at the somata of DHC, occurred in adolescent but not adult neurons. Plasticity, measured at the somata of adult DHC neurons, could be induced by manipulation of membrane potential in the depolarizing direction, which appears to be, at least partially, dependent on activation of L-type channels. Manipulation of membrane potential in the opposite (hyperpolarizing) direction is capable of blocking plasticity for adult VHC neurons. In contrast, recordings from adolescent DHC, VHC (Fig. 2), and middle hippocampal neurons (Narayanan et al. 2010) show that SD h plasticity occurs regardless of differences in membrane potential. Additionally, Narayanan et al. (2010) showed that blockade of L- and T-type calcium channels, with a combination of nimodipine and NiCl2, did not influence plasticity in adolescent animals. Together, this suggests that an L-type channel component of the SD h-plasticity mechanism emerges between adolescence and adulthood. Although we are unaware of direct comparisons of L-type channel expression between adolescence and adulthood, age-dependent increases in L-type channel activity have been described with age for middle-aged rats and older (Gant et al. 2006; Thibault and Landfield 1996).
L-Type channels have several established links with store-dependent processes. The frequency of store-dependent Ca2+ events or “puffs” in the dendrites of pyramidal neurons is changed with membrane voltage and L-type channel activation (Manita and Ross 2009). The function of these events is not clear, although they undergo age-dependent changes in magnitude and kinetics (Miyazaki et al. 2012). L-Type channels were also recently linked with SOC entry. L-Type VGCCs are suppressed by Orai1-STIM1 channel complexes through a structural relationship (Park et al. 2010; Wang et al. 2010), but the age dependence of this phenomenon has not been investigated. Future experiments could explore if this suppression is specific to adolescents, thereby leading to a role for L-type VGCC in adult SD h plasticity but not in adolescents.
Implications for Disease Vulnerability and ER Stress
The UPR is an extensively studied response initiated with ER stress. The UPR activates transcription of genes that enhance protein folding, inhibit translation, and promote degradation of misfolded proteins. The UPR is adaptive, but if ER stress is prolonged, then the UPR signaling pathway will revert to apoptosis and trigger cell death (Verkhratsky 2005). The adaptive components of the UPR signaling pathway are reduced with age (Gavilán et al. 2009; Hussain and Ramaiah 2007; Kesslak et al. 1995; Naidoo et al. 2008; Novati et al. 2011); it is suggested that age-dependent modification of the UPR is responsible for susceptibilities to disease and cellular death (Brown and Naidoo 2012; Saxena and Caroni 2011; Schipanski et al. 2013). If SD h plasticity is an alternate form of adaptation to ER stress, as has been proposed (Narayanan et al. 2010), then the findings of the present study could provide a basis for changes in ER stress-related vulnerabilities with age.
Conclusions
In conclusion, we have demonstrated that plasticity of h channels in response to depletion of calcium stores from the ER is significantly different between adolescence and adulthood and that these differences were highly dependent on the dorsoventral location in the hippocampus. We saw a shift in plasticity location from a perisomatic expression in adolescent DHC and VHC neurons to a dendritic and somatodendritic expression of plasticity for adult DHC and VHC neurons. We propose that this age-dependent shift in plasticity location may function specifically to alter the throughput of synapses that may be causing overexcitation and cellular stress. Furthermore, we observed an age-dependent introduction of L-type channels to the plasticity mechanism that appeared to underlie the differences in plasticity observed at the soma and dendrites of adult DHC and VHC neurons. We postulate that the present observations are relevant for age- and region-dependent disease vulnerabilities in the mammalian brain.
GRANTS
Support for this study was provided by the National Institute of Mental Health (grant MH048432) and National Institute of Neurological Disorders and Stroke (grant NS077477) to D. Johnston.
DISCLOSURES
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Author contributions: A.M.C. and D.J. conception and design of research; A.M.C. performed experiments; A.M.C. analyzed data; A.M.C. and D.J. interpreted results of experiments; A.M.C. prepared figures; A.M.C. drafted manuscript; A.M.C. and D.J. edited and revised manuscript; A.M.C. and D.J. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Rishikesh Narayanan and members of the D. Johnston laboratory for helpful comments and discussion of the manuscript.
REFERENCES
- Alberts B. Molecular Biology of the Cell. New York: Garland, 2008 [Google Scholar]
- Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci 10: 410–422, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery RB, Johnston D. Multiple channel types contribute to the low-voltage-activated calcium current in hippocampal CA3 pyramidal neurons. J Neurosci 16: 5567–5582, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badawy RA, Vogrin SJ, Lai A, Cook MJ. Patterns of cortical hyperexcitability in adolescent/adult-onset generalized epilepsies. Epilepsia 54: 871–878, 2013 [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Rawlins JN, McHugh SB, Deacon RM, Yee BK, Bast T, Zhang WN, Pothuizen HH, Feldon J. Regional dissociations within the hippocampus—memory and anxiety. Neurosci Biobehav Rev 28: 273–283, 2004 [DOI] [PubMed] [Google Scholar]
- Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32: 235–249, 2002 [DOI] [PubMed] [Google Scholar]
- Brenhouse HC, Andersen SL. Developmental trajectories during adolescence in males and females: a cross-species understanding of underlying brain changes. Neurosci Biobehav Rev 35: 1687–1703, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MK, Naidoo N. The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol 3: 263, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD. STIMulating store-operated Ca(2+) entry. Nat Cell Biol 11: 669–677, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac E, Daoudal G, Ankri N, Debanne D. Downregulation of dendritic Ih in CA1 pyramidal neurons after LTP. J Neurosci 28: 8635–8643, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KA, Islam T, Johnston D. Intrinsic excitability of CA1 pyramidal neurones from the rat dorsal and ventral hippocampus. J Physiol 590: 5707–5722, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KA, Nicholson DA, Diaz L, Buss EW, Neuman KM, Chetkovich DM, Johnston D. Differential expression of HCN subunits alters voltage-dependent gating of h-channels in CA1 pyramidal neurons from dorsal and ventral hippocampus. J Neurophysiol 109: 1940–1953, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Deng P, Wang YC, Lu HC, Xu ZC, Schulz PE. Transient cerebral ischemia increases CA1 pyramidal neuron excitability. Exp Neurol 212: 415–421, 2008 [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65: 7–19, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci 26: 3482–3490, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavilán MP, Pintado C, Gavilán E, Jiménez S, Ríos RM, Vitorica J, Castaño A, Ruano D. Dysfunction of the unfolded protein response increases neurodegeneration in aged rat hippocampus following proteasome inhibition. Aging Cell 8: 654–665, 2009 [DOI] [PubMed] [Google Scholar]
- Grigoryan G, Korkotian E, Segal M. Selective facilitation of LTP in the ventral hippocampus by calcium stores. Hippocampus 22: 1635–1644, 2012 [DOI] [PubMed] [Google Scholar]
- Harte MK, Powell SB, Swerdlow NR, Geyer MA, Reynolds GP. Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J Neural Transm 114: 893–898, 2007 [DOI] [PubMed] [Google Scholar]
- Henke PG. Hippocampal pathway to the amygdala and stress ulcer development. Brain Res Bull 25: 691–695, 1990 [DOI] [PubMed] [Google Scholar]
- Hertle DN, Yeckel MF. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience 150: 625–638, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GL. Epilepsy in the developing brain: lessons from the laboratory and clinic. Epilepsia 38: 12–30, 1997 [DOI] [PubMed] [Google Scholar]
- Hussain SG, Ramaiah KVA. Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem Biophys Res Commun 355: 365–370, 2007 [DOI] [PubMed] [Google Scholar]
- Hutcheon B, Miura RM, Puil E. Models of subthreshold membrane resonance in neocortical neurons. J Neurophysiol 76: 698–714, 1996 [DOI] [PubMed] [Google Scholar]
- Jacob SN, Choe CU, Uhlen P, DeGray B, Yeckel MF, Ehrlich BE. Signaling microdomains regulate inositol 1,4,5-trisphosphate-mediated intracellular calcium transients in cultured neurons. J Neurosci 25: 2853–2864, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Jones TD, Lugo JN, Sheerin AH, Miller JW, D'Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci 27: 13012–13021, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesslak JP, Yuan D, Neeper S, Cotman CW. Vulnerability of the hippocampus to kainate excitotoxicity in the aged, mature and young adult rat. Neurosci Lett 188: 117–120, 1995 [DOI] [PubMed] [Google Scholar]
- Kjelstrup KG, Tuvnes FA, Steffenach HA, Murison R, Moser EI, Moser MB. Reduced fear expression after lesions of the ventral hippocampus. Proc Natl Acad Sci USA 99: 10825–10830, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klejman ME, Gruszczynska-Biegala J, Skibinska-Kijek A, Wisniewska MB, Misztal K, Blazejczyk M, Bojarski L, Kuznicki J. Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca(2+) store in neurons. Neurochem Int 54: 49–55, 2009 [DOI] [PubMed] [Google Scholar]
- Koss DJ, Riedel G, Bence K, Platt B. Store-operated Ca2+ entry in hippocampal neurons: regulation by protein tyrosine phosphatase PTP1B. Cell Calcium 53: 125–138, 2013 [DOI] [PubMed] [Google Scholar]
- Magee JC, Avery RB, Christie BR, Johnston D. Dihydropyridine-sensitive, voltage-gated Ca2+ channels contribute to the resting intracellular Ca2+ concentration of hippocampal CA1 pyramidal neurons. J Neurophysiol 76: 3460–3470, 1996 [DOI] [PubMed] [Google Scholar]
- Manita S, Ross WN. Synaptic activation and membrane potential changes modulate the frequency of spontaneous elementary Ca2+ release events in the dendrites of pyramidal neurons. J Neurosci 29: 7833–7845, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelin B, Lugo JN, Brewster AL, Liu Z, Lewis AS, McClelland S, Chetkovich DM, Baram TZ, Anderson AE, Becker A, Esclapez M, Bernard C. Differential dorso-ventral distributions of Kv4.2 and HCN proteins confer distinct integrative properties to hippocampal CA1 pyramidal cell distal dendrites. J Biol Chem 287: 17656–17661, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwiese BJ, Acheson SK, Levin ED, Wilson WA, Swartzwelder HS. Differential effects of ethanol on memory in adolescent and adult rats. Alcohol Clin Exp Res 22: 416–421, 1998 [PubMed] [Google Scholar]
- McDermott CM, LaHoste GJ, Chen C, Musto A, Bazan NG, Magee JC. Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J Neurosci 23: 9687–9695, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Manita S, Ross WN. Developmental profile of localized spontaneous Ca(2+) release events in the dendrites of rat hippocampal pyramidal neurons. Cell Calcium 52: 422–432, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser MB, Moser EI, Forrest E, Andersen P, Morris RG. Spatial learning with a minislab in the dorsal hippocampus. Proc Natl Acad Sci USA 92: 9697–9701, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo N. ER and aging-protein folding and the ER stress response. Ageing Res Rev 8: 150–159, 2009 [DOI] [PubMed] [Google Scholar]
- Naidoo N, Ferber M, Master M, Zhu Y, Pack AI. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J Neurosci 28: 6539–6548, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Dougherty KJ, Johnston D. Calcium store depletion induces persistent perisomatic increases in the functional density of h channels in hippocampal pyramidal neurons. Neuron 68: 921–935, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Johnston D. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron 56: 1061–1075, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Johnston D. The h channel mediates location dependence and plasticity of intrinsic phase response in rat hippocampal neurons. J Neurosci 28: 5846–5860, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novati A, Hulshof HJ, Koolhaas JM, Lucassen PJ, Meerlo P. Chronic sleep restriction causes a decrease in hippocampal volume in adolescent rats, which is not explained by changes in glucocorticoid levels or neurogenesis. Neuroscience 190: 145–155, 2011 [DOI] [PubMed] [Google Scholar]
- Pal S, Sun D, Limbrick D, Rafiq A, DeLorenzo RJ. Epileptogenesis induces long-term alterations in intracellular calcium release and sequestration mechanisms in the hippocampal neuronal culture model of epilepsy. Cell Calcium 30: 285–296, 2001 [DOI] [PubMed] [Google Scholar]
- Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330: 101–105, 2010 [DOI] [PubMed] [Google Scholar]
- Pascual R, Zamora-León P, Catalán-Ahumada M, Valero-Cabré A. Early social isolation decreases the expression of calbindin D-28k and dendritic branching in the medial prefrontal cortex of the rat. Int J Neurosci 117: 465–476, 2007 [DOI] [PubMed] [Google Scholar]
- Paz Gavilán M, Vela J, Castaño A, Ramos B, del Río JC, Vitorica J, Ruano D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging 27: 973–982, 2006 [DOI] [PubMed] [Google Scholar]
- Rall W, Segev I. Space-clamp problems when voltage clamping branched neurons with intracellular microelectrodes. In: Voltage and Patch Clamping with Microelectrodes. Berlin: Springer, 1985, p. 191–215 [Google Scholar]
- Rathour RK, Narayanan R. Influence fields: a quantitative framework for the representation and analysis of active dendrites. J Neurophysiol 107: 2313–2334, 2012 [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519–529, 2007 [DOI] [PubMed] [Google Scholar]
- Sánchez-Alonso JL, Halliwell JV, Colino A. ZD 7288 inhibits T-type calcium current in rat hippocampal pyramidal cells. Neurosci Lett 439: 275–280, 2008 [DOI] [PubMed] [Google Scholar]
- Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71: 35–48, 2011 [DOI] [PubMed] [Google Scholar]
- Schipanski A, Lange S, Segref A, Gutschmidt A, Lomas DA, Miranda E, Schweizer M, Hoppe T, Glatzel M. A novel interaction between aging and ER overload in a protein conformational dementia. Genetics 193: 865–876, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, Ehrlich BE. Unloading intracellular calcium stores reveals regionally specific functions. Neuron 68: 806–808, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Jaffe DB, Williams SH, Johnston D. Voltage- and space-clamp errors associated with the measurement of electrotonically remote synaptic events. J Neurophysiol 70: 781–802, 1993 [DOI] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science 272: 1017–1020, 1996 [DOI] [PubMed] [Google Scholar]
- Varlinskaya EI, Spear LP. Social interactions in adolescent and adult Sprague-Dawley rats: impact of social deprivation and test context familiarity. Behav Brain Res 188: 398–405, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 85: 201–279, 2005 [DOI] [PubMed] [Google Scholar]
- Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330: 105–109, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Xu NL, Wu CP, Duan S, Poo MM. Bidirectional changes in spatial dendritic integration accompanying long-term synaptic modifications. Neuron 37: 463–472, 2003 [DOI] [PubMed] [Google Scholar]
- Xu C. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115: 2656–2664, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci 4: 885–900, 2003 [DOI] [PubMed] [Google Scholar]